 Xiao-Ming Meng
Xiao-Ming Meng Patrick Ming-Kuen Tang
Patrick Ming-Kuen Tang Jun Li1
Jun Li1 Hui Yao Lan
Hui Yao Lan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 19 March 2015
Sec. Renal Physiology and Pathophysiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00082
This article is part of the Research Topic Advances in mechanisms of renal fibrosis View all 10 articles
TGF-β (transforming growth factor-β) is well identified as a central mediator in renal fibrosis. TGF-β initiates canonical and non-canonical pathways to exert multiple biological effects. Among them, Smad signaling is recognized as a major pathway of TGF-β signaling in progressive renal fibrosis. During fibrogenesis, Smad3 is highly activated, which is associated with the down-regulation of an inhibitory Smad7 via an ubiquitin E3-ligases-dependent degradation mechanism. The equilibrium shift between Smad3 and Smad7 leads to accumulation and activation of myofibroblasts, overproduction of ECM (extracellular matrix), and reduction in ECM degradation in the diseased kidney. Therefore, overexpression of Smad7 has been shown to be a therapeutic agent for renal fibrosis in various models of kidney diseases. In contrast, another downstream effecter of TGF-β/Smad signaling pathway, Smad2, exerts its renal protective role by counter-regulating the Smad3. Furthermore, recent studies demonstrated that Smad3 mediates renal fibrosis by down-regulating miR-29 and miR-200 but up-regulating miR-21 and miR-192. Thus, overexpression of miR-29 and miR-200 or down-regulation of miR-21 and miR-192 is capable of attenuating Smad3-mediated renal fibrosis in various mouse models of chronic kidney diseases (CKD). Taken together, TGF-β/Smad signaling plays an important role in renal fibrosis. Targeting TGF-β/Smad3 signaling may represent a specific and effective therapy for CKD associated with renal fibrosis.
The TGF-β superfamily consists of highly pleiotropic molecules including activins, inhibins, BMPs (Bone morphogenic proteins), GDFs (Growth differentiation factors) and GDNFs (Glial-derived neurotrophic factors), and exerts multiple biological functions in renal inflammation, fibrosis, cell apoptosis, and proliferation (Massague and Wotton, 2000). Among them, TGF-β is known as a key pro-fibrotic mediator in fibrotic diseases. Three isoforms of TGF-β have been identified in mammals, termed TGF-β1, 2, and 3, of which TGF-β1 is the most abundant isoform and can be produced by all types of renal resident cells. After synthesis, TGF-β1 is released in association with LAP (latency-associated peptide) as a latent form of TGF-β1 which binds to LTBP (Latent TGF-β-binding protein) in the target tissues. When exposed to multiple types of stimuli, including ROS (Reactive oxygen species), plasmin and acid (Lyons et al., 1990; Munger et al., 1999; Meng et al., 2013), TGF-β1 can be released from the LAP and LTBP and becomes active. The active TGF-β1 then binds to TβRII (Type II TGF-β receptor), a constitutively active kinase, which recruits TβRI (Type I TGF-β receptor) and phosphorylates the downstream receptor-associated Smads (R-Smads) i.e., Smad2 and Smad3 (Wrana et al., 1994). Then the phosphorylated Smad2 and Smad3 form an oligomeric complex with a common Smad, Smad4, and translocates into the nucleus to regulate the transcription of target genes in collaboration with various co-activators and co-repressors. It is interesting that an inhibitory Smad, Smad7, can be induced in a Smad3-dependent manner. Smad7 consequently competes with the R-Smads for binding to the activated receptors, in order to exert its negative effect on TGF-β/Smad signaling (Shi and Massague, 2003). Additionally, TGF-β1 is able to function through the Smad-independent pathways, including p38, ERK (Extracellular-signal-regulated kinase), MAPK, Rho-GTPases, Rac, Cdc42, ILK (Integrin linked kinase) (Attisano and Wrana, 2002; Derynck and Zhang, 2003; Li et al., 2009; Loeffler and Wolf, 2014). In this review, we focus on the pathological roles of TGF-β/Smad signaling in renal fibrosis.
Renal fibrosis, characterized by excessive deposition of ECM (Extracellular matrix), is recognized as a common pathological feature of CKD (Chronic kidney diseases) which leads to the development of ESRD (End-stage renal disease), accompanied by a progression of renal malfunctions (Eddy and Neilson, 2006). Although effective therapy for renal fibrosis is still lacking, a number of studies demonstrated that TGF-β is the key mediator in CKD associated with progressive renal fibrosis. It is well documented that TGF-β1 has multiple biological properties including cell proliferation, differentiation, apoptosis, autophagy, production of ECM, etc. (Meng et al., 2013). Considerable evidence revealed that TGF-β is substantially upregulated in the injured kidney on both patients and animal disease models (Yamamoto et al., 1996; Bottinger and Bitzer, 2002). It is also showed that the urinary levels of TGF-β are significantly increased in patients with various renal diseases, which is positively correlated with the degree of renal fibrosis (Murakami et al., 1997). Moreover, the importance of TGF-β1 in renal fibrosis is further supported by the findings that overexpression of active TGF-β1 in rodent liver is capable of inducing the fibrotic response in kidney; whereas blocking TGF-β with neutralizing antibody, antisense oligonucleotides, inhibitors, or genetic deletion of receptors can attenuate kidney fibrosis in vivo and in vitro (Sanderson et al., 1995; Kopp et al., 1996; Border and Noble, 1998; Moon et al., 2006; Petersen et al., 2008; Meng et al., 2012a). In contrast to the active form of TGF-β1, the latent form of TGF-β1 can protect the kidney against fibrosis and inflammation by upregulating Smad7 that is observed in the latent TGF-β transgenic mice received with UUO-induced nephropathy or anti-GBM-induced glomerulonephritis (Huang et al., 2008a,b). Taken together, TGF-β exerts profibrotic effects on the kidney through several possible mechanisms: (1) TGF-β1 directly induces the production of ECM, including collagen I and fibronectin, through the Smad3-dependent or -independent mechanisms (Samarakoon et al., 2012); (2) TGF-β1 suppresses the degradation of ECM by inhibiting MMPs (Matrix metalloproteinases) but inducing TIMPs (Tissue inhibitor of metalloproteinase) and the natural inhibitor of MMPs; (3) TGF-β1 is believed to play critical roles in the transdifferentiation toward myofibroblast of several types of cells, including epithelial cells, endothelial cells, and pericytes, although the origin of myofibroblast is still undefined (Meng et al., 2013; Wu et al., 2013); (4) TGF-β1 acts directly on different types of renal resident cells, for example: it can promote the proliferation of mesangial cells in order to increase matrix production, or induce the elimination of tubular epithelial cells and podocytes which may lead to a deterioration of renal injury and incur more severe renal fibrosis (Bottinger and Bitzer, 2002; Lopez-Hernandez and Lopez-Novoa, 2012) (Figure 1).
