 Yael Yaniv
Yael Yaniv Kenta Tsutsui
Kenta Tsutsui Edward G. Lakatta
Edward G. Lakatta
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Physiol. , 23 February 2015
Sec. Cardiac Electrophysiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00047
This article is part of the Research Topic Remodeling of cardiac passive electrical properties and susceptibility to ventricular and atrial arrhythmias View all 13 articles
The heart's regular electrical activity is initiated by specialized cardiac pacemaker cells residing in the sinoatrial node. The rate and rhythm of spontaneous action potential firing of sinoatrial node cells are regulated by stochastic mechanisms that determine the level of coupling of chemical to electrical clocks within cardiac pacemaker cells. This coupled-clock system is modulated by autonomic signaling from the brain via neurotransmitter release from the vagus and sympathetic nerves. Abnormalities in brain-heart clock connections or in any molecular clock activity within pacemaker cells lead to abnormalities in the beating rate and rhythm of the pacemaker tissue that initiates the cardiac impulse. Dysfunction of pacemaker tissue can lead to tachy-brady heart rate alternation or exit block that leads to long atrial pauses and increases susceptibility to other cardiac arrhythmia. Here we review evidence for the idea that disturbances in the intrinsic components of pacemaker cells may be implemented in arrhythmia induction in the heart.
Normal cardiac impulse initiation and conduction are generated by specialized, self-excitable, pacemaker cells residing in the sinoatrial node (SAN). Defects in these cell-intrinsic capacities to elicit spontaneous action potentials (APs) can lead to disturbances of the rate and rhythm of heart beats, and can induce numerous clinical arrhythmia syndromes: (i) SAN dysfunction has been postulated to be a source of sinus nodal re-entry (i.e., reciprocal beats between SAN and atrium) tachyarrhythmia (Birchfield et al., 1957) which accounts for 2–17% of all arrhythmias (Cossu and Steinberg, 1998). While diagnosis of this arrhythmia is difficult, due to electrocardiographic similarity of the P-wave to the normal sinus rhythm (Gomes et al., 1995), microelectrode studies in isolated rabbit hearts (Han et al., 1968) and later in humans (Childers et al., 1973) indeed demonstrate that SAN is the source that induces re-entry. (ii) Sick sinus syndrome, characterized by symptomatic dysfunction of the SAN (reviewed in Dobrzynski et al., 2007), can be manifested as sinus bradycardia, sinus arrest, or SAN block, and in some cases supraventricular tachyarrhythmias (“tachy-brady” syndrome), atrial flutter or atrial fibrillation. In mice with an inducible phenotype that mimics sick sinus syndrome, heart beating intervals (BIs) were completely irregular both in vivo and in the isolated Langendroff perfused model (no brain-heart signaling) (Herrmann et al., 2007). Telemetric ECG recordings revealed a variety of arrhythmias: sino-atrial arrhythmia, sino-atrial pause and supraventricular or ventricular tachycardia. (iii) It has been suggested that abnormal stretch of the rat atria that accompanies many heart diseases (De Jong et al., 2013) and occurs even in transplanted human hearts (Slovut et al., 1998) (no brain-heart connection) induces respiratory sinus arrhythmia.
Here we review evidence for the idea that changes in the membrane and sarcoplasmic reticulum (SR) components of pacemaker cells may be implicated in arrhythmia induction in the heart.
To understand abnormal SAN function it is essential to first understand the normal function of intrinsic properties of pacemaker cells and their modulation by brain-heart signaling. Experimental and theoretical data over the past two decades indicate that pacemaker cells residing in the SAN entrain their AP BI variability (BIV) by regulation of intracellular electric and mechanical coupling (reviewed in Yaniv et al., 2013a).
The coupled-clock system (Yaniv et al., 2013b; Maltsev et al., 2014) that controls the pacemaker cell beating rate and rhythm consists of an intracellular “Ca2+ clock” and “M clock.” The sarcoplasmic reticulum Ca2+ pump and ryanodine channels act as a “Ca2+ clock,” discharging local Ca2+ releases (LCRs) close to the cell surface membrane; LCRs activate membrane electrogenic clock molecules (“M clock”), mainly the Na+/Ca2+ exchanger. Na+-Ca2+ exchange current, the f-channel current, and K+ channel current, other components of the M clock, concurrently drive the diastolic membrane depolarization to ignite the next AP. The Ca2+ and M clocks entrain each other through electrical and chemical signaling: Ca2+ activation of calmodulin -adenylyl cyclase (AC)-dependent protein kinase A (PKA) and Ca2+/calmodulin-dependent protein kinase II (CaMKII). Both of these signaling pathways affect phosphorylation of proteins of both clocks [i.e., phospholamban (PLB) and ryanodine receptors (Ca2+ clock) and L type and K+ channels (M clock)]. Additionally, cAMP positively shifts the f-channel activation curve. Based on the coupled-clock theory, a change in the activity or in the quantity of every molecule within the M or the Ca2+ clock or a change in the chemical coupling signaling of both clocks will perturb the function of the other clock, and thus alter the degree of entrainment between them. For example, in rabbit pacemaker cells, reduction in Ifactivity or in Ca2+ clock proteins leads to reduction in the coupled-clock phosphorylation activity and LCR signal (Yaniv et al., 2014b). Thus, a reduction of internal signaling within one clock can lead to a reduction in the degree of clock coupling and changes in the function of the other clock that change automaticity.
Even under normal conditions, spontaneous AP BIs of mammals including human pacemaker cells are not constant, but vary around the average AP BI, due to stochastic properties of intrinsic mechanisms of the coupled-clock system (Verheijck et al., 1998; Rocchetti et al., 2000; Zaza and Lombardi, 2001; Monfredi et al., 2013; Papaioannou et al., 2013; Yaniv et al., 2014b). The degree of AP BIV is related to variability in both the timing of LCR occurrence during the diastolic depolarization, and to the ensemble LCR Ca2+ signal: an increase in LCR variability is associated with a reduced ensemble LCR Ca2+ signal that occurs later in diastole (i.e., prolonging the next AP ignition). Based on the coupled-clock theory, the stochasticity of LCR periods (i.e., the times of LCR occurrences following the prior AP) not only depends upon stochasticity of spontaneous RyR activation, but also upon stochastic sarcolemmal ion channel openings and closings that regulate the cell Ca2+ balance. The amplitude and timing of LCR Ca2+ signals to M clock proteins report the efficiency of clock coupling, i.e., a weaker LCR signal to M clock proteins that occurs later in time reports less-efficient clock coupling. Consequently, changes in the steady-state AP BI and the BIV embody contributions of both clocks. Reduction in the degree of synchronization between the clocks disturbs the ability to maintain the basal average AP BI, leading not only to a reduction in the average AP BI, but also to an increase in variability around the prolonged average AP BI.
