 Yan Yu
Yan Yu Daolin Tang
Daolin Tang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 07 April 2015
Sec. Redox Physiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00093
High mobility group box 1 (HMGB1) is a widely-expressed and highly-abundant protein that acts as an extracellular signal upon active secretion by immune cells or passive release by dead, dying, and injured cells. Both intracellular and extracellular HMGB1 play pivotal roles in regulation of the cellular response to stress. Targeting the translocation, release, and activity of HMGB1 can limit inflammation and reduce tissue damage during infection and sterile inflammation. Although the mechanisms contributing to HMGB1 biology are still under investigation, it appears that oxidative stress is a central regulator of HMGB1's translocation, release, and activity in inflammation and cell death (e.g., necrosis, apoptosis, autophagic cell death, pyroptosis, and NETosis). Thus, targeting HMGB1 with antioxidant compounds may be an attractive therapeutic strategy for inflammation-associated diseases such as sepsis, ischemia and reperfusion injury, arthritis, diabetes, and cancer.
High mobility group box 1 (HMGB1), a widely-expressed and highly-abundant protein, plays multiple roles in physiological and pathological processes and is implicated in human health and diseases (Kang et al., 2014a), especially aging (Huang et al., 2014b) and inflammation-associated diseases (Andersson and Tracey, 2011; Harris et al., 2012). HMGB1 was first identified in 1973 as a non-histone nuclear protein containing two DNA-binding HMG box domains (N-terminal A and central B) and an acidic C-terminal tail (Goodwin et al., 1973). Nuclear HMGB1, as an architectural factor, sustains chromosome structure and stability. Loss of HMGB1 in vitro and/or in vivo leads to a number of abnormalities in nuclear structure and function, such as genomic instability (Giavara et al., 2005), abnormal gene transcription (Rowell et al., 2012), impaired DNA damage response (Lange et al., 2008), impaired genome chromatinization (Celona et al., 2011), and inflammatory nucleosome release (Kang et al., 2014c). Under stress conditions, HMGB1 translocates from the nucleus to the cytosol and then releases into the extracellular space (Zhang et al., 2013). Cytosolic HMGB1, as a Beclin-1-binding protein, promotes autophagy, an evolutionarily-conserved and strictly-regulated lysosomal degradation pathway (Tang et al., 2010b). In some cases, HMGB1 presents in the cell membrane, contributing to neurite outgrowth, platelet activation, and cell adhesion. HMGB1 has not only intracellular functions, but also many extracellular functions such as cytokine and chemokine activity, which are mediated by HMGB1 receptors [e.g., receptor for advanced glycation end products (RAGE) (Hori et al., 1995), toll-like receptors (TLRs, such as TLR2, TLR4, and TLR9) (Park et al., 2004; Tian et al., 2007)] or endocytic HMGB1 uptake (Kang et al., 2014b; Xu et al., 2014a) to activate the downstream signaling pathway (e.g., nuclear factor-κB, interferon regulatory factor-3 and phosphatidylinositol 3-kinase) (Lotze and Tracey, 2005). Other aspects of HMGB1, such as post-translational modification [e.g., acetylation (Bonaldi et al., 2003) and phosphorylation (Youn and Shin, 2006), cleavage (Leblanc et al., 2014), and degradation (Li et al., 2011), as well as its binding partners (Bianchi, 2009), are also involved in the regulation of extracellular HMGB1 activity. HMGB1 global knockout mice die shortly after birth (Calogero et al., 1999), whereas HMGB1 conditional knockout/knockin mice exhibit a tissue-dependent phenotype under various stressors (Kitahara et al., 2008; Yanai et al., 2013; Huang et al., 2014a; Huebener et al., 2014; Kang et al., 2014c). Collectively, these findings suggest that HMGB1 plays a location- and modification-dependent role with fine-tuned mechanisms (Andersson et al., 2014).
Although the mechanisms for regulating HMGB1 release and activity vary depending on context, experimental studies reveal that oxidative stress is likely a common mechanism regulating HMGB1 translocation, release, and activity (Tang et al., 2011c). Oxidative stress is defined as an imbalance between the production of reactive oxygen species (ROS) and antioxidant defenses. Mild oxidative stress has been demonstrated to promote cell survival, whereas severe oxidative stress has been demonstrated to cause oxidative injury and even death. ROS, including superoxide anion (O−2), hydroxyl radical (OH), hydrogen peroxide (H2O2), and singlet oxygen (1O2), are primarily generated by mitochondria and NADPH oxidases (NOXs). Mitochondrion is not only an ROS production source, but also contains antioxidants. Mitochondrial dysfunction and NOX abnormality have been implicated in the etiology of numerous common diseases and aging (Finkel and Holbrook, 2000). This review describes recent advances in our understanding of oxidative stress-mediated regulation of HMGB1 biology in inflammation and cell death (Green et al., 2009; Linkermann et al., 2014).
Sepsis is a systemic inflammatory response syndrome often caused by microbial infection or an inflammatory insult that induces the release of early and late response pro-inflammatory cytokines from immune (e.g., macrophages and monocytes) and non-immune (e.g., endothelial) cells (Angus and Van Der Poll, 2013). Early response cytokines [e.g., tumor necrosis factor and interleukin (IL)-1β] peak within the first hours after infection; circulatory levels then revert to near baseline in 3–4 h (Tracey and Cerami, 1993). HMGB1 is one of the delayed response cytokines, secreted by immune cells 20 h after activation with lipopolysaccharide (Wang et al., 1999). In vivo, HMGB1 is first detectable in the circulation 8 h after the onset of lethal endotoxemia and sepsis, subsequently increasing to plateau levels from 16 to 32 h (Wang et al., 1999). Targeting HMGB1 rather than early response cytokines provides a wider time window for clinical intervention to prevent lethal sepsis (Wang et al., 2008, 2014).
HMGB1 cannot be actively secreted via the classical endoplasmic reticulum-Golgi secretory pathway due to lack of a leader signal sequence. Instead, several mechanisms have been reported to be involved in active HMGB1 secretion in activated immune cells. These mechanisms include chromosome region maintenance 1 (CRM1)-mediated nuclear export (Bonaldi et al., 2003), the lysosome-mediated secretory pathway (Gardella et al., 2002), pyruvate kinase M2 isoform-mediated metabolism reprogramming (Yang et al., 2014), and double-stranded RNA-activated protein kinase (PKR)-mediated inflammasome activation (Lu et al., 2012), as well as oxidative stress-mediated mitogen-activated protein kinases and nuclear factor-κB pathways (Tang et al., 2007b). Antioxidants such as quercetin (Tang et al., 2009), edaravone (Kato et al., 2009), epigallocatechin gallate (Li et al., 2007), and resveratrol (Xu et al., 2014b) significantly inhibit HMGB1 release in animal models of sepsis, suggesting that oxidative stress mediates HMGB1 secretion during infection. Indeed, H2O2 at nontoxic doses (e.g., 0.0125–0.125 mM) can directly induce HMGB1 cytoplasmic translocation and activate release in macrophages and monocytes (Tang et al., 2007b). However, the effects of other ROS resources on HMGB1 release remain unexplained.