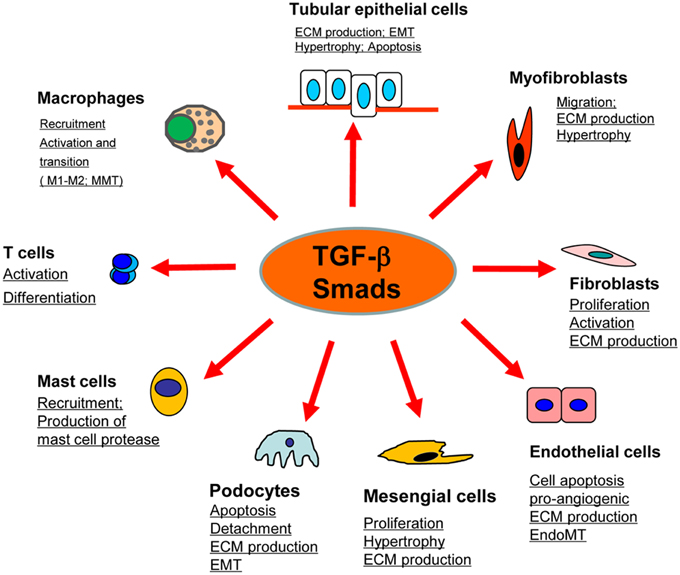
Figure 1. Role of TGF-β/Smad signaling in kidney disease. TGF-β1 signals through the downstream mediators to exert its biological activities on different cell types of kidney cells during renal inflammation and fibrosis.
It is consistently demonstrated that Smad2 and Smad3 are extensively activated in the fibrotic kidney in patients and animal models with CKD (Meng et al., 2013). Although Smad2 and Smad3 share more than 90% similarity in their amino acid sequences, their functional roles in renal fibrosis are distinct. It is well documented that Smad3 is pathogenic since knockout of Smad3 gene inhibits fibrosis in obstructive nephropathy (Sato et al., 2003), diabetic nephropathy (Fujimoto et al., 2003), hypertensive nephropathy (Liu et al., 2012), and drug-toxicity-related nephropathy (Zhou et al., 2010). Of note, Smad3 promotes renal fibrosis by directly binding to the promoter region of collagens to trigger their production (Vindevoghel et al., 1998; Chen et al., 1999), and inhibiting the ECM degradation via induction of TIMP-1 while reducing MMP-1 activities in fibroblasts (Yuan and Varga, 2001). In contrast to Smad3, Smad2 is unable to directly bind to the genomic DNA (Dennler et al., 1998). Previous study suggested that roles of Smad2 and Smad3 might be different in fibrotic diseases (Piek et al., 2001; Yang et al., 2003b; Phanish et al., 2006). Consistent with the finding that the endogenous ratio of Smad2 and Smad3 may ultimately influence the cytostatic function of Smad3 (Kim et al., 2005), results from our recent study demonstrated that conditional knockout of Smad2 from tubular epithelial cells enhances Smad3-mediated renal fibrosis in vivo and in vitro, which is associated with the increase in phosphorylation and nuclear translocation of Smad3, promotion of the Smad3 responsive promoter activity, and binding of Smad3 to Col1A2 promoter (Meng et al., 2010).
As a common Smad for TGF-β/BMP signaling, Smad4 plays a critical role in nucleocytoplasmic shuttling of Smad2/3 and Smad1/5/8 complexes (Massague and Wotton, 2000). It has been demonstrated that loss of Smad4 in mesangial cells inhibits TGF-β1-induced ECM deposition (Tsuchida et al., 2003), which is further confirmed by our recent finding that specific deletion of Smad4 from renal tubular epithelial cells attenuates the UUO-induced renal fibrosis by suppressing Smad3 responsive promoter activity and decreasing the binding of Smad3 to the target genes independent of its phosphorylation and nuclear translocation (Meng et al., 2012b).
As an inhibitory regulator in the TGF-β/Smad signaling pathway, Smad7 can be induced by a Smad3-dependent mechanism, which in turn blocks the signal transduction of TGF-β1 via its negative feedback loop (Afrakhte et al., 1998; Zhu et al., 1999; Kavsak et al., 2000; Ebisawa et al., 2001). Moreover, the regulatory mechanism of Smad7 on TGF-β signaling occurs in an elegant manner, i.e., TGF-β not only induces Smad7 transcription, but also promotes the degradation of Smad7 by activating the Smad3-dependent Smurfs/arkadia-mediated ubiquitin–proteasome degradation pathway (Kavsak et al., 2000; Ebisawa et al., 2001; Fukasawa et al., 2004; Liu et al., 2008). In this setting, the level of Smad7 protein is significantly reduced in response to the high level of active TGF-β1 in CKD. Most importantly, the functional role of Smad7 is further defined by the findings that deletion of Smad7 accelerates renal fibrogenesis in obstructive nephropathy, diabetic nephropathy as well as hypertensive nephropathy (Chung et al., 2009; Chen et al., 2011; Liu et al., 2013), suggesting Smad7 as a therapeutic agent for treatment of CKD (Lan et al., 2003; Hou et al., 2005; Ka et al., 2007, 2012; Chen et al., 2011; Liu et al., 2014).
Collectively, compelling evidence indicates that hyperactivation of Smad3 associated with progressive degradation of Smad7, is a key feature of renal fibrotic diseases. More importantly, the imbalance of Smad3 and Smad7 was proved to be one of the major mechanisms in mediating the fibrotic response. In this regard, rebalancing the disturbed Smad3/Smad7 ratio, through downregulating Smad3 and upregulating Smad7 simultaneously, seems to be an effective strategy for treatment of renal fibrosis.
