
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 17 March 2015
Sec. Cardiac Electrophysiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00076
This article is part of the Research TopicCa2+ Signaling and Heart RhythmView all 13 articles
 Yanwen Wang1
Yanwen Wang1 Hoyee Tsui2
Hoyee Tsui2 Emma L. Bolton1
Emma L. Bolton1 Xin Wang2
Xin Wang2 Christopher L.-H. Huang3
Christopher L.-H. Huang3 R. John Solaro4
R. John Solaro4 Yunbo Ke4*
Yunbo Ke4* Ming Lei1*
Ming Lei1*A series of recent studies report novel roles for Pak1, a key member of the highly conserved family of serine-threonine protein kinases regulated by Ras-related small G-proteins, Cdc42/Rac1, in cardiac physiology and cardioprotection. Previous studies had identified Pak1 in the regulation of hypertrophic remodeling that could potentially lead to heart failure. This article provides a review of more recent findings on the roles of Pak1 in cardiac Ca2+ homeostasis. These findings identified crucial roles for Pak1 in cardiomyocyte Ca2+ handling and demonstrated that it functions through unique mechanisms involving regulation of the post-transcriptional activity of key Ca2+-handling proteins, including the expression of Ca2+-ATPase SERCA2a, along with the speculative possibility of an involvement in the maintenance of transverse (T)-tubular structure. They highlight important regulatory functions of Pak1 in Ca2+ homeostasis in cardiac cells, and identify novel potential therapeutic strategies directed at manipulation of Pak1 signaling for the management of cardiac disease, particularly heart failure.
Protein kinases are versatile signaling molecules involved in the regulation of a wide range of physiological processes. Of these, the p21-activated kinases (Paks) form a group of serine/threonine protein kinases activated by Cdc42 and Rac1, and were first discovered in rat brain tissue two decades ago (Manser et al., 1994). The structure, substrate-specificity and functional role of Paks are evolutionarily conserved from protozoa to mammals. Vertebrate Paks are particularly important in cytoskeletal remodeling and the assembly of focal adhesion structures, thereby contributing to the modulation of dynamic processes such as cell migration and synaptic plasticity (Manser and Lim, 1999; Hofmann et al., 2004; Zhao and Manser, 2005). Over the past decade, significant progress has been made in understanding the functions of Pak1, a key member of the Pak family, in particular, its roles in the regulation of excitability and contractility of the heart (Ke et al., 2004, 2007, 2009). The present review now provides an updated account of these recent findings regarding additional roles of Pak1 in Ca2+ homeostasis and Ca2+ handling in cardiac cells.
The role of pak1 in the regulation of ion channel activity in cardiomyocytes was first demonstrated in isolated guinea pig sinoatrial node (SAN) pacemaker cells (Ke et al., 2007). In cultured guinea pig SAN cells, where active Pak1 expression was induced through infection with recombinant adenovirus expressing constitutively active Pak1 (CA-Pak1), responses of both L-type Ca2+ channel (LTCC) and delayed rectifier K+ channel currents to β-adrenergic stimulation by isoproterenol were depressed in comparison to SAN cells infected with control virus, Ad-LacZ. Similarly chronotropic responses to isoproterenol stimulation, reflected in repetitive action potential frequency were depressed in both intact SAN tissue and isolated SAN cells expressing active Pak1, when compared to controls expressing Ad-LacZ (Ke et al., 2007). In contrast, Pak1 deletion in cardiac specific conditional knockout (Pak1cko) or in total knockout mice resulted in an increased SAN driven heart rate (Wang et al., 2014a). These modified responses to isoproterenol stimulation in SAN tissue and cells infected by CA-Pak1 likely reflect alterations in protein phosphorylation which modulate LTCC and delayed rectifier K+ channel activity, and their responses to β-adrenergic stimulation.
Our further studies focussing on LTCC regulation in SAN cells implicated Pak1 as a regulator of protein phosphatase 2A (PP2A). Immunoprecipitation studies indicated that Pak1 and PP2A form a complex, leading to the hypothesis that Pak1 may regulate LTCC activity through PP2A action (Ke et al., 2007). This hypothesis was substantiated by studying the influence of the PP2A inhibitor okadaic acid (OA) on the effects of isoproterenol on LTCC activity in Ad-Pak1–infected cells. OA partially reversed the suppressant effect of active Pak1 on the response of LTCCs to isoproterenol in Ad-Pak1–infected cells. This suggests that Pak1 acts by increasing PP2A activity. Conversely, our recent study demonstrated that mice with a cardiomyocyte-specific Pak1 deletion (Pak1cko) showed higher heart rates than their control littermates (Pak1f/f), although Pak1cko and control Pak1f/f mice showed similar baseline electrocardiographic P wave durations and P-R, QRS and QT intervals (Wang et al., 2014a).