Nuclear factor (erythroid-derived 2)-like 2 (NRF2) was recently identified as a general regulator of the anti-oxidative stress response by inducing the expression of a number of antioxidant response element-dependent genes such as heme oxygenase-1 (HO-1), a rate-limiting enzyme in the catabolism of heme. Suppression of NRF2 or HO-1 significantly increases oxidative injury and HMGB1 release, whereas upregulation of HO-1 inhibits HMGB1 release in infection (Kawahara et al., 2009; Takamiya et al., 2009; Tsoyi et al., 2009) (Figure 1A). Induction of HO-1 by NRF2 requires activation of p38 mitogen-activated protein kinases, confirming crosstalk between inflammation and the antioxidative response (Park et al., 2013). Thus, activation of the NRF2 pathway may have a therapeutic effect in inflammatory disease.
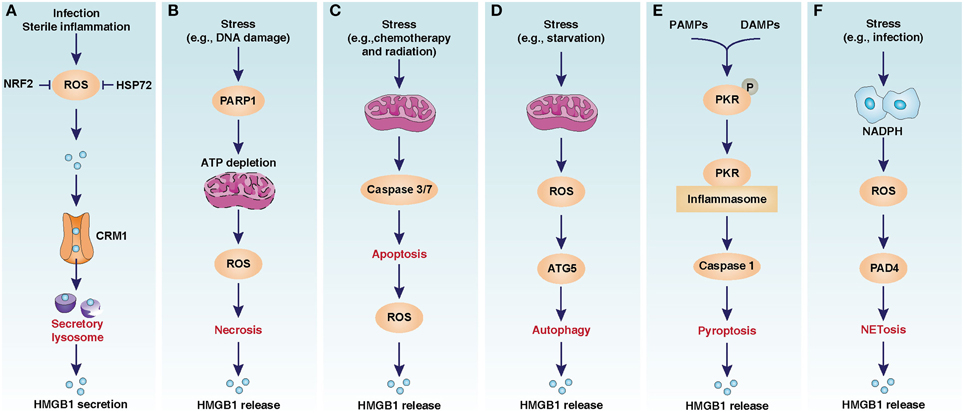
Figure 1. Oxidative stress-mediated HMGB1 release in infection, inflammation, and cell death. (A) CRM1-mediated nuclear export of HMGB1 in activated immune cells. (B) PARP1-medaited HMGB1 release in necrosis. (C) Caspase3/7-medaited HMGB1 release in apoptosis. (D) ATG5-medaited HMGB1 release in autophagy. (E) PKR-mediated HMGB1 release in pyroptosis. (F) PAD4-mediated HMGB1 release in NETosis. The image is modified from Kang et al. (2013).
Heat shock proteins (HSPs) are highly-conserved and present in cells under normal conditions, but are overexpressed under some specific stress conditions such as temperature jump and infection. Induction of the heat shock response can suppress lipopolysaccharide-induced HMGB1 release in macrophages (Tang et al., 2005). HSP72, the main stress-induced HSP, inhibits oxidative stress-induced HMGB1 release by direct protein-to-protein interaction in macrophages (Tang et al., 2007a) (Figure 1A). Although these findings identify an essential role for HSP72 in blocking inflammation and preventing HMGB1 release in activated macrophages, it is not known whether an impaired heat shock response contributes to HMGB1 release in other cells.
Inflammation after tissue injury (e.g., ischemic, trauma, and transplant) in the absence of pathogens has been recognized for a long time and involves many changes, including release of endogenous damage-associated molecular patterns (DAMPs) (Tsung et al., 2014). Compared with pathogen-associated molecular patterns (PAMPs), which are non-self-molecules, DAMPs are self-molecules with the ability to activate inflammation via pattern recognition receptors. HMGB1 is a typical DAMP and mediates the sterile inflammatory response in response to ischemia/reperfusion (I/R) injury to multiple tissues such as liver (Tsung et al., 2005), heart (Andrassy et al., 2008), kidney (Wu et al., 2010), brain (Kim et al., 2006), and intestine (He et al., 2012). I/R injury is tissue damage caused when blood supply returns to tissue after a period of ischemia or lack of oxygen. Unlike in sepsis, HMGB1 levels are increased as early as 1 h after I/R injury and then increase in a time-dependent manner for up to 24 h (Tsung et al., 2005). Although several receptors (e.g., TLRs and RAGE) contribute to HMGB1 activity in I/R injury, TLR4 appears to be the dominant receptor for HMGB1-induced sterile inflammation in the liver (Tsung et al., 2005).
The release of HMGB1 during liver I/R is not only mainly caused by cell death, but may also be an active process regulated by ROS signaling (Tsung et al., 2007). Antioxidants N-acetyl-L-cysteine or Trolox inhibit hypoxic (1% oxygen)- or H2O2-induced HMGB1 release in hepatocytes in vitro and in vivo (Tsung et al., 2007). Remarkably, the calcium/calmodulin-dependent protein kinase type IV- and β-mediated calcium-dependent signaling pathways are downstream of oxidative stress, allowing HMGB1 release in I/R. Genetic and pharmacologic inhibition of calcium/calmodulin-dependent protein kinases inhibits HMGB1 release and protects against liver I/R injury (Tsung et al., 2007). Inhibition of HMGB1 activity by injection of neutralizing antibody partially abolishes the increase in liver manganese superoxide dismutase, a key mitochondrial antioxidant enzyme, after I/R (Pardo et al., 2008). In addition to inhibiting HMGB1 release in sepsis, activation of the NRF2-HO1 pathway also limits HMGB1 release and protects against I/R injury and other sterile inflammatory injuries (Yun et al., 2010; Wang et al., 2013) (Figure 1A). Considering these findings together, oxidative stress-induced calcium signaling facilitates HMGB1 release in sterile inflammation.
In its default form, necrosis is a type of unexpected and accidental cell damage lacking the morphological characteristics of apoptosis or autophagy (Galluzzi et al., 2012). A very large number of chemicals or physical stimuli (e.g., toxins, radiation, heat, trauma, alcohol, drugs, and I/R injury) can cause necrosis. The release of intracellular contents, including HMGB1, after cellular membrane damage is the cause of inflammation in necrosis (Scaffidi et al., 2002). It is assumed that adenosine triphosphate (ATP) depletion from overactivation of ADP ribose polymerase (PARP) causes necrosis (Ha and Snyder, 1999). The best known member of the PARP family is a DNA nick sensor enzyme that becomes activated by DNA breaks. Excessive DNA damage during oxidative injury triggers PARP-mediated necrosis. Knockout of PARP1 in cells inhibits oxidative injury-induced HMGB1 release following necrosis in response to DNA damage (Ditsworth et al., 2007) (Figure 1B). PARP1 deficiency in mice and PARP inhibitors protect against stroke, I/R injury, sepsis, diabetes, and pancreatitis, suggesting that PARP1 is a potential target in inflammatory disease (Giansanti et al., 2010; Luo and Kraus, 2012).