Emerging evidence suggests that the accumulation of myofibroblasts, a predominant source for ECM production, is a critical step in the progression of renal fibrosis (Wynn and Ramalingam, 2012). However, the origin of myofibroblast is still controversial. It has been reported that myofibroblasts may be derived from the resident fibroblasts, pericytes, bone marrow cells (e.g., fibrocytes), epithelial cells (Epithelial–mesenchymal transition, EMT), and endothelial cells (Endothelial–mesenchymal transition, EndMT) (Allison, 2013; LeBleu et al., 2013; Meng et al., 2014). Our latest data also revealed that bone marrow-derived macrophages were capable of becoming myofibroblast phenotype via a process of macrophage-myofibroblast transition (MMT) in patients and UUO model with active renal fibrosis (Nikolic-Paterson et al., 2014). In addition, it is generally accepted that local fibroblasts can differentiate into myofibroblast under the stimulation of TGF-β (Evans et al., 2003; Midgley et al., 2013). Increasing evidence indicates that fibrocytes can produce a large amount of collagens directly in response to the stimulus such as TGF-β (Hong et al., 2007; Wada et al., 2007). Administration of TGF-β promotes the transdifferentiation of epithelial cells and endothelial cells toward myofibroblast-like cells, whereas, blockade of TGF-β/Smad signaling with inhibitors or antagonists attenuates or reverses the process of EMT or EndMT (Fan et al., 1999; Zeisberg et al., 2003, 2008; Li et al., 2010; Liu, 2010; Yang et al., 2010; Xavier et al., 2014). In addition, TGF-β1 can promote renal fibrosis via the cell-cell interaction mechanism as TGF-β1 released from the injured epithelium is able to activate pericyte-myofibroblast transition (Wu et al., 2013). Moreover, we also identify that advanced glycation end products (AGEs) and angiotensin II are capable of activating Smad3 to mediate the process of EMT under diabetes and hypertension conditions (Li et al., 2003, 2004; Wang et al., 2006b; Yang et al., 2009, 2010; Chung et al., 2010b).
Increasing evidence demonstrates that TGF-β1 can also regulate several miRNAs to facilitate renal fibrogenesis. As illustrated in Figure 2, TGF-β1 up-regulates miR-21, miR192, miR-377, miR-382, and miR-491-5p, but down-regulates miR-29 and miR-200 families during renal fibrosis (Kantharidis et al., 2011; Kriegel et al., 2012; Lan and Chung, 2012; Chung et al., 2013a,b). In fibrotic kidneys, the level of miR-21 is highly induced (Godwin et al., 2010; Zhong et al., 2011, 2013; Chau et al., 2012; Xu et al., 2012; Wang et al., 2013), whereas inhibition of miR-21 attenuates deposition of ECM and halts the progression of renal fibrosis (Zhong et al., 2011, 2013; Chau et al., 2012). Role of miR-192 in fibrosis is still controversial. It is reported that miR-192 is elevated in fibrotic mouse models and TGF-β1-treated murine cells (Kato et al., 2007; Chung et al., 2010a; Putta et al., 2012). Knockout or knockdown of miR-192 largely attenuated renal fibrosis possibly through induction of ZEB1/2 in vivo and in vitro. However, a recent study indicated that TGF-β1 reduces miR-192 expression in human TECs and deficiency of miR-192 accelerates renal fibrosis in diabetic nephropathy (Krupa et al., 2010), which is further evident by the results from the renal biopsy of diabetic patients with lower level of miR-192 (Wang et al., 2010). The discrepancy in these studies suggests the complexity of miR-192 in renal fibrogenesis. The miR-29 and miR-200 are TGF-β1-dependent anti-fibrotic miRNAs that are extensively suppressed in the diseased kidneys (Qin et al., 2011). Of note, more than 20 ECM-related genes, including collagens, are potential targets for miR-29 where some of them are regulated by the TGF-β signaling (van Rooij et al., 2008; Xiao et al., 2012). Overexpression of miR-29 attenuates renal fibrosis in vivo in obstructive and diabetic nephropathies and suppresses the fibrotic genes in vitro in response to various stimuli including TGF-β1, high glucose or salt-induced hypertensive conditions (Du et al., 2010; Liu et al., 2010; Qin et al., 2011; Chen et al., 2014). The miR-200 family contains miR-200a, miR-200b, miR-200c, miR-429, and miR-141 (Howe et al., 2012). Downregulation of miR-200a and miR-141 are detected in the fibrotic kidneys of obstructive and diabetic nephropathies (Wang et al., 2011; Xiong et al., 2012). As miR-200 has a major role in maintaining the epithelial differentiation, delivery of miR-200b significantly reduces renal fibrotic response by suppressing the transcriptional repressors of E-cadherin ZEB1 and ZEB2 (Korpal et al., 2008; Oba et al., 2010).
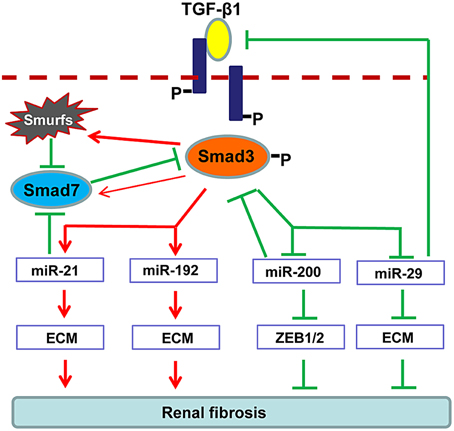
Figure 2. Regulation of TGF-β/Smad3 in fibrosis-related microRNAs during renal fibrosis. TGF-β1 activates Smad3 that binds directly to a number of microRNAs to either negatively or positively regulate their expression and function in renal fibrosis.
The therapeutic potential of anti-TGF-β1 therapy has been widely tested according to the pathogenic role of TGF-β1 in fibrogenesis. It has been shown that TGF-β neutralizing antibodies, antisense TGF-β oligodeoxynucleotides, soluble human TβRII (sTβRII.Fc) and specific inhibitors to TβR kinases (such as GW788388 and IN-1130) can effectively halt the progression of renal fibrosis in a number of experimental kidney disease models. A recent study also demonstrated that blockade of TGF-β1 receptor posttranslational core fucosylation can attenuate renal interstitial fibrosis (Shen et al., 2013). In addition, some TGF-β inhibitors have been further tested in preclinical or clinical trials (Tampe and Zeisberg, 2014). For an instance, treatment with Pirfenidone, a small molecule that blocks TGF-β1 promoter, can prevent the decline of eGFR (estimated glomerular filtration rate) in patients with focal segmental glomerulosclerosis (FSGS) or diabetic nephropathy (Cho et al., 2007; Sharma et al., 2011). In addition, Fresolimumab and LY2382770, neutralizers for TGF-β1 activity, are also tested in FSGS and diabetic kidney diseases in human (Trachtman et al., 2011; Choi et al., 2012; Tampe and Zeisberg, 2014). However, the major obstacle and risk for these potential therapies by generally blocking TGF-β signaling may be related to the abrogation of its anti-inflammatory and anti-tumorigenesis property. Nevertheless, it should be mentioned that TGF-β1 may also serve as a potential biomarker for renal fibrosis, since significant upregulation of urine TGF-β1 have been detected in progressive renal diseases (Tsakas and Goumenos, 2006).
