 Rebecca A. Capel
Rebecca A. Capel Derek A. Terrar
Derek A. Terrar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 25 March 2015
Sec. Cardiac Electrophysiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00080
This article is part of the Research TopicCa2+ Signaling and Heart RhythmView all 13 articles
Mechanisms underlying pacemaker activity in the sinus node remain controversial, with some ascribing a dominant role to timing events in the surface membrane (“membrane clock”) and others to uptake and release of calcium from the sarcoplasmic reticulum (SR) (“calcium clock”). Here we discuss recent evidence on mechanisms underlying pacemaker activity with a particular emphasis on the many roles of calcium. There are particular areas of controversy concerning the contribution of calcium spark-like events and the importance of I(f) to spontaneous diastolic depolarisation, though it will be suggested that neither of these is essential for pacemaking. Sodium-calcium exchange (NCX) is most often considered in the context of mediating membrane depolarisation after spark-like events. We present evidence for a broader role of this electrogenic exchanger which need not always depend upon these spark-like events. Short (milliseconds or seconds) and long (minutes) term influences of calcium are discussed including direct regulation of ion channels and NCX, and control of the activity of calcium-dependent enzymes (including CaMKII, AC1, and AC8). The balance between the many contributory factors to pacemaker activity may well alter with experimental and clinical conditions, and potentially redundant mechanisms are desirable to ensure the regular spontaneous heart rate that is essential for life. This review presents evidence that calcium is central to the normal control of pacemaking across a range of temporal scales and seeks to broaden the accepted description of the “calcium clock” to cover these important influences.
The aim of this review is to discuss the many different roles of Ca2+ in regulating pacemaker function in the sino-atrial node (SAN). The major determinants of pacemaker activity remain controversial, as illustrated by reviews from the Lakatta and DiFrancesco groups (Lakatta and DiFrancesco, 2009; DiFrancesco and Noble, 2012a,b; Lakatta and Maltsev, 2012; Maltsev and Lakatta, 2012). Other important reviews have been published in the last 10 years (Dobrzynski et al., 2007; Imtiaz et al., 2007; Wu and Anderson, 2014). A comprehensive review from Mangoni and Nargeot also presents a valuable overview of pacemaker mechanisms, particularly with respect to conclusions drawn from genetic abnormalities and genetic manipulations (Mangoni and Nargeot, 2008). The starting point for the discussion here will be the broadly excellent review by Irisawa et al. (1993) (see also Irisawa (1978) and Noma (1996)) which is a very comprehensive in its discussion of surface membrane currents. Irisawa et al. make little or no inclusion of the possible influence of cytosolic Ca2+, particularly that released from the sarcoplasmic reticulum (SR), since little was known on this aspect of pacemaker mechanisms at the time the review was written. In recent years much of the debate concerning the origin of pacemaker activity in the heart has been presented as a choice between two alternative mechanisms, a “membrane clock” in which I(f) activated by hyperpolarization plays the dominant role or a “Ca2+ clock” in which the timing of uptake and release of Ca2+ by the SR is the major determinant of the cardiac rhythm (DiFrancesco and Noble, 2012a,b; Lakatta and Maltsev, 2012; Maltsev and Lakatta, 2012).
This review seeks to discuss broader aspects of the influence of Ca2+ on pacemaker activity than are frequently considered in debates on the relative importance of Ca2+ and membrane clocks. The evidence discussed below supports the view that a variety of ionic currents in addition to I(f) can contribute to the timing of the membrane clock, that these events are potentially modulated by intracellular Ca2+ in a number of ways and that the relative importance of these pathways might vary under different physiological and clinical conditions. We consider data relating to the role of the Ca2+ clock under a range of conditions, and discuss whether such a clock needs to depend solely on spontaneous Ca2+ sparks or local calcium releases (LCRs) or whether other rhythmic Ca2+-dependent mechanisms should also be taken into account to form a complete picture. It appears that the Ca2+ clock could play a fundamentally important role for the timing mechanism of the cardiac pacemaker under particular conditions, but in many circumstances might play a cooperative interacting role with the membrane clock.
Timing mechanisms for different sorts of pacemaker activity have been discussed in many different tissues including oscillations in smooth muscle, interstitial cells, brain and heart (e.g., Berridge and Galione, 1988). Mechanisms include what have been called membrane oscillators and cytosolic Ca2+ oscillators in smooth muscle and brain, and ideas concerning a Ca2+ clock are not unique to the heart (Imtiaz et al., 2006; McHale et al., 2006; Berridge, 2008; Imtiaz, 2012).
In the heart, a key feature that distinguishes pacemaker tissue from surrounding atrial muscle is the absence of the stabilizing influence of IK1. Other important characteristics are the presence of the connexin protein Cx45 (Coppen et al., 1999) and I(f) (Biel et al., 2002) and lack of Cx43 (ten Velde et al., 1995), but the lack of IK1 is particularly functionally important for the following reasons. The presence of IK1 channels in atrial and ventricular myocytes is responsible for the ~−90 mV resting membrane potential in these cells, dominated by the equilibrium potential for potassium ions in physiological solutions. In the absence of IK1 the SAN membrane potential is not forced to “rest” at this potential. In addition, lack of the IK1 conducting pathway leads to a greatly increased membrane resistance (reduced conductance) in SAN cells in comparison to atrial and ventricular myocytes and this allows very small ionic currents to exert a profound influence on membrane potential. In this regard, it is also relevant to consider that SAN myocytes exhibit small cell capacitance (of the order of 30–40 pF) in turn requiring only small currents to charge or discharge the membrane capacitance. The significance of the lack of IK1 in mammalian SAN pacemaker tissue was first demonstrated in an important paper from Irisawa (Noma et al., 1984) (see also Shibata and Giles, 1985 for similar observations in amphibian pacemaker tissue). The susceptibility to oscillations causing spontaneous activity when IK1 is suppressed in ventricular tissue was also shown by Miake et al. (2002).
Although there is no “resting” potential in a pacemaker cell showing continuous electrical activity, an important observation is that when pacemaker activity is arrested (for example by the L-type Ca2+ channel blocker nifedipine (Kodama et al., 1997), or by blockers of voltage-gated potassium channels (Lei et al., 2001) the membrane adopts a potential at least for a period of seconds at approximately −30 to −40 mV. A similar potential is adopted when spontaneous activity is stopped by chelation of cytosolic Ca2+ with intracellular BAPTA (Capel and Terrar, this issue, and see later). A “resting potential” of −38 mV was also described in rabbit SA node by Noma and Irisawa (1975). Again, a similar potential is recorded in amphibian sinus venosus when spontaneous activity is “arrested” by nifedipine (Bramich et al., 1993). Verheijck et al. (1995) also described a “background current with a reversal potential of −32 mV in rabbit SA node in the presence of nifedipine and E-4031.”
With this “background” conductance as a starting point, a very simple pacemaker can be constructed in which an action potential upstroke carried by calcium ions leads to a depolarisation that then activates a voltage-gated potassium conductance and this in turn brings about repolarisation. Potassium channel de-activation will then lead to a removal of hyperpolarizing influence and allow the membrane to move back toward its “resting” level as a consequence of the influence of the background conductance pathway (Figure 1A). Early modeling work suggested that this mechanism is capable of sustaining spontaneous action potential generation (Hauswirth et al., 1968) and see (Noble et al., 1992).
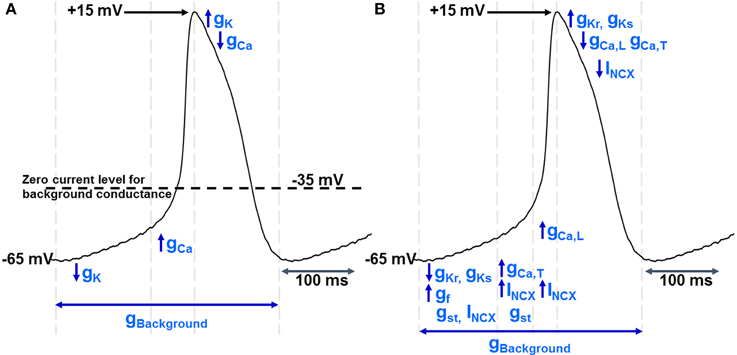
Figure 1. (A) shows a simple model of pacemaker function in which there is a “background” current/conductance with a zero current level between −30 and −40 mV. There is extensive evidence for such a pathway, although as discussed in the text the nature of the conductance(s) contributing to this pathway remain poorly understood. However, given the existence of this pathway, pacemaker activity can be maintained by sequential activation of voltage-gated K+ and Ca2+ ion channels, noting that de-activation of K+ conductance (gK) after repolarization will be associated with the “diastolic” or pacemaker depolarization as the potential moves toward the zero current level for the “background” conductance. When the membrane potential reaches the threshold for voltage-gated Ca2+ channels, activation of these channels (increasing gCa) will lead to the upstroke of the action potential and the depolarization will activate voltage-gated potassium channels to complete the cycle of repetitive activity. The vertical dotted lines show an approximate division of the time period of the pacemaker cycle to represent these phases of channel activation and de-activation. The experimental record that is shown to illustrate these phases was recorded from a guinea-pig SAN myocyte in our laboratory and is similar to records in Figures 2, 5 (and also to records in Capel and Terrar in this issue). A formal mathematical representation of these changes in conductance was not constructed, but the analysis is broadly similar to the principles used for the basic mathematical model presented by Hauswirth, Noble, and Tsien in 1968 to account for spontaneous activity in Purkinje fibers (Hauswirth et al., 1968).(B) shows a more comprehensive description of the ionic conductances and currents underlying pacemaker activity, including gCaL and gCaT (conductance provided by the two varieties of voltage-gated Ca2+ channel in cardiac muscle), gKs and gKr (the slowly and rapidly activating voltage-gated K+ channels), gf (the conductance associated with channels activated by hyperpolarization and carrying the “funny” current), gst (the sustained inward current channels), and INCX (the NCX current thought to flow throughout the cardiac cycle as outlined in more detail in Figure 2). Again, the vertical dotted lines show an approximate division of the time period of the pacemaker cycle to represent different phases of channel activation and de-activation.
A more comprehensive model that will be used as a framework for later discussion is shown for comparison in Figure 1B.
Even in a review with an emphasis on the many roles for Ca2+, the existence of a background current with a reversal potential in the region of −30 to −40 mV is so fundamentally important for pacemaker mechanisms that it deserves further discussion. It is also conceivable that this poorly understood pathway is itself Ca2+-dependent. The first question that arises from the simple model is how the “pseudo resting” level of −30 to −40 mV is determined and what, in turn, is the selectivity of the membrane to different ions at such a “pseudo resting” potential when voltage-gated channels are not active.
Another way of phrasing the question in the previous paragraph is what is the “background current,” or perhaps better what is the background conductance because little or no net current will flow at the “pseudo resting” potential. Although the evidence presented above in favor of the existence of a background conductance is compelling, there is surprisingly little evidence or agreement on the ion conducting pathways that give rise to this conductance. One approach is to block everything we think we understand and to label what is left as background current e.g., Hagiwara et al. (1992), and this approach leads to the suggestion that background conductance is determined by a balance between potassium conductance (which if dominant would lead to a membrane potential close to the potassium equilibrium potential of perhaps −90 mV) and sodium and/or Ca2+ conductances (that if dominant would take the potential to more positive values determined by their equilibrium potentials of ~+60 mV in the case of Na+ and more positive than +100 mV in the case of Ca2+). This balance could lead to a “pseudo resting” potential of −30 to −40 mV. Chloride ions cannot be excluded and might also contribute [while a contribution of Cl− to “background current” was not detected by Hagiwara et al. (1992), other observations from Seyama (1979) and from Huang (Huang et al., 2009) support a role for Cl−].
At rest, single channels with a zero current potential close to the “pseudo resting” level have not been described. Ito et al. (1994) observed activity of IKACh channels in the absence of ACh. These were the only K+ channels recorded “at rest” in 105 experiments on SA node cells. These authors concluded that an additional pathway carrying inward current at −50 mV must also exist. These channels could be difficult to find and record from (if they were too scarce, low conductance or too fast and short lived to allow easy detection). One possibility is that a major contributor to background conductance is not an ion channel but NCX in a leak mode as first suggested by Kang and Hilgemann (2004), see later.
Another relevant pathway is the sustained inward current described by Noma and colleagues (Mitsuiye et al., 2000). This pathway cannot contribute to the “pseudo resting” level when nifedipine is used to cause cessation of pacemaker activity, since this drug blocks I(st) in addition to blocking L-type Ca2+ channels. However, I(st) is carried by channels that allow passage of monovalent cations and the kinetics of this time and voltage-dependent pathway are sufficiently slow that it could play a role similar to that proposed for the “leak” or background conductance pathway. Under physiological conditions this pathway allows passage of both sodium and potassium ions, with a reversal potential for the current of approximately −20 mV. The single channel conductance of the I(st) pathway has been reported to be 13 pS, and to have an open probability that is regulated by protein kinase A (PKA). In rabbit SAN this pathway gives rise to an approximately 50 pA current at −50 mV in an isolated SAN myocyte (Mitsuiye et al., 1999).
The I(f) current activated by hyperpolarization will be discussed in more detail later, but it is worth mentioning that its kinetics are also slow at potentials close to −60 mV and it could feasibly contribute to conductance over many seconds as well as playing a role in “pacemaker depolarization” during a single cardiac cycle. In this context, Verkerk and Wilders have argued that de-activation of I(f) near the overshoot of the action potential, though fast, is not instantaneous, and significant I(f) can be present in their models at the start of diastolic depolarization, persisting from activation of this pathway during preceding action potentials (Verkerk and Wilders, 2013). In addition, Proenza et al. (2002) suggested that an “instantaneous” current (that could contribute to steady current) might flow via HCN2 channels.
Store Operated Ca2+ Channels (STOCCs) could influence heart rate through the generation of a background current that is modulated through beat to beat SR Ca2+ content changes (Ju and Allen, 2007) and (Liu et al., 2015). Roles have also been suggested for TRPM4 (Hof et al., 2013) and TRPM7 (Sah et al., 2013).
Other channels that might contribute include BK channels (Lai et al., 2014), SK channels (Chen et al., 2013) and Ca2+ activated Cl channels (Verkerk et al., 2002).