In addition to the classic, unregulated form of necrosis, a form of regulated cell death, namely necroptosis, also exists (Galluzzi et al., 2012). The core components of the necroptosis machinery include receptor-interacting protein kinase (RIPK)-1, RIPK3, and the mixed lineage kinase domain-like protein (MLKL) (Linkermann and Green, 2014). Upon ligand binding, RIPK1 is recruited to tumor necrosis factor receptor superfamily and TLR complexes, promoting pro-survival and inflammatory signaling. RIPK1 also directly regulates caspase-8-mediated apoptosis or, if caspase-8 activity is blocked, RIPK3-MLKL-dependent necroptosis. Loss of RIPK1, RIPK3, and MLKL in mice protects against acute inflammation injury associated with decreased serum HMGB1 levels (Kaczmarek et al., 2013; Lau et al., 2013; Dannappel et al., 2014; Murakami et al., 2014). Oxidative stress is involved in execution of necroptosis downstream of RIPK1 activation (Vanden Berghe et al., 2014). Necrostatin 1, an inhibitor of necroptosis, inhibits ROS accumulation and HMGB1 release/activity in experimental animal models of sepsis and I/R injury (Duprez et al., 2011; Zhang et al., 2014). Moreover, the activity of PARP1 is regulated by RIPK1/RIPK3 in necroptosis, suggesting that interplay between PARP and RIPK can control necroptosis (Jouan-Lanhouet et al., 2012). One of the most active pursuits of cell death research is to elucidate the necroptotic pathway in the regulation of DAMP release, as well as the inflammatory response.
Apoptosis is the process of programmed cell death that may occur in a wide variety of physiological and pathological situations. It is often stated that apoptotic cell death doesn't provoke inflammation due to the cell membrane remaining intact and because apoptotic cells are rapidly phagocytosed by macrophages. Interestingly, HMGB1 was originally identified as a marker to distinguish between necrosis and apoptosis because HMGB1 is only released in necrosis, but not apoptosis, even after undergoing secondary necrosis and partial autolysis (Scaffidi et al., 2002). However, recent studies suggest that HMGB1 can be released in apoptosis by macrophages, cancer cells, and other cells (Bell et al., 2006; Jiang et al., 2007). DNA fragmentation is a marker of apoptosis that allows the release of nucleosomes (histones and DNA) and HMGB1 into the cytoplasm and extracellular space. Moreover, HMGB1 release is observed in macrophages during phagocytic clearance of apoptotic cells in sepsis (Qin et al., 2006). HMGB1 released from apoptotic cell death is an important mediator of immunogenic cell death in cancer therapy by TLR4 (Apetoh et al., 2007).
The activity of HMGB1 release in apoptosis is context-dependent. Oxidative stress is likely to diminish the immune activity of HMGB1 in apoptosis. Caspases, a family of cysteine proteases, are the central regulators of apoptosis. Activated caspase-3 and -7 in the mitochondrial apoptotic pathway can cleave NADH dehydrogenase Fe-S protein-1, the 75 kDa subunit of respiratory complex I. This process leads to a loss of ATP and the generation of ROS. As a redox-sensitive protein, HMGB1 contains three cysteines: C23, C45, and C106. The oxidation of Cys106 in apoptosis abolishes the ability of HMGB1 to activate dendritic cells, the specialized antigen-presenting cells that initiate and direct T-cell immunity (Kazama et al., 2008). In contrast, mutation of a single amino acid in the caspase cleavage site of NADH dehydrogenase Fe-S protein-1 can inhibit ROS production and restore the immune activity of released HMGB1 during apoptosis (Kazama et al., 2008) (Figure 1C). Thus, the redox status of HMGB1 in apoptosis appears to determine whether cell death is tolerogenic or immunogenic.
Autophagy is a conserved, intracellular, lysosome-dependent degradation process that allows the degradation and recycling of cellular components such as proteins and organelles. Autophagy-related genes (Atg) are essential regulators of autophagy in development and most other stages of adult life. Homozygous genetic knockouts of most Atg genes (e.g., Atg3, Atg5, Atg6, Atg7, Atg9, and Atg16L1) in mice are developmentally lethal (Mizushima and Levine, 2010). In addition to physiological function, dysfunction of autophagy is implicated in human diseases such as autoimmune diseases, inflammatory diseases, and cancer. Autophagy is upregulated in response to various stressors, including oxidative stress. In many cases, upregulated autophagy promotes cell survival during various stress conditions, while excessive or uncontrolled autophagy promotes cell death in some cases (Kroemer et al., 2010). The concept of autophagic cell death was first presented based on observations of increased morphological features (e.g., accumulation of autophagic vesicles) in dying cells (Galluzzi et al., 2012). Moreover, selective overexpression of autophagy can cause a special type of regulated cell death termed “autosis,” which is mediated by the Na+, K+-ATPase pump (Liu et al., 2013). This report has led to increasing concerns about the pathogenic role of autophagic cell death, although controversy still exists.
HMGB1 plays location-dependent roles in the induction of autophagy. Cytoplasmic HMGB1 promotes starvation- and oxidative stress-induced autophagy by binding Beclin-1 (Tang et al., 2010b), a critical regulator of autophagy, as well as apoptosis (Kang et al., 2011). Extracellular HMGB1 induces autophagy by binding its receptor RAGE (Tang et al., 2010a). Nuclear HMGB1 regulates autophagy by inducing expression of heat shock protein β-1, which allows membrane dynamic trafficking during autophagy and mitophagy (Tang et al., 2011a). Like other ATGs, it was recently suggested that HMGB1-independent autophagy exists in some tissues. The mechanisms of the HMGB1-dependent and -independent autophagic signaling pathways remain to be defined.
HMGB1 release is observed in macrophages, fibroblasts, and cancer cells during autophagy and autophagic cell death (Thorburn et al., 2009; Tang et al., 2010b; Dupont et al., 2011) (Figure 1D). Superoxide dismutases (SOD), including SOD1, SOD2, and SOD3, can efficiently catalyze the dismutation of superoxide anions. Antioxidant (e.g., N-acetyl-L-cysteine) SOD1 RNAi, and SOD2 RNAi limit the translocation and release of HMGB1 in starvation-induced autophagy, suggesting that oxidative stress is involved in starvation-mediated HMGB1 release (Tang et al., 2010b). Moreover, H2O2 and loss of SOD1-mediated oxidative stress promote cytosolic HMGB1 expression and extracellular release (Tang et al., 2011b). Inhibition of HMGB1 release or loss of HMGB1 decrease the number of autolysosomes and autophagic flux in human and mouse cell lines under conditions of oxidative stress (Tang et al., 2011b). Collectively, HMGB1 appears to serve as an important autophagic sensor in oxidative stress.
Pyroptosis is a caspase-1 or caspase-11-dependent regulated type of cell death that plays a central role in inflammation and immunity. According to their function, caspases are divided into apoptotic (caspase-3, -6, -7, -8, and -9 in mammals) and inflammatory (caspase-1, -4, -5, -12 in humans and caspase-1, -11, and -12 in mice) families (McIlwain et al., 2013). Compared with caspase-11-dependent pyroptosis (Broz and Monack, 2013), caspase-1-dependent pyroptosis is well-characterized (Bergsbaken et al., 2009). Caspase-1 is activated during pyroptosis by inflammasome, a large supramolecular complex largely composed of dimers of the adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain). Four subfamilies of inflammasome are defined; namely, NLRP1, NLRP3, NLRC4, and absent in melanoma 2 (AIM2). These inflammasomes detect PAMPs and DAMPs in infection and sterile inflammation (Schroder and Tschopp, 2010). Mitochondrial and NOX-mediated oxidative stress is an initial signal to induce inflammasome activation. In particular, the generation of ROS via NOX causes thioredoxin-interacting protein to associate with NLRP3, which facilitates inflammasome formation (Zhou et al., 2010). Finally, caspaspe-1 is activated by inflammasome and then promotes the procession and release of the highly pro-inflammatory cytokines IL-1β and IL-18, as well as HMGB1. Inhibition of inflammasome activation decreases serum HMGB1 level and protects against liver ischemic injury and pancreatitis (Kamo et al., 2013). Interestingly, HMGB1 can activate NLRP3 and AIM2 inflammasome after binding various DNA in a TLR9- or RAGE-dependent manner, suggesting a feedback loop between HMGB1 and inflammasome (Tian et al., 2007). Alternatively, HMGB1 endocytosis results in NLRP3 inflammasome-independent and lysosome-dependent caspase-1 activation (Xu et al., 2014a). Thus, HMGB1 acting through different signaling pathways sequentially induces pyroptosis.