Accumulating evidence implicates a coordinated interplay between the activities of kinases and phosphatases in modulation of LTCC-mediated Ca2+ influx even in the absence of humoral stimulation. For example, in the well-known β-adrenergic receptor/protein kinase A (PKA) cascade, inhibitor-1 is a downstream PKA target whose activation results in an attenuation of protein phosphatase 1 (PP1) activity (Gupta et al., 1996). Santana et al. (2002) showed that the phosphatase calcineurin opposes PKA action in mouse ventricular myocytes. Application of the PP2A inhibitor OA can activate LTCC (duBell and Rogers, 2004). Calyculin A, which inhibits both PP1 and PP2A, increases contractility in ventricular myocytes by increasing LTCC activity (duBell et al., 2002). A complementary study by duBell et al reported that addition of exogenous PP2A decreased LTCC currents in rat ventricular myocytes (duBell et al., 1996).
These results together suggested an existence of a dynamic regulatory balance between kinase and phosphatase activity in regulating the LTCC and delayed rectifier K+ channel activity in cardiac cells that may be important in controlling cardiac pacemaker activity in response to autonomic and humoral stimulation.
In parallel with Pak1 action in SAN myocytes, enhanced Pak1 function brought about by Pak1 activating peptide, PAP (Wang et al., 2014b) in ventricular tissue prevented hypertrophic associated ventricular arrhythmias, and Pak1 deletion in Pak1cko or in knockout mice increased the risks of ventricular alternans and arrhythmias compared to WT mice. These findings went with a co-immunoprecipitation of Pak1 and PP2A suggesting a complex formation in ventricular myocytes, in common with SAN cells (Ke et al., 2007). Recent studies have also suggested regulatory roles of Pak1 in Ca2+ homeostasis in ventricular myocytes (Sheehan et al., 2009; DeSantiago et al., 2013; Wang et al., 2014a). CA-Pak1 over-expression altered Ca2+ transient decay constants (τCa) (Sheehan et al., 2009) and antagonized adrenergic signaling by attenuating isoproterenol-induced increases in the activity ofLTCCs and other proteins regulating Ca2+ handling (Sheehan et al., 2009). In contrast, ventricular myocytes from Pak1cko mice with a cardiomyocyte-specific Pak1 knockout showed abnormal Ca2+ homeostasis including increased diastolic [Ca2+]i, as well as decreased sarcoplasmic reticular (SR) Ca2+ content and decreased SERCA function, particularly during β-adrenergic stress (Wang et al., 2014a). Significant differences in Ca2+ homeostasis were observed between isolated Pak1cko and wild type, Pak1f/f, ventricular myocytes. Diastolic [Ca2+]i was higher in Pak1cko than Pak1f/f myocytes under both baseline and chronic β-adrenergic stress conditions. Pak1cko myocytes showed more frequent irregular, alternating Ca2+ transients and/or Ca2+ waves at increased stimulation frequencies than Pak1f/f myocytes under both baseline and chronic β-adrenergic stress conditions during 0.5, 1, and 3 Hz field stimulation. This abnormal Ca2+ homeostasis in Pak1cko myocytes correlated with differences in evoked cytosolic and SR Ca2+ responses between Pak1f/f and Pak1cko myocytes, in both the absence and presence of chronic β-adrenergic stress (Wang et al., 2014a). Thus, time constants for decays of the Na+-Ca2+ exchange (NCX) current (INCX) following INCX induction by 10 mM caffeine were significantly greater in Pak1cko than Pak1f/f myocytes under chronic β-adrenergic stress. SR Ca2+content, measured by integration of the INCX records, was reduced in Pak1cko compared to Pak1f/f myocytes in both the absence and presence of chronic β-adrenergic stress. The decay rate constants of systolic Ca2+ transients in Pak1cko myocytes kSERCA, representing the kinetics of cytosolic Ca2+ removal brought about by both SERCA and NCX, was significantly lower than that shown by Pak1f/f myocytes in both the absence and presence of chronic β-adrenergic stress (Wang et al., 2014a). The rate constant kSERCA was reduced by chronic β-adrenergic stress in both the Pak1f/f and Pak1cko myocytes but more severely so in the Pak1cko myocytes. Finally, peak systolic [Ca2+]i, estimated from the differences between peaks and baselines of systolic Ca2+ transients obtained during regular stimulation, was indistinguishable between Pak1f/f and Pak1cko under baseline conditions, but was significantly reduced by chronic β-adrenergic stress in both Pak1f/f and Pak1cko, and again more severely so in the Pak1cko (Wang et al., 2014a). In contrast, LTCC activity was indistinguishable between Pak1cko and Pak1f/f under both baseline and chronic β-adrenergic stress conditions, but chronic β-adrenergic stress reduced LTCC current in both Pak1cko and Pak1f/f ventricular myocytes, which suggests that genetic deletion of Pak1 did not alter the expression and activity of LTCC in these cells. This result contrasts with the effect of CA-Pak1 in SAN cells, and may reflect differing effects of Pak1 on ion channels in different cell types or physiological conditions, and requires further investigation (Wang et al., 2014a). These alterations in Ca2+ homeostasis were associated with an increased incidence of ventricular arrhythmias and electrophysiological instability during either acute or chronic β-adrenergic challenge induced by isoproterenol in Pak1cko hearts. Hence modulation of Pak1 activity modified Ca2+ handling under both physiological and β-adrenergic challenge conditions.