In order to avoid the side effects caused by complete blockade of TGF-β1 signaling, more focus has been paid on inhibiting the downstream targets of this signaling pathway including Smad3, Smad7, and Smad-dependent miRNAs (Ng et al., 2009). As shown in Figure 3, SIS3, a specific inhibitor of Smad3 phosphorylation, can attenuate renal fibrosis in diabetic nephropathy (Li et al., 2010). Accumulated evidence shows that targeting Smad3 by overexpressing renal Smad7 produces inhibitory effects on both renal inflammation and fibrosis in a variety of kidney disease models (Hou et al., 2005; Ka et al., 2007, 2012; Chen et al., 2011; Liu et al., 2014). Moreover, recent studies also revealed that overexpression of miR-29, miR-200 or inhibition of miR-21 and miR-192 can effectively decelerate the progression of renal fibrosis (Oba et al., 2010; Chung et al., 2010a; Qin et al., 2011; Zhong et al., 2011, 2013; Chen et al., 2014) (Figure 3).
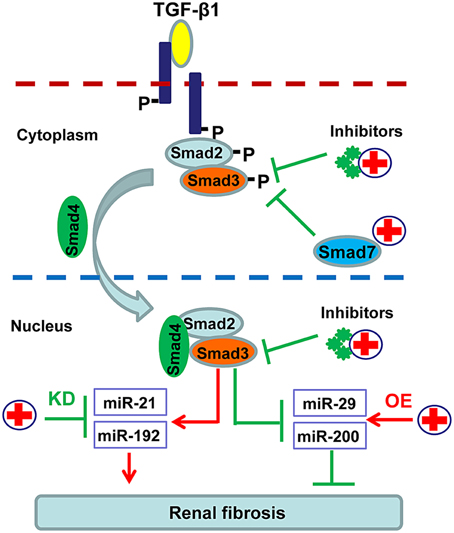
Figure 3. Potential therapeutic strategies for renal fibrosis by specifically targeting downstream TGF-β/Smad signaling. Since renal fibrosis is mediated positively by Smad3 but negatively by Smad7, treatment for renal fibrosis can target Smad3 with specific inhibitors or Smad3-dependent microRNAs that regulate fibrosis, and/or by promoting Smad7 with gene therapy or specific agonists.
BMP-7 is a natural antagonist for TGF-β through inhibiting the TGF-β/Smad3, which has been demonstrated on various renal disease models (Hruska et al., 2000; Zeisberg et al., 2003; Wang et al., 2006a; Sugimoto et al., 2007; Luo et al., 2010; Meng et al., 2013). Klotho, a single-pass transmembrane protein predominantly expressed in renal tubular epithelial cells, is capable of suppressing renal fibrosis by directly binding to type II TGF-β receptor to block the TGF-β-initiated signaling (Doi et al., 2011). Most recently, a study showed that an adaptor protein, Kindlin-2, recruits Smad3 to TGF-β type I receptor, therefore contributing to TGF-β/Smad3-mediating renal interstitial fibrosis (Wei et al., 2013). In addition, two well-known Smad transcriptional co-repressors Ski (Sloan-Kettering Institute proto-oncogene) and SnoN (Ski-related novel gene, non Alu-containing), elicit their anti-fibrotic effects on TGF-β by antagonizing Smad-mediated gene transcription (Yang et al., 2003a). Moreover, GQ5, a small molecular phenolic compound extracted from dried resin of Toxicodendron vernicifluum, has been shown to inhibit the interaction between TGF-β type I receptor and Smad3 through interfering the binding of Smad3 to SARA, thereby reducing the phosphorylation of Smad3 and downregulating the transcription of downstream fibrotic indexes including α-SMA, collagen I and fibronectin in vivo and in vitro (Ai et al., 2014). Furthermore, a number of miRNAs, such as let-7b and miR-29, are capable of regulating TGF-β signaling and altering the progression of renal fibrosis (Kato et al., 2011; Xiao et al., 2012; Wang et al., 2014).
An equilibrium shift of TGF-β/Smad signaling due to the hyperactivation of Smad3 but reduction of Smad7 may be a key pathological mechanism leading to renal fibrogenesis. Thus, rebalancing the TGF-β/Smad signaling by targeting Smad3 activity, up-regulating Smad7, as well as specifically modulating Smad3-dependent miRNAs related to fibrosis may represent an effective therapy for CKD associated with progressive real fibrosis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by the Major State Basic Research Development Program of China (973 program, 2012CB517705), the Research Grant Council of Hong Kong (CUHK3/CRF/12R, T12-402/13-N), the Focused Investment Scheme A and B from Chinese University of Hong Kong (HL), and the National Natural Science Foundation of China (No. 81300580).
Afrakhte, M., Moren, A., Jossan, S., Itoh, S., Sampath, K., Westermark, B., et al. (1998). Induction of inhibitory Smad6 and Smad7 mRNA by TGF-beta family members. Biochem. Biophys. Res. Commun. 249, 505–511. doi: 10.1006/bbrc.1998.9170
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ai, J., Nie, J., He, J., Guo, Q., Li, M., Lei, Y., et al. (2014). GQ5 hinders renal fibrosis in obstructive nephropathy by selectively inhibiting TGF-beta-induced Smad3 phosphorylation. J. Am. Soc. Nephrol. doi: 10.1681/ASN.2014040363. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Allison, S. J. (2013). Fibrosis: the source of myofibroblasts in kidney fibrosis. Nat. Rev. Nephrol. 9, 494. doi: 10.1038/nrneph.2013.141
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Attisano, L., and Wrana, J. L. (2002). Signal transduction by the TGF-beta superfamily. Science 296, 1646–1647. doi: 10.1126/science.1071809
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Border, W. A., and Noble, N. A. (1998). Evidence that TGF-beta should be a therapeutic target in diabetic nephropathy. Kidney Int. 54, 1390–1391. doi: 10.1046/j.1523-1755.1998.00127.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bottinger, E. P., and Bitzer, M. (2002). TGF-beta signaling in renal disease. J. Am. Soc. Nephrol. 13, 2600–2610. doi: 10.1097/01.ASN.0000033611.79556.AE
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chau, B. N., Xin, C., Hartner, J., Ren, S., Castano, A. P., Linn, G., et al. (2012). MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 4, 121ra118. doi: 10.1126/scitranslmed.3003205
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, H. Y., Huang, X. R., Wang, W., Li, J. H., Heuchel, R. L., Chung, A. C., et al. (2011). The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes 60, 590–601. doi: 10.2337/db10-0403
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, H. Y., Zhong, X., Huang, X. R., Meng, X. M., You, Y., Chung, A. C., et al. (2014). MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol. Ther. 22, 842–853. doi: 10.1038/mt.2013.235
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, S. J., Yuan, W., Mori, Y., Levenson, A., Trojanowska, M., and Varga, J. (1999). Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: involvement of Smad 3. J. Invest. Dermatol. 112, 49–57. doi: 10.1046/j.1523-1747.1999.00477.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cho, M. E., Smith, D. C., Branton, M. H., Penzak, S. R., and Kopp, J. B. (2007). Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2, 906–913. doi: 10.2215/CJN.01050207
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Choi, M. E., Ding, Y., and Kim, S. I. (2012). TGF-beta signaling via TAK1 pathway: role in kidney fibrosis. Semin. Nephrol. 32, 244–252. doi: 10.1016/j.semnephrol.2012.04.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chung, A. C., Dong, Y., Yang, W., Zhong, X., Li, R., and Lan, H. Y. (2013a). Smad7 suppresses renal fibrosis via altering expression of TGF-beta/Smad3-regulated microRNAs. Mol. Ther. 21, 388–398. doi: 10.1038/mt.2012.251
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chung, A. C., Huang, X. R., Meng, X., and Lan, H. Y. (2010a). miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 21, 1317–1325. doi: 10.1681/ASN.2010020134