Autonomic neural input can entrain the rate and rhythm of electrical impulses that are generated by SAN tissue of mammals (Difrancesco, 1993; Boyett et al., 2000; Monfredi et al., 2014; Yaniv et al., 2014a). The balance between sympathetic to parasympathetic stimulation has a role in synchronizing intrinsic clock periods of individual pacemaker cells. β adrenergic receptor stimulation increases synchronization of intrinsic clock mechanisms leading to a decrease of both BI and BIV of pacemaker cells (Figures 1B,C). Moreover, β adrenergic receptor stimulation of a single pacemaker cell increases the probability that the beating intervals exhibit fractal-like behavior. Cholinergic receptor stimulation of pacemaker cells, on the other hand, decreases synchronization of intrinsic clock mechanisms, leading to an increase of both the average BI and BIV (Figures 1B,C).
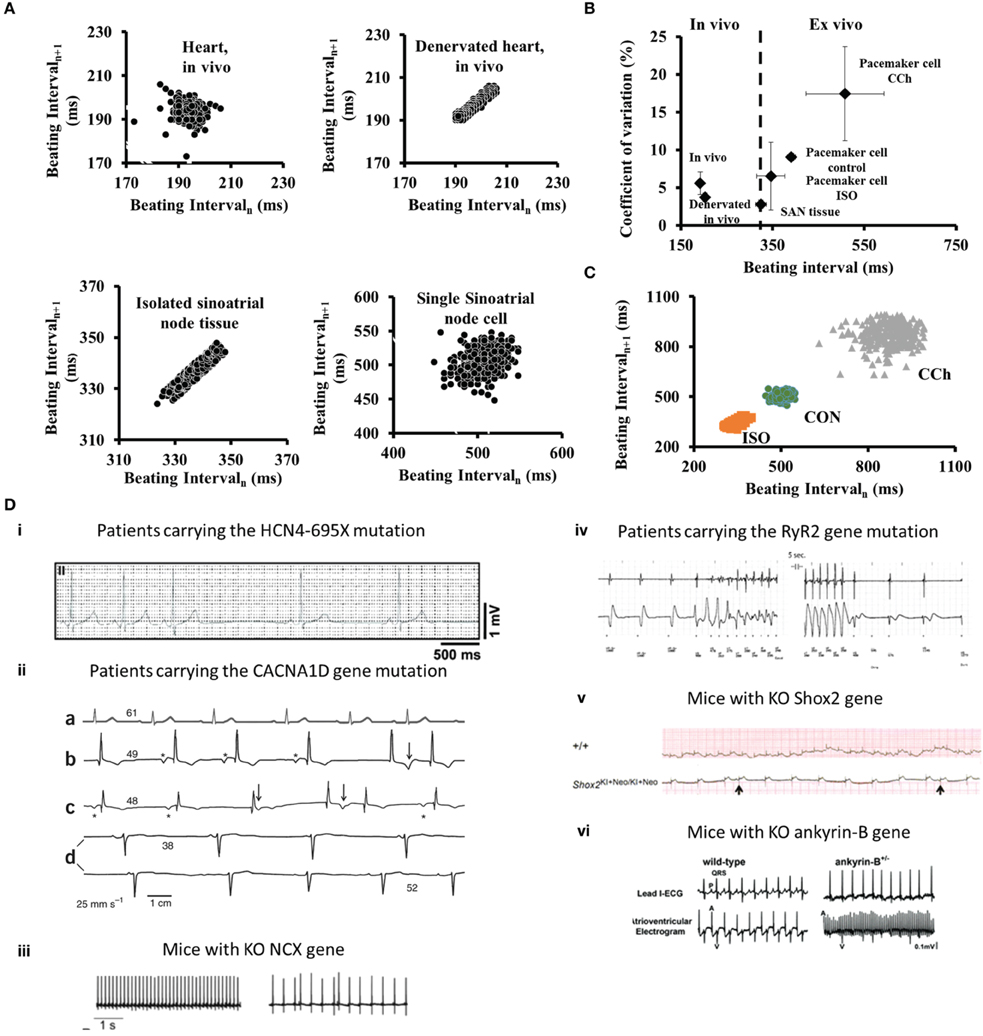
Figure 1. (A) Poincaré plots of the beating interval at different levels of integration from the heart in vivo to single isolated pacemaker cells. (B) The relationship between beating intervals and coefficients of variation at different levels of integration from the heart in vivo to single pacemaker cells in isolation. (C) Poincaré plots of the beating interval variability in single SANC under control (CON), β-adrenergic receptor stimulation (ISO) or cholinergic receptor stimulation (CCh). Modified from Yaniv et al. (2014a). (D) Examples of arrhythmias associated with changes in intrinsic clock mechanisms recorded in vivo: (i) in patients carrying the HCN4-695X mutation (adapted from Schweizer et al., 2010); (ii) (a–d) ECG recordings from an unaffected person (a) and three individuals who are homozygous for the CACNA1D mutation (b–d) (adapted from Baig et al., 2011); (iii) Cardiac arrhythmias in freely moving NCX KO mice (adapted from Herrmann et al., 2013); (iv) A rapid and presumably polymorphic ventricular tachycardia in patents with mutation in RyR2 (adapted from Bhuiyan et al., 2007); (v) ECG recording demonstrate the occurrence of arrhythmia in Shox2 KO mice (adapted from Liu et al., 2011); (vi) ECG recording from ankyrin-B KO mice (adapted from Cunha et al., 2011).
Although we have focused here upon synchronization of mechanisms intrinsic to pacemaker cells, cell-to-cell interactions (electrotonic and mechanical) of pacemaker cells residing in SAN tissue also entrain the rate and rhythm of electrical impulses that emanate from the SAN (Jalife, 1984; Watanabe et al., 1995). These interactions have a role in synchronizing the intrinsic clock periods of individual cells (Sheikh et al., 2013), because the average range of basal AP BI and AP BIV of single isolated pacemaker cells is well above their range when they reside in rabbit SAN tissue (Yaniv et al., 2014a) (Figure 1A). When pacemaker cells are embedded within SAN tissue, those with the shortest AP BI create a primary pacemaker area within the SAN, leading to the origin of an electrotonic force that spreads to other SAN cells, resulting in the emanation of an electric impulse that excites the rest of the heart (Anumonwo et al., 1991). This impulse controls the heart rate and rhythm. When rabbit pacemaker cells are isolated from SAN tissue, their beating interval entropy increases dramatically compared to that when these cells reside in SAN tissue (Figure 1B), and fractal-like behavior of AP BIs, a feature that characterizes AP BI of SAN tissue, is absent in isolated single pacemaker cells (Yaniv et al., 2014a).