Our further molecular studies associated these physiological findings with an impaired SERCA2a function and down-regulation of SERCA2a mRNA and protein expression in Pak1cko hearts (Wang et al., 2014a). Further exploration of this altered transcriptional regulation in cultured neonatal rat cardiomyocytes (NRCMs) demonstrated that exposure to control Ad-shC2 virus infection increased SERCA2a protein and mRNA levels following phenylephrine challenge (Wang et al., 2014a). This was abolished by the Pak1-knockdown in Ad-shPak1-infected NRCMs and increased by constitutive over-expression of active Pak1 (Ad-CAPak1) (Wang et al., 2014a). This regulation appeared to involve activation of serum response factor (SRF), a transcriptional factor well-known for its vital role in regulation of cardiogenesis genes in the Pak1-dependent regulation of SERCA2a (Wang et al., 2014a).
The above results indicate that modulation of Pak1 activity in ventricular myocytes can have a significant impact on Ca2+ handling in these cells under both baseline physiological and β-adrenergic challenge conditions.
Preliminary evidence (DeSantiago et al., 2013) suggests structural roles of Pak1, in addition to the above functional role of Pak1 in maintaining transverse (T)-tubular structure, which is altered in hypertrophic remodeling. Pak1−/− ventricular myocytes showed decreased cell capacitances compared to WT suggesting decreased T-tubular density. Under these conditions, cells showed comparable SR Ca loads and phospholamban phosphorylation, while their systolic Ca2+ transients showed decreased amplitudes and delayed rise times, consistent with a reduced coupling between LTCC-mediated Ca2+ influx and RyR2-mediated Ca2+-induced Ca2+ release (CICR); changes which were not observed in Pak1−/− atrial myocytes. Such findings are consistent with the central role of T-tubules in triggering and synchronizing excitation–contraction coupling, and merit further exploration. T-tubular remodeling has been reported in other cardiac pathologies, and could well be involved in the associated alterations in cellular processes, and hence cardiac function.
In summary, as illustrated in Figure 1, these novel Pak1 effects on Ca2+ homestasis complement previously established actions upon PP2A and the resulting balance between kinase and phosphatase activity in controlling cardiac ion channel activity and rhythmic Ca2+ cycling.
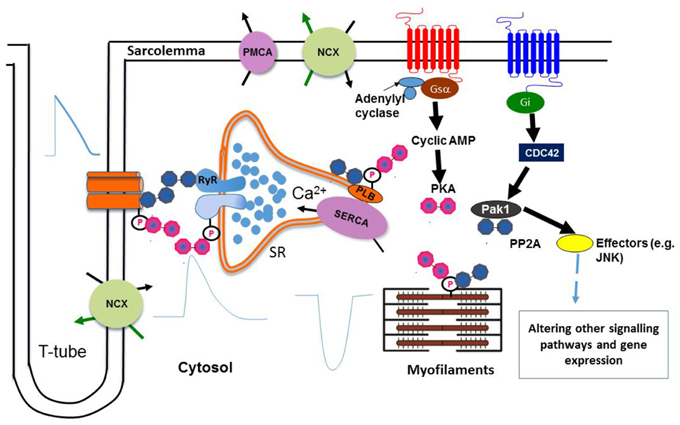
Figure 1. Regulation of Ca2+ homeostasis by Pak1. Protein kinase PKA and phosphatase PP2A are associated with key Ca2+ handling and regulatory proteins, which in turn are linked to upstream signaling cascades. A balance of protein kinase and phosphatase activities is required to maintain normal cardiac functions. Pak1 also regulates hypertrophic signaling and gene expression of SERCA2a through other signaling pathways by activating its downstream effectors (e.g., JNK). (Liu et al., 2011; Wang et al., 2014a) (NCX: Na+-Ca2+ exchanger, PMCA: Plasma membrane Ca2+ ATPase, JNK: c-Jun N-terminal Kinase).