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chung, A. C., Huang, X. R., Zhou, L., Heuchel, R., Lai, K. N., and Lan, H. Y. (2009). Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol. Dial. Transplant 24, 1443–1454. doi: 10.1093/ndt/gfn699
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chung, A. C., Yu, X., and Lan, H. Y. (2013b). MicroRNA and nephropathy: emerging concepts. Int. J. Nephrol. Renovasc. Dis. 6, 169–179. doi: 10.2147/IJNRD.S37885
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chung, A. C., Zhang, H., Kong, Y. Z., Tan, J. J., Huang, X. R., Kopp, J. B., et al. (2010b). Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J. Am. Soc. Nephrol. 21, 249–260. doi: 10.1681/ASN.2009010018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dennler, S., Itoh, S., Vivien, D., Ten Dijke, P., Huet, S., and Gauthier, J. M. (1998). Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17, 3091–3100. doi: 10.1093/emboj/17.11.3091
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584. doi: 10.1038/nature02006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Doi, S., Zou, Y., Togao, O., Pastor, J. V., John, G. B., Wang, L., et al. (2011). Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 286, 8655–8665. doi: 10.1074/jbc.M110.174037
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Du, B., Ma, L. M., Huang, M. B., Zhou, H., Huang, H. L., Shao, P., et al. (2010). High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 584, 811–816. doi: 10.1016/j.febslet.2009.12.053
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ebisawa, T., Fukuchi, M., Murakami, G., Chiba, T., Tanaka, K., Imamura, T., et al. (2001). Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 276, 12477–12480. doi: 10.1074/jbc.C100008200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Eddy, A. A., and Neilson, E. G. (2006). Chronic kidney disease progression. J. Am. Soc. Nephrol. 17, 2964–2966. doi: 10.1681/ASN.2006070704
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Evans, R. A., Tian, Y. C., Steadman, R., and Phillips, A. O. (2003). TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp. Cell Res. 282, 90–100. doi: 10.1016/S0014-4827(02)00015-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fan, J. M., Ng, Y. Y., Hill, P. A., Nikolic-Paterson, D. J., Mu, W., Atkins, R. C., et al. (1999). Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 56, 1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fujimoto, M., Maezawa, Y., Yokote, K., Joh, K., Kobayashi, K., Kawamura, H., et al. (2003). Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem. Biophys. Res. Commun. 305, 1002–1007. doi: 10.1016/S0006-291X(03)00885-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fukasawa, H., Yamamoto, T., Togawa, A., Ohashi, N., Fujigaki, Y., Oda, T., et al. (2004). Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc. Natl. Acad. Sci. U.S.A. 101, 8687–8692. doi: 10.1073/pnas.0400035101
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Godwin, J. G., Ge, X., Stephan, K., Jurisch, A., Tullius, S. G., and Iacomini, J. (2010). Identification of a microRNA signature of renal ischemia reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 107, 14339–14344. doi: 10.1073/pnas.0912701107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hong, K. M., Belperio, J. A., Keane, M. P., Burdick, M. D., and Strieter, R. M. (2007). Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 282, 22910–22920. doi: 10.1074/jbc.M703597200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hou, C. C., Wang, W., Huang, X. R., Fu, P., Chen, T. H., Sheikh-Hamad, D., et al. (2005). Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am. J. Pathol. 166, 761–771. doi: 10.1016/S0002-9440(10)62297-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Howe, E. N., Cochrane, D. R., and Richer, J. K. (2012). The miR-200 and miR-221/222 microRNA families: opposing effects on epithelial identity. J. Mammary Gland Biol. Neoplasia 17, 65–77. doi: 10.1007/s10911-012-9244-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hruska, K. A., Guo, G., Wozniak, M., Martin, D., Miller, S., Liapis, H., et al. (2000). Osteogenic protein-1 prevents renal fibrogenesis associated with ureteral obstruction. Am. J. Physiol. Renal Physiol. 279, F130–F143.
Huang, X. R., Chung, A. C., Wang, X. J., Lai, K. N., and Lan, H. Y. (2008a). Mice overexpressing latent TGF-beta1 are protected against renal fibrosis in obstructive kidney disease. Am. J. Physiol. Renal Physiol. 295, F118–F127. doi: 10.1152/ajprenal.00021.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, X. R., Chung, A. C., Zhou, L., Wang, X. J., and Lan, H. Y. (2008b). Latent TGF-beta1 protects against crescentic glomerulonephritis. J. Am. Soc. Nephrol. 19, 233–242. doi: 10.1681/ASN.2007040484
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ka, S. M., Huang, X. R., Lan, H. Y., Tsai, P. Y., Yang, S. M., Shui, H. A., et al. (2007). Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J. Am. Soc. Nephrol. 18, 1777–1788. doi: 10.1681/ASN.2006080901
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ka, S. M., Yeh, Y. C., Huang, X. R., Chao, T. K., Hung, Y. J., Yu, C. P., et al. (2012). Kidney-targeting Smad7 gene transfer inhibits renal TGF-beta/MAD homologue (SMAD) and nuclear factor kappaB (NF-kappaB) signalling pathways, and improves diabetic nephropathy in mice. Diabetologia 55, 509–519. doi: 10.1007/s00125-011-2364-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kantharidis, P., Wang, B., Carew, R. M., and Lan, H. Y. (2011). Diabetes complications: the microRNA perspective. Diabetes 60, 1832–1837. doi: 10.2337/db11-0082
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kato, M., Arce, L., Wang, M., Putta, S., Lanting, L., and Natarajan, R. (2011). A microRNA circuit mediates transforming growth factor-beta1 autoregulation in renal glomerular mesangial cells. Kidney Int. 80, 358–368. doi: 10.1038/ki.2011.43
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kato, M., Zhang, J., Wang, M., Lanting, L., Yuan, H., Rossi, J. J., et al. (2007). MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. U.S.A. 104, 3432–3437. doi: 10.1073/pnas.0611192104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kavsak, P., Rasmussen, R. K., Causing, C. G., Bonni, S., Zhu, H., Thomsen, G. H., et al. (2000). Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 6, 1365–1375. doi: 10.1016/S1097-2765(00)00134-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kim, S. G., Kim, H. A., Jong, H. S., Park, J. H., Kim, N. K., Hong, S. H., et al. (2005). The endogenous ratio of Smad2 and Smad3 influences the cytostatic function of Smad3. Mol. Biol. Cell 16, 4672–4683. doi: 10.1091/mbc.E05-01-0054
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kopp, J. B., Factor, V. M., Mozes, M., Nagy, P., Sanderson, N., Bottinger, E. P., et al. (1996). Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab. Invest. 74, 991–1003.