There are at least three possible ways in which electrogenic NCX could contribute to pacemaker activity (Figure 2). The most discussed mechanism, often given so much prominence that other possibilities could be overlooked, is the suggestion that there are there are spontaneous events resulting from Ca2+ release from the SR [sometimes referred to as Ca2+ “sparks” (Huser et al., 2000) or local Ca2+ release events (LCRs) (Bogdanov et al., 2001; Vinogradova et al., 2005)] and that the accompanying rises in subsarcolemmal Ca2+ cause local depolarizations arising from Ca2+ extrusion by electrogenic NCX. These sparks/LCRs can occur in the later stages of the pacemaker depolarization preceding the upstroke of the action potential (Huser et al., 2000; Bogdanov et al., 2001; Vinogradova et al., 2002a, 2005). Although it seems very likely that this is an important component of electrogenic NCX, evidence supports other possibilities listed below that might be equally or under some conditions more important.
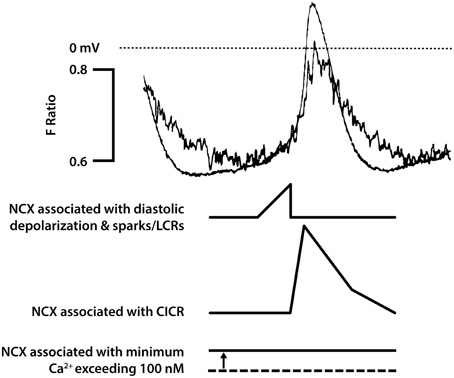
Figure 2. This figure seeks to show in cartoon form the various contributions of NCX to pacemaker activity [that are not confined just to rises in subsarcolemmal Ca2+ associated with spontaneous Ca2+ release from the SR described as Ca2+ sparks or local calcium release events (LCRs)]. The magnitude of the NCX currents will broadly follow the magnitude of the Ca2+ transient (though if there are sufficient changes in subsarcolemmal Ca2+ and Na+, these changes will influence the equilibrium potentials for these ions and therefore the driving force for NCX). The time course of the Ca2+ transient as reported by the Ca2+ probe indo-1 (fluorescence ratio with emission at 405 and 485 nm) is shown overlaid with an action potential in a pacemaker cell in the upper pair of traces. It is recognized that the indo-1 fluorescence ratio (noisy trace) will show a lag as a consequence of the kinetics of Ca2+ binding to the probe, and there may be a delay in the decline in Ca2+ as a consequence of slow dissociation of Ca2+ from the probe (although this delay seems not to be severely limiting since isoprenaline was able to speed the Ca2+ signals recorded with this probe, see Figure 5). There is a rise in Ca2+ that precedes the rapid upstroke of the action potential, and under conditions in which spontaneous Ca2+ sparks/LCRs occur these events will contribute to the early phase of NCX. This is followed by CICR triggered by Ca2+ entry largely through ICaL, and the accompanying large rise in subsarcolemmal Ca2+ will be associated with a substantial additional component of NCX. It is thought that the cytosolic and subsarcolemmal Ca2+ concentrations remain greater than 100 nM even at the most negative membrane potential between action potentials and this is associated with the third component of NCX represented in the bottom trace, with a dotted line to represent zero NCX current that would be expected to occur at approximately 100 nM Ca2+. Although the components of NCX are shown as separate in this diagrammatic representation to emphasize the many roles of NCX, it is recognized that in reality these components run together as a continuum and cannot easily be dissected experimentally. In addition to these three components of electrogenic NCX with a 3 Ca2+:1 Na+ stoichiometry, there may be an additional “leakage” component (see text). The experimental traces are from Rigg et al. (2000).
In addition to the spontaneous Ca2+ release events toward the end of the pacemaker depolarization, there will be the rise in cytosolic Ca2+ that is the result of Ca2+ entry through Ca2+channels during the upstroke of the action potential, and the consequent Ca2+-induced Ca2+ release (CICR) from the SR. CICR occurs as a global release event (meaning a release that occurs synchronously across all of the SR) arising from ryanodine receptor Ca2+ release channels. The global, substantial rise in subsarcolemmal Ca2+ accompanying CICR during the upstroke of the action potential will nevertheless be accompanied by a major component of Ca2+ extrusion through electrogenic NCX (Figure 2). Even when SR function is suppressed, the substantial influx of Ca2+ through L-type channels during the upstroke of the action potential will be accompanied by extrusion of Ca2+ and at least some electrogenic extrusion could be fast enough to re-inforce this action potential upstroke. Note that ryanodine has been reported to reduce the maximum rate of rise of the pacemaker action potential, consistent with a contribution of NCX associated with CICR in addition to the charge carried by L-type Ca2+ channels (Rigg and Terrar, 1996; Rigg et al., 2000), although other Ca2+ dependent mechanisms such as effects of CaMKII on L-type channels could also contribute to this.
The third aspect of possible contribution of NCX to pacemaker activity is a consequence of slower changes in Ca2+concentration. It is clear that the level of Ca2+ activity in SAN cells between beats does not typically fall to the ~100 nM that is measured in quiescent ventricular or atrial myocytes (e.g., Cannell et al., 1987; Schaub et al., 2006), and has been reported as 225 nM (Sanders et al., 2006). If the level of Ca2+ concentration between beats were approximately 200 nM, there would be continuous extrusion of Ca2+ even during the intervals between beats. Ca2+ balance must be maintained in the steady state, but this does not necessitate discrete entry and extrusion phases. Net Ca2+ entry is clearly exhibited during the later stages of pacemaker depolarisation and upstroke of the action potential but Sanders et al suggest that the balancing role of Ca2+ extrusion (mainly through NCX) occurs throughout the cycle including the most negative potential between beats. Thus, there could be a maintained depolarizing influence of NCX (Figure 2).
The three aspects of electrogenic 3:1 NCX have been discussed as separate entities above, in order to emphasize their contributions in different parts of the pacemaker cycle, but it is recognized that in reality they represent a continuum of activity that overlap and cannot be easily distinguished. The clear point is that electrogenic 3:1 NCX can play a role throughout the cardiac cycle and will be dependent on the subsarcolemmal Ca2+concentration under the control of both local and global events.
The average current through the combined contribution of these three components of NCX is substantial since NCX is thought to be the major method of Ca2+ extrusion. Every Ca2+ ion that enters the cell via a Ca2+ channel (L-type or T-type) adds two charges to the cell interior, while extrusion of each Ca2+ by NCX adds one charge (with three sodium ions entering in exchange for each divalent Ca2+), and it therefore follows that if the major component of Ca2+ extrusion is via NCX then the average depolarizing current through NCX in the steady state throughout the cardiac cycle must be approximately half that through the total Ca2+ entry mechanisms.
A fourth mechanism by which NCX could contribute to pacemaker activity is the leak pathway proposed by Kang and Hilgemann (2004) that was briefly mentioned above in the context of “background current.” The Kang and Hilgemann model was based on extensive experimental evidence using conventional voltage-clamp methods to measure current and voltage across “macro” patches combined with the use of ion sensitive electrodes to give information concerning the sodium and Ca2+ gradients close to these membranes. The comprehensive experimental data collected in this way led to the conclusion that the stoichiometry of NCX was 3.2 sodium ions to 1 Ca2+, rather than exactly 3:1. The explanation of a 3.2:1 stoichiometry was that there were additional “modes” of NCX. In particular, a mode with slower kinetics that allowed a single external Ca2+ together with a single external sodium ion to be exchanged for a single internal Ca2+, resulting in electrogenic sodium ion leak without net flux of Ca2+, was suggested as a possible contributor to “background current” in pacemaker cells.
The sodium “leak” mode of NCX proposed by Kang and Hilgemann requires the presence of Ca2+on both sides of the membrane, but since there is no net movement of Ca2+ this mode is not dependent on the driving force for Ca2+ entry into the cell. It seems likely, however, that such a pathway could not operate if the cytosolic Ca2+ were too low to occupy the internal site on the NCX protein.
The discussion above focuses on the importance of NCX as a direct contributor to membrane currents. However, in addition to its direct effects as a charge carrier, NCX may have additional functionally important roles in the context of pacemaker mechanisms. In particular these roles include influencing the overall Ca2+ balance of the cell (including Ca2+ content of the SR), and controlling Ca2+ concentrations in cytosolic microdomains within the cell that might influence the behavior/sensitivity of intracellular release channels such as ryanodine receptors (or perhaps IP3 receptors see Ju et al., 2011) or Ca2+-dependent proteins (see below). Relevant to the present discussion is the thoughtful review by Ottolia et al. (2013) on the importance of NCX in ventricular myocytes. One aspect concerns the balance between NCX and SERCA in determining the amount of Ca2+ loaded into the SR which in turn will have an influence on oscillatory Ca2+ mechanisms and therefore on Ca2+-dependent currents (whether global or local). Although the interplay between NCX and degree of Ca2+ loading of the SR has not yet been studied in detail in pacemaker cells, it seems very likely that the mechanisms that operate in ventricular myocytes will have a close parallel in cardiac pacemaker tissue.
A final point that is relevant here is that NCX might be regulated by protein kinases and these could include Ca2+-sensitive enzymes (Zhang and Hancox, 2009) as discussed below.
It is difficult to determine the various contributions of NCX to pacemaker activity by experimental study. There are two problems concerning the use of drugs. The first is selectivity, but even if a drug were available with perfect selectivity for NCX over other cellular targets, the importance of NCX as the major mechanism for Ca2+ extrusion under normal conditions means that blockade of NCX would lead to rises in intracellular Ca2+concentrations that would have extensive secondary effects. As discussed below, cytosolic Ca2+ influences so many ion channels and other aspects of cell function it is difficult to separate primary and secondary effects of NCX suppression. An “ideal” experiment would require instantaneous blockade of NCX so that effects can be observed before substantial secondary effects have time to occur.
Rapid block of NCX can be achieved by reduction of the extracellular solution by replacing some or all of the Na+ with Li+ (which can pass through most of the ion channels that are permeated by sodium ions but which do not substitute for sodium in NCX). Switch to low sodium does suppress the initiation of spontaneous action potentials, but this suppression can take many beats to establish in toad sinus venosus (Ju and Allen, 1998) and rabbit SA node cells (Bogdanov et al., 2001) when the speed of solution change is modest. When Bogdanov et al caused a rapid but transient solution exchange lasting less than 1 s using a “picospritzer,” they showed that replacement of Na+ with Li+ could suppress a single action potential (while leaving a slightly depressed Ca2+ transient with approximately the same timing as the missing action potential). Sanders et al. (2006) used a rapid switch system that could exchange the solution flowing over individual pacemaker cells in less than 1 s and maintain the flow of this solution. Using this system in guinea-pig SA node cells, rapid switch to low sodium caused immediate cessation of spontaneous activity (both of action potentials and spontaneous Ca2+ transients), and this cessation was maintained for the full several seconds of exposure. During this time cytosolic Ca2+ as measured by the fluorescent probe fluo-5F fell rather than rose (presumably as a consequence of Ca2+ uptake by the SR; see Figure 2 in Sanders et al., 2006).
A possible criticism of sodium replacement experiments is that if the reduction in sodium is very large (e.g., to 14.5 mM or approximately 10%) there may be outward currents through NCX that could complicate interpretation. However, immediate cessation of activity was also seen with smaller reductions of extracellular sodium to 50 mM and even 75 mM (Sanders et al., 2006).
Cessation of SAN myocyte AP firing by low-sodium switch is demonstrably not solely due to suppression of LCR-mediated depolarisation events. Inhibition of Ca2+ uptake into the SR using cyclopiazonic acid (CPA), which would be expected after prolonged exposure to abolish SR Ca2+ loading, slowed but did not abolish pacemaker activity, and switch to low sodium under these conditions again caused immediate cessation (Sanders et al., 2006). This observation demonstrates the fundamental importance of NCX under these conditions even though the SR is not functional.
Another experimental point arising from the rapid switch experiments is that immediately after the switch back from low sodium to normal sodium the spontaneous rate was restored, but was transiently greater than that observed under control conditions (see Figure 1 in Sanders et al., 2006). This would be consistent with an influence of increased intracellular Ca2+ (perhaps an increase in the SR Ca2+ load) during the period of exposure to low sodium. In the absence of CPA, when the SR was functional, Sanders et al consistently observed a reduction in cytosolic Ca2+ when beating was stopped by the switch to low sodium even though the major mechanism for Ca2+ extrusion was inhibited, and this was thought to be accompanied by SR Ca2+ uptake (see later).
The observed effects of several drugs that block NCX in causing cessation of pacemaker activity are also consistent with the view that this pathway exerts a profound influence and is probably essential for pacemaking under most conditions (e.g., rapid switch to 5 μM KB-R7943 caused cessation of beating in approximately 20 s, perhaps as a consequence of the time taken to block NCX Sanders et al., 2006), but these observations must be treated with caution because of off target effects of the compounds.
The importance of NCX for pacemaking has also been investigated using genetic approaches to suppress expression of NCX proteins. A knockout of NCX does not survive because it is lethal at the embryonic stage (Cho et al., 2000; Koushik et al., 2001; Reuter et al., 2002). Conditional knockouts in which NCX is selectively suppressed in atrial or SA node tissue have been made. As is the case of drugs, the difficulty of separating direct and indirect effects of NCX suppression applies, and there are compensatory changes that may occur. One recent paper concludes that genetic inhibition (not complete abolition) of NCX1 disables the ability of the SA node to show its normal increase in rate as a “fight or flight” response, but that resting heart rate continues unchanged (Gao et al., 2013). Herrmann et al. (2013) used an inducible SA-node specific Cre transgene to create mice lacking NCX1 in the pacemaker region and showed that ablation of NCX1 was accompanied by a progressive slowing of heart rate and severe arrhythmias. Another recent paper concludes that complete atrial-specific knockout of NCX1 eliminates SA node pacemaker activity (Groenke et al., 2013). In this case the mice were able to survive with the AV node taking over the normal pacemaker function, while the atria appeared to be quiescent. Further, SAN myoyctes isolated when NCX was eliminated also failed to show pacemaker activity.
Taken together the observations above using low sodium, drugs and genetic approaches are consistent with the view that NCX is very important and probably essential for pacemaker mechanisms in the SA node.