Like all caspases, caspase-1 can recognize a unique cleavage site in substrate. A recent study identified that HMGB1 is a direct substrate of caspase-1, but not other caspases (-2, -3, -5, -7, -9, or -11) (Leblanc et al., 2014). In contrast to HMGB1 oxidization-mediated immune tolerance in apoptosis following caspase-3 and -7 activation (Kazama et al., 2008), HMGB1 cleavage by caspapse-1 generates an active A-box peptide and then induces inflammation by RAGE (Leblanc et al., 2014). These findings suggest that activation of different caspapses determines the immune activity of HMGB1 in death. In addition to the classical inflammasome components, PKR is a novel binding protein of inflammasome to regulate inflammation. Inhibition of PKR expression or activity inhibits inflammasome activation and HMGB1 release in macrophages (Lu et al., 2012) (Figure 1E). Mitophagy or autophagy generally inhibits inflammasome activation by inhibition of mitochondrial ROS production or direct degradation of the core components of inflammasome, respectively (Nakahira et al., 2011; Deretic et al., 2013). Correlations between autophagy, pyroptosis, and DAMP release have not yet been studied in human disease.
NETosis is a antimicrobial cell death that occurs primarily in polymorphonuclear leukocytes or neutrophils in response to infection (Remijsen et al., 2011). The discovery of neutrophil extracellular traps (NETs), DNA-protein structures released by neutrophils (Brinkmann et al., 2004), was originally studied. NETs can capture and kill invading microorganisms and pathogens without using the mechanism of phagocytosis. In addition to infection and PAMPs (e.g., lipopolysaccharide), several cytokines (e.g., IL-8 and tumor necrosis factor) and DAMPs (e.g., uric acid) can induce NET formation in neutrophils and immune cells (Brinkmann and Zychlinsky, 2012; Yipp and Kubes, 2013). Accumulating evidence indicates that NETs are released in the context of cell death, a process called NETosis (Steinberg and Grinstein, 2007). NETosis not only involves the innate host defense, but also promotes thrombosis and I/R injury (Martinod and Wagner, 2014; Savchenko et al., 2014). Importantly, tumor-induced neutrophils are more sensitive to NETosis than normal neutrophils, suggesting that NETosis is involved in cancer progression (Demers et al., 2012).
In addition to histones and DNA, HMGB1 is a nuclear component of NETs regulated by several different mechanisms, including autophagy (Mitroulis et al., 2011). Extracellular HMGB1 can also induce NET formation by autophagy (Tadie et al., 2013; Maugeri et al., 2014a) and exhibits bacterial killing ability (Zetterstrom et al., 2006). ROS production by activation of NOX contributes to NETosis (Figure 1F). Interestingly, extracellular HMGB1 may directly induce NOX activation in neutrophils and mediate hemorrhagic shock in a TLR4-dependent manner (Fan et al., 2007). Thus, release of HMGB1 in inflammation may amplify oxidative stress and NETosis. Although eptidylarginine deiminase 4-mediated histone citrullination is a critical step to regulate histone release in NETosis and other types of cell death, future work will address whether HMGB1 is a regulator of eptidylarginine deiminase 4, which is required for NET-mediated innate immunity and thrombosis.
In inflammation, extracellular HMGB1 has the ability to stimulate cell migration and cytokine production in receptor-dependent and -independent manners. This HMGB1 activity is determined by its redox status of cysteines (C23, C45, and C106). Three redox forms of HMGB1 have been identified: all-cysteine-reduced HMGB1, disulfide HMGB1, and all-cysteine-oxidized HMGB1. All-cysteine-reduced HMGB1 forms a heterocomplex with C-X-C motif chemokine 12 and then binds to C-X-C chemokine receptor type 4 to induce cell migration and inflammatory cell recruitment (Venereau et al., 2012). Surprisingly, this all-cysteine-reduced HMGB1 can't induce cytokine production in immune cells. In contrast, reduced C106 is required for binding of HMGB1 to TLR4 to trigger cytokine release in macrophages (Yang et al., 2010). Subsequent studies demonstrated that a disulfide bond between C23 and C45 is also required for HMGB1's cytokine-inducing activity (Venereau et al., 2012; Yang et al., 2012). Finally, all-cysteine-oxidized HMGB1 does not show cytokine or chemotactic activity (Venereau et al., 2012). However, other studies have revealed that oxidized HMGB1 still has the ability to activate neutrophils and vascular endothelial cells and trigger age-related inflammation (Davalos et al., 2013; Maugeri et al., 2014b). The reasons for the discrepancies between studies are unclear, but may be the result of differences in purity of HMGB1 redox protein preparations and the used dose. More convincing evidence is needed to develop specific neutralizing antibody to recognize these three different redox HMGB1 proteins or use mice with HMGB1 cysteine genetic mutation. Overall, the question of whether different redox forms of HMGB1 are good or bad is inadequately framed.
In addition to inflammation, redox modifications are crucial for the function of HMGB1 in DNA repair and autophagy. Compared with oxidized HMGB1, reduced HMGB1 exhibits a stronger affinity for distorted DNA structures, but their functional roles are not yet clear (Park and Lippard, 2011). Replacement of Cys23 and/or 45 with serines did not affect the nuclear distribution of the mutant proteins, while C106S and triple cysteine mutations impaired nuclear localization of HMGB1 (Hoppe et al., 2006), allowing entry of some of the protein into the cytosol to induce autophagy (Tang et al., 2010b). A disulfide bond between C23 and C45 is required for binding of HMGB1 to Beclin-1 to induce autophagy (Tang et al., 2010b). Moreover, extracellularly-reduced HMGB1 promotes autophagy in a RAGE-dependent manner, whereas oxidized HMGB1 triggers caspase-dependent apoptosis in cancer cells (Tang et al., 2010a). Whether and how these redox modifications of HMGB1 can affect cell-to-cell communication in the tumor microenvironment remain to be elucidated (Kang et al., 2013).