An increase in pacemaker cells AP BIV, or in mathematical terms coupled-clock-system entropy, above a certain threshold leads to abnormal impulse generation by the SAN that is defined as arrhythmia. Two regimes of heart rate variability (HRV) are analyzed in patients with arrhythmogenic events: during the events when the entropy of the system increases (Costa et al., 2008), and before the occurrence of arrhythmia. The occurrence of major arrhythmic events in patients with right ventricular cardiomyopathy is associated with a reduced BIV (Battipaglia et al., 2012). Interestingly, heart rate variability indexes decrease just prior to an arrhythmogenic event (Postolache et al., 2011). As we summarized here, the degree of synchronization of intrinsic mechanisms to pacemaker cells and the degree of synchronization among pacemaker cells within the SAN are determinants of the heart rate and rhythm. Autonomic receptors on pacemaker cells respond to the imbalances of autonomic impulses associated with cardiac diseases. Specifically, autonomic receptor stimulation of single pacemaker cells alters their beat-to-beat variability. Thus, intrinsic pacemaker mechanisms may contribute to HRV in vivo. Although mechanisms of HRV may vary from one patient to another, documentation of the relationship between HRV and different arrhythmias in human patients (Table 1) is an important initial step to conceptually link intrinsic pacemaker mechanisms to arrhythmogenic events. Note, that this sort of evidence, however, does not prove that altered synchronization of pacemaker clock mechanisms residing within the SAN are the sole cause of all patient arrhythmias that may be linked to changes in HRV.
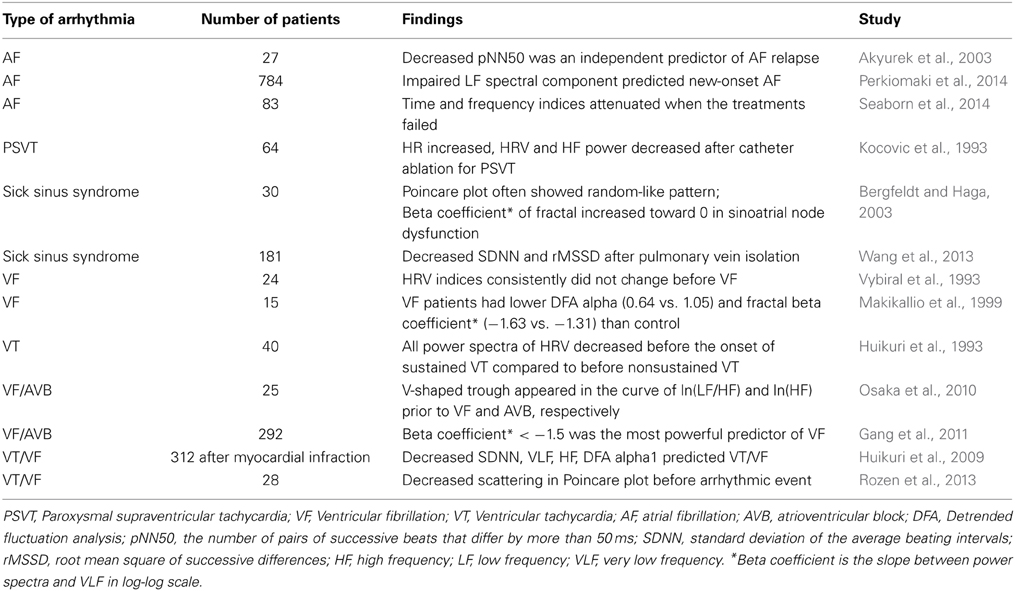
Table 1. Summary of primary studies assessing the changes of heart rate variability when arrhythmia occurs.
Direct pharmacological inhibition of coupled-clock proteins can induce arrhythmias. For example: (i) caging of intracellular Ca2+ by NP-EGTA in isolated rabbit pacemaker cells induces an increase in LCR variability and AP BI bradycardia together with arrhythmic events (Yaniv et al., 2011). (ii) A sudden increase in stochastic ryanodine receptor open probability, elicited by caffeine spritz in isolated rabbit pacemaker cells, induces tachycardia, together with arrhythmic events (Yaniv et al., 2013d). (iii) Specific PKA inhibitors (Younes et al., 2008) or CaMKII inhibitors (Yaniv et al., 2013c) superfused onto isolated rabbit pacemaker cells induce AP BI bradycardia together with arrhythmic events. (iv) Perturbing clock coupling in rabbit pacemaker cells by directly inhibiting either the M (ivabradine, an If inhibitor) or Ca2+ clock (cyclopiazonic acid, a SR Ca2+ pump inhibitor) produces increases in AP BI and AP BIV that are related to increases in LCR period and LCR period variability (Yaniv et al., 2014b). These results provide evidence that supports the coupled-clock theory, demonstrating the ability of the LCR Ca2+ signal to report the degree of synchronization between the two clocks, and how changes in the degree of synchronization lead to changes in AP BI and AP BIV.
Similar to direct pharmacological inhibitors of coupled-clock proteins, mutation and genetically induced gene deletion of different components of the coupled-clock system are associated with arrhythmias in vivo.
The hyperpolarization-activated channel (If) consists of three HCN members (HCN1, HCN2, and HCN4) (Ludwig et al., 1998). HCN4 comprises the major fraction (70–80%) of SAN If. Various mutations of human HCN4 channels are associated with arrhythmias, and with bradycardia in particular (Yeh et al., 2009; Schweizer et al., 2010; Duhme et al., 2013). Interestingly, the spontaneous cardiac beating rate of HCN4-knockout embryos is significantly slower than that of wild-type, but no arrhythmic events are observed. These results are in contrast to the conditional deletion of HCN4 in adult animals, where bradycardia is not evident but sinus pauses are detected (Stieber et al., 2004; Herrmann et al., 2007; Yeh et al., 2009) (Figure 1D). Other HCN transcripts that compose funny current channel in the mouse are HCN2, and a low level of HCN1. HCN2-deficient mice display mild cardiac dysrhythmia, both in the presence and absence of autonomic control of the heart rate (Stieber et al., 2004; Herrmann et al., 2007). Similarly, HCN1-deficient mice exhibit sinus dysrhythmia in vivo and in single isolated cells (Fenske et al., 2013). Interestingly, HCN1-deficient mice exhibit high beat-to-beat dispersion (quantified by Poincaré plots) that is typically observed in SAN dysfunction (Fenske et al., 2013). Therefore, HCN4 apparently is required to protect the heart from severe bradycardia and HCN2 and HCN1 are required to prevent arrhythmias.
Ivabradine is a specific If blocker that reduces the heart rate in patients, specifically patients with inappropriate sinus tachycardia to eliminate arrhythmia (Cappato et al., 2012). However, a recent study in isolated rabbit pacemaker cells demonstrated that ivabradine, even at a concentration that specifically inhibits If, but does not directly suppress L-type current, SR Ca2+ cycling and other surface membrane ion channels, indirectly suppresses intracellular Ca2+ cycling (Yaniv et al., 2012, 2013b). The reduction in arrhythmic events is therefore likely due to drug effect on synchronization of functions within the coupled-clock system, and not simply to Ifinhibition, per se (Yaniv and Lakatta, 2013).