The “funny” current, I(f) was first described by DiFrancesco and reviewed in DiFrancesco (2010). Similar currents [normally designated I(h)] have been described in neurons showing bursting activity (He et al., 2014). The ion channels carrying the current (HCN channels) are activated by hyperpolarization and the magnitude of the currents in the physiological range is increased when the cyclic nucleotide, cAMP, is bound to the channels [since the activation kinetics are quickened (Wainger et al., 2001) and the activation curve is shifted to less negative potentials, (DiFrancesco and Mangoni, 1994)]. Of the four HCN subtypes, HCN4 is the predominant form in the pacemaker region of the heart, though HCN1 and HCN2 also contribute, and there are species differences between the balance of contributions from different subtypes (Baruscotti and Difrancesco, 2004; Biel et al., 2009; DiFrancesco, 2010).
To answer the question whether or not I(f) is essential for pacemaking, it would be helpful either to block the current pharmacologically, or use the techniques of molecular biology to knock down expression of the channel.
In the case of pharmacological approaches there are a variety of drugs including ZD7288, zetabradine and ivabradine (Baruscotti et al., 2005). Cesium ions have also been used to block the currents (Denyer and Brown, 1990). In all cases the drugs slow but do not completely stop pacemaking. In an early study on SAN myocytes isolated from rabbit SA node 2 mM Cs+ was observed to cause close to complete blockade of I(f) without effects on voltage-gated Ca2+ and potassium currents. This concentration reduced pacemaker rate by about 30% but did not stop the initiation of action potentials (Denyer and Brown, 1990). The authors concluded that I(f) is not essential, and that there must be an additional “background” current during the pacemaker depolarization (see above discussion). The more recent drugs ZD7288 and ivabradine also slow but do not stop pacemaker activity (BoSmith et al., 1993; Baruscotti et al., 2005). It is, however, difficult to exclude the argument that blockade of I(f) by the drugs is incomplete under the conditions of these experiments, and that only a very small residual current would be necessary for pacemaking. The use of greater concentrations would not be persuasive because of possible non-specific effects such as blockade of L-type Ca2+ channels or voltage-gated potassium channels, either or both of which can cause cessation of pacemaker activity.
There has been discussion in the past about whether the I(f) current is sufficiently large to make an important contribution to pacemaking, particularly in the context of conventional experiments in which the cell is held at a constant negative potential (commonly −40 mV) and hyperpolarizing voltage steps are applied. Although large currents are recorded when the cell is hyperpolarized to potentials that are not often experienced in normal physiology (such as −80 or −100 mV), the currents recorded at potentials less negative than −70 mV are small. Convincing evidence for a role of I(f) under conditions that approximate those of normal pacemaker activity is provided by “action potential clamp” experiments (Zaza et al., 1997). The essence of this approach is to record and store the action potential waveform from a single myocyte, and then re-apply this waveform under voltage-clamp conditions. In a single cell the net current under control conditions is zero (since ionic currents charge and discharge the membrane capacitance), but interesting information is provided when blockers are applied to reveal a “difference current” that reflects the particular current pathways that are suppressed by the blocker. Zaza et al. used 2 mM Cs+ to block I(f), and concluded that I(f) currents played an important role during the pacemaker depolarization, and that these currents were larger than would have been predicted by conventional voltage-clamp experiments applying only hyperpolarizing pulses, but that other inward currents besides I(f) contribute during this phase.
In the case of genetic experiments, several studies show that suppression of expression of HCN4 proteins also causes a slowing without cessation of activity in the SAN. The most recent uses a conditional knockout system in which HCN4 expression is suppressed only in cardiac tissue of the mouse (Mesirca et al., 2014a). The reduction in spontaneous rate of beating in the SAN reached plateau of about 50% as the I(f) was progressively reduced to very low levels. Interestingly, while the rate reduction in the SAN was reduced by ~50%, there was a cessation of activity in the AV node leading to heart block and death. The apparently greater sensitivity of AV conduction to I(f) suppression seems surprising, but is consistent with the effect of the I(f) blocker ZD7288, which also stops murine AV function at a concentration of 3 μM while reducing spontaneous activity in the SAN by ~50% (Yuill and Hancox, 2002).
In summary, both genetic studies to reduce expression of HCN channels and the effects of selective blockers of I(f) in the mouse show similar effects in that as the severity of action increases to a maximal level the effect on spontaneous rate in the SAN appears to plateau at about a 50% reduction in rate.
There may be other functions of I(f) current in the SAN than determining rate. HCN1 and HCN2 proteins are also present in SAN, and show different kinetics of activation and de-activation compared to HCN4 (Wahl-Schott et al., 2014). Recently knockdown of HCN1 proteins has been shown to influence the stability of pacemaking (and the pacemaker region, Fenske et al., 2013) but not to be essential for pacemaker activity.
Observations from Hagiwara and Irisawa (1989) provide very convincing evidence that that the magnitude of I(f) is influenced by cytosolic Ca2+. These authors found that buffering Ca2+ at 10−10 M caused a substantial (approximately 75%) decrease in current amplitudes, while under the conditions of their experiments 10−7 M caused an increase. In experiments in which the cytosol was perfused with different concentrations of Ca2+, it appeared that a log(concentration)-response curve could be established showing increases in the amplitude of I(f) over the range of Ca2+ concentrations 10−10–10−6 M with the steepest part of the curve covering the range 10−8–10−7 M. Although the authors concluded that there was a direct effect of Ca2+ on I(f) since the effects of Ca2+ were not abolished by the calmodulin inhibitor calmidazolium, it seems likely that the concentration of this compound (10−6 M) was not sufficient to cause effective inhibition. This view is supported by the observations of Zaza et al. (1991) who demonstrated that there were no direct effects of Ca2+ on I(f) in experiments on inside out patches. Rigg et al. (2003) provided evidence that I(f) is regulated by a Ca2+-calmodulin pathway that could at least in part result from a Ca2+-calmodulin regulation of adenylyl cyclase, and this possibility is discussed in more detail below.
The importance of T-type Ca2+ channels for cardiac pacemaker activity has recently been reviewed by Mesirca et al. (2014b). The first evidence for a role for T-type Ca2+ channels during pacemaking activity in the SA node was provided by drugs with moderate selectivity, and it was concluded that blockade of these channels caused a slowing but not cessation of rate (Hagiwara et al., 1998). Huser et al. (2000) emphasized a role for T-type channels in triggering Ca2+ sparks, though others report that Ca2+ sparks can occur without involvement of these channels (Vinogradova et al., 2002a). Genetic studies, particularly concerning CaV3.1 which is the dominant form of T-type Ca2+ channel in the adult SA node, show that suppression of this protein causes only modest reduction in heart rate (Mangoni et al., 2006b). It appears from these observations that T-type Ca2+ channels can contribute to pacemaker depolarization but make only a modest contribution to pacemaker rate under most conditions.
Huser et al. (2000) first suggested a pacemaker role for Ca2+ sparks and associated NCX currents during the later stage of the pacemaker depolarization that precedes the upstroke of the action potential in both subsidiary pacemaker and normal pacemaker myocytes from the cat. Extensive work from the Lakatta laboratory on rabbit SA node cells also shows LCRs as key players in pacemaking (Vinogradova et al., 2002a,b, 2004, 2005, 2006, 2008, 2010; Vinogradova and Lakatta, 2009). Although we have consistently argued for a role of SR Ca2+ release in pacemaking (Rigg and Terrar, 1996; Rigg et al., 2000), and this could include an important role for Ca2+ sparks/LCRs, it is interesting to note that in our experiments on Ca2+ signals in guinea-pig SAN myocytes using both linescan and Nipkow disk techniques (Rakovic et al., 2001; Jackson et al., 2002) we frequently observe cells with apparently normal spontaneous activity that appear to lack detectable Ca2+ sparks/LCRs (although the fraction of cells showing this behavior must be regarded as unpublished observations). In addition, it is observed that under most conditions ryanodine slows but does not stop pacemaker activity and as expected this also suppresses LCRs (Vinogradova et al., 2002a). CPA also abolishes LCRs but does not normally cause cessation of pacemaker activity. It therefore appears that Ca2+ sparks/LCRs are not essential for pacemaker activity, though when they occur the associated electrogenic Ca2+ extrusion via NCX can contribute to the later stages of diastolic depolarization.
A Ca2+ clock mechanism allows for the possibility that there can be generalized Ca2+ releases from the SR alternating with periods of Ca2+ uptake. Although such a mechanism is normally discussed in the context of LCRs, it is not a requirement for a Ca2+ clock mechanisms to have separate local Ca2+ events since the release of Ca2+ from the SR could be synchronized over all or a large part of the cell when the Ca2+ within the SR reaches a critical concentration (leading to a “global” Ca2+ release event similar to the CICR described above but arising from events/conditions within the SR). Note that when membrane potential is clamped at a fixed level close to the most negative potential following a period of normal spontaneous beating it is observed that Ca2+ transients can occur with a timing and frequency which is close to the frequency and timing that would have been adopted by spontaneous action potentials if firing had not been interrupted (Vinogradova et al., 2004). The spatial dimensions of these spontaneous releases are frequently much greater than those of LCRs, and it seems possible that these large events arise spontaneously as a consequence of properties of the Ca2+ release mechanism in the SR. The timing will depend on the kinetics of Ca2+ uptake by SERCA and the time it takes for the Ca2+ concentration within the SR to reach a critical level for synchronous Ca2+ release. The suggestion of the requirement to reach a critical level of Ca2+ within the SR is one way in which such a Ca2+ clock could operate, though other timing mechanisms are possible for example arising from the time constants of activation/de-activation of critical proteins in the uptake-release sequence controlling Ca2+ handling by the SR. The “global” Ca2+ clock (whether arising from the time for Ca2+ to reach a critical level or from other kinetic properties of the uptake/release process) could provide a driving mechanism for pacemaker activity, but may not be essential for pacemaker activity. The observation that spontaneous pacemaker activity in the form of action potentials is slowed but not stopped (Rigg and Terrar, 1996; Rigg et al., 2000; Bogdanov et al., 2006) when SR function is suppressed with ryanodine and/or CPA shows that synchronized spontaneous Ca2+ release from the Ca2+ clock is not always essential for pacemaker activity.
Although under most conditions, ryanodine does not stop spontaneous activity, we have sometimes observed cessation when we have used Ca2+ probes with high Ca2+ affinity, such as indo-1 (perhaps associated with the additional Ca2+ buffering properties of such probes, Rigg et al., 2000). Figure 6 from Rigg et al. is reproduced here showing the progressive development of the effect of ryanodine with a high concentration of isoprenaline present (Figure 3). After 4 min exposure to ryanodine, there is oscillating electrical activity that is so small that it must be subthreshold for the normal action potential mechanism but which nevertheless shows a repeating activity with the same period as the underlying Ca2+ signal (and is slightly slowed compared with spontaneous action potentials recorded in the same cell). The observed oscillations recorded during the development of the effect are best explained by a Ca2+ clock that is driving electrical activity presumably through NCX.
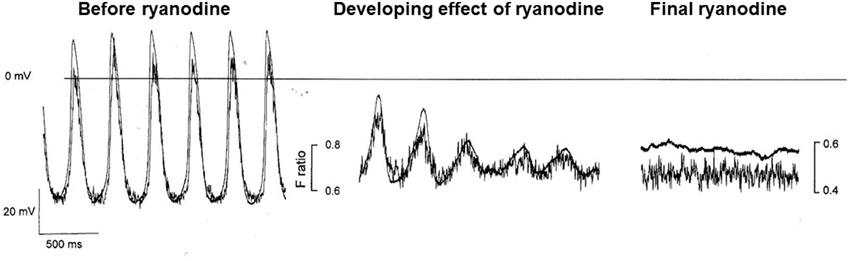
Figure 3. This figure shows the effect of ryanodine in a pacemaker cell in the presence of the calcium probe indo-1 and isoprenaline. Although in our experience, ryanodine slows but does not normally stop spontaneous activity, we have observed cessation of activity after ryanodine when calcium probes are present, particularly those with a high Ca2+ affinity, and we have attributed this to the additional Ca2+ buffering effects of the probe (see text). Despite these limitations, the trace is instructive in that before cessation of activity (which occurs at a potential close to that shown in Figure 1A and described in the text as the zero current level for “background” conductance, see pair of traces on right) subthreshold oscillations occur (middle pair of traces) with a period that is not substantially different from full action potentials at an earlier stage of ryanodine effect. These membrane potential fluctuations are mirrored by corresponding fluctuations in the Ca2+ fluorescence signal. The experimental traces are from Rigg et al. (2000).
There is a very impressive body of evidence showing that the timing of the Ca2+ clock (as judged from the frequency of spontaneous Ca2+ events when the membrane potential is held at a constant membrane potential) is correlated with the timing of the action potentials on the surface membrane recorded when spontaneous electrical activity is allowed to occur (Vinogradova et al., 2002a,b, 2004, 2005, 2006, 2008, 2010; Bogdanov et al., 2006; Vinogradova and Lakatta, 2009). This has been examined over a broad range of frequencies when the rate is either increased by adrenoceptor agonists or decreased with muscarinic agonists, and the correlation between membrane and Ca2+ oscillations remains very strong throughout the range of frequencies studied. There is a parallel correlation between timing of the Ca2+ clock and the degree of phosphorylation and dephosphorylation of functionally important proteins such as L-type Ca2+ channels and phospholamban (Vinogradova et al., 2006). Although the correlation between the timing of the Ca2+ clock and the frequency of spontaneous action potentials is impressive, this does not establish that the relationship is always causal.
It could be argued that the timing of release and uptake of Ca2+ by the SR needs to keep pace with events in the surface membrane in order for a pacemaker mechanism driven primarily by the surface membrane to be maintained in an efficient way, without any disturbance that might otherwise arise from competition with these SR-dependent mechanisms. In other words, the primary mechanism driving the timing of the SA node pacemaker could still depend on the kinetics of activation and deactivation of membrane proteins making up the membrane clock. However, a coupled clock mechanism (Imtiaz et al., 2006, 2007, 2010; Yaniv et al., 2011, 2013; Imtiaz, 2012; Maltsev et al., 2013, 2014) in which both membrane and Ca2+ clocks exert mutually interdependent influences also seems plausible, and perhaps more likely under many circumstances because the system will function most efficiently when the surface membrane and “Ca2+ clock” mechanisms are not competing against one another. The two clocks would normally be synchronized, and this may be the optimal configuration. When the Ca2+ clock does play a role, there are a variety of ways that cytosolic Ca2+ will influence the periodicity of the clock, including influencing the rate of uptake of Ca2+ by SERCA and in turn the time taken to reach a critical level within the SR. Other longer term mechanisms for Ca2+ to influence the clock by actions on Ca2+-dependent enzymes are considered below. However, it is clear from the discussion above, and particularly from the observation that pacemaker activity can occur without a functional SR, that the Ca2+ clock is not essential for spontaneous pacemaker activity supported by a membrane clock.