Since the discovery of HMGB1 in 1973 (Goodwin et al., 1973), the multi-faceted HMGB1 has recently been identified as an important mediator of infection and sterile inflammation, which has initiated a new field of translational medicine for targeting HMGB1 in disease (Kang et al., 2014a). What lies ahead? An improved understanding of the underlying regulation mechanisms of HMGB1 expression, translocation, and release; how HMGB1 contributes to the pathophysiology of human health and diseases; and the development of more specific and less toxic compounds will benefit many more patients and improve their quality of life. In particular, oxidative stress-associated human aging and many diseases have been increasingly recognized as central regulators of HMGB1 biology in infection, sterile inflammation, and cell death types such as necrosis, apoptosis, autophagic cell death, pyroptosis, and NETosis (Tang et al., 2011c). Different stressors may cause different stress responses, cell deaths, and immune responses. Activation of autophagy possibly mediates the crosstalk between cell death, inflammation, and DAMP release during stress (Levine et al., 2011; Deretic et al., 2013; Zhang et al., 2013; Green and Levine, 2014). Targeting HMGB1 via antioxidant compounds such as N-acetyl-cysteine protects against inflammation-associated diseases in animal experiments. However, these studies seem to contradict human clinical trials in which antioxidant therapy proved largely ineffective in treating various pathologies. One possible reason for this is that the reactivity of ROS is orders of magnitude too fast. This leaves a narrow therapeutic window for administration of antioxidant treatment, and antioxidant therapies are not effective when delivered after the acute cytokine response has occurred. Alternatively, reducing HMGB1 release and the inflammatory response through bolstering intracellular enzymatic antioxidants (e.g., HO-1, SODs, and catalase) or blocking their enzymatic production may be an attractive strategy. In addition to blocking HMGB1 release, HMGB1 neutralizing antibody and A box protein can block activity of extracellular HMGB1 in animal experiments. Further investigation is needed to evaluate these therapies and their possible roles in clinical practice. Although much attention has been paid to the different modifications of HMGB1 within the past 5 years, monitoring the intracellular and extracellular redox statuses of HMGB1 in disease still presents a major challenge (Antoine et al., 2014; Janko et al., 2014). Additionally, much remains unknown about how individual signal transduction pathways are regulated upon activation by different redox forms of HMGB1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to the researchers who were not referenced due to space limitations. We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript. This work was supported by the National Institutes of Health (NIH) (R01CA160417 to D.T.), a 2013 Pancreatic Cancer Action Network-AACR Career Development Award (Grant Number 13-20-25-TANG), and a grant from the National Natural Sciences Foundation of China (81270616 to Y.Y.). Work performed in support of findings reviewed in this manuscript was aided by the Core Support of the University of Pittsburgh Cancer Institute (P30CA047904).
Andersson, U., Antoine, D. J., and Tracey, K. J. (2014). The functions of HMGB1 depend on molecular localization and post-translational modifications. J. Intern. Med. 276, 420–424. doi: 10.1111/joim.12309
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Andersson, U., and Tracey, K. J. (2011). HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29, 139–162. doi: 10.1146/annurev-immunol-030409-101323
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Andrassy, M., Volz, H. C., Igwe, J. C., Funke, B., Eichberger, S. N., Kaya, Z., et al. (2008). High-mobility group box-1 in ischemia-reperfusion injury of the heart. Circulation 117, 3216–3226. doi: 10.1161/CIRCULATIONAHA.108.769331
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Angus, D. C., and Van Der Poll, T. (2013). Severe sepsis and septic shock. N. Engl. J. Med. 369, 840–851. doi: 10.1056/NEJMra1208623
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Antoine, D. J., Harris, H. E., Andersson, U., Tracey, K. J., and Bianchi, M. E. (2014). A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol. Med. 20, 135–137. doi: 10.2119/molmed.2014.00022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Apetoh, L., Ghiringhelli, F., Tesniere, A., Obeid, M., Ortiz, C., Criollo, A., et al. (2007). Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059. doi: 10.1038/nm1622
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bell, C. W., Jiang, W., Reich, C. F. III, and Pisetsky, D. S. (2006). The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 291, C1318–C1325. doi: 10.1152/ajpcell.00616.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bianchi, M. E. (2009). HMGB1 loves company. J. Leukoc. Biol. 86, 573–576. doi: 10.1189/jlb.1008585
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bonaldi, T., Talamo, F., Scaffidi, P., Ferrera, D., Porto, A., Bachi, A., et al. (2003). Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo J. 22, 5551–5560. doi: 10.1093/emboj/cdg516
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. doi: 10.1126/science.1092385
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brinkmann, V., and Zychlinsky, A. (2012). Neutrophil extracellular traps: is immunity the second function of chromatin? J. Cell Biol. 198, 773–783. doi: 10.1083/jcb.201203170
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Broz, P., and Monack, D. M. (2013). Noncanonical inflammasomes: caspase-11 activation and effector mechanisms. PLoS Pathog. 9:e1003144. doi: 10.1371/journal.ppat.1003144
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Calogero, S., Grassi, F., Aguzzi, A., Voigtlander, T., Ferrier, P., Ferrari, S., et al. (1999). The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 22, 276–280. doi: 10.1038/10338
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Celona, B., Weiner, A., Di Felice, F., Mancuso, F. M., Cesarini, E., Rossi, R. L., et al. (2011). Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol. 9:e1001086. doi: 10.1371/journal.pbio.1001086
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dannappel, M., Vlantis, K., Kumari, S., Polykratis, A., Kim, C., Wachsmuth, L., et al. (2014). RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94. doi: 10.1038/nature13608
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Davalos, A. R., Kawahara, M., Malhotra, G. K., Schaum, N., Huang, J., Ved, U., et al. (2013). p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201, 613–629. doi: 10.1083/jcb.201206006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Demers, M., Krause, D. S., Schatzberg, D., Martinod, K., Voorhees, J. R., Fuchs, T. A., et al. (2012). Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. U.S.A. 109, 13076–13081. doi: 10.1073/pnas.1200419109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deretic, V., Saitoh, T., and Akira, S. (2013). Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737. doi: 10.1038/nri3532
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ditsworth, D., Zong, W. X., and Thompson, C. B. (2007). Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J. Biol. Chem. 282, 17845–17854. doi: 10.1074/jbc.M701465200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dupont, N., Jiang, S., Pilli, M., Ornatowski, W., Bhattacharya, D., and Deretic, V. (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. Embo J. 30, 4701–4711. doi: 10.1038/emboj.2011.398
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duprez, L., Takahashi, N., Van Hauwermeiren, F., Vandendriessche, B., Goossens, V., Vanden Berghe, T., et al. (2011). RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35, 908–918. doi: 10.1016/j.immuni.2011.09.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fan, J., Li, Y., Levy, R. M., Fan, J. J., Hackam, D. J., Vodovotz, Y., et al. (2007). Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J. Immunol. 178, 6573–6580. doi: 10.4049/jimmunol.178.10.6573