Earlier reports (Liu et al., 2011) had identified Pak1 as a novel regulator of hypertrophic remodeling whose cardiomyocyte-specific deletion exacerbated cardiac hypertrophy leading to heart failure, following transverse aortic constriction. These features were prevented by the non-selective Pak1 activator FTY720 in wild-type but not Pak1cko mice (Liu et al., 2013). Such cardiac hypertrophy, with improved cardiac function and decreased myocyte apoptosis compared to WT, was reduced in mice over-expressing Pak1, with improved cardiac function and decreased myocyte apoptosis compared to WT (Mao et al., 2009). A recent study identified Pak1 as one of the significant genes on the core networks for dilated cardiomyopathies by pathway analyses in a Genome wide association study (GWAS) dataset of patients suffering from DCM (Backes et al., 2014). Over the past decade, significant progress has also been made in understanding of additional roles of Pak1 in the regulation of Ca2+ homeostasis and Ca2+ handling in turn regulating cardiomyocyte excitability and contractility (Ke et al., 2004, 2007, 2009). There have also been speculative suggestions for a role of Pak1 in the maintenance of transverse (T) tubule structure. The present review now provides an updated account of these recent findings. Thus, Pak1 may thus offer a novel therapeutic target for modulation of Ca2+ handling in cardiac disease conditions.
The work was supported by the Medical Research Council (G10002647:ML, XW, EJC, JRS, YBK), the British Heart Foundation (PG/11/59/29006; PG/12/21/29473: ML, XW), The MacVeigh benefaction (CLH)” and a Chinese Nature Science Foundation Grant (31171085: ML), NIH Grant HL 064035 (JRS), PO1 HL 062426 (JRS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Backes, C., Ruhle, F., Stoll, M., Haas, J., Frese, K., Franke, A., et al. (2014). Systematic permutation testing in gwas pathway analyses: identification of genetic networks in dilated cardiomyopathy and ulcerative colitis. BMC Genomics 15:622. doi: 10.1186/1471-2164-15-622
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
DeSantiago, J., Bare, D. J., Ke, Y., Sheehan, K. A., Solaro, R. J., and Banach, K. (2013). Functional integrity of the t-tubular system in cardiomyocytes depends on p21-activated kinase 1. J. Mol. Cell. Cardiol. 60, 121–128. doi: 10.1016/j.yjmcc.2013.04.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
duBell, W. H., Gigena, M. S., Guatimosim, S., Long, X., Lederer, W. J., and Rogers, T. B. (2002). Effects of pp1/pp2a inhibitor calyculin a on the e-c coupling cascade in murine ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 282, H38–H48. doi: 10.1152/ajpheart.00536.2001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
duBell, W. H., and Rogers, T. B. (2004). Protein phosphatase 1 and an opposing protein kinase regulate steady-state l-type Ca2+ current in mouse cardiac myocytes. J. Physiol. 556, 79–93. doi: 10.1113/jphysiol.2003.059329
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
duBell, W., Lederer, W., and Rogers, T. (1996). Dynamic modulation of excitation-contraction coupling by protein phosphatases in rat ventricular myocytes. J. Physiol. 493, 793–800. doi: 10.1113/jphysiol.1996.sp021423
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gupta, R. C., Neumann, J., Watanabe, A. M., Lesch, M., and Sabbah, H. N. (1996). Evidence for presence and hormonal regulation of protein phosphatase inhibitor-1 in ventricular cardiomyocyte. Am. J. Physiol. Heart Circ. Physiol. 270, H1159–H1164.