If the Ca2+ handling properties of the SR can provide an important timing mechanism that under some conditions drives the generation of action potentials in the SA node, it might be speculated that defects in the function of the SR could lead to rhythm abnormalities.
An abnormality concerning ryanodine receptor Ca2+ release channels in the SR membrane supports such a possibility (Neco et al., 2012). This was studied in mice carrying the catecholaminergic polymorphic ventricular tachycardia–linked mutation of ryanodine receptor (RyR2R4496C). The SAN region was studied using confocal microscopy and the Ca2+ probe fluo-4 at room temperature. The SAN from RyR2R4496C mice showed slowed pacemaker activity and reduced response to catecholamines as compared to myocytes from WT. There were also frequent pauses in the generation of spontaneous action potentials, while the overall frequency of Ca2+ sparks/LCRs was increased. Experiments were also carried in SAN preparations loaded with rhod-2 to measure Ca2+ and di-4-ANEPPS to measure membrane potential. Pauses interspersed with trains of spontaneous events were observed for both the Ca2+ and voltage signals. The pauses were associated with unusually high cytosolic Ca2+ concentrations, and it was suggested that the pauses may have resulted from inactivation of the L-type Ca2+ channels.
A recent paper from the Federov laboratory (Glukhov et al., 2015) also shows that pacemaker activity is altered when Ca2+ handling is modified in mice lacking the SR protein calsequestrin (Casq2−/−). These mice showed bradycardia and beat to beat heart rate variability. SAN myocytes isolated from Casq2−/− mice showed an unstable rate with frequent pauses between “trains” of action potentials and the abnormal behavior was associated with abnormal Ca2+ release from the SR. In particular the pauses were accompanied by elevated levels of diastolic Ca2+ concentration (Glukhov et al., 2015). The effects of calsequestrin lack are complex reflecting the central role of calsequestrin not just as a Ca2+ buffer, but as a protein that together with junction and triadin, appears to regulate Ca2+ release via ryanodine receptor releases channels in the SR membrane. The responses to isoprenaline were also abnormal for both the chronotropic effect in whole animals and for the change in rate in isolated SAN myocytes. The authors proposed that “the observed pauses in between rhythmic Ca2+ transients in Casq2−/− SAN cells are caused by excessive diastolic SR Ca2+ release in turn resulting in (i) inhibition of the SAN upstroke ICaL current from partially inactivated L-type Ca2+ channels, and/or (ii) decrease in the SR Ca2+ content below a threshold level required for the generation of spontaneous SR Ca2+ releases.”
Another important point in this context is a possible parallel between a Ca2+ clock in pacemaker tissue and ventricular arrhythmias in which Ca2+ release from the SR is proposed to occur when the Ca2+ concentration within the SR reaches a specific level detected by a “calcium sensing gate” of the ryanodine receptor (RyR2). This “store overload-induced Ca2+ release” (SOICR) is modified in ventricular myocytes when a point mutation is introduced in the proposed calcium sensing gate (Chen et al., 2014) [and see (Sitsapesan and Williams, 1994, 1997) for evidence concerning the regulation of RyR2 gating by SR luminal Ca2+]. Carvedilol and a structural analog which lacks ability to block β-adrenoceptors both block SOICR (Zhou et al., 2011; Maruyama et al., 2013). The possible similarity between a pacemaker Ca2+ clock and SOICR has received support from another paper involving the same authors in experiments showing that the carvedilol analog and ryanodine both reduced spontaneous rate and also reduced the positive chronotropic effect of isoprenaline (Shinohara et al., 2014).
Taken together, the observations above show very clearly that pacemaker activity is disrupted when Ca2+ handling by the SR is abnormal and demonstrate that at least under some conditions the SR can play a central role.
Most of the pacemaker mechanisms described above play a role over times of seconds or less. However, there are important effects of Ca2+ that may take much longer to take effect, including activation of a variety of Ca2+ dependent enzymes.
An important Ca2+-dependent enzyme for which there is very convincing evidence concerning a role during pacemaking is CaMKII. Evidence using selective drugs supports an important role for CaMKII in maintaining and regulating pacemaker activity in the rabbit SA node (Vinogradova et al., 2000). It was emphasized that CaMKII has properties to function as a “frequency detector” and is therefore well suited to the regulation of pacemaker activity. The CaMKII inhibitor KN93 (but not the inactive analog KN92) substantially slowed or even stopped pacemaker activity. Similar observations were made using the peptide inhibitor AIP. The protein targets for CaMKII activated by Ca2+ were shown to include L-type Ca2+ channels. Phospholamban regulating the uptake of Ca2+ into the SR is another important target. Observations concerning the effects of KN93, KN92, and autocamtide inhibitor peptide (AIP) on spontaneous action potentials in guinea pig SAN myocytes (Xie et al., 2015) were broadly similar to those in the rabbit.
Genetic approaches also provide evidence for the influence of CaMKII on pacemaking at least under stress conditions. One interesting approach is to overexpress an inhibitor peptide for CaMKII (Vinogradova et al., 2000; Chen et al., 2009; Wu et al., 2009; Gao et al., 2011). A “control” is also provided by overexpression of a modified inhibitor protein that lacks ability to suppress CaMKII. In these experiments overexpression of the peptide inhibitor for CaMKII in mouse had little or no effect on resting heart rate (for experiments in vivo, in isolated Langendorff perfused hearts and in isolated SA node myocytes), but suppression of CaMKII activity by the peptide did cause a substantial reduction of the positive chronotropic response to isoprenaline. The responses that were reduced by inhibition of CaMKII included the amount of Ca2+ loaded in to the SR, the frequency of Ca2+ sparks/LCRs and the slope of the diastolic potential. However, interestingly the enhancement of L-type Ca2+ current amplitude by isoprenaline was maintained when CaMKII activity was suppressed by the inhibitor peptide. Another mouse model also showed little or no effect of CaMKII knockout on resting rate (Wu et al., 2009) but the positive chronotropic response to isoprenaline was again reduced (and indeed even a slowing of heart rate was seen in the CaMKII KO mice exposed to isoprenaline). These observations support a role for CaMKII in making a major contribution to the “fight or flight” mechanisms in which pacemaker activity is speeded up by adrenoceptor mediated cell signaling. It is clear that CaMKII is a Ca2+ stimulated enzyme that can influence pacemaker activity, even if the role of this enzyme under resting conditions remains to be fully established. The importance of CaMKII for pacemaker activity in the SA node has been recently reviewed (Wu and Anderson, 2014).
There are important differences between ventricular myocytes and those in the atria and SA node in terms of the subtypes of adenylyl cyclases, and of the basal activity of these enzymes. In particular it has been shown that actions of ACh at rest in the atria are consistent with inhibition of ongoing basal activity of ACs, while in the ventricle inhibitory actions of ACh are readily detected when adenylyl cyclases are stimulated by aderenoceptor activation but not in the absence of this stimulation (Fischmeister and Hartzell, 1986; Hescheler et al., 1986; Fischmeister and Shrier, 1989; Petit-Jacques et al., 1993). One possibility in the context of the importance of Ca2+ for pacemaking mechanisms is the possibility that Ca2+-sensitive adenylyl cyclases are preferentially expressed in the atria and SA node as compared with the ventricle. Evidence that I(f) is regulated by a Ca2+-calmodulin dependent mechanism that is not CaMKII was presented by Rigg et al. (2003) who also speculated that Ca2+ regulated adenylyl cyclase could provide such a mechanism. Application of the Ca2+ chelator BAPTA reduced I(f) as did calmodulin inhibitors including W7. Although W7 is known to have non-specific effects (Chatelier et al., 2005), these seem unlikely to be solely responsible for the observations since effects of BAPTA on I(f) were no longer seen in the presence of W7. More direct evidence for the presence of Ca2+-stimulated adenylyl cyclases was presented by Mattick et al. (2007). The Ca2+-stimulated adenylyl cyclase AC1 was shown to be expressed in the SAN by RT-PCR methods to detect mRNA as well as by immunoblotting with a specific antibody. Confocal microscopy also showed staining with the specific antibody in SAN myocytes. In addition to AC1, another Ca2+-stimulated adenylyl cyclase AC8, was detected by immunocytochemistry in the SAN myocytes. Functional evidence using BAPTA and selective inhibitors was also presented to show the importance of this pathway for regulating I(f) although it was also pointed out that such a pathway might well have additional effects via PKA and phosphorylation of additional protein targets. Younes et al. (2008) provided further evidence using RT-PCR for the presence of Ca2+-stimulated AC1 and AC8, in addition to AC types 2, 5, and 6 in SAN myocytes. These experiments also demonstrated the functional importance of the high basal activity of adenylyl cyclase in SA node myocytes for the regulation of a variety of proteins phosphorylated by PKA.
In addition, Kryukova et al. (2012) carried out experiments in which AC1 or AC6 were co-expressed with HCN2 in neonatal rat ventricular myocytes that lack normally AC1, and concluded from their observations that AC1 could play an important Ca2+-dependent role in regulating HCN2.
The importance of the Ca2+ stimulated adenylyl cyclases (AC1 and/or AC8) for the regulation of L-type Ca2+ currents has recently been demonstrated in atrial myocytes, and the observations may have relevance to SAN function (Collins and Terrar, 2012). In these experiments on atrial myocytes it was observed that buffering of cytosolic Ca2+ with BAPTA reduced Ca2+ current amplitude, as did inhibition of Ca2+ release from the SR using ryanodine and thapsigargin. Interestingly, the effects of BAPTA were prevented by inclusion of cAMP in the patch pipette. The observations are therefore consistent with a role for Ca2+ (including those released from the SR) in stimulating adenylyl cyclases and regulating the magnitude of L-type Ca2+ currents. It is speculated that similar mechanisms may operate in the SA node.
A variety of other Ca2+ dependent enzymes have the potential to influence pacemaker activity but have not yet been studied in detail. These include phophodiesterases, phosphatases and P21-activated kinase (PAK) enzymes that regulate phosphatases (Ke et al., 2007; Chen et al., 2014; Wang et al., 2014). There may also be effects of Ca2+ on NO synthase(s) in SAN myocytes since evidence supports actions of NO donors on pacemaker function (e.g., Han et al., 1995; Yoo et al., 1998; Musialek et al., 2000).
The importance of voltage-gated potassium channels, not only for repolarization of the action potential but also as a contributor to pacemaker depolarization as a consequence of time-dependent de-activation, was emphasized at the start of this review (and see Clark et al., 2004). Evidence that the contribution of potassium channels is subject to regulation by cytosolic Ca2+ was provided by Zaza et al. (1997) using the “action potential” clamp methods in rabbit SA node mentioned above in relation to I(f). Zaza et al. (1997) concluded that Ca2+ influx during the pacemaker cycle also increases a potassium conductance. This potassium conductance could arise from a single channel component or multiple components.
Xie et al. (2015) have recently presented evidence that the slow component of the delayed rectifier current (IKs) is regulated by a Ca2+-dependent process thought to be CaMKII in guinea-pig SA node. This current was found to be decreased when the cytosolic Ca2+ concentration was reduced from 10−7 to 10−10 M (buffered by EGTA in a ruptured patch conditions). When the cytosolic Ca2+ concentration was maintained at 10−7 M, addition of calmodulin via the patch pipette increased the current. Inhibitors of CaMKII (autocamtide-2 inhibitor peptide and the less selective KN-93) substantially reduced the current, while in the presence of KN-93 there was no effect of calmodulin addition. Taken together these observations provide convincing evidence that IKs is modulated by cytosolic Ca2+ and that CaMKII plays an important role in this modulation.
Might other channel components be involved? The action potential clamp experiments of Zaza et al. mentioned above were carried out in rabbit SA node, and in this species the rapidly activating IKr is thought to provide the major voltage-gated potassium current pathway (Verheijck et al., 1995; Lei and Brown, 1996). Although under the conditions of the action potential clamp experiments, a Ca2+ dependence of IKr was not detected, applications of nifedipine to block Ca2+ entry may have been too brief (less than 10s in Figure 2 of Zaza et al., 1997 while the full effect of Ca2+ on an enzyme pathway might take a period of minutes to develop). More evidence against Ca2+ regulation of IKr is provided by Wu and Anderson (2014). However, it has been shown that IKr in guinea pig ventricular cells can be influenced by cytosolic Ca2+ (Heath and Terrar, 2000) in a complex pathway perhaps involving PKC that depends on Ca2+ entry via L-type Ca2+ channels leading changes in the amplitude and rectification/inactivation of the channel. It therefore seems premature to exclude an effect of Ca2+ on IKr in SA node at this stage.
An interesting additional point in the debate on Ca2+ regulation of potassium conductance in the SA node concerns the possible contribution of the Ca2+ activated potassium channel BK (Imlach et al., 2010; Lai et al., 2014) and SK (Chen et al., 2013).
As was mentioned above, membrane and Ca2+ clocks might normally be synchronized, and consequently CICR could occur at (or very close to) a time when “global” spontaneous Ca2+ release would also have occurred. However, it is also possible that CICR can, and perhaps often does, occur without the accompaniment of spontaneous Ca2+ release. In this case Ca2+ released by CICR from the SR would be expected to drive a significant component of NCX that is independent of the Ca2+ clock.
Ca2+ released from the SR by CICR that is independent of the Ca2+ clock could also drive Ca2+-dependent enzymes. These include CaMKII, AC1 and AC8 as described above. A role for SR-released Ca2+ (sensitive to ryanodine and thapsigargin) leading to increased L-type Ca2+ currents by a mechanism involving Ca2+-stimulated adenylyl cyclases was recently demonstrated in atrial myocytes (Collins and Terrar, 2012), and it seems very likely that a similar mechanism operates to control pacemaker activity in SAN myocytes. Such a Ca2+-induced stimulation of adenylyl cyclases that is at least partly dependent on Ca2+ released from the SR by CICR could also contribute to stimulation of I(f) (by elevation of cAMP) and other ion channels such as I(st) and voltage-gated potassium channels (regulated by PKA).