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Finkel, T., and Holbrook, N. J. (2000). Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247. doi: 10.1038/35041687
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., et al. (2012). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120. doi: 10.1038/cdd.2011.96
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gardella, S., Andrei, C., Ferrera, D., Lotti, L. V., Torrisi, M. R., Bianchi, M. E., et al. (2002). The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 3, 995–1001. doi: 10.1093/embo-reports/kvf198
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giansanti, V., Dona, F., Tillhon, M., and Scovassi, A. I. (2010). PARP inhibitors: new tools to protect from inflammation. Biochem. Pharmacol. 80, 1869–1877. doi: 10.1016/j.bcp.2010.04.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giavara, S., Kosmidou, E., Hande, M. P., Bianchi, M. E., Morgan, A., D'adda Di Fagagna, F., et al. (2005). Yeast Nhp6A/B and mammalian Hmgb1 facilitate the maintenance of genome stability. Curr. Biol. 15, 68–72. doi: 10.1016/j.cub.2004.12.065
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goodwin, G. H., Sanders, C., and Johns, E. W. (1973). A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur. J. Biochem. 38, 14–19. doi: 10.1111/j.1432-1033.1973.tb03026.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Green, D. R., Ferguson, T., Zitvogel, L., and Kroemer, G. (2009). Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363. doi: 10.1038/nri2545
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Green, D. R., and Levine, B. (2014). To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157, 65–75. doi: 10.1016/j.cell.2014.02.049
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ha, H. C., and Snyder, S. H. (1999). Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. U.S.A. 96, 13978–13982. doi: 10.1073/pnas.96.24.13978
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Harris, H. E., Andersson, U., and Pisetsky, D. S. (2012). HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 8, 195–202. doi: 10.1038/nrrheum.2011.222
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
He, G. Z., Zhou, K. G., Zhang, R., Wang, Y. K., and Chen, X. F. (2012). Impact of intestinal ischemia/reperfusion and lymph drainage on distant organs in rats. World J. Gastroenterol. 18, 7271–7278. doi: 10.3748/wjg.v18.i48.7271
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hoppe, G., Talcott, K. E., Bhattacharya, S. K., Crabb, J. W., and Sears, J. E. (2006). Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp. Cell Res. 312, 3526–3538. doi: 10.1016/j.yexcr.2006.07.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hori, O., Brett, J., Slattery, T., Cao, R., Zhang, J., Chen, J. X., et al. (1995). The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 270, 25752–25761. doi: 10.1074/jbc.270.43.25752
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, H., Nace, G. W., McDonald, K. A., Tai, S., Klune, J. R., Rosborough, B. R., et al. (2014a). Hepatocyte specific HMGB1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular HMGB1 in cellular protection. Hepatology 59, 1984–1997. doi: 10.1002/hep.26976
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, J., Xie, Y., Sun, X., Zeh, H. J. 3rd., Kang, R., Lotze, M. T., et al. (2014b). DAMPs, ageing, and cancer: the ‘DAMP Hypothesis’. Ageing Res. Rev. doi: 10.1016/j.arr.2014.10.004. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huebener, P., Gwak, G. Y., Pradere, J. P., Quinzii, C. M., Friedman, R., Lin, C. S., et al. (2014). High-mobility group box 1 is dispensable for autophagy, mitochondrial quality control, and organ function in vivo. Cell Metab. 19, 539–547. doi: 10.1016/j.cmet.2014.01.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Janko, C., Filipovic, M., Munoz, L. E., Schorn, C., Schett, G., Ivanovic-Burmazovic, I., et al. (2014). Redox modulation of HMGB1-related signaling. Antioxid. Redox Signal. 20, 1075–1085. doi: 10.1089/ars.2013.5179
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jiang, W., Bell, C. W., and Pisetsky, D. S. (2007). The relationship between apoptosis and high-mobility group protein 1 release from murine macrophages stimulated with lipopolysaccharide or polyinosinic-polycytidylic acid. J. Immunol. 178, 6495–6503. doi: 10.4049/jimmunol.178.10.6495
Jouan-Lanhouet, S., Arshad, M. I., Piquet-Pellorce, C., Martin-Chouly, C., Le Moigne-Muller, G., Van Herreweghe, F., et al. (2012). TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 19, 2003–2014. doi: 10.1038/cdd.2012.90
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaczmarek, A., Vandenabeele, P., and Krysko, D. V. (2013). Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223. doi: 10.1016/j.immuni.2013.02.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamo, N., Ke, B., Ghaffari, A. A., Shen, X. D., Busuttil, R. W., Cheng, G., et al. (2013). ASC/caspase-1/IL-1beta signaling triggers inflammatory responses by promoting HMGB1 induction in liver ischemia/reperfusion injury. Hepatology 58, 351–362. doi: 10.1002/hep.26320
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, R., Chen, R., Zhang, Q., Hou, W., Wu, S., Cao, L., et al. (2014a). HMGB1 in health and disease. Mol. Aspects Med. 40, 1–116. doi: 10.1016/j.mam.2014.05.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, R., Tang, D., Schapiro, N. E., Loux, T., Livesey, K. M., Billiar, T. R., et al. (2014b). The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 33, 567–577. doi: 10.1038/onc.2012.631
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580. doi: 10.1038/cdd.2010.191
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, R., Zhang, Q., Hou, W., Yan, Z., Chen, R., Bonaroti, J., et al. (2014c). Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 146, 1097–1107. doi: 10.1053/j.gastro.2013.12.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, R., Zhang, Q., Zeh, H. J. III, Lotze, M. T., and Tang, D. (2013). HMGB1 in cancer: good, bad, or both? Clin. Cancer Res. 19, 4046–4057. doi: 10.1158/1078-0432.CCR-13-0495
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kato, S., Hussein, M. H., Kakita, H., Goto, T., Daoud, G. A., Kato, T., et al. (2009). Edaravone, a novel free radical scavenger, reduces high-mobility group box 1 and prolongs survival in a neonatal sepsis model. Shock 32, 586–592. doi: 10.1097/SHK.0b013e3181a2b886
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kawahara, K., Hashiguchi, T., Masuda, K., Saniabadi, A. R., Kikuchi, K., Tancharoen, S., et al. (2009). Mechanism of HMGB1 release inhibition from RAW264.7 cells by oleanolic acid in Prunus mume Sieb. et Zucc. Int. J. Mol. Med. 23, 615–620. doi: 10.3892/ijmm_00000172
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kazama, H., Ricci, J. E., Herndon, J. M., Hoppe, G., Green, D. R., and Ferguson, T. A. (2008). Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29, 21–32. doi: 10.1016/j.immuni.2008.05.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, J. B., Sig Choi, J., Yu, Y. M., Nam, K., Piao, C. S., Kim, S. W., et al. (2006). HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 26, 6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kitahara, T., Takeishi, Y., Harada, M., Niizeki, T., Suzuki, S., Sasaki, T., et al. (2008). High-mobility group box 1 restores cardiac function after myocardial infarction in transgenic mice. Cardiovasc. Res. 80, 40–46. doi: 10.1093/cvr/cvn163
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kroemer, G., Marino, G., and Levine, B. (2010). Autophagy and the integrated stress response. Mol. Cell 40, 280–293. doi: 10.1016/j.molcel.2010.09.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lange, S. S., Mitchell, D. L., and Vasquez, K. M. (2008). High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc. Natl. Acad. Sci. U.S.A. 105, 10320–10325. doi: 10.1073/pnas.0803181105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lau, A., Wang, S., Jiang, J., Haig, A., Pavlosky, A., Linkermann, A., et al. (2013). RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transplant. 13, 2805–2818. doi: 10.1111/ajt.12447
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Leblanc, P. M., Doggett, T. A., Choi, J., Hancock, M. A., Durocher, Y., Frank, F., et al. (2014). An immunogenic peptide in the A-box of HMGB1 protein reverses apoptosis-induced tolerance through RAGE receptor. J. Biol. Chem. 289, 7777–7786. doi: 10.1074/jbc.M113.541474