Korpal, M., Lee, E. S., Hu, G., and Kang, Y. (2008). The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 283, 14910–14914. doi: 10.1074/jbc.C800074200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kriegel, A. J., Liu, Y., Cohen, B., Usa, K., and Liang, M. (2012). MiR-382 targeting of kallikrein 5 contributes to renal inner medullary interstitial fibrosis. Physiol. Genomics 44, 259–267. doi: 10.1152/physiolgenomics.00173.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Krupa, A., Jenkins, R., Luo, D. D., Lewis, A., Phillips, A., and Fraser, D. (2010). Loss of MicroRNA-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Nephrol. 21, 438–447. doi: 10.1681/ASN.2009050530
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lan, H. Y., and Chung, A. C. (2012). TGF-beta/Smad signaling in kidney disease. Semin. Nephrol. 32, 236–243. doi: 10.1016/j.semnephrol.2012.04.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lan, H. Y., Mu, W., Tomita, N., Huang, X. R., Li, J. H., Zhu, H. J., et al. (2003). Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J. Am. Soc. Nephrol. 14, 1535–1548. doi: 10.1097/01.ASN.0000067632.04658.B8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
LeBleu, V. S., Taduri, G., O'Connell, J., Teng, Y., Cooke, V. G., Woda, C., et al. (2013). Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053. doi: 10.1038/nm.3218
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, J. H., Huang, X. R., Zhu, H. J., Johnson, R., and Lan, H. Y. (2003). Role of TGF-beta signaling in extracellular matrix production under high glucose conditions. Kidney Int. 63, 2010–2019. doi: 10.1046/j.1523-1755.2003.00016.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, J. H., Huang, X. R., Zhu, H. J., Oldfield, M., Cooper, M., Truong, L. D., et al. (2004). Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 18, 176–178. doi: 10.1096/fj.02-1117fje
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, J., Qu, X., Yao, J., Caruana, G., Ricardo, S. D., Yamamoto, Y., et al. (2010). Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 59, 2612–2624. doi: 10.2337/db09-1631
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, Y., Tan, X., Dai, C., Stolz, D. B., Wang, D., and Liu, Y. (2009). Inhibition of integrin-linked kinase attenuates renal interstitial fibrosis. J. Am. Soc. Nephrol. 20, 1907–1918. doi: 10.1681/ASN.2008090930
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, F. Y., Li, X. Z., Peng, Y. M., Liu, H., and Liu, Y. H. (2008). Arkadia regulates TGF-beta signaling during renal tubular epithelial to mesenchymal cell transition. Kidney Int. 73, 588–594. doi: 10.1038/sj.ki.5002713
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, G. X., Li, Y. Q., Huang, X. R., Wei, L., Chen, H. Y., Shi, Y. J., et al. (2013). Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-beta/Smad3-NF.kappaB-dependent mechanisms in mice. PLoS ONE 8:e53573. doi: 10.1371/journal.pone.0053573
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, G. X., Li, Y. Q., Huang, X. R., Wei, L. H., Zhang, Y., Feng, M., et al. (2014). Smad7 inhibits AngII-mediated hypertensive nephropathy in a mouse model of hypertension. Clin. Sci. 127, 195–208. doi: 10.1042/CS20130706
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, Y. (2010). New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 21, 212–222. doi: 10.1681/ASN.2008121226
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, Y., Taylor, N. E., Lu, L., Usa, K., Cowley, A. W. Jr., Ferreri, N. R., et al. (2010). Renal medullary microRNAs in Dahl salt-sensitive rats: miR-29b regulates several collagens and related genes. Hypertension 55, 974–982. doi: 10.1161/HYPERTENSIONAHA.109.144428
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, Z., Huang, X. R., and Lan, H. Y. (2012). Smad3 mediates ANG II-induced hypertensive kidney disease in mice. Am. J. Physiol. Renal Physiol. 302, F986–F997. doi: 10.1152/ajprenal.00595.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Loeffler, I., and Wolf, G. (2014). Transforming growth factor-beta and the progression of renal disease. Nephrol. Dial. Transplant. 29(Suppl. 1), i37–i45. doi: 10.1093/ndt/gft267
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lopez-Hernandez, F. J., and Lopez-Novoa, J. M. (2012). Role of TGF-beta in chronic kidney disease: an integration of tubular, glomerular and vascular effects. Cell Tissue Res. 347, 141–154. doi: 10.1007/s00441-011-1275-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luo, D. D., Phillips, A., and Fraser, D. (2010). Bone morphogenetic protein-7 inhibits proximal tubular epithelial cell Smad3 signaling via increased SnoN expression. Am. J. Pathol. 176, 1139–1147. doi: 10.2353/ajpath.2010.090459
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lyons, R. M., Gentry, L. E., Purchio, A. F., and Moses, H. L. (1990). Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J. Cell Biol. 110, 1361–1367. doi: 10.1083/jcb.110.4.1361
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Massague, J., and Wotton, D. (2000). Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 19, 1745–1754. doi: 10.1093/emboj/19.8.1745
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meng, X. M., Chung, A. C., and Lan, H. Y. (2013). Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin. Sci. 124, 243–254. doi: 10.1042/CS20120252
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meng, X. M., Huang, X. R., Chung, A. C., Qin, W., Shao, X., Igarashi, P., et al. (2010). Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 21, 1477–1487. doi: 10.1681/ASN.2009121244
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meng, X. M., Huang, X. R., Xiao, J., Chen, H. Y., Zhong, X., Chung, A. C., et al. (2012a). Diverse roles of TGF-beta receptor II in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 227, 175–188. doi: 10.1002/path.3976
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meng, X. M., Huang, X. R., Xiao, J., Chung, A. C., Qin, W., Chen, H. Y., et al. (2012b). Disruption of Smad4 impairs TGF-beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 81, 266–279. doi: 10.1038/ki.2011.327
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Meng, X. M., Nikolic-Paterson, D. J., and Lan, H. Y. (2014). Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 10, 493–503. doi: 10.1038/nrneph.2014.114
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Midgley, A. C., Rogers, M., Hallett, M. B., Clayton, A., Bowen, T., Phillips, A. O., et al. (2013). Transforming growth factor-beta1 (TGF-beta1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 288, 14824–14838. doi: 10.1074/jbc.M113.451336
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Moon, J. A., Kim, H. T., Cho, I. S., Sheen, Y. Y., and Kim, D. K. (2006). IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 70, 1234–1243. doi: 10.1038/sj.ki.5001775