Voltage-gated L-type Ca2+ channels are a major component of the membrane oscillator providing explosive positive feedback for the upstroke of the action potential which in turn initiates repolarization as a consequence of activation of voltage-gated K+ channels. The positive feedback for activation of L-type Ca2+ channels underlies the fast upstroke and rapid spread of the action potential that synchronizes Ca2+ entry and CICR across the cell. The Ca2+ entry via L-type Ca2+ channels is a major determinant of the degree of Ca2+ loading of the SR. The action potential signal is essential for conduction of the pacemaker activity to neighboring myocytes. In addition, it appears that current through CaV1.3 channels can contribute to the late stage of diastolic depolarization (Zhang et al., 2002; Mangoni et al., 2003, 2006a; Christel et al., 2012). It is also important to note that Ca2+ ions entering the cell via L-type Ca2+ channels, at any stage during the cardiac cycle but particularly during the rapid upstroke, will drive NCX and cause the extensive secondary effects discussed elsewhere in this review, including enhancement of the activities of PKA and CaMKII (see later).
At the present time, it is probably only possible to investigate the effects of Ca2+ chelators in single cells, since in multicellular preparations it is difficult to be sure that uptake of Ca2+ chelator is complete. It is recognized that isolated myocytes might behave differently from those in the intact myocardium for a variety of reasons including different mechanical influences, lack of paracrine influences from neighboring cells and lack of electrical connections to adjacent myocytes.
In the case of isolated myocytes, Zaza et al. (1997) reported that “under buffering of intracellular Ca2+ (ruptured patch, 10 mM intracellular EGTA), spontaneous activity became unstable and ceased in the majority of cells several minutes after achieving the whole-cell configuration.” Another approach to apply the Ca2+ chelator is to expose the cells to the acetoxymethyl (AM) ester of the Ca2+ chelator applied in the extracellular solution. The membrane permeant ester enters the cell and the active chelator is liberated following the action of esterases in the cytosol. Application of EGTA-AM or BAPTA-AM in this way causes cessation of beating (e.g., Sanders et al., 2006), although it is possible that unspecific effects such as blockade of IKrcould contribute to these actions (Tang et al., 2007). Loading of SAN myocytes with AM esters of Ca2+ probes such as indo-1 can also lead to cessation of activity, and it was suggested that this could arise from Ca2+ chelation by the probe (Rigg et al., 2000). One important exception concerning effects of BAPTA on pacemaker activity is the paper by Himeno et al. (2011) showing that pacemaker activity in the form of action potentials can continue while contraction of the cells is suppressed by application of cytosolic BAPTA. However, these authors also noted that “action potential generation became irregular or stopped completely ~5 min after the application of BAPTA in our experiments.” What is the significance of the observations very soon after breakthrough? In support of their contention that buffering is effective even 20 s after breakthrough the authors show a prolongation of action potentials (thought to arise from slowing of inactivation of Ca2+ currents) that would be consistent with the chelating effects of BAPTA. Maltsev et al. (Maltsev et al., 2011; Yaniv et al., 2011) have challenged the significance of these observations with the suggestion that breakthrough from perforated patch to “whole” cell patch weakens the “seal” around the electrode noting that such a weakened seal could allow a “leakage” current that could substitute for a physiological current that is blocked by BAPTA. In other words they maintain that spontaneous activity can continue with little change after chelation of BAPTA since leakage current via a weakened “seal” around the patch pipette can replace the depolarizing influence that would normally be supplied by NCX (or any other Ca2+-sensitive “background” current that occurs when Ca2+ is maintained at a physiological level). In support of this suggestion they note that in Figure 5 of Himeno et al. there is an upward deflection on the contraction trace following breakthrough that would be consistent with a small developing contraction associated with entry of Ca2+ from the extracellular solution via the leaky seal into the cytosol. Contribution of leakage current around a seal in sustaining pacemaker activity has been considered by other authors, and can make an important contribution under some conditions (Denyer and Brown, 1990), though these authors concluded from observations comparing the effects of Cs+ under perforated patch conditions and in unpatched cells that the effects of the leakage current can be negligibly small if the seal is sufficiently robust.
This point is addressed in another paper in this issue of Frontiers in Physiology (Capel and Terrar, 2015). It is reported that under the conditions of these experiments, rupture of the membrane beneath a patch pipette to apply BAPTA to the cytosol can cause rapid cessation of activity. When conventional whole-cell patch recording was used with 10 mM BAPTA in the patch pipette solution, this cessation of spontaneous activity was almost immediate (less than 10 s). When the same rupture of the membrane beneath the pipette was made without BAPTA present the spontaneous activity appeared not to be reduced throughout the period of recording (approximately 3 min). When action potentials were first recorded with permeabilised patch (with amphotericin in the pipette to approximate the conditions of Himeno et al.) followed by rupture of the patch membrane, the disruption of spontaneous activity after cytosolic application of 10 mM BAPTA was still clearly observed but slower to develop (perhaps because of effects of amphotericin or its solvent). The speed of disruption depended on the concentration of BAPTA, progressively decreasing over the range 10, 1, and 0.1 mM BAPTA.
The many effects of Ca2+ on the mechanisms underlying pacemaker activity and the time windows within which they are thought to act are summarized in Figure 4.
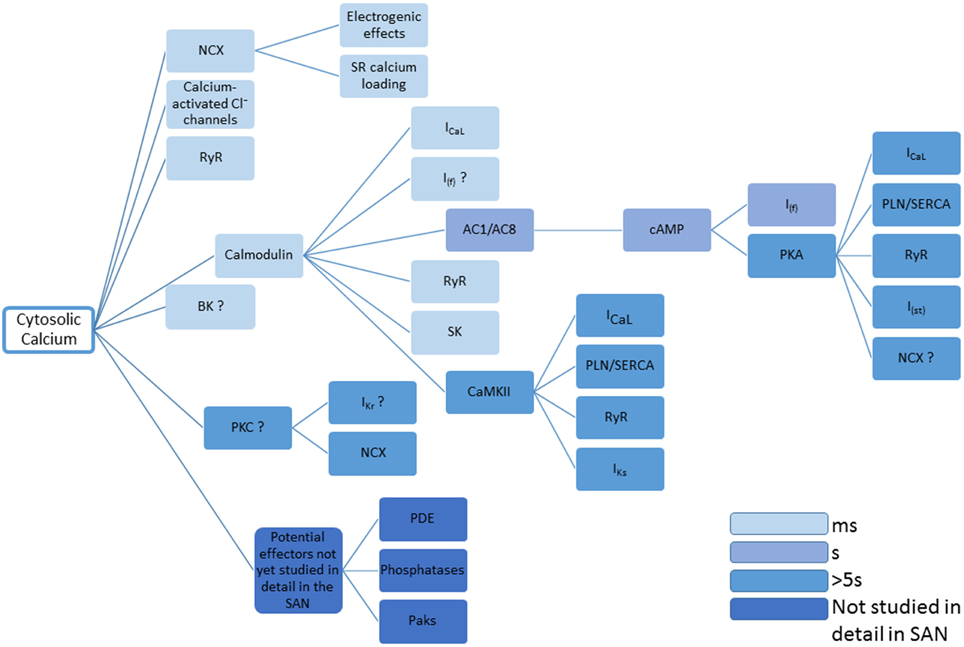
Figure 4. This figure summarizes the many disparate roles for Ca2+ in controlling the activity of proteins that determine pacemaker activity. Question marks represent cases in which there is currently limited evidence, and references to support the actions shown can be found in the main text. The entries are color coded to give an approximate indication concerning time taken for changes in cytosolic Ca2+ to take effect. The figure concentrates on the effects of cytosolic Ca2+, though it will be the case that Ca2+ within the SR will have additional effects, for example through calsequestrin or RyR2 (either via a possible Ca2+ sensor within the RyR structure, or through inactivation of RyRs that might be influenced by SR Ca2+). Also omitted are the possible roles of NO produced via Ca2+-stimulated nNOS and eNOS. Small molecules controlling Ca2+ signaling (e.g., IP3, see Ju et al., 2011) are also not included.
Evidence concerning the mechanisms underlying the positive chronotropic actions of isoprenaline have been recently discussed with particular reference to computer models of pacemaker activity (Zhang et al., 2012). In the context of the review presented here, the focus will be on the importance of the many Ca2+-dependent mechanisms that are enhanced during β-adrenoceptor function. The importance of the SR in contributing to the positive chronotropic actions of isoprenaline was first demonstrated by Rigg et al. (2000) in experiments on both intact SA node and isolated SAN myocytes. After exposure to ryanodine, the log(concentration)-response curve for the effects of isoprenaline on intact node was depressed with a reduced slope and maximum effect.
Often effects of isoprenaline (isoproterenol) acting on β-adrenoceptors and of sympathetic nerve stimulation are treated as equivalent, although some have argued that this is over-simplistic (Bramich et al., 1993; Choate et al., 1993; Bramich and Cousins, 1999). There seems justification in this point of view, though a detailed discussion is beyond the scope of this review except to point out that the effects of nerve released noradrenaline on α and β adrenoceptors, as well as the effects of co-transmitters such as ATP and peptides (e.g., Herring, 2014) might have additional effects on Ca2+-dependent pathways.
A major effect of β-adrenoceptor stimulation by isoprenaline is to increase Ca2+ currents. Many authors agree that this effect is a major factor in the positive chronotropic effect (Brown et al., 1975; Zhang et al., 2012), and an interesting possibility is that there may be important effects of β-adrenoceptor stimulation to increase current through CaV1.3 channels that appear to contribute to the later stages of diastolic depolarization (Zhang et al., 2002; Mangoni et al., 2003, 2006a). However, as mentioned above, the effects of β-adrenoceptor stimulation cannot result from the direct effects on the upstroke of the action potential alone as a consequence of increased L-type Ca2+ currents. This is because the upstroke makes up only about 10% or less of the cardiac cycle, and so even if the upstroke became infinitely fast (in other words effectively vertical), the pacemaker period would only be reduced to 90% of the value before the increase in Ca2+ current (or a rate increase of 1/0.9 giving close to a 10% increase in rate). The increase in upstroke velocity would still be functionally useful in increasing the rate of conduction of the pacemaker message in the form of the action potential to surrounding tissue. However, in terms of the rate change it seems very likely that an important aspect of the β-adrenoceptor mediated increase in Ca2+ current is to increase entry of Ca2+ which in turn influences many of the Ca2+-stimulated processes that have been described above.
The most obvious of these is NCX. Broadly if Ca2+ entry were to double, the cell would need, in the steady state, to remove twice as much Ca2+ and if the major mechanism for achieving this were NCX in its normal 3:1 mode then NCX would also double (and the total charge moved by NCX would remain approximately half that of the total Ca2+ entry through channels). Although this seems very likely to be the case, one additional point concerns another effect of β-adrenoceptor stimulation via cAMP and PKA. This is phosphorylation of phospholamban which will lead to increased Ca2+ uptake by SERCA into the SR. This might lead to a greater fraction of the Ca2+ to be taken up by the SR (or at least a greater rate of uptake into the SR depending on how quickly the Ca2+ is released), but in the steady state increased Ca2+ entry through the surface membrane must be accompanied by increased extrusion. In terms of the Ca2+ clock, the kinetics of Ca2+ uptake are expected to have a profound effect, and this might be one factor underlying the reduced effect of isoprenaline after ryanodine (Rigg et al., 2000), though other factors might also contribute as discussed below.
Consistent with the above suggestions of increased Ca2+ transients in single SAN myocytes, Rigg et al. (2000) recorded substantial increases in the amplitude as well as decay of Ca2+ transients recorded with the Ca2+ probe indo-1 in the presence of isoprenaline (see Figure 5). The effects of autonomic transmitters on Ca2+ transients and action potentials in single SAN myocytes have also been reported by Vinogradova et al. (2002a) and van Borren et al. (2010).
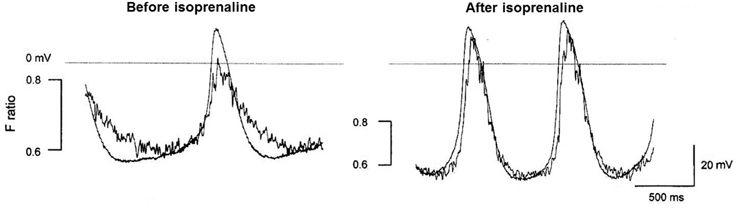
Figure 5. This figure shows the Ca2+ transient reported by indo-1 (fluorescence ratio with emission at 405 and 485 nm) accompanying spontaneous action potentials in the same cell in the presence and absence of isoprenaline. It can be seen that the increase in rate of occurrence of spontaneous action potentials caused by isoprenaline was accompanied by an increased amplitude of the Ca2+ transient, an increased rate of rise of the Ca2+ transient and an increased rate of decay of the transient. The same limitations outlined in Figure 2 apply here concerning a possible lag in the response of the Ca2+ probe to a rise in Ca2+ and possible slowing of the time course (though clearly any such delays do not prevent the detection of the speedier decay of the Ca2+ transient in the presence of isoprenaline). The experimental traces are from Rigg et al. (2000).
The additional Ca2+ both entering through L-type channels, and released from the SR (in individual releases and even more as a time average over many beats) could drive many Ca2+-dependent processes, such as Ca2+-stimulated enzymes and these are expected to contribute to the positive chronotropic effect. The Ca2+-stimulated enzymes include CaMKII as discussed by Wu and Anderson (2014), as well as the Ca2+-stimulated adenylyl cyclases, AC1 and AC8. As discussed above, Ca2+-activation of these enzymes is expected to increase I(f), I(st), IKs, IKr and possibly NCX, as well as ICaL.
Effects of acetylcholine include both inhibition of adenylyl cyclase and opening of IKACh channels. The effects on I KACh channels are blocked by tertiapin Q. During parasympathetic nerve stimulation there is a rapid phase of slowing that is sensitive to tertiapin Q and therefore IKACh (Bolter and Turner, 2009). The additional effects of ACh to inhibit adenylyl cyclase are in many respects the opposite of β-adrenoceptor stimulation discussed above. Consequently the inhibition of L-type Ca2+ current will again be very important, not so much for direct effect on the rate of rise of the action potential, but for the consequences of reduced Ca2+ entry on the many Ca2+-dependent processes discussed above. In the context of Ca2+-dependent mechanisms and in particular the Ca2+ clock Lyashkov et al. (2009) show that ACh reduced the number and size of LCRs, and these effects were correlated with the reduction in beating rate. ACh also reduced cAMP levels and the log(concentration)-response curves for reduction in rate and inhibition of phosphorylation of phospholamban were very similar. The authors conclude that there is a tight coupling between suppression of PKA-dependent Ca2+ signaling, IKACh activation and reduction of spontaneous rate. van Borren et al. (2010) also investigate the effects of ACh on Ca2+ transient, cAMP production and pacemaker frequency. They found that when I(f) was inhibited by 2 mM Cs+, and IKACh was inhibited by tertiapin Q, 1 μM ACh was still able to reduce pacemaker frequency by 72%. Under these conditions, there was a good correlation between the reduction in beating rate and the amplitude of the Ca2+ transient. In addition ryanodine and exposure to BAPTA-AM both facilitated ACh mediated slowing. They also showed that inhibition of the Ca2+ transient by ryanodine (3 μM) or BAPTA-AM (25 μM) exaggerated the ACh-mediated inhibition of cAMP content, consistent with the proposal that Ca2+ affects cAMP production in SAN cells. There may be additional effects of ACh on phosphodiesterases (e.g., Han et al., 1995, for a review of PDE subtypes in SAN see Hua et al., 2012) and phosphatases (Ke et al., 2007).