Voltage-gated Cav1 channels (Cav1.2 and Cav1.3) mediate L-type Ca2+ channels that play distinct roles in mediating Ca2+ balance in the pacemaker cell. Both Cav1.2 and Cav1.3 channels are expressed in SAN, and Cav1.3 expression in the atria and SAN cells is higher than in ventricular myocytes (Marger et al., 2011). Cav1.3 current is activated faster and at more negative voltages than Cav1.2 current and, therefore, in mice can contribute earlier during the diastolic depolarization (Christel et al., 2012). Bradycardia and arrhythmia are particularly prominent in Cav1.3 knockout mouse pacemaker cells (Mangoni et al., 2003). Loss of function of Cav1.3 both in mice and humans causes sick sinus syndrome (see above) and is characterized by severe bradycardia (Platzer et al., 2000; Baig et al., 2011). Moreover, patients with a mutation in the CACNA1D gene, which encodes the pore-forming α1 subunit of Cav1.3, experience pronounced bradycardia in 12–24-h ECG recordings, and their HRV time-domain indices are increased (Baig et al., 2011) (Figure 1D). Note that functional significance of Cav1.3 in large mammals has yet to be demonstrated.
The Na+/Ca2+ exchanger has two important roles in pacemaker cells: it not only maintains the cell Ca2+ balance by matching Ca2+ efflux to Ca2+ influx through the L-type Ca2+ current, but also contributes to diastolic depolarization (Maltsev et al., 2014). Specific knockout of SAN Na+/Ca2+ exchanger mice induces bradycardia and increases BIV in the proportion of mycoytes that express arrhythmic AP BI compared to control mice (Herrmann et al., 2013) (Figure 1D). Interestingly, numerical model simulations predict that only a reduction in Na+/Ca2+ exchanger density to below a specific threshold is accompanied by arrhythmic AP BI (Maltsev et al., 2013).
TRPM4 is a monovalent nonselective cation channel permeable to Na+, K+, and Li+, but not to Ca2+ (Launay et al., 2002). Activation of TRPM4 channels that exist in murine pacemaker cells is achieved by both membrane depolarization and by a rise in intracellular Ca2+ (Hof et al., 2013). Although TRPM4 KO mice have heart rates similar to those of their controls, they exhibit a higher incidence of sinus pauses (Hof et al., 2013).
The stochasticity of spontaneous RyR activation determines the diastolic LCR Ca2+ signal and therefore the degree of synchronization of intracellular function of the coupled-clock system. A mutation in RyR, exon-3, in patients with catecholaminergic polymorphic ventricular tachycardia, is associated with arrhythmias (Bhuiyan et al., 2007) (Figure 1D). Isolated pacemaker cells from mice that express this mutation have a prolonged average AP BI with pauses between AP BIs together with an impaired chronotropic response to β adrenergic stimulation (Neco et al., 2012). Similarly, in inducible, heart tissue-specific RyR2 knockout mice, both in vivo ECG telemetry and in vitro isolated perfused heart, demonstrated bradycardic BI and arrhythmia (Bround et al., 2012).
The Shox2 transcriptional factor has been identified as a key regulator in pacemaker formation and differentiation (Liu et al., 2011). Shox2 gene KO mice have a significantly reduced heart beat rate and increased number of arrhythmic events (Liu et al., 2011) (Figure 1D). Moreover, deficient Shox2 transcription factor during development may cause abnormal of mouse SAN development associated with severe arrhythmias (Hoffmann et al., 2013). Therefore, the Shox2 gene also appears to be critical for normal pacemaking function. Other transcription factors than Shox2 are involved in pacemaker function. In this regard, expression of Tbx18 in guinea pig has been shown as an essential gene whose expression can convert quiescent cardiomyocytes to pacemaker cells (Kapoor et al., 2013), therefore, increasing the pacemaker-induced spontaneous beating rate of the cells and decreasing their BIV. Interestingly, Tbx18 transduction to the guinea pig embryonic cell lineage inhibits Cx43 expression, leading to significant electrical uncoupling (Kapoor et al., 2011).
Ankyrins are adaptor proteins that are required for targeting channels and transporters in pacemaker cells to the membranes in which they function. Human patients with ankyrin-B-deficiency have highly penetrant sinus node dysfunction coupled with increased susceptibility to spontaneous and inducible atrial fibrillation (Le Scouarnec et al., 2008). Interestingly, ankyrin-B -deficient mice also have reduced expression of Na+/Ca2+ exchanger and Na+/K+ ATPase (Le Scouarnec et al., 2008). Finally, cells isolated from ankyrin-B-deficient mice have increased BIV (Cunha et al., 2011) (Figure 1D). Thus, down regulation of ankyrin-B induces abnormal membrane organization that is implicated in a reduced efficiency of pacemaker clock coupling that causes abnormal electrical activity within SAN.
As described above, the BIs of pacemaker cells residing in the SAN become entrained by electrotonic and mechanical cell-to-cell interactions within the tissue (Jalife, 1984; Watanabe et al., 1995). Numerical model simulations predict that cardiac arrhythmias can occur when normal coupling between pacemaker cells in SAN tissue is perturbed (Ostborn et al., 2001). Cardiac diseases, and specifically sick sinus syndrome, are associated with reduction in cell-cell junctional proteins (Dobrzynski et al., 2007). ECG of mice with a cardiac conduction-specific knockout of desmoplakin, a protein that affects mechanical cell-to-cell interaction in the cardiac conduction system, exhibits sinus arrhythmias characterized by a strikingly increased number of sinus pauses compared to wild-type mice (Sheikh et al., 2013).
Although connexin43 is absent in the center of the pacemaker tissue, it is expressed in the peripheral area. A reduction in connexin 43 in aged guinea pig SAN is associated with a reduction in heart rate and an increase in arrhythmogenic events (Jones et al., 2004). Moreover, pacemaker tissue contains functional gap junctions and connecting cardiac fibroblasts (Camelliti et al., 2004). Because an increase in fibroblasts expression can slow the generation of pacemaker excitability, it may be involved in bradycardia and sick sinus syndrome (for review see Kohl et al., 2005). A detailed review of the role of passive pacemaker tissue properties on its electrical conductance is present in this issue (Unudurthi et al., 2014).
High-resolution optical mapping of SAN tissue has helped to resolve how reduced synchronization of activity across populations of cells within the SAN can induce arrhythmia. In this regard, different intrinsic mechanisms can be involved in tachy-brady heart-rate alteration and exit block that leads to long sinus pauses and increases susceptibility to cardiac arrhythmias: (i) an increase in adenosine level in human SAN tissue, an endogenous metabolite of the heart, through adenosine A1 receptor upregulating, can lead to SAN dysfunction (Lou et al., 2013, 2014); (ii) an increase in B-type and C-type natriuretic peptides increase the mice SAN conduction velocity and shift the initial exit site (Azer et al., 2014). (iii) Mutation in Ca2+-binding protein calsequestrin 2 is associated with different cardiac diseases. In calsequestrin knockout mice the SAN exhibits bradycardia, conduction abnormalities and increase beat-to-beat variability (Glukhov et al., 2013). (iv) Ganglion nerve plexi can stimulate the intrinsic cardiac nervous system. In mice pulmonary vein ganglia stimulation shifts the origin of SAN discharges and decreases the heart rate (Zarzoso et al., 2013). (v) Impaired SR function in canine failing hearts results in an impaired shift in the location of the pacemaker site in response to β-AR stimulation (Shinohara et al., 2010).