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Munger, J. S., Huang, X., Kawakatsu, H., Griffiths, M. J., Dalton, S. L., Wu, J., et al. (1999). The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328. doi: 10.1016/S0092-8674(00)80545-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murakami, K., Takemura, T., Hino, S., and Yoshioka, K. (1997). Urinary transforming growth factor-beta in patients with glomerular diseases. Pediatr. Nephrol. 11, 334–336. doi: 10.1007/s004670050289
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ng, Y. Y., Chen, Y. M., Tsai, T. J., Lan, X. R., Yang, W. C., and Lan, H. Y. (2009). Pentoxifylline inhibits transforming growth factor-beta signaling and renal fibrosis in experimental crescentic glomerulonephritis in rats. Am. J. Nephrol. 29, 43–53. doi: 10.1159/000150600
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nikolic-Paterson, D. J., Wang, S., and Lan, H. Y. (2014). Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. 4, 34–38. doi: 10.1038/kisup.2014.7
Oba, S., Kumano, S., Suzuki, E., Nishimatsu, H., Takahashi, M., Takamori, H., et al. (2010). miR-200b precursor can ameliorate renal tubulointerstitial fibrosis. PLoS ONE 5:e13614. doi: 10.1371/journal.pone.0013614
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petersen, M., Thorikay, M., Deckers, M., van Dinther, M., Grygielko, E. T., Gellibert, F., et al. (2008). Oral administration of GW788388, an inhibitor of TGF-beta type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 73, 705–715. doi: 10.1038/sj.ki.5002717
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Phanish, M. K., Wahab, N. A., Colville-Nash, P., Hendry, B. M., and Dockrell, M. E. (2006). The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem. J. 393, 601–607. doi: 10.1042/BJ20051106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Piek, E., Ju, W. J., Heyer, J., Escalante-Alcalde, D., Stewart, C. L., Weinstein, M., et al. (2001). Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J. Biol. Chem. 276, 19945–19953. doi: 10.1074/jbc.M102382200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Putta, S., Lanting, L., Sun, G., Lawson, G., Kato, M., and Natarajan, R. (2012). Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 23, 458–469. doi: 10.1681/ASN.2011050485
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qin, W., Chung, A. C., Huang, X. R., Meng, X. M., Hui, D. S., Yu, C. M., et al. (2011). TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 22, 1462–1474. doi: 10.1681/ASN.2010121308
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Samarakoon, R., Overstreet, J. M., Higgins, S. P., and Higgins, P. J. (2012). TGF-beta1 –> SMAD/p53/USF2 –> PAI-1 transcriptional axis in ureteral obstruction-induced renal fibrosis. Cell Tissue Res. 347, 117–128. doi: 10.1007/s00441-011-1181-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sanderson, N., Factor, V., Nagy, P., Kopp, J., Kondaiah, P., Wakefield, L., et al. (1995). Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc. Natl. Acad. Sci. U.S.A. 92, 2572–2576. doi: 10.1073/pnas.92.7.2572
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sato, M., Muragaki, Y., Saika, S., Roberts, A. B., and Ooshima, A. (2003). Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 112, 1486–1494. doi: 10.1172/JCI200319270
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sharma, K., Ix, J. H., Mathew, A. V., Cho, M., Pflueger, A., Dunn, S. R., et al. (2011). Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 22, 1144–1151. doi: 10.1681/ASN.2010101049
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shen, N., Lin, H., Wu, T., Wang, D., Wang, W., Xie, H., et al. (2013). Inhibition of TGF-beta1-receptor posttranslational core fucosylation attenuates rat renal interstitial fibrosis. Kidney Int. 84, 64–77. doi: 10.1038/ki.2013.82
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shi, Y., and Massague, J. (2003). Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113, 685–700. doi: 10.1016/S0092-8674(03)00432-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sugimoto, H., Grahovac, G., Zeisberg, M., and Kalluri, R. (2007). Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes 56, 1825–1833. doi: 10.2337/db06-1226
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tampe, D., and Zeisberg, M. (2014). Potential approaches to reverse or repair renal fibrosis. Nat. Rev. Nephrol. 10, 226–237. doi: 10.1038/nrneph.2014.14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Trachtman, H., Fervenza, F. C., Gipson, D. S., Heering, P., Jayne, D. R., Peters, H., et al. (2011). A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 79, 1236–1243. doi: 10.1038/ki.2011.33
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsakas, S., and Goumenos, D. S. (2006). Accurate measurement and clinical significance of urinary transforming growth factor-beta1. Am. J. Nephrol. 26, 186–193. doi: 10.1159/000093178
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsuchida, K., Zhu, Y., Siva, S., Dunn, S. R., and Sharma, K. (2003). Role of Smad4 on TGF-beta-induced extracellular matrix stimulation in mesangial cells. Kidney Int. 63, 2000–2009. doi: 10.1046/j.1523-1755.2003.00009.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van Rooij, E., Sutherland, L. B., Thatcher, J. E., Dimaio, J. M., Naseem, R. H., Marshall, W. S., et al. (2008). Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. U.S.A. 105, 13027–13032. doi: 10.1073/pnas.0805038105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vindevoghel, L., Lechleider, R. J., Kon, A., de Caestecker, M. P., Uitto, J., Roberts, A. B., et al. (1998). SMAD3/4-dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor beta. Proc. Natl. Acad. Sci. U.S.A. 95, 14769–14774. doi: 10.1073/pnas.95.25.14769
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wada, T., Sakai, N., Matsushima, K., and Kaneko, S. (2007). Fibrocytes: a new insight into kidney fibrosis. Kidney Int. 72, 269–273. doi: 10.1038/sj.ki.5002325
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, B., Herman-Edelstein, M., Koh, P., Burns, W., Jandeleit-Dahm, K., Watson, A., et al. (2010). E-cadherin expression is regulated by miR-192/215 by a mechanism that is independent of the profibrotic effects of transforming growth factor-beta. Diabetes 59, 1794–1802. doi: 10.2337/db09-1736
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, B., Jha, J. C., Hagiwara, S., McClelland, A. D., Jandeleit-Dahm, K., Thomas, M. C., et al. (2014). Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 85, 352–361. doi: 10.1038/ki.2013.372