The above discussion leads to the view that pacemaker activity in the heart normally requires L-type Ca2+ channels and at least one type of voltage-gated potassium channel, while the evidence seems to support the hypothesis that NCX is also essential whether or not it plays a role in determining the poorly understood “background” conductance pathway. The influence of this background conductance pathway allows de-activation of K+ channels to cause depolarization toward the threshold for voltage-gated Ca2+ channels, and future identification of this pathway will be important for our understanding of pacemaker mechanisms. Pacemaker depolarization is enhanced by activation of I(f). The idea that the I(f) pathway is essential for pacemaker depolarization cannot be completely excluded because of the uncertainties concerning whether the pathway is totally suppressed in experiments using drug blockade or techniques of genetic modification, but it seems likely that I(f) is more an important modulator than an essential component of the timing mechanism. Ca2+ sparks/LCRs can sometimes contribute to the later stages of pacemaker depolarization preceding the action potential but are not essential for pacemaking. However, “global” properties of Ca2+ uptake and release by the SR across the whole myocyte (or at least a substantial fraction of the SR) can give rise to a “Ca2+ clock” (as a consequence of the SR filling with Ca2+ to a critical level or from other kinetic properties of the proteins involved in uptake and release of Ca2+). The “Ca2+ clock” arising in this way frequently plays a modulatory rather than essential role, though there may be conditions in which it is absolutely necessary to re-initiate spontaneous activity if this were to stop. This would mean that Ca2+ released from the SR and driving electrogenic NCX may be essential to re-start spontaneous activity under particular conditions. Although a Ca2+ clock is not essential for pacemaker activity, it seems probable that under normal conditions there will be a cooperative interaction between membrane and Ca2+ clocks that has been referred to as a coupled clock mechanism. Under pathological conditions when the Ca2+ clock is not synchronized with the membrane clock the Ca2+ clock can lead to disturbed pacemaker rhythms. In normal physiology, Ca2+ is thought to drive a depolarizing influence of NCX throughout the cardiac cycle, including during the most negative potential of the pacemaker myocytes, at least under most conditions when the cytosolic Ca2+ concentration remains substantially higher than 100 nM. Ca2+ also plays important roles in maintaining and regulating pacemaker activity by activating a variety of Ca2+-dependent enzymes including CaMKII, AC1, and AC8. Ca2+ seems to exert a direct or indirect influence on most if not all of the proteins providing ionic pathways in the surface membrane.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Pacemaking work in the DAT laboratory has been supported for many years by the British Heart Foundation and Wellcome Trust. RAC was supported by a British Heart Foundation Graduate Studentship and, thereafter, a British Heart Foundation research grant.
Baruscotti, M., Bucchi, A., and Difrancesco, D. (2005). Physiology and pharmacology of the cardiac pacemaker (“funny”) current. Pharmacol. Ther. 107, 59–79. doi: 10.1016/j.pharmthera.2005.01.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baruscotti, M., and Difrancesco, D. (2004). Pacemaker channels. Ann. N.Y. Acad. Sci. 1015, 111–121. doi: 10.1196/annals.1302.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Berridge, M. J. (2008). Smooth muscle cell calcium activation mechanisms. J. Physiol. 586, 5047–5061. doi: 10.1113/jphysiol.2008.160440
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Biel, M., Schneider, A., and Wahl, C. (2002). Cardiac HCN channels: structure, function, and modulation. Trends Cardiovasc. Med. 12, 206–212. doi: 10.1016/S1050-1738(02)00162-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Biel, M., Wahl-schott, C., Michalakis, S., and Zong, X. (2009). Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847–885. doi: 10.1152/physrev.00029.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bogdanov, K. Y., Maltsev, V. A., Vinogradova, T. M., Lyashkov, A. E., Spurgeon, H. A., Stern, M. D., et al. (2006). Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ. Res. 99, 979–987. doi: 10.1161/01.RES.0000247933.66532.0b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bogdanov, K. Y., Vinogradova, T. M., and Lakatta, E. G. (2001). Sinoatrial nodal cell ryanodine receptor and Na(+)-Ca(2+) exchanger: molecular partners in pacemaker regulation. Circ. Res. 88, 1254–1258. doi: 10.1161/hh1201.092095
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bolter, C. P., and Turner, M. J. (2009). Tertiapin-Q removes a large and rapidly acting component of vagal slowing of the guinea-pig cardiac pacemaker. Auton. Neurosci. 150, 76–81. doi: 10.1016/j.autneu.2009.05.244
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
BoSmith, R. E., Briggs, I., and Sturgess, N. C. (1993). Inhibitory actions of ZENECA ZD7288 on whole-cell hyperpolarization activated inward current (If) in guinea-pig dissociated sinoatrial node cells. Br. J. Pharmacol. 110, 343–349. doi: 10.1111/j.1476-5381.1993.tb13815.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bramich, N. J., Brock, J. A., Edwards, F. R., and Hirst, G. D. (1993). Responses to sympathetic nerve stimulation of the sinus venosus of the toad. J. Physiol. 461, 403–430. doi: 10.1113/jphysiol.1993.sp019520
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bramich, N. J., and Cousins, H. M. (1999). Effects of sympathetic nerve stimulation on membrane potential, [Ca2+]i, and force in the toad sinus venosus. Am. J. Physiol. 276, H115–H128.
Brown, H. F., McNaughton, P. A., Noble, D., and Noble, S. J. (1975). Adrenergic control of cardian pacemaker currents. Philos. Trans. R. Soc. Lond. B Biol. Sci. 270, 527–537. doi: 10.1098/rstb.1975.0029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cannell, M. B., Berlin, J. R., and Lederer, W. J. (1987). Intracellular calcium in cardiac myocytes: calcium transients measured using fluorescence imaging. Soc. Gen. Physiol. Ser. 42, 201–214.
Capel, R. A., and Terrar, D. A. (2015). Cytosolic calcium ions exert a major influence on the firing rate and maintenance of pacemaker activity in guinea-pig sinus node. Front. Physiol. 6:23. doi: 10.3389/fphys.2015.00023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chatelier, A., Renaudon, B., Bescond, J., El Chemaly, A., Demion, M., and Bois, P. (2005). Calmodulin antagonist W7 directly inhibits f-type current in rabbit sino-atrial cells. Eur. J. Pharmacol. 521, 29–33. doi: 10.1016/j.ejphar.2005.08.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, B., Wu, Y., Mohler, P. J., Anderson, M. E., and Song, L. S. (2009). Local control of Ca2+-induced Ca2+ release in mouse sinoatrial node cells. J. Mol. Cell. Cardiol. 47, 706–715. doi: 10.1016/j.yjmcc.2009.07.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, W., Wang, R., Chen, B., Zhong, X., Kong, H., Bai, Y., et al. (2014). The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat. Med. 20, 184–192. doi: 10.1038/nm.3440
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, W. T., Chen, Y. C., Lu, Y. Y., Kao, Y. H., Huang, J. H., Lin, Y. K., et al. (2013). Apamin modulates electrophysiological characteristics of the pulmonary vein and the Sinoatrial Node. Eur. J. Clin. Invest. 43, 957–963. doi: 10.1111/eci.12125
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cho, C. H., Kim, S. S., Jeong, M. J., Lee, C. O., and Shin, H. S. (2000). The Na+ -Ca2+ exchanger is essential for embryonic heart development in mice. Mol. Cells 10, 712–722. doi: 10.1007/s100590000034
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Choate, J. K., Edwards, F. R., Hirst, G. D., and O'Shea, J. E. (1993). Effects of sympathetic nerve stimulation on the sino-atrial node of the guinea-pig. J. Physiol. 471, 707–727. doi: 10.1113/jphysiol.1993.sp019924
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Christel, C. J., Cardona, N., Mesirca, P., Herrmann, S., Hofmann, F., Striessnig, J., et al. (2012). Distinct localization and modulation of Cav1.2 and Cav1.3 L-type Ca2+ channels in mouse sinoatrial node. J. Physiol. 590, 6327–6342. doi: 10.1113/jphysiol.2012.239954
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clark, R. B., Mangoni, M. E., Lueger, A., Couette, B., Nargeot, J., and Giles, W. R. (2004). A rapidly activating delayed rectifier K+ current regulates pacemaker activity in adult mouse sinoatrial node cells. Am. J. Physiol. Heart Circ. Physiol. 286, H1757–H1766. doi: 10.1152/ajpheart.00753.2003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Collins, T. P., and Terrar, D. A. (2012). Ca(2+)-stimulated adenylyl cyclases regulate the L-type Ca(2+) current in guinea-pig atrial myocytes. J. Physiol. 590, 1881–1893. doi: 10.1113/jphysiol.2011.227066
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Coppen, S. R., Kodama, I., Boyett, M. R., Dobrzynski, H., Takagishi, Y., Honjo, H., et al. (1999). Connexin45, a major connexin of the rabbit sinoatrial node, is co-expressed with connexin43 in a restricted zone at the nodal-crista terminalis border. J. Histochem. Cytochem. 47, 907–918. doi: 10.1177/002215549904700708
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Denyer, J. C., and Brown, H. F. (1990). Pacemaking in rabbit isolated sino-atrial node cells during Cs+ block of the hyperpolarization-activated current if. J. Physiol. 429, 401–409. doi: 10.1113/jphysiol.1990.sp018264
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DiFrancesco, D. (2010). The role of the funny current in pacemaker activity. Circ. Res. 106, 434–446. doi: 10.1161/CIRCRESAHA.109.208041
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DiFrancesco, D., and Mangoni, M. (1994). Modulation of single hyperpolarization-activated channels (i(f)) by cAMP in the rabbit sino-atrial node. J. Physiol. 474, 473–482. doi: 10.1113/jphysiol.1994.sp020038
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DiFrancesco, D., and Noble, D. (2012a). The funny current has a major pacemaking role in the sinus node. Heart Rhythm 9, 299–301. doi: 10.1016/j.hrthm.2011.09.021
DiFrancesco, D., and Noble, D. (2012b). Rebuttal: “The funny current in the context of the coupled clock pacemaker cell system.” Heart Rhythm 9, 457–458. doi: 10.1016/j.hrthm.2011.09.023
Dobrzynski, H., Boyett, M. R., and Anderson, R. H. (2007). New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115, 1921–1932. doi: 10.1161/CIRCULATIONAHA.106.616011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fenske, S., Krause, S. C., Hassan, S. I., Becirovic, E., Auer, F., Bernard, R., et al. (2013). Sick sinus syndrome in HCN1-deficient mice. Circulation 128, 2585–2594. doi: 10.1161/CIRCULATIONAHA.113.003712
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fischmeister, R., and Hartzell, H. C. (1986). Mechanism of action of acetylcholine on calcium current in single cells from frog ventricle. J. Physiol. 376, 183–202. doi: 10.1113/jphysiol.1986.sp016148
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fischmeister, R., and Shrier, A. (1989). Interactive effects of isoprenaline, forskolin and acetylcholine on Ca2+ current in frog ventricular myocytes. J. Physiol. 417, 213–239. doi: 10.1113/jphysiol.1989.sp017798
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, Z., Rasmussen, T. P., Li, Y., Kutschke, W., Koval, O. M., Wu, Y., et al. (2013). Genetic inhibition of Na+-Ca2+ exchanger current disables fight or flight sinoatrial node activity without affecting resting heart rate. Circ. Res. 112, 309–317. doi: 10.1161/CIRCRESAHA.111.300193
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, Z., Singh, M. V., Hall, D. D., Koval, O. M., Luczak, E. D., Joiner, M. L., et al. (2011). Catecholamine-independent heart rate increases require Ca2+/calmodulin-dependent protein kinase II. Circ. Arrhythm. Electrophysiol. 4, 379–387. doi: 10.1161/CIRCEP.110.961771
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Glukhov, A. V., Kalyanasundaram, A., Lou, Q., Hage, L. T., Hansen, B. J., Belevych, A. E., et al. (2015). Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex. Eur. Heart J. 36, 686–697. doi: 10.1093/eurheartj/eht452
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Groenke, S., Larson, E. D., Alber, S., Zhang, R., Lamp, S. T., Ren, X., et al. (2013). Complete atrial-specific knockout of sodium-calcium exchange eliminates sinoatrial node pacemaker activity. PLoS ONE 8:e81633. doi: 10.1371/journal.pone.0081633
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hagiwara, N., and Irisawa, H. (1989). Modulation by intracellular Ca2+ of the hyperpolarization-activated inward current in rabbit single sino-atrial node cells. J. Physiol. 409, 121–141. doi: 10.1113/jphysiol.1989.sp017488
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hagiwara, N., Irisawa, H., and Kameyama, M. (1998). Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J. Physiol. 395, 233–253.
Hagiwara, N., Irisawa, H., Kasanuki, H., and Hosoda, S. (1992). Background current in sino-atrial node cells of the rabbit heart. J. Physiol. 448, 53–72. doi: 10.1113/jphysiol.1992.sp019029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Han, X., Shimoni, Y., and Giles, W. R. (1995). A cellular mechanism for nitric oxide-mediated cholinergic control of mammalian heart rate. J. Gen. Physiol. 106, 45–65. doi: 10.1085/jgp.106.1.45
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hauswirth, O., Noble, D., and Tsien, R. W. (1968). Adrenaline: mechanism of action on the pacemaker potential in cardiac Purkinje fibers. Science 162, 916–917. doi: 10.1126/science.162.3856.916
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
He, C., Chen, F., Li, B., and Hu, Z. (2014). Neurophysiology of HCN channels: from cellular functions to multiple regulations. Prog. Neurobiol. 112, 1–23. doi: 10.1016/j.pneurobio.2013.10.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Heath, B. M., and Terrar, D. A. (2000). Protein kinase C enhances the rapidly activating delayed rectifier potassium current, IKr, through a reduction in C-type inactivation in guinea-pig ventricular myocytes. J. Physiol. 522(Pt 3), 391–402. doi: 10.1111/j.1469-7793.2000.t01-2-00391.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Herring, N. (2014). Autonomic control of the heart: going beyond the classical neurotransmitters. Exp. Physiol. doi: 10.1113/expphysiol.2014.080184. [Epub ahead of print].