Hofmann, C., Shepelev, M., and Chernoff, J. (2004). The genetics of pak. J. Cell Sci. 117, 4343–4354. doi: 10.1242/jcs.01392
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ke, Y., Lei, M., Collins, T. P., Rakovic, S., Mattick, P. A., Yamasaki, M., et al. (2007). Regulation of l-type calcium channel and delayed rectifier potassium channel activity by p21-activated kinase-1 in guinea pig sinoatrial node pacemaker cells. Circ. Res. 100, 1317–1327. doi: 10.1161/01.RES.0000266742.51389.a4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ke, Y., Lei, M., and Solaro, R. J. (2009). Regulation of cardiac excitation and contraction by p21 activated kinase-1. Prog. Biophys. Mol. Biol. 98, 238–250. doi: 10.1016/j.pbiomolbio.2009.01.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ke, Y., Wang, L., Pyle, W. G., de Tombe, P. P., and Solaro, R. J. (2004). Intracellular localization and functional effects of p21-activated kinase-1 (pak1) in cardiac myocytes. Circ. Res. 94, 194–200. doi: 10.1161/01.RES.0000111522.02730.56
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, W., Zi, M., Naumann, R., Ulm, S., Jin, J., Taglieri, D. M., et al. (2011). Pak1 as a novel therapeutic target for antihypertrophic treatment in the heart. Circulation 124, 2702–2715. doi: 10.1161/CIRCULATIONAHA.111.048785
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, W., Zi, M., Tsui, H., Chowdhury, S. K., Zeef, L., Meng, Q.-J., et al. (2013). A novel immunomodulator, fty-720 reverses existing cardiac hypertrophy and fibrosis from pressure overload by targeting nfat (nuclear factor of activated t-cells) signaling and periostin. Circ. Heart Fail. 6, 833–844. doi: 10.1161/CIRCHEARTFAILURE.112.000123
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Manser, E., Leung, T., Salihuddin, H., Zhao, Z.-S., and Lim, L. (1994). A brain serine/threonine protein kinase activated by cdc42 and rac1. Nature 367, 40–46. doi: 10.1038/367040a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Manser, E., and Lim, L. (1999). Roles of pak family kinases. Prog. Mol. Subcell. Biol. 22, 115–133. doi: 10.1007/978-3-642-58591-3_6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mao, K., Healy, C., Timm, D., Kobayashi, S., Volden, P., Chernoff, J., et al. (2009). P21 activated kinase 1 (pak1) protects heart from pressure overload induced pathological cardiac remodeling. FASEB J. 23(Meeting Abstract Supplement), 362.5.
Santana, L. F., Chase, E. G., Votaw, V. S., Nelson, M. T., and Greven, R. (2002). Functional coupling of calcineurin and protein kinase a in mouse ventricular myocytes. J. Physiol. 544, 57–69. doi: 10.1113/jphysiol.2002.020552
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sheehan, K. A., Ke, Y., Wolska, B. M., and Solaro, R. J. (2009). Expression of active p21-activated kinase-1 (pak1) induces ca2+-flux modification with altered regulatory protein phosphorylation in cardiac myocytes. Am. J. Physiol. Cell Physiol. 296, C47–C58. doi: 10.1152/ajpcell.00012.2008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, R., Wang, Y., Lin, K., Zhang, Y., Liu, W., Huang, K., et al. (2014b). Inhibition of angiotensin ii-induced cardiac hypertrophy and associated ventricular arrhythmias by a p21 activated kinase 1 bioactive peptide. PLoS ONE 9:e101974. doi: 10.1371/journal.pone.0101974
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Tsui, H., Ke, Y., Shi, Y., Li, Y., Davies, L., et al. (2014a). Pak1 is required to maintain ventricular Ca2+ homeostasis and electrophysiological stability through serca2a regulation in mice. Circ. Arrhythm. Electrophysiol. 7, 938–948. doi: 10.1161/CIRCEP.113.001198
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhao, Z., and Manser, E. (2005). Pak and other rho-associated kinases–effectors with surprisingly diverse mechanisms of regulation. Biochem. J. 386, 201–214. doi: 10.1042/BJ20041638
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Pak1, Ca2+ homeostasis, heart
Citation: Wang Y, Tsui H, Bolton EL, Wang X, Huang CL-H, Solaro RJ, Ke Y and Lei M (2015) Novel insights into mechanisms for Pak1-mediated regulation of cardiac Ca2+ homeostasis. Front. Physiol. 6:76. doi: 10.3389/fphys.2015.00076
Received: 20 December 2014; Accepted: 25 February 2015;
Published: 17 March 2015.
Edited by:
Zhilin Qu, University of California, Los Angeles, USAReviewed by:
David R. Van Wagoner, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USACopyright © 2015 Wang, Tsui, Bolton, Wang, Huang, Solaro, Ke and Lei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunbo Ke, Department of Physiology and Biophysics, University of Illinois, 835 S. Wolcott Ave., M/C 901, Chicago, IL 60612, USAeWtlQHVpYy5lZHU=;
Ming Lei, Department of Pharmacology, University of Oxford, Mansfield Road, Oxford OX1 3QT, UKbWluZy5sZWlAcGhhcm0ub3guYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.