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, B., Koh, P., Winbanks, C., Coughlan, M. T., McClelland, A., Watson, A., et al. (2011). miR-200a Prevents renal fibrogenesis through repression of TGF-beta2 expression. Diabetes 60, 280–287. doi: 10.2337/db10-0892
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J., Gao, Y., Ma, M., Li, M., Zou, D., Yang, J., et al. (2013). Effect of miR-21 on renal fibrosis by regulating MMP-9 and TIMP1 in kk-ay diabetic nephropathy mice. Cell Biochem. Biophys. 67, 537–546. doi: 10.1007/s12013-013-9539-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, S., de Caestecker, M., Kopp, J., Mitu, G., Lapage, J., and Hirschberg, R. (2006a). Renal bone morphogenetic protein-7 protects against diabetic nephropathy. J. Am. Soc. Nephrol. 17, 2504–2512. doi: 10.1681/ASN.2006030278
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, W., Huang, X. R., Canlas, E., Oka, K., Truong, L. D., Deng, C., et al. (2006b). Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 98, 1032–1039. doi: 10.1161/01.RES.0000218782.52610.dc
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wei, X., Xia, Y., Li, F., Tang, Y., Nie, J., Liu, Y., et al. (2013). Kindlin-2 mediates activation of TGF-beta/Smad signaling and renal fibrosis. J. Am. Soc. Nephrol. 24, 1387–1398. doi: 10.1681/ASN.2012101041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wrana, J. L., Attisano, L., Wieser, R., Ventura, F., and Massague, J. (1994). Mechanism of activation of the TGF-beta receptor. Nature 370, 341–347. doi: 10.1038/370341a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, C. F., Chiang, W. C., Lai, C. F., Chang, F. C., Chen, Y. T., Chou, Y. H., et al. (2013). Transforming growth factor beta-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am. J. Pathol. 182, 118–131. doi: 10.1016/j.ajpath.2012.09.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wynn, T. A., and Ramalingam, T. R. (2012). Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040. doi: 10.1038/nm.2807
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xavier, S., Vasko, R., Matsumoto, K., Zullo, J. A., Chen, R., Maizel, J., et al. (2014). Curtailing endothelial TGF-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J. Am. Soc. Nephrol. doi: 10.1681/ASN.2013101137. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xiao, J., Meng, X. M., Huang, X. R., Chung, A. C., Feng, Y. L., Hui, D. S., et al. (2012). miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol. Ther. 20, 1251–1260. doi: 10.1038/mt.2012.36
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xiong, M., Jiang, L., Zhou, Y., Qiu, W., Fang, L., Tan, R., et al. (2012). The miR-200 family regulates TGF-beta1-induced renal tubular epithelial to mesenchymal transition through Smad pathway by targeting ZEB1 and ZEB2 expression. Am. J. Physiol. Renal Physiol. 302, F369–F379. doi: 10.1152/ajprenal.00268.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xu, X., Kriegel, A. J., Liu, Y., Usa, K., Mladinov, D., Liu, H., et al. (2012). Delayed ischemic preconditioning contributes to renal protection by upregulation of miR-21. Kidney Int. 82, 1167–1175. doi: 10.1038/ki.2012.241
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yamamoto, T., Noble, N. A., Cohen, A. H., Nast, C. C., Hishida, A., Gold, L. I., et al. (1996). Expression of transforming growth factor-beta isoforms in human glomerular diseases. Kidney Int. 49, 461–469. doi: 10.1038/ki.1996.65
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, F., Chung, A. C., Huang, X. R., and Lan, H. Y. (2009). Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 54, 877–884. doi: 10.1161/HYPERTENSIONAHA.109.136531
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, F., Huang, X. R., Chung, A. C., Hou, C. C., Lai, K. N., and Lan, H. Y. (2010). Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J. Pathol. 221, 390–401. doi: 10.1002/path.2721
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, J., Zhang, X., Li, Y., and Liu, Y. (2003a). Downregulation of Smad transcriptional corepressors SnoN and Ski in the fibrotic kidney: an amplification mechanism for TGF-beta1 signaling. J. Am. Soc. Nephrol. 14, 3167–3177. doi: 10.1097/01.ASN.0000099373.33259.B2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yang, Y. C., Piek, E., Zavadil, J., Liang, D., Xie, D., Heyer, J., et al. (2003b). Hierarchical model of gene regulation by transforming growth factor beta. Proc. Natl. Acad. Sci. U.S.A. 100, 10269–10274. doi: 10.1073/pnas.1834070100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yuan, W., and Varga, J. (2001). Transforming growth factor-beta repression of matrix metalloproteinase-1 in dermal fibroblasts involves Smad3. J. Biol. Chem. 276, 38502–38510. doi: 10.1074/jbc.M107081200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zeisberg, E. M., Potenta, S. E., Sugimoto, H., Zeisberg, M., and Kalluri, R. (2008). Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 19, 2282–2287. doi: 10.1681/ASN.2008050513
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zeisberg, M., Hanai, J., Sugimoto, H., Mammoto, T., Charytan, D., Strutz, F., et al. (2003). BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968. doi: 10.1038/nm888
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhong, X., Chung, A. C., Chen, H. Y., Dong, Y., Meng, X. M., Li, R., et al. (2013). miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia 56, 663–674. doi: 10.1007/s00125-012-2804-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhong, X., Chung, A. C., Chen, H. Y., Meng, X. M., and Lan, H. Y. (2011). Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 22, 1668–1681. doi: 10.1681/ASN.2010111168
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, L., Fu, P., Huang, X. R., Liu, F., Chung, A. C., Lai, K. N., et al. (2010). Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am. J. Physiol. Renal Physiol. 298, F1006–F1017. doi: 10.1152/ajprenal.00675.2009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhu, H. J., Iaria, J., and Sizeland, A. M. (1999). Smad7 differentially regulates transforming growth factor beta-mediated signaling pathways. J. Biol. Chem. 274, 32258–32264. doi: 10.1074/jbc.274.45.32258
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: TGF-ß, Smads mediators, renal fibrosis, therapeutics, mechanisms
Citation: Meng X-M, Tang PM-K, Li J and Lan HY (2015) TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 6:82. doi: 10.3389/fphys.2015.00082
Received: 15 January 2015; Accepted: 03 March 2015;
Published: 19 March 2015.
Edited by:
David Nikolic-Paterson, Monash Medical Centre, AustraliaReviewed by:
Steven Condliffe, University of Otago, New ZealandCopyright © 2015 Meng, Tang, Li and Lan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Yao Lan, Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong, ChinaaHlsYW5AY3Voay5lZHUuaGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.