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Herrmann, S., Lipp, P., Wiesen, K., Stieber, J., Nguyen, H., Kaiser, E., et al. (2013). The cardiac sodium-calcium exchanger NCX1 is a key player in the initiation and maintenance of a stable heart rhythm. Cardiovasc. Res. 99, 780–788. doi: 10.1093/cvr/cvt154
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hescheler, J., Kameyama, M., and Trautwein, W. (1986). On the mechanism of muscarinic inhibition of the cardiac Ca current. Pflugers Arch. 407, 182–189. doi: 10.1007/BF00580674
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Himeno, Y., Toyoda, F., Satoh, H., Amano, A., Cha, C. Y., Matsuura, H., et al. (2011). Minor contribution of cytosolic Ca2+ transients to the pacemaker rhythm in guinea pig sinoatrial node cells. Am. J. Physiol. Heart Circ. Physiol. 300, H251–H261. doi: 10.1152/ajpheart.00764.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hof, T., Simard, C., Rouet, R., Salle, L., and Guinamard, R. (2013). Implication of the TRPM4 nonselective cation channel in mammalian sinus rhythm. Heart Rhythm 10, 1683–1689. doi: 10.1016/j.hrthm.2013.08.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hua, R., Adamczyk, A., Robbins, C., Ray, G., and Rose, R. A. (2012). Distinct patterns of constitutive phosphodiesterase activity in mouse sinoatrial node and atrial myocardium. PLoS ONE 7:e47652. doi: 10.1371/journal.pone.0047652
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huang, Z. M., Prasad, C., Britton, F. C., Ye, L. L., Hatton, W. J., and Duan, D. (2009). Functional role of CLC-2 chloride inward rectifier channels in cardiac sinoatrial nodal pacemaker cells. J. Mol. Cell. Cardiol. 47, 121–132. doi: 10.1016/j.yjmcc.2009.04.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Huser, J., Blatter, L. A., and Lipsius, S. L. (2000). Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J. Physiol. 524(Pt 2), 415–422. doi: 10.1111/j.1469-7793.2000.00415.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imlach, W. L., Finch, S. C., Miller, J. H., Meredith, A. L., and Dalziel, J. E. (2010). A role for BK channels in heart rate regulation in rodents. PLoS ONE 5:e8698. doi: 10.1371/journal.pone.0008698
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imtiaz, M. S. (2012). Calcium oscillations and pacemaking. Adv. Exp. Med. Biol. 740, 511–520. doi: 10.1007/978-94-007-2888-2_22
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imtiaz, M. S., Katnik, C. P., Smith, D. W., and van Helden, D. F. (2006). Role of voltage-dependent modulation of store Ca2+ release in synchronization of Ca2+ oscillations. Biophys. J. 90, 1–23. doi: 10.1529/biophysj.104.058743
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imtiaz, M. S., von der Weid, P. Y., Laver, D. R., and van Helden, D. F. (2010). SR Ca2+ store refill–a key factor in cardiac pacemaking. J. Mol. Cell. Cardiol. 49, 412–426. doi: 10.1016/j.yjmcc.2010.03.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Imtiaz, M. S., Zhao, J., Hosaka, K., von der Weid, P. Y., Crowe, M., and van Helden, D. F. (2007). Pacemaking through Ca2+ stores interacting as coupled oscillators via membrane depolarization. Biophys. J. 92, 3843–3861. doi: 10.1529/biophysj.106.095687
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Irisawa, H. (1978). Comparative physiology of the cardiac pacemaker mechanism. Physiol. Rev. 58, 461–498.
Irisawa, H., Brown, H. F., and Giles, W. (1993). Cardiac pacemaking in the sinoatrial node. Physiol. Rev. 73, 197–227.
Ito, H., Ono, K., and Noma, A. (1994). Background conductance attributable to spontaneous opening of muscarinic K+ channels in rabbit sino-atrial node cells. J. Physiol. 476, 55–68.
Jackson, V. M., Rakovic, S., and Terrar, D. A. (2002). Effects of isoproterenol on calcium transients and calcium sparks in guinea-pig sino-atrial node cells imaged by confocal microscopy. Biophys. J. 82, 648A.
Ju, Y. K., and Allen, D. G. (1998). Intracellular calcium and Na+-Ca2+ exchange current in isolated toad pacemaker cells. J. Physiol. 508(Pt 1), 153–166. doi: 10.1111/j.1469-7793.1998.153br.x
Ju, Y. K., and Allen, D. G. (2007). Store-operated Ca2+ entry and TRPC expression; possible roles in cardiac pacemaker tissue. Heart Lung Circ. 16, 349–355. doi: 10.1016/j.hlc.2007.07.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ju, Y. K., Liu, J., Lee, B. H., Lai, D., Woodcock, E. A., Lei, M., et al. (2011). Distribution and functional role of inositol 1,4,5-trisphosphate receptors in mouse sinoatrial node. Circ. Res. 109, 848–857. doi: 10.1161/CIRCRESAHA.111.243824
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kang, T. M., and Hilgemann, D. W. (2004). Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature 427, 544–548. doi: 10.1038/nature02271
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ke, Y., Lei, M., Collins, T. P., Rakovic, S., Mattick, P. A., Yamasaki, M., et al. (2007). Regulation of L-type calcium channel and delayed rectifier potassium channel activity by p21-activated kinase-1 in guinea pig sinoatrial node pacemaker cells. Circ. Res. 100, 1317–1327. doi: 10.1161/01.RES.0000266742.51389.a4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kodama, I., Nikmaram, M. R., Boyett, M. R., Suzuki, R., Honjo, H., and Owen, J. M. (1997). Regional differences in the role of the Ca2+ and Na+ currents in pacemaker activity in the sinoatrial node. Am. J. Physiol. 272, H2793–H2806.
Koushik, S. V., Wang, J., Rogers, R., Moskophidis, D., Lambert, N. A., Creazzo, T. L., et al. (2001). Targeted inactivation of the sodium-calcium exchanger (Ncx1) results in the lack of a heartbeat and abnormal myofibrillar organization. FASEB J. 15, 1209–1211. doi: 10.1096/fj.00-0696fje
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kryukova, Y. N., Protas, L., and Robinson, R. B. (2012). Ca2+-activated adenylyl cyclase 1 introduces Ca2+-dependence to beta-adrenergic stimulation of HCN2 current. J. Mol. Cell. Cardiol. 52, 1233–1239. doi: 10.1016/j.yjmcc.2012.03.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lai, M. H., Wu, Y., Gao, Z., Anderson, M. E., Dalziel, J. E., and Meredith, A. L. (2014). BK channels regulate sinoatrial node firing rate and cardiac pacing in vivo. Am. J. Physiol. Heart Circ. Physiol. 307, H1327–H1338. doi: 10.1152/ajpheart.00354.2014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lakatta, E. G., and DiFrancesco, D. (2009). What keeps us ticking: a funny current, a calcium clock, or both? J. Mol. Cell. Cardiol. 47, 157–170. doi: 10.1016/j.yjmcc.2009.03.022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lakatta, E. G., and Maltsev, V. A. (2012). Rebuttal: what I(f) the shoe doesn't fit? “The funny current has a major pacemaking role in the sinus node.” Heart Rhythm 9, 459–460. doi: 10.1016/j.hrthm.2011.09.024
Lei, M., and Brown, H. F. (1996). Two components of the delayed rectifier potassium current, IK, in rabbit sino-atrial node cells. Exp. Physiol. 81, 725–741. doi: 10.1113/expphysiol.1996.sp003972
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lei, M., Honjo, H., Kodama, I., and Boyett, M. R. (2001). Heterogeneous expression of the delayed-rectifier K+ currents i(K,r) and i(K,s) in rabbit sinoatrial node cells. J. Physiol. 535, 703–714. doi: 10.1111/j.1469-7793.2001.t01-1-00703.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, J., Xin, L., Benson, V. L., Allen, D. G., and Ju, Y. K. (2015). Store-operated calcium entry and the localization of STIM1 and Orai1 proteins in isolated mouse sinoatrial node cells. Front. Physiol. 6:69. doi: 10.3389/fphys.2015.00069
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lyashkov, A. E., Vinogradova, T. M., Zahanich, I., Li, Y., Younes, A., Nuss, H. B., et al. (2009). Cholinergic receptor signaling modulates spontaneous firing of sinoatrial nodal cells via integrated effects on PKA-dependent Ca(2+) cycling and I(KACh). Am. J. Physiol. Heart Circ. Physiol. 297, H949–H959. doi: 10.1152/ajpheart.01340.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maltsev, A. V., Yaniv, Y., Stern, M. D., Lakatta, E. G., and Maltsev, V. A. (2013). RyR-NCX-SERCA local cross-talk ensures pacemaker cell function at rest and during the fight-or-flight reflex. Circ. Res. 113, e94–e100. doi: 10.1161/CIRCRESAHA.113.302465
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maltsev, V. A., and Lakatta, E. G. (2012). The funny current in the context of the coupled-clock pacemaker cell system. Heart Rhythm 9, 302–307. doi: 10.1016/j.hrthm.2011.09.022
Maltsev, V. A., Vinogradova, T. M., Stern, M. D., and Lakatta, E. G. (2011). Letter to the editor: “Validating the requirement for beat-to-beat coupling of the Ca2+ clock and M clock in pacemaker cell normal automaticity.” Am. J. Physiol. Heart Circ. Physiol. 300, H2323–H2324; author reply H2325–H2326. doi: 10.1152/ajpheart.00110.2011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maltsev, V. A., Yaniv, Y., Maltsev, A. V., Stern, M. D., and Lakatta, E. G. (2014). Modern perspectives on numerical modeling of cardiac pacemaker cell. J. Pharmacol. Sci. 125, 6–38. doi: 10.1254/jphs.13R04CR
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mangoni, M. E., Couette, B., Bourinet, E., Platzer, J., Reimer, D., Striessnig, J., et al. (2003). Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. U.S.A. 100, 5543–5548. doi: 10.1073/pnas.0935295100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mangoni, M. E., Couette, B., Marger, L., Bourinet, E., Striessnig, J., and Nargeot, J. (2006a). Voltage-dependent calcium channels and cardiac pacemaker activity: from ionic currents to genes. Prog. Biophys. Mol. Biol. 90, 38–63. doi: 10.1016/j.pbiomolbio.2005.05.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mangoni, M. E., and Nargeot, J. (2008). Genesis and regulation of the heart automaticity. Physiol. Rev. 88, 919–982. doi: 10.1152/physrev.00018.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mangoni, M. E., Traboulsie, A., Leoni, A. L., Couette, B., Marger, L., Le Quang, K., et al. (2006b). Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/alpha1G T-type calcium channels. Circ. Res. 98, 1422–1430. doi: 10.1161/01.RES.0000225862.14314.49
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maruyama, M., Xiao, J., Zhou, Q., Vembaiyan, K., Chua, S. K., Rubart-Von Der Lohe, M., et al. (2013). Carvedilol analogue inhibits triggered activities evoked by both early and delayed afterdepolarizations. Heart Rhythm 10, 101–107. doi: 10.1016/j.hrthm.2012.09.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mattick, P., Parrington, J., Odia, E., Simpson, A., Collins, T., and Terrar, D. (2007). Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I(f) pacemaker current. J. Physiol. 582, 1195–1203. doi: 10.1113/jphysiol.2007.133439
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
McHale, N., Hollywood, M., Sergeant, G., and Thornbury, K. (2006). Origin of spontaneous rhythmicity in smooth muscle. J. Physiol. 570, 23–28. doi: 10.1113/jphysiol.2005.098376
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mesirca, P., Alig, J., Torrente, A. G., Muller, J. C., Marger, L., Rollin, A., et al. (2014a). Cardiac arrhythmia induced by genetic silencing of ‘funny’ (f) channels is rescued by GIRK4 inactivation. Nat. Commun. 5, 4664. doi: 10.1038/ncomms5664
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mesirca, P., Torrente, A. G., and Mangoni, M. E. (2014b). T-type channels in the sino-atrial and atrioventricular pacemaker mechanism. Pflugers Arch. 466, 791–799. doi: 10.1007/s00424-014-1482-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miake, J., Marban, E., and Nuss, H. B. (2002). Biological pacemaker created by gene transfer. Nature 419, 132–133. doi: 10.1038/419132b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mitsuiye, T., Guo, J., and Noma, A. (1999). Nicardipine-sensitive Na+-mediated single channel currents in guinea-pig sinoatrial node pacemaker cells. J. Physiol. 521(Pt 1), 69–79. doi: 10.1111/j.1469-7793.1999.00069.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mitsuiye, T., Shinagawa, Y., and Noma, A. (2000). Sustained inward current during pacemaker depolarization in mammalian sinoatrial node cells. Circ. Res. 87, 88–91. doi: 10.1161/01.RES.87.2.88
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Musialek, P., Rigg, L., Terrar, D. A., Paterson, D. J., and Casadei, B. (2000). Role of cGMP-inhibited phosphodiesterase and sarcoplasmic calcium in mediating the increase in basal heart rate with nitric oxide donors. J. Mol. Cell. Cardiol. 32, 1831–1840. doi: 10.1006/jmcc.2000.1216
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neco, P., Torrente, A. G., Mesirca, P., Zorio, E., Liu, N., Priori, S. G., et al. (2012). Paradoxical effect of increased diastolic Ca(2+) release and decreased sinoatrial node activity in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation 126, 392–401. doi: 10.1161/CIRCULATIONAHA.111.075382
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Noble, D., Denyer, J. C., Brown, H. F., and Difrancesco, D. (1992). Reciprocal role of the inward currents ib, Na and i(f) in controlling and stabilizing pacemaker frequency of rabbit sino-atrial node cells. Proc. Biol. Sci. 250, 199–207. doi: 10.1098/rspb.1992.0150
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Noma, A. (1996). Ionic mechanisms of the cardiac pacemaker potential. Jpn. Heart J. 37, 673–682. doi: 10.1536/ihj.37.673
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Noma, A., and Irisawa, H. (1975). Effects of Na+ and K+ on the resting membrane potential of the rabbit sinoatrial node cell. Jpn. J. Physiol. 25, 207–302. doi: 10.2170/jjphysiol.25.287
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Noma, A., Nakayama, T., Kurachi, Y., and Irisawa, H. (1984). Resting K conductances in pacemaker and non-pacemaker heart cells of the rabbit. Jpn. J. Physiol. 34, 245–254. doi: 10.2170/jjphysiol.34.245
Ottolia, M., Torres, N., Bridge, J. H., Philipson, K. D., and Goldhaber, J. I. (2013). Na/Ca exchange and contraction of the heart. J. Mol. Cell. Cardiol. 61, 28–33. doi: 10.1016/j.yjmcc.2013.06.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petit-Jacques, J., Bois, P., Bescond, J., and Lenfant, J. (1993). Mechanism of muscarinic control of the high-threshold calcium current in rabbit sino-atrial node myocytes. Pflugers Arch. 423, 21–27. doi: 10.1007/BF00374956
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Proenza, C., Angoli, D., Agranovich, E., Macri, V., and Accili, E. A. (2002). Pacemaker channels produce an instantaneous current. J. Biol. Chem. 277, 5101–5109. doi: 10.1074/jbc.M106974200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rakovic, S., Rigg, L., Ishida, H., and Terrar, D. A. (2001). Calcium transients in guinea-pig sino-atrial node cells imaged by confocal microscopy. Biophys. J. 80, 350A.