Changes in heart rate and rhythm are harbingers of the appearance of arrhythmogenic events. Reduction in the degree of synchronization of any intrinsic clock functions of pacemaker cells or in the synchronization among pacemaker cells residing in the SAN can be associated with arrhythmia occurrence. The extent to which restoring normal synchronization of intrinsic clock periods within pacemaker cells and among pacemaker cells can prevent arrhythmogenic events awaits further elucidation. In our opinion, future work requires a focus on the connection between dysfunction of inherent intrinsic mechanisms associated with different cardiac diseases and cardiac arrhythmias. This connection can be explored in genetically manipulated mouse models, in animals like rabbit, dog and sheep with heart failure or atrial fibrillation, and in human-derived cardiomyocytes or human SAN. This knowledge will contribute greatly to our understanding of cardiac impulse initiation in health and in cardiac disease.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was partially supported by the Intramural Research Program of the NIH, National Institute on Aging, by Technion VPR Fund –Krbling Biomedical Engineering Research Fund (YY) and by NSFC-ISF joint research program, No. 398/14 (YY).
Akyurek, O., Diker, E., Guldal, M., and Oral, D. (2003). Predictive value of heart rate variability for the recurrence of chronic atrial fibrillation after electrical cardioversion. Clin. Cardiol. 26, 196–200. doi: 10.1002/clc.4960260411
Anumonwo, J. M., Delmar, M., Vinet, A., Michaels, D. C., and Jalife, J. (1991). Phase resetting and entrainment of pacemaker activity in single sinus nodal cells. Circ. Res. 68, 1138–1153. doi: 10.1161/01.RES.68.4.1138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Azer, J., Hua, R., Krishnaswamy, P. S., and Rose, R. A. (2014). Effects of natriuretic peptides on electrical conduction in the sinoatrial node and atrial myocardium of the heart. J. Physiol. 592, 1025–1045. doi: 10.1113/jphysiol.2013.265405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baig, S. M., Koschak, A., Lieb, A., Gebhart, M., Dafinger, C., Nurnberg, G., et al. (2011). Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 14, 77–84. doi: 10.1038/nn.2694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Battipaglia, I., Scalone, G., Macchione, A., Pinnacchio, G., Laurito, M., Milo, M., et al. (2012). Association of heart rate variability with arrhythmic events in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ. J. 76, 618–623. doi: 10.1253/circj.CJ-11-1052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bergfeldt, L., and Haga, Y. (2003). Power spectral and Poincare plot characteristics in sinus node dysfunction. J. Appl. Physiol. (1985). 94, 2217–2224. doi: 10.1152/japplphysiol.01037.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhuiyan, Z. A., Van Den Berg, M. P., Van Tintelen, J. P., Bink-Boelkens, M. T., Wiesfeld, A. C., Alders, M., et al. (2007). Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation 116, 1569–1576. doi: 10.1161/CIRCULATIONAHA.107.711606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Birchfield, R. I., Menefee, E. E., and Bryant, G. D. (1957). Disease of the sinoatrial node associated with bradycardia, asystole, syncope, and paroxysmal atrial fibrillation. Circulation 16, 20–26. doi: 10.1161/01.CIR.16.1.20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boyett, M. R., Honjo, H., and Kodama, I. (2000). The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res. 47, 658–687. doi: 10.1016/S0008-6363(00)00135-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bround, M. J., Asghari, P., Wambolt, R. B., Bohunek, L., Smits, C., Philit, M., et al. (2012). Cardiac ryanodine receptors control heart rate and rhythmicity in adult mice. Cardiovasc. Res. 96, 372–380. doi: 10.1093/cvr/cvs260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Camelliti, P., Green, C. R., Legrice, I., and Kohl, P. (2004). Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 94, 828–835. doi: 10.1161/01.RES.0000122382.19400.14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cappato, R., Castelvecchio, S., Ricci, C., Bianco, E., Vitali-Serdoz, L., Gnecchi-Ruscone, T., et al. (2012). Clinical efficacy of ivabradine in patients with inappropriate sinus tachycardia: a prospective, randomized, placebo-controlled, double-blind, crossover evaluation. J. Am. Coll. Cardiol. 60, 1323–1329. doi: 10.1016/j.jacc.2012.06.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Childers, R. W., Arnsdorf, M. F., De La Fuente, D. J., Gambetta, M., and Svenson, R. (1973). Sinus nodal echoes. Clinical case report and canine studies. Am. J. Cardiol. 31, 220–231. doi: 10.1016/0002-9149(73)91035-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christel, C. J., Cardona, N., Mesirca, P., Herrmann, S., Hofmann, F., Striessnig, J., et al. (2012). Distinct localization and modulation of Cav1.2 and Cav1.3 L-type Ca2+ channels in mouse sinoatrial node. J. Physiol. 590, 6327–6342. doi: 10.1113/jphysiol.2012.239954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cossu, S. F., and Steinberg, J. S. (1998). Supraventricular tachyarrhythmias involving the sinus node: clinical and electrophysiologic characteristics. Prog. Cardiovasc. Dis. 41, 51–63. doi: 10.1016/S0033-0620(98)80022-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Costa, M. D., Peng, C. K., and Goldberger, A. L. (2008). Multiscale analysis of heart rate dynamics: entropy and time irreversibility measures. Cardiovasc. Eng. 8, 88–93. doi: 10.1007/s10558-007-9049-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cunha, S. R., Hund, T. J., Hashemi, S., Voigt, N., Li, N., Wright, P., et al. (2011). Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 124, 1212–1222. doi: 10.1161/CIRCULATIONAHA.111.023986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Jong, A. M., Maass, A. H., Oberdorf-Maass, S. U., De Boer, R. A., Van Gilst, W. H., and Van Gelder, I. C. (2013). Cyclical stretch induces structural changes in atrial myocytes. J. Cell. Mol. Med. 17, 743–753. doi: 10.1111/jcmm.12064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Difrancesco, D. (1993). Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 55, 455–472. doi: 10.1146/annurev.ph.55.030193.002323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dobrzynski, H., Boyett, M. R., and Anderson, R. H. (2007). New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115, 1921–1932. doi: 10.1161/CIRCULATIONAHA.106.616011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Duhme, N., Schweizer, P. A., Thomas, D., Becker, R., Schroter, J., Barends, T. R., et al. (2013). Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur. Heart J. 34, 2768–2775. doi: 10.1093/eurheartj/ehs391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fenske, S., Krause, S. C., Hassan, S. I., Becirovic, E., Auer, F., Bernard, R., et al. (2013). Sick sinus syndrome in HCN1-deficient mice. Circulation 128, 2585–2594. doi: 10.1161/CIRCULATIONAHA.113.003712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gang, U. J., Jons, C., Jorgensen, R. M., Abildstrom, S. Z., Messier, M. D., Haarbo, J., et al. (2011). Risk markers of late high-degree atrioventricular block in patients with left ventricular dysfunction after an acute myocardial infarction: a CARISMA substudy. Europace 13, 1471–1477. doi: 10.1093/europace/eur165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Glukhov, A. V., Kalyanasundaram, A., Lou, Q., Hage, L. T., Hansen, B. J., Belevych, A. E., et al. (2013). Calsequestrin 2 deletion causes sinoatrial node dysfunction and atrial arrhythmias associated with altered sarcoplasmic reticulum calcium cycling and degenerative fibrosis within the mouse atrial pacemaker complex. Eur. Heart J. doi: 10.1093/eurheartj/eht452. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gomes, J. A., Mehta, D., and Langan, M. N. (1995). Sinus node reentrant tachycardia. Pacing Clin. Electrophysiol. 18, 1045–1057. doi: 10.1111/j.1540-8159.1995.tb04747.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, J., Malozzi, A. M., Moe, G. K. (1968). Sino-atrial reciprocation in the isolated rabbit heart. Circ. Res. 22, 355–362.