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Levine, B., Mizushima, N., and Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature 469, 323–335. doi: 10.1038/nature09782
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, W., Ashok, M., Li, J., Yang, H., Sama, A. E., and Wang, H. (2007). A major ingredient of green tea rescues mice from lethal sepsis partly by inhibiting HMGB1. PLoS ONE 2:e1153. doi: 10.1371/journal.pone.0001153
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, W., Zhu, S., Li, J., Assa, A., Jundoria, A., Xu, J., et al. (2011). EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem. Pharmacol. 81, 1152–1163. doi: 10.1016/j.bcp.2011.02.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Linkermann, A., and Green, D. R. (2014). Necroptosis. N. Engl. J. Med. 370, 455–465. doi: 10.1056/NEJMra1310050
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Linkermann, A., Stockwell, B. R., Krautwald, S., and Anders, H. J. (2014). Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat. Rev. Immunol. 14, 759–767. doi: 10.1038/nri3743
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, Y., Shoji-Kawata, S., Sumpter, R. M. Jr., Wei, Y., Ginet, V., Zhang, L., et al. (2013). Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. U.S.A. 110, 20364–20371. doi: 10.1073/pnas.1319661110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lotze, M. T., and Tracey, K. J. (2005). High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5, 331–342. doi: 10.1038/nri1594
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lu, B., Nakamura, T., Inouye, K., Li, J., Tang, Y., Lundback, P., et al. (2012). Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674. doi: 10.1038/nature11290
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luo, X., and Kraus, W. L. (2012). On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 26, 417–432. doi: 10.1101/gad.183509.111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Martinod, K., and Wagner, D. D. (2014). Thrombosis: tangled up in NETs. Blood 123, 2768–2776. doi: 10.1182/blood-2013-10-463646
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maugeri, N., Campana, L., Gavina, M., Covino, C., De Metrio, M., Panciroli, C., et al. (2014a). Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 12, 2074–2088. doi: 10.1111/jth.12710
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maugeri, N., Rovere-Querini, P., Baldini, M., Baldissera, E., Sabbadini, M. G., Bianchi, M. E., et al. (2014b). Oxidative stress elicits platelet/leukocyte inflammatory interactions via HMGB1: a candidate for microvessel injury in sytemic sclerosis. Antioxid. Redox Signal. 20, 1060–1074. doi: 10.1089/ars.2013.5298
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McIlwain, D. R., Berger, T., and Mak, T. W. (2013). Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 5:a008656. doi: 10.1101/cshperspect.a008656
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mitroulis, I., Kambas, K., Chrysanthopoulou, A., Skendros, P., Apostolidou, E., Kourtzelis, I., et al. (2011). Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS ONE 6:e29318. doi: 10.1371/journal.pone.0029318
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mizushima, N., and Levine, B. (2010). Autophagy in mammalian development and differentiation. Nat. Cell Biol. 12, 823–830. doi: 10.1038/ncb0910-823
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murakami, Y., Matsumoto, H., Roh, M., Giani, A., Kataoka, K., Morizane, Y., et al. (2014). Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 21, 270–277. doi: 10.1038/cdd.2013.109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pardo, M., Budick-Harmelin, N., Tirosh, B., and Tirosh, O. (2008). Antioxidant defense in hepatic ischemia-reperfusion injury is regulated by damage-associated molecular pattern signal molecules. Free Radic. Biol. Med. 45, 1073–1083. doi: 10.1016/j.freeradbiomed.2008.06.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, E. J., Kim, Y. M., Park, S. W., Kim, H. J., Lee, J. H., Lee, D. U., et al. (2013). Induction of HO-1 through p38 MAPK/Nrf2 signaling pathway by ethanol extract of Inula helenium L. reduces inflammation in LPS-activated RAW 264.7 cells and CLP-induced septic mice. Food Chem. Toxicol. 55, 386–395. doi: 10.1016/j.fct.2012.12.027
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, J. S., Svetkauskaite, D., He, Q., Kim, J. Y., Strassheim, D., Ishizaka, A., et al. (2004). Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279, 7370–7377. doi: 10.1074/jbc.M306793200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Park, S., and Lippard, S. J. (2011). Redox state-dependent interaction of HMGB1 and cisplatin-modified DNA. Biochemistry 50, 2567–2574. doi: 10.1021/bi2000214
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qin, S., Wang, H., Yuan, R., Li, H., Ochani, M., Ochani, K., et al. (2006). Role of HMGB1 in apoptosis-mediated sepsis lethality. J. Exp. Med. 203, 1637–1642. doi: 10.1084/jem.20052203
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Remijsen, Q., Kuijpers, T. W., Wirawan, E., Lippens, S., Vandenabeele, P., and Vanden Berghe, T. (2011). Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 18, 581–588. doi: 10.1038/cdd.2011.1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rowell, J. P., Simpson, K. L., Stott, K., Watson, M., and Thomas, J. O. (2012). HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure 20, 2014–2024. doi: 10.1016/j.str.2012.09.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Savchenko, A. S., Borissoff, J. I., Martinod, K., De Meyer, S. F., Gallant, M., Erpenbeck, L., et al. (2014). VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood 123, 141–148. doi: 10.1182/blood-2013-07-514992
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. doi: 10.1038/nature00858
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Steinberg, B. E., and Grinstein, S. (2007). Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci. STKE 2007:pe11. doi: 10.1126/stke.3792007pe11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tadie, J. M., Bae, H. B., Jiang, S., Park, D. W., Bell, C. P., Yang, H., et al. (2013). HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Lung Cell. Mol. Physiol. 304, L342–L349. doi: 10.1152/ajplung.00151.2012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takamiya, R., Hung, C. C., Hall, S. R., Fukunaga, K., Nagaishi, T., Maeno, T., et al. (2009). High-mobility group box 1 contributes to lethality of endotoxemia in heme oxygenase-1-deficient mice. Am. J. Respir. Cell. Mol. Biol. 41, 129–135. doi: 10.1165/rcmb.2008-0331OC
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, D., Kang, R., Cheh, C. W., Livesey, K. M., Liang, X., Schapiro, N. E., et al. (2010a). HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–5310. doi: 10.1038/onc.2010.261
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, D., Kang, R., Livesey, K. M., Cheh, C. W., Farkas, A., Loughran, P., et al. (2010b). Endogenous HMGB1 regulates autophagy. J. Cell Biol. 190, 881–892. doi: 10.1083/jcb.200911078
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, D., Kang, R., Livesey, K. M., Kroemer, G., Billiar, T. R., Van Houten, B., et al. (2011a). High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 13, 701–711. doi: 10.1016/j.cmet.2011.04.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, D., Kang, R., Livesey, K. M., Zeh, H. J. 3rd, and Lotze, M. T. (2011b). High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid. Redox Signal. 15, 2185–2195. doi: 10.1089/ars.2010.3666
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, D., Kang, R., Xiao, W., Jiang, L., Liu, M., Shi, Y., et al. (2007a). Nuclear heat shock protein 72 as a negative regulator of oxidative stress (hydrogen peroxide)-induced HMGB1 cytoplasmic translocation and release. J. Immunol. 178, 7376–7384. doi: 10.4049/jimmunol.178.11.7376
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, D., Kang, R., Xiao, W., Zhang, H., Lotze, M. T., Wang, H., et al. (2009). Quercetin prevents LPS-induced high-mobility group box 1 release and proinflammatory function. Am. J. Respir. Cell Mol. Biol. 41, 651–660. doi: 10.1165/rcmb.2008-0119OC