Reuter, H., Henderson, S. A., Han, T., Ross, R. S., Goldhaber, J. I., and Philipson, K. D. (2002). The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ. Res. 90, 305–308. doi: 10.1161/hh0302.104562
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rigg, L., Heath, B. M., Cui, Y., and Terrar, D. A. (2000). Localisation and functional significance of ryanodine receptors during beta-adrenoceptor stimulation in the guinea-pig sino-atrial node. Cardiovasc. Res. 48, 254–264. doi: 10.1016/S0008-6363(00)00153-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rigg, L., Mattick, P. A., Heath, B. M., and Terrar, D. A. (2003). Modulation of the hyperpolarization-activated current (I(f)) by calcium and calmodulin in the guinea-pig sino-atrial node. Cardiovasc. Res. 57, 497–504. doi: 10.1016/S0008-6363(02)00668-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rigg, L., and Terrar, D. A. (1996). Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp. Physiol. 81, 877–880. doi: 10.1113/expphysiol.1996.sp003983
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sah, R., Mesirca, P., Van den Boogert, M., Rosen, J., Mably, J., Mangoni, M. E., et al. (2013). Ion channel-kinase TRPM7 is required for maintaining cardiac automaticity. Proc. Natl. Acad. Sci. U.S.A. 110, E3037–E3046. doi: 10.1073/pnas.1311865110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sanders, L., Rakovic, S., Lowe, M., Mattick, P. A., and Terrar, D. A. (2006). Fundamental importance of Na+-Ca2+ exchange for the pacemaking mechanism in guinea-pig sino-atrial node. J. Physiol. 571, 639–649. doi: 10.1113/jphysiol.2005.100305
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schaub, M. C., Hefti, M. A., and Zaugg, M. (2006). Integration of calcium with the signaling network in cardiac myocytes. J. Mol. Cell. Cardiol. 41, 183–214. doi: 10.1016/j.yjmcc.2006.04.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Seyama, I. (1979). Characteristics of the anion channel in the sino-atrial node cell of the rabbit. J. Physiol. 294, 447–460. doi: 10.1113/jphysiol.1979.sp012940
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shibata, E. F., and Giles, W. R. (1985). Ionic currents that generate the spontaneous diastolic depolarization in individual cardiac pacemaker cells. Proc. Natl. Acad. Sci. U.S.A. 82, 7796–7800. doi: 10.1073/pnas.82.22.7796
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shinohara, T., Kim, D., Joung, B., Maruyama, M., Vembaiyan, K., Back, T. G., et al. (2014). Carvedilol analog modulates both basal and stimulated sinoatrial node automaticity. Heart Vessels 29, 396–403. doi: 10.1007/s00380-013-0378-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sitsapesan, R., and Williams, A. J. (1994). Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+. J. Membr. Biol. 137, 215–226. doi: 10.1007/BF00232590
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sitsapesan, R., and Williams, A. J. (1997). Regulation of current flow through ryanodine receptors by luminal Ca2+. J. Membr. Biol. 159, 179–185. doi: 10.1007/s002329900281
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tang, Q., Jin, M. W., Xiang, J. Z., Dong, M. Q., Sun, H. Y., Lau, C. P., et al. (2007). The membrane permeable calcium chelator BAPTA-AM directly blocks human ether a-go-go-related gene potassium channels stably expressed in HEK 293 cells. Biochem. Pharmacol. 74, 1596–1607. doi: 10.1016/j.bcp.2007.07.042
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
ten Velde, I., de Jonge, B., Verheijck, E. E., van Kempen, M. J., Analbers, L., Gros, D., et al. (1995). Spatial distribution of connexin43, the major cardiac gap junction protein, visualizes the cellular network for impulse propagation from sinoatrial node to atrium. Circ. Res. 76, 802–811. doi: 10.1161/01.RES.76.5.802
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
van Borren, M. M., Verkerk, A. O., Wilders, R., Hajji, N., Zegers, J. G., Bourier, J., et al. (2010). Effects of muscarinic receptor stimulation on Ca2+ transient, cAMP production and pacemaker frequency of rabbit sinoatrial node cells. Basic Res. Cardiol. 105, 73–87. doi: 10.1007/s00395-009-0048-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verheijck, E. E., van Ginneken, A. C., Bourier, J., and Bouman, L. N. (1995). Effects of delayed rectifier current blockade by E-4031 on impulse generation in single sinoatrial nodal myocytes of the rabbit. Circ. Res. 76, 607–615. doi: 10.1161/01.RES.76.4.607
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verkerk, A. O., and Wilders, R. (2013). Hyperpolarization-activated current, If, in mathematical models of rabbit sinoatrial node pacemaker cells. Biomed Res. Int. 2013, 872454. doi: 10.1155/2013/872454
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Verkerk, A. O., Wilders, R., Zegers, J. G., van Borren, M. M., Ravesloot, J. H., and Verheijck, E. E. (2002). Ca(2+)-activated Cl(-) current in rabbit sinoatrial node cells. J. Physiol. 540, 105–117. doi: 10.1113/jphysiol.2001.013184
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., Bogdanov, K. Y., and Lakatta, E. G. (2002a). beta-Adrenergic stimulation modulates ryanodine receptor Ca(2+) release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ. Res. 90, 73–79. doi: 10.1161/hh0102.102271
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., Bogdanov, K. Y., and Lakatta, E. G. (2002b). Novel perspectives on the beating rate of the heart. Circ. Res. 91, e3. doi: 10.1161/01.RES.0000031164.28289.55
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., Brochet, D. X., Sirenko, S., Li, Y., Spurgeon, H., and Lakatta, E. G. (2010). Sarcoplasmic reticulum Ca2+ pumping kinetics regulates timing of local Ca2+ releases and spontaneous beating rate of rabbit sinoatrial node pacemaker cells. Circ. Res. 107, 767–775. doi: 10.1161/CIRCRESAHA.110.220517
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., and Lakatta, E. G. (2009). Regulation of basal and reserve cardiac pacemaker function by interactions of cAMP-mediated PKA-dependent Ca2+ cycling with surface membrane channels. J. Mol. Cell. Cardiol. 47, 456–474. doi: 10.1016/j.yjmcc.2009.06.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., Lyashkov, A. E., Zhu, W., Ruknudin, A. M., Sirenko, S., Yang, D., et al. (2006). High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ. Res. 98, 505–514. doi: 10.1161/01.RES.0000204575.94040.d1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., Maltsev, V. A., Bogdanov, K. Y., Lyashkov, A. E., and Lakatta, E. G. (2005). Rhythmic Ca2+ oscillations drive sinoatrial nodal cell pacemaker function to make the heart tick. Ann. N.Y. Acad. Sci. 1047, 138–156. doi: 10.1196/annals.1341.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., Sirenko, S., Lyashkov, A. E., Younes, A., Li, Y., Zhu, W., et al. (2008). Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ. Res. 102, 761–769. doi: 10.1161/CIRCRESAHA.107.161679
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., Zhou, Y. Y., Bogdanov, K. Y., Yang, D., Kuschel, M., Cheng, H., et al. (2000). Sinoatrial node pacemaker activity requires Ca(2+)/calmodulin-dependent protein kinase II activation. Circ. Res. 87, 760–767. doi: 10.1161/01.RES.87.9.760
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vinogradova, T. M., Zhou, Y. Y., Maltsev, V., Lyashkov, A., Stern, M., and Lakatta, E. G. (2004). Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ. Res. 94, 802–809. doi: 10.1161/01.RES.0000122045.55331.0F
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wahl-Schott, C., Fenske, S., and Biel, M. (2014). HCN channels: new roles in sinoatrial node function. Curr. Opin. Pharmacol. 15, 83–90. doi: 10.1016/j.coph.2013.12.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wainger, B. J., Degennaro, M., Santoro, B., Siegelbaum, S. A., and Tibbs, G. R. (2001). Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411, 805–810. doi: 10.1038/35081088
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Tsui, H., Ke, Y., Shi, Y., Li, Y., Davies, L., et al. (2014). Pak1 is required to maintain ventricular Ca(2)(+) homeostasis and electrophysiological stability through SERCA2a regulation in mice. Circ. Arrhythm. Electrophysiol. 7, 938–948. doi: 10.1161/CIRCEP.113.001198
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, Y., and Anderson, M. E. (2014). CaMKII in sinoatrial node physiology and dysfunction. Front. Pharmacol. 5:48. doi: 10.3389/fphar.2014.00048
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wu, Y., Gao, Z., Chen, B., Koval, O. M., Singh, M. V., Guan, X., et al. (2009). Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc. Natl. Acad. Sci. U.S.A. 106, 5972–5977. doi: 10.1073/pnas.0806422106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Xie, Y., Ding, W. G., and Matsuura, H. (2015). Ca2+/calmodulin potentiates I Ks in sinoatrial node cells by activating Ca2+/calmodulin-dependent protein kinase II. Pflugers Arch. 467, 241–251. doi: 10.1007/s00424-014-1507-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yaniv, Y., Maltsev, V. A., Escobar, A. L., Spurgeon, H. A., Ziman, B. D., Stern, M. D., et al. (2011). Beat-to-beat Ca(2+)-dependent regulation of sinoatrial nodal pacemaker cell rate and rhythm. J. Mol. Cell. Cardiol. 51, 902–905. doi: 10.1016/j.yjmcc.2011.08.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yaniv, Y., Stern, M. D., Lakatta, E. G., and Maltsev, V. A. (2013). Mechanisms of beat-to-beat regulation of cardiac pacemaker cell function by Ca(2)(+) cycling dynamics. Biophys. J. 105, 1551–1561. doi: 10.1016/j.bpj.2013.08.024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yoo, S., Lee, S. H., Choi, B. H., Yeom, J. B., Ho, W. K., and Earm, Y. E. (1998). Dual effect of nitric oxide on the hyperpolarization-activated inward current (I(f)) in sino-atrial node cells of the rabbit. J. Mol. Cell. Cardiol. 30, 2729–2738. doi: 10.1006/jmcc.1998.0845
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Younes, A., Lyashkov, A. E., Graham, D., Sheydina, A., Volkova, M. V., Mitsak, M., et al. (2008). Ca(2+) -stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J. Biol. Chem. 283, 14461–14468. doi: 10.1074/jbc.M707540200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yuill, K. H., and Hancox, J. C. (2002). Characteristics of single cells isolated from the atrioventricular node of the adult guinea-pig heart. Pflugers Arch. 445, 311–320. doi: 10.1007/s00424-002-0932-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zaza, A., Maccaferri, G., Mangoni, M., and Difrancesco, D. (1991). Intracellular calcium does not directly modulate cardiac pacemaker (if) channels. Pflugers Arch. 419, 662–664. doi: 10.1007/BF00370312
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zaza, A., Micheletti, M., Brioschi, A., and Rocchetti, M. (1997). Ionic currents during sustained pacemaker activity in rabbit sino-atrial myocytes. J. Physiol. 505(Pt 3), 677–688. doi: 10.1111/j.1469-7793.1997.677ba.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, H., Butters, T., Adeniran, I., Higham, J., Holden, A. V., Boyett, M. R., et al. (2012). Modeling the chronotropic effect of isoprenaline on rabbit sinoatrial node. Front. Physiol. 3:241. doi: 10.3389/fphys.2012.00241
Zhang, Y. H., and Hancox, J. C. (2009). Regulation of cardiac Na+-Ca2+ exchanger activity by protein kinase phosphorylation–still a paradox? Cell Calcium 45, 1–10. doi: 10.1016/j.ceca.2008.05.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhang, Z., Xu, Y., Song, H., Rodriguez, J., Tuteja, D., Namkung, Y., et al. (2002). Functional Roles of Ca(v)1.3 (alpha(1D)) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ. Res. 90, 981–987. doi: 10.1161/01.RES.0000018003.14304.E2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, Q., Xiao, J., Jiang, D., Wang, R., Vembaiyan, K., Wang, A., et al. (2011). Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat. Med. 17, 1003–1009. doi: 10.1038/nm.2406
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: sino-atrial node, cardiac, pacemaking, cytosolic calcium, calcium clock, membrane clock
Citation: Capel RA and Terrar DA (2015) The importance of Ca2+-dependent mechanisms for the initiation of the heartbeat. Front. Physiol. 6:80. doi: 10.3389/fphys.2015.00080
Received: 09 January 2015; Accepted: 02 March 2015;
Published: 25 March 2015.
Edited by:
Carol Ann Remme, University of Amsterdam, NetherlandsCopyright © 2015 Capel and Terrar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Derek A. Terrar, Department of Pharmacology, University of Oxford, British Heart Foundation Centre of Research Excellence, Mansfield Road, Oxford, Oxon, OX1 3QT, UKZGVyZWsudGVycmFyQHBoYXJtLm94LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.