Herrmann, S., Lipp, P., Wiesen, K., Stieber, J., Nguyen, H., Kaiser, E., et al. (2013). The cardiac sodium-calcium exchanger NCX1 is a key player in the initiation and maintenance of a stable heart rhythm. Cardiovasc. Res. 99, 780–788. doi: 10.1093/cvr/cvt154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herrmann, S., Stieber, J., Stockl, G., Hofmann, F., and Ludwig, A. (2007). HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 26, 4423–4432. doi: 10.1038/sj.emboj.7601868
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hof, T., Simard, C., Rouet, R., Salle, L., and Guinamard, R. (2013). Implication of the TRPM4 nonselective cation channel in mammalian sinus rhythm. Heart Rhythm 10, 1683–1689. doi: 10.1016/j.hrthm.2013.08.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoffmann, S., Berger, I. M., Glaser, A., Bacon, C., Li, L., Gretz, N., et al. (2013). Islet1 is a direct transcriptional target of the homeodomain transcription factor Shox2 and rescues the Shox2-mediated bradycardia. Basic Res. Cardiol. 108, 339. doi: 10.1007/s00395-013-0339-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huikuri, H. V., Raatikainen, M. J., Moerch-Joergensen, R., Hartikainen, J., Virtanen, V., Boland, J., et al. (2009). Prediction of fatal or near-fatal cardiac arrhythmia events in patients with depressed left ventricular function after an acute myocardial infarction. Eur. Heart J. 30, 689–698. doi: 10.1093/eurheartj/ehn537
Huikuri, H. V., Valkama, J. O., Airaksinen, K. E., Seppanen, T., Kessler, K. M., Takkunen, J. T., et al. (1993). Frequency domain measures of heart rate variability before the onset of nonsustained and sustained ventricular tachycardia in patients with coronary artery disease. Circulation 87, 1220–1228. doi: 10.1161/01.CIR.87.4.1220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jalife, J. (1984). Mutual entrainment and electrical coupling as mechanisms for synchronous firing of rabbit sino-atrial pace-maker cells. J. Physiol. 356, 221–243. doi: 10.1113/jphysiol.1984.sp015461
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, S. A., Lancaster, M. K., and Boyett, M. R. (2004). Ageing-related changes of connexins and conduction within the sinoatrial node. J. Physiol. 560, 429–437. doi: 10.1113/jphysiol.2004.072108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kapoor, N., Galang, G., Marban, E., and Cho, H. C. (2011). Transcriptional suppression of connexin43 by TBX18 undermines cell-cell electrical coupling in postnatal cardiomyocytes. J. Biol. Chem. 286, 14073–14079. doi: 10.1074/jbc.M110.185298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kapoor, N., Liang, W., Marban, E., and Cho, H. C. (2013). Direct conversion of quiescent cardiomyocytes to pacemaker cells by expression of Tbx18. Nat. Biotechnol. 31, 54–62. doi: 10.1038/nbt.2465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kocovic, D. Z., Harada, T., Shea, J. B., Soroff, D., and Friedman, P. L. (1993). Alterations of heart rate and of heart rate variability after radiofrequency catheter ablation of supraventricular tachycardia. Delineation of parasympathetic pathways in the human heart. Circulation 88, 1671–1681. doi: 10.1161/01.CIR.88.4.1671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kohl, P., Camelliti, P., Burton, F. L., and Smith, G. L. (2005). Electrical coupling of fibroblasts and myocytes: relevance for cardiac propagation. J. Electrocardiol. 38, 45–50. doi: 10.1016/j.jelectrocard.2005.06.096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Launay, P., Fleig, A., Perraud, A. L., Scharenberg, A. M., Penner, R., and Kinet, J. P. (2002). TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 109, 397–407. doi: 10.1016/S0092-8674(02)00719-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Le Scouarnec, S., Bhasin, N., Vieyres, C., Hund, T. J., Cunha, S. R., Koval, O., et al. (2008). Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc. Natl. Acad. Sci. U.S.A. 105, 15617–15622. doi: 10.1073/pnas.0805500105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, H., Chen, C. H., Espinoza-Lewis, R. A., Jiao, Z., Sheu, I., Hu, X., et al. (2011). Functional redundancy between human SHOX and mouse Shox2 genes in the regulation of sinoatrial node formation and pacemaking function. J. Biol. Chem. 286, 17029–17038. doi: 10.1074/jbc.M111.234252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lou, Q., Glukhov, A. V., Hansen, B., Hage, L., Vargas-Pinto, P., Billman, G. E., et al. (2013). Tachy-brady arrhythmias: the critical role of adenosine-induced sinoatrial conduction block in post-tachycardia pauses. Heart Rhythm 10, 110–118. doi: 10.1016/j.hrthm.2012.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lou, Q., Hansen, B. J., Fedorenko, O., Csepe, T. A., Kalyanasundaram, A., Li, N., et al. (2014). Upregulation of adenosine A1 receptors facilitates sinoatrial node dysfunction in chronic canine heart failure by exacerbating nodal conduction abnormalities revealed by novel dual-sided intramural optical mapping. Circulation 130, 315–324. doi: 10.1161/CIRCULATIONAHA.113.007086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ludwig, A., Zong, X., Jeglitsch, M., Hofmann, F., and Biel, M. (1998). A family of hyperpolarization-activated mammalian cation channels. Nature 393, 587–591. doi: 10.1038/31255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Makikallio, T. H., Koistinen, J., Jordaens, L., Tulppo, M. P., Wood, N., Golosarsky, B., et al. (1999). Heart rate dynamics before spontaneous onset of ventricular fibrillation in patients with healed myocardial infarcts. Am. J. Cardiol. 83, 880–884. doi: 10.1016/S0002-9149(98)01068-6