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, D., Kang, R., Zeh, H. J. III, and Lotze, M. T. (2011c). High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox Signal. 14, 1315–1335. doi: 10.1089/ars.2010.3356
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, D., Shi, Y., Jang, L., Wang, K., Xiao, W., and Xiao, X. (2005). Heat shock response inhibits release of high mobility group box 1 protein induced by endotoxin in murine macrophages. Shock 23, 434–440. doi: 10.1097/01.shk.0000159556.95285.df
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, D., Shi, Y., Kang, R., Li, T., Xiao, W., Wang, H., et al. (2007b). Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc. Biol. 81, 741–747. doi: 10.1189/jlb.0806540
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thorburn, J., Horita, H., Redzic, J., Hansen, K., Frankel, A. E., and Thorburn, A. (2009). Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 16, 175–183. doi: 10.1038/cdd.2008.143
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tian, J., Avalos, A. M., Mao, S. Y., Chen, B., Senthil, K., Wu, H., et al. (2007). Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487–496. doi: 10.1038/ni1457
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tracey, K. J., and Cerami, A. (1993). Tumor necrosis factor, other cytokines and disease. Annu. Rev. Cell Biol. 9, 317–343. doi: 10.1146/annurev.cb.09.110193.001533
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsoyi, K., Lee, T. Y., Lee, Y. S., Kim, H. J., Seo, H. G., Lee, J. H., et al. (2009). Heme-oxygenase-1 induction and carbon monoxide-releasing molecule inhibit lipopolysaccharide (LPS)-induced high-mobility group box 1 release in vitro and improve survival of mice in LPS- and cecal ligation and puncture-induced sepsis model in vivo. Mol. Pharmacol. 76, 173–182. doi: 10.1124/mol.109.055137
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsung, A., Klune, J. R., Zhang, X., Jeyabalan, G., Cao, Z., Peng, X., et al. (2007). HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 204, 2913–2923. doi: 10.1084/jem.20070247
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsung, A., Sahai, R., Tanaka, H., Nakao, A., Fink, M. P., Lotze, M. T., et al. (2005). The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 201, 1135–1143. doi: 10.1084/jem.20042614
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsung, A., Tohme, S., and Billiar, T. R. (2014). High-mobility group box-1 in sterile inflammation. J. Intern. Med. 276, 425–443. doi: 10.1111/joim.12276
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vanden Berghe, T., Linkermann, A., Jouan-Lanhouet, S., Walczak, H., and Vandenabeele, P. (2014). Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15, 135–147. doi: 10.1038/nrm3737
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Venereau, E., Casalgrandi, M., Schiraldi, M., Antoine, D. J., Cattaneo, A., De Marchis, F., et al. (2012). Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 209, 1519–1528. doi: 10.1084/jem.20120189
Wang, H., Bloom, O., Zhang, M., Vishnubhakat, J. M., Ombrellino, M., Che, J., et al. (1999). HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251. doi: 10.1126/science.285.5425.248
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, H., Ward, M. F., and Sama, A. E. (2014). Targeting HMGB1 in the treatment of sepsis. Expert Opin. Ther. Targets 18, 257–268. doi: 10.1517/14728222.2014.863876
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, H., Zhu, S., Zhou, R., Li, W., and Sama, A. E. (2008). Therapeutic potential of HMGB1-targeting agents in sepsis. Expert Rev. Mol. Med. 10, e32. doi: 10.1017/S1462399408000884
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J., Yang, H., Hu, X., Fu, W., Xie, J., Zhou, X., et al. (2013). Dobutamine-mediated heme oxygenase-1 induction via PI3K and p38 MAPK inhibits high mobility group box 1 protein release and attenuates rat myocardial ischemia/reperfusion injury in vivo. J. Surg. Res. 183, 509–516. doi: 10.1016/j.jss.2013.02.051
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, H., Ma, J., Wang, P., Corpuz, T. M., Panchapakesan, U., Wyburn, K. R., et al. (2010). HMGB1 contributes to kidney ischemia reperfusion injury. J. Am. Soc. Nephrol. 21, 1878–1890. doi: 10.1681/ASN.2009101048
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, J., Jiang, Y., Wang, J., Shi, X., Liu, Q., Liu, Z., et al. (2014a). Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 21, 1229–1239. doi: 10.1038/cdd.2014.40
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, W., Lu, Y., Yao, J., Li, Z., Chen, Z., Wang, G., et al. (2014b). Novel role of resveratrol: suppression of high-mobility group protein box 1 nucleocytoplasmic translocation by the upregulation of sirtuin 1 in sepsis-induced liver injury. Shock 42, 440–447. doi: 10.1097/SHK.0000000000000225
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yanai, H., Matsuda, A., An, J., Koshiba, R., Nishio, J., Negishi, H., et al. (2013). Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 110, 20699–20704. doi: 10.1073/pnas.1320808110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, H., Hreggvidsdottir, H. S., Palmblad, K., Wang, H., Ochani, M., Li, J., et al. (2010). A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U.S.A. 107, 11942–11947. doi: 10.1073/pnas.1003893107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, H., Lundback, P., Ottosson, L., Erlandsson-Harris, H., Venereau, E., Bianchi, M. E., et al. (2012). Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol. Med. 18, 250–259. doi: 10.2119/molmed.2011.00389
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, L., Xie, M., Yang, M., Yu, Y., Zhu, S., Hou, W., et al. (2014). PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 5, 4436. doi: 10.1038/ncomms5436
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yipp, B. G., and Kubes, P. (2013). NETosis: how vital is it? Blood 122, 2784–2794. doi: 10.1182/blood-2013-04-457671
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Youn, J. H., and Shin, J. S. (2006). Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J. Immunol. 177, 7889–7897. doi: 10.4049/jimmunol.177.11.7889
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yun, N., Eum, H. A., and Lee, S. M. (2010). Protective role of heme oxygenase-1 against liver damage caused by hepatic ischemia and reperfusion in rats. Antioxid. Redox Signal. 13, 1503–1512. doi: 10.1089/ars.2009.2873
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zetterstrom, C. K., Strand, M. L., and Soder, O. (2006). The high mobility group box chromosomal protein 1 is expressed in the human and rat testis where it may function as an antibacterial factor. Hum. Reprod. 21, 2801–2809. doi: 10.1093/humrep/del256
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, A., Mao, X., Li, L., Tong, Y., Huang, Y., Lan, Y., et al. (2014). Necrostatin-1 inhibits Hmgb1-IL-23/IL-17 pathway and attenuates cardiac ischemia reperfusion injury. Transpl. Int. 27, 1077–1085. doi: 10.1111/tri.12349
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, Q., Kang, R., Zeh, H. J. 3rd, Lotze, M. T., and Tang, D. (2013). DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy 9, 451–458. doi: 10.4161/auto.23691
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, R., Tardivel, A., Thorens, B., Choi, I., and Tschopp, J. (2010). Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140. doi: 10.1038/ni.1831
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: HMGB1, ROS, inflammation, necrosis, apoptosis, autophagy, pyroptosis, NETosis
Citation: Yu Y, Tang D and Kang R (2015) Oxidative stress-mediated HMGB1 biology. Front. Physiol. 6:93. doi: 10.3389/fphys.2015.00093
Received: 03 December 2014; Accepted: 11 March 2015;
Published: 07 April 2015.
Edited by:
Narasaiah Kolliputi, University of South Florida, USAReviewed by:
Deepesh Pandey, Johns Hopkins University, USACopyright © 2015 Yu, Tang and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daolin Tang and Rui Kang, Department of Surgery, Hillman Cancer Center, 5117 Centre Avenue, Pittsburgh, PA 15213, USAa2FuZ3JAdXBtYy5lZHU=;dGFuZ2QyQHVwbWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.