Maltsev, A. V., Yaniv, Y., Stern, M. D., Lakatta, E. G., and Maltsev, V. A. (2013). RyR-NCX-SERCA local cross-talk ensures pacemaker cell function at rest and during the fight-or-flight reflex. Circ. Res. 113, e94–e100. doi: 10.1161/CIRCRESAHA.113.302465
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maltsev, V. A., Yaniv, Y., Maltsev, A. V., Stern, M. D., and Lakatta, E. G. (2014). Modern perspectives on numerical modeling of cardiac pacemaker cell. J. Pharmacol. Sci. 125, 6–38. doi: 10.1254/jphs.13R04CR
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mangoni, M. E., Couette, B., Bourinet, E., Platzer, J., Reimer, D., Striessnig, J., et al. (2003). Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc. Natl. Acad. Sci. U.S.A. 100, 5543–5548. doi: 10.1073/pnas.0935295100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marger, L., Mesirca, P., Alig, J., Torrente, A., Dubel, S., Engeland, B., et al. (2011). Functional roles of Ca(v)1.3, Ca(v)3.1 and HCN channels in automaticity of mouse atrioventricular cells: insights into the atrioventricular pacemaker mechanism. Channels (Austin) 5, 251–261. doi: 10.4161/chan.5.3.15266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monfredi, O., Lyashkov, A. E., Johnsen, A. B., Inada, S., Schneider, H., Wang, R., et al. (2014). Biophysical characterization of the underappreciated and important relationship between heart rate variability and heart rate. Hypertension 64, 1334–1343. doi: 10.1161/HYPERTENSIONAHA.114.03782
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monfredi, O., Maltseva, L. A., Spurgeon, H. A., Boyett, M. R., Lakatta, E. G., and Maltsev, V. A. (2013). Beat-to-beat variation in periodicity of local calcium releases contributes to intrinsic variations of spontaneous cycle length in isolated single sinoatrial node cells. PLoS ONE 8:e67247. doi: 10.1371/journal.pone.0067247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neco, P., Torrente, A. G., Mesirca, P., Zorio, E., Liu, N., Priori, S. G., et al. (2012). Paradoxical effect of increased diastolic Ca(2+) release and decreased sinoatrial node activity in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation 126, 392–401. doi: 10.1161/CIRCULATIONAHA.111.075382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Osaka, M., Watanabe, E., Murata, H., Fuwamoto, Y., Nanba, S., Sakai, K., et al. (2010). V-shaped trough in autonomic activity is a possible precursor of life-threatening cardiac events. Circ. J. 74, 1906–1915. doi: 10.1253/circj.CJ-09-0935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ostborn, P., Wohlfart, B., Ohlén, G. (2001). Arrhythmia as a result of poor intercellular coupling in the sinus node: a simulation study. J. Theor. Biol. 211, 201–217. doi: 10.1006/jtbi.2001.2339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Papaioannou, V. E., Verkerk, A. O., Amin, A. S., and De Bakker, J. M. (2013). Intracardiac origin of heart rate variability, pacemaker funny current and their possible association with critical illness. Curr. Cardiol. Rev. 9, 82–96. doi: 10.2174/157340313805076359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perkiomaki, J., Ukkola, O., Kiviniemi, A., Tulppo, M., Ylitalo, A., Kesaniemi, Y. A., et al. (2014). Heart rate variability findings as a predictor of atrial fibrillation in middle-aged population. J. Cardiovasc. Electrophysiol. 25, 719–724. doi: 10.1111/jce.12402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Platzer, J., Engel, J., Schrott-Fischer, A., Stephan, K., Bova, S., Chen, H., et al. (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102, 89–97. doi: 10.1016/S0092-8674(00)00013-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Postolache, G., Oliveira, M., Rocha, I., Girao, P. S., and Postolache, O. (2011). New insight into arrhythmia onset using HRV and BPV analysis. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2011, 2691–2694. doi: 10.1109/IEMBS.2011.6090739
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rocchetti, M., Malfatto, G., Lombardi, F., and Zaza, A. (2000). Role of the input/output relation of sinoatrial myocytes in cholinergic modulation of heart rate variability. J. Cardiovasc. Electrophysiol. 11, 522–530. doi: 10.1111/j.1540-8167.2000.tb00005.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rozen, G., Kobo, R., Beinart, R., Feldman, S., Sapunar, M., Luria, D., et al. (2013). Multipole analysis of heart rate variability as a predictor of imminent ventricular arrhythmias in ICD patients. Pacing Clin. Electrophysiol. 36, 1342–1347. doi: 10.1111/pace.12180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schweizer, P. A., Duhme, N., Thomas, D., Becker, R., Zehelein, J., Draguhn, A., et al. (2010). cAMP sensitivity of HCN pacemaker channels determines basal heart rate but is not critical for autonomic rate control. Circ. Arrhythm. Electrophysiol. 3, 542–552. doi: 10.1161/CIRCEP.110.949768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seaborn, G. E., Todd, K., Michael, K. A., Baranchuk, A., Abdollah, H., Simpson, C. S., et al. (2014). Heart rate variability and procedural outcome in catheter ablation for atrial fibrillation. Ann. Noninvasive Electrocardiol. 19, 23–33. doi: 10.1111/anec.12098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sheikh, F., Mezzano, V., Wright, A., Zanella, F., Lyon, R. S., Gu, Y., et al. (2013). A novel role for mechanical cell-cell junction proteins in sinus node function. J. Mol. Cell. Cardiol. 65.
Shinohara, T., Park, H. W., Han, S., Shen, M. J., Maruyama, M., Kim, D., et al. (2010). Ca2+ clock malfunction in a canine model of pacing-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 299, H1805–H1811. doi: 10.1152/ajpheart.00723.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Slovut, D. P., Wenstrom, J. C., Moeckel, R. B., Wilson, R. F., Osborn, J. W., and Abrams, J. H. (1998). Respiratory sinus dysrhythmia persists in transplanted human hearts following autonomic blockade. Clin. Exp. Pharmacol. Physiol. 25, 322–330. doi: 10.1111/j.1440-1681.1998.tb02358.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stieber, J., Hofmann, F., and Ludwig, A. (2004). Pacemaker channels and sinus node arrhythmia. Trends Cardiovasc. Med. 14, 23–28. doi: 10.1016/j.tcm.2003.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Unudurthi, S. D., Wolf, R. M., and Hund, T. J. (2014). Role of sinoatrial node architecture in maintaining a balanced source-sink relationship and synchronous cardiac pacemaking. Front. Physiol. 5:446. doi: 10.3389/fphys.2014.00446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verheijck, E. E., Wilders, R., Joyner, R. W., Golod, D. A., Kumar, R., Jongsma, H. J., et al. (1998). Pacemaker synchronization of electrically coupled rabbit sinoatrial node cells. J. Gen. Physiol. 111, 95–112. doi: 10.1085/jgp.111.1.95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vybiral, T., Glaeser, D. H., Goldberger, A. L., Rigney, D. R., Hess, K. R., Mietus, J., et al. (1993). Conventional heart rate variability analysis of ambulatory electrocardiographic recordings fails to predict imminent ventricular fibrillation. J. Am. Coll. Cardiol. 22, 557–565. doi: 10.1016/0735-1097(93)90064-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, K., Chang, D., Chu, Z., Yang, Y., Gao, L., Zhang, S., et al. (2013). Denervation as a common mechanism underlying different pulmonary vein isolation strategies for paroxysmal atrial fibrillation: evidenced by heart rate variability after ablation. Sci. World J. 2013:569564. doi: 10.1155/2013/569564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watanabe, E. I., Honjo, H., Anno, T., Boyett, M. R., Kodama, I., and Toyama, J. (1995). Modulation of pacemaker activity of sinoatrial node cells by electrical load imposed by an atrial cell model. Am. J. Physiol. 269, H1735–H1742.
Yaniv, Y., Ahmet, I., Liu, J., Lyashkov, A. E., Guiriba, T. R., Okamoto, Y., et al. (2014a). Synchronization of sinoatrial node pacemaker cell clocks and its autonomic modulation impart complexity to heart beating intervals. Heart Rhythm 11, 1210–1219. doi: 10.1016/j.hrthm.2014.03.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yaniv, Y., and Lakatta, E. G. (2013). Pacemaker gene mutations, bradycardia, arrhythmias and the coupled clock theory. J. Cardiovasc. Electrophysiol. 24, E28–E29. doi: 10.1111/jce.12236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yaniv, Y., Lyashkov, A. E., and Lakatta, E. G. (2013a). The fractal-like complexity of heart rate variability beyond neurotransmitters and autonomic receptors: signaling intrinsic to sinoatrial node pacemaker cells. Cardiovasc. Pharmacol. 2:111. doi: 10.4172/2329-6607.1000111
Yaniv, Y., Lyashkov, A. E., Sirenko, S., Okamoto, Y., Guiriba, T. R., Ziman, B. D., et al. (2014b). Stochasticity intrinsic to coupled-clock mechanisms underlies beat-to-beat variability of spontaneous action potential firing in sinoatrial node pacemaker cells. J. Mol. Cell. Cardiol. 77, 1–10. doi: 10.1016/j.yjmcc.2014.09.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yaniv, Y., Maltsev, V. A., Escobar, A. L., Spurgeon, H. A., Ziman, B. D., Stern, M. D., et al. (2011). Beat-to-beat Ca(2+)-dependent regulation of sinoatrial nodal pacemaker cell rate and rhythm. J. Mol. Cell. Cardiol. 51, 902–905. doi: 10.1016/j.yjmcc.2011.08.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yaniv, Y., Maltsev, V. A., Ziman, B. D., Spurgeon, H. A., and Lakatta, E. G. (2012). The “Funny” current inhibition by ivabradine at membrane potentials encompassing spontaneous depolarization in pacemaker cells. Molecules 17, 8241–8254. doi: 10.3390/molecules17078241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yaniv, Y., Sirenko, S., Ziman, B. D., Spurgeon, H. A., Maltsev, V. A., and Lakatta, E. G. (2013b). New evidence for coupled clock regulation of the normal automaticity of sinoatrial nodal pacemaker cells: bradycardic effects of ivabradine are linked to suppression of intracellular Ca(2)(+) cycling. J. Mol. Cell. Cardiol. 62, 80–89. doi: 10.1016/j.yjmcc.2013.04.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yaniv, Y., Spurgeon, H. A., Ziman, B. D., and Lakatta, E. G. (2013c). Ca(2)+/calmodulin-dependent protein kinase II (CaMKII) activity and sinoatrial nodal pacemaker cell energetics. PLoS ONE 8:e57079. doi: 10.1371/journal.pone.0057079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yaniv, Y., Stern, M. D., Lakatta, E. G., and Maltsev, V. A. (2013d). Mechanisms of beat-to-beat regulation of cardiac pacemaker cell function by Ca(2)(+) cycling dynamics. Biophys. J. 105, 1551–1561. doi: 10.1016/j.bpj.2013.08.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yeh, Y. H., Burstein, B., Qi, X. Y., Sakabe, M., Chartier, D., Comtois, P., et al. (2009). Funny current downregulation and sinus node dysfunction associated with atrial tachyarrhythmia: a molecular basis for tachycardia-bradycardia syndrome. Circulation 119, 1576–1585. doi: 10.1161/CIRCULATIONAHA.108.789677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Younes, A., Lyashkov, A. E., Graham, D., Sheydina, A., Volkova, M. V., Mitsak, M., et al. (2008). Ca(2+) -stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J. Biol. Chem. 283, 14461–14468. doi: 10.1074/jbc.M707540200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zarzoso, M., Rysevaite, K., Milstein, M. L., Calvo, C. J., Kean, A. C., Atienza, F., et al. (2013). Nerves projecting from the intrinsic cardiac ganglia of the pulmonary veins modulate sinoatrial node pacemaker function. Cardiovasc. Res. 99, 566–575. doi: 10.1093/cvr/cvt081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zaza, A., and Lombardi, F. (2001). Autonomic indexes based on the analysis of heart rate variability: a view from the sinus node. Cardiovasc. Res. 50, 434–442. doi: 10.1016/S0008-6363(01)00240-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: arrhythmias, atrial fibrillation, coupled-clock pacemaker system, heart rate variability, sinus node disease
Citation: Yaniv Y, Tsutsui K and Lakatta EG (2015) Potential effects of intrinsic heart pacemaker cell mechanisms on dysrhythmic cardiac action potential firing. Front. Physiol. 6:47. doi: 10.3389/fphys.2015.00047
Received: 15 November 2014; Accepted: 03 February 2015;
Published online: 23 February 2015.
Edited by:
George E. Billman, The Ohio State University, USAReviewed by:
Vadim V. Fedorov, The Ohio State University, USACopyright © 2015 Yaniv, Tsutsui and Lakatta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yael Yaniv, Biomedical Engineering Faculty, Technion-Israel Institute of Technology, 3200003 Haifa, Israel e-mail:eWFlbHlAYm0udGVjaG5pb24uYWMuaWw=;
Edward G. Lakatta, Laboratory of Cardiovascular Science, Biomedical Research Center, Intramural Research Program, National Institute on Aging, NIH, 251 Bayview Blvd, Baltimore, 21224 MD, USA e-mail:bGFrYXR0YWVAZ3JjLm5pYS5uaWguZ292
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.