 Xun Ai
Xun Ai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 19 February 2015
Sec. Cardiac Electrophysiology
Volume 6 - 2015 | https://doi.org/10.3389/fphys.2015.00046
This article is part of the Research TopicCa2+ Signaling and Heart RhythmView all 13 articles
Atrial fibrillation (AF) is the most common sustained arrhythmia. It is associated with a markedly increased risk of premature death due to embolic stroke and also complicates co-existing cardiovascular diseases such as heart failure. The prevalence of AF increases dramatically with age, and aging has been shown to be an independent risk of AF. Due to an aging population in the world, a growing body of AF patients are suffering a diminished quality of life and causing an associated economic burden. However, effective pharmacologic treatments and prevention strategies are lacking due to a poor understanding of the molecular and electrophysiologic mechanisms of AF in the failing and/or aged heart. Recent studies suggest that altered atrial calcium handling contributes to the onset and maintenance of AF. Here we review the role of stress-response kinases and calcium handling dysfunction in AF genesis in the aged and failing heart.
Clinical studies have shown that atrial fibrillation (AF) is the most common cardiac arrhythmia and has an associated high risk of mortality and morbidity (such as stroke and heart failure) in the aging population (Benjamin et al., 1994; Psaty et al., 1997; Podrid, 1999; Go et al., 2001; Miyasaka et al., 2006; Rich, 2009). Both heart failure (HF) and aging have been shown to be independent risk factors for AF (Benjamin et al., 1994; Kannel et al., 1998; Ehrlich et al., 2002; Neuberger et al., 2007). HF affects nearly 15 million people worldwide (Cowie et al., 1997; Hershberger et al., 2003). One third to one half of patients with HF develop AF (Markides and Peters, 2002). New-onset AF among HF patients has consistently been associated with a 2-fold increase in all-cause mortality. Due to an aging population, the prevalence of both AF and HF is predicted to more than double by 2050 (Linne et al., 2000; Di Lenarda et al., 2003). The high prevalence of these multiple co-morbidities (Wang et al., 2003) (e.g., very frequent co-existence of HF, AF, with aging) has tremendous impact on the quality of life and daily functioning of elderly individuals, and is a significant financial burden worldwide (Linne et al., 2000; Di Lenarda et al., 2003). However, pharmacological treatment and prevention strategies remain ineffective due to the incomplete understanding of the underlying molecular and electrophysiologic mechanisms of AF genesis and development.
Accumulating evidence suggests that intrinsic stress (e.g., oxidative stress and chronic inflammatory stress) are markedly enhanced in aging, HF, and AF, while the aged and pathologically altered hearts have been shown to exhibit a higher susceptibility to extrinsic stress stimuli (Belmin et al., 1995; Beckman and Ames, 1998; Juhaszova et al., 2005; Neuman et al., 2007; He et al., 2011; Ismahil et al., 2014). The mitogen-activated protein kinase (MAPK) cascade is composed of a family of signaling cascades, which act as critical regulators of cell survival and growth in response to both intrinsic and extrinsic stress challenges. The three MAPK subfamilies c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinases (ERKs), and p38 MAPKs have been the focus of extensive studies to uncover their roles in cardiac disease development (Davis, 2000; Karin and Gallagher, 2005; Ramos, 2008; Rose et al., 2010). The impacts of these stress-response kinases on sarcoplasmic reticulum (SR) calcium (Ca) handling proteins have begun to be revealed (Ho et al., 1998, 2001; Takahashi et al., 2004; Hagiwara et al., 2007; Scharf et al., 2013; Huang et al., 2014). Extensive studies suggest that alterations of Ca handling proteins including RyR2, phospholamban [PLB, an inhibitory protein of SR Ca pump (SERCA2)], and L-type Ca channels (Cav1.2) contribute to changed intracellular Ca transients and diastolic SR Ca release that in turn lead to Ca-triggered ventricular and atrial arrhythmogenesis (Schulman et al., 1992; Wu et al., 1999; DeSantiago et al., 2002). Thus, this review focuses on the recent progress in understanding the role of stress-response kinases and calcium signaling dysfunction in AF genesis in the aged and failing heart.
It is generally believed that abnormal triggers initiate AF, while an arrhythmogenic substrate sustains it (Nattel et al., 2008). While reentry circuits due to the formation of arrhythmogenic substrate including molecular and structural remodeling have been demonstrated to be important in AF development (Allessie et al., 1976; Mandapati et al., 2000), the underlying mechanisms of AF initiated by abnormal ectopic trigger activities remain unclear. Extensive studies in ventricular myocytes have shown that ectopic activities can occur by prolonged action potential duration (APD) causing early afterdepolarizations (EADs) and by spontaneous SR Ca releases leading to delayed afterdepolarizations (DADs) (Nattel et al., 2008). EADs normally occur with abnormal depolarization during phase 2 or phase 3 of the action potential (AP). While ventricular myocytes can only develop phase 2 EADs, atrial myocytes do not produce phase 2 EAD but may produce late phase 3 EADs with an abbreviation of the atrial APD (Burashnikov and Antzelevitch, 2003; Patterson et al., 2005). Studies suggest that electrical remodeling of atrial membrane ion channels (e.g., Ca and potassium channels) leads to altered APD and atrial effective refractory period (AERP); both have been found to be associated with the development of AF (Marx et al., 2000; Christ et al., 2004; Nattel et al., 2007). Before the onset of AF, shorter AERPs were associated with a higher inducibility of AF, while longer AERPs and slowing atrial conduction velocity, which may cause a pro-arrhythmogeinc shortening of the conduction wavelength, Rensma et al. (1988) were found to be linked to AF development in HF patients and animals (Huang et al., 2003; Sanders et al., 2003). In aged rabbit left atrium, we found that a slight reduction in AERP and unchanged action potential duration (APD30 and APD60; pacing cycle length = 200 ms) were associated with slowed conduction velocity and a markedly increased pacing induced AF compared to that of young controls (Figure 1)(Yan et al., 2013). Although similar results of slightly altered APD and AERP were also reported in aged canine and rat atria, Anyukhovsky et al. (2005) and Huang et al. (2006) studies from coronary artery bypass graft (CABG) surgery patients suggest that AERP was positively correlated with age (Sakabe et al., 2003). However, the molecular and electrophysiological properties of human hearts are known to be varied and complicated, especially when co-existing pathological conditions (such as HF or myocardial infarction) are present. While these results need to be further confirmed in healthy aging human donor hearts and further validated in other animal aging models, studies suggest that atrial electrical remodeling was found to occur long before the first occurrence of AF, and was not always correlated with the occurrence of sustained AF in patients and animal models (van der Velden et al., 2000; Kanagaratnam et al., 2008). In addition, late-phase 3 EADs have only been shown to be responsible for the immediate initiation of AF following termination of paroxysmal AF, but not in the case of newly onset AF or reoccurrence of AF that has been terminated for a long time (Timmermans et al., 1998; Oral et al., 2003). Thus, other features of the arrhythmogenic substrate such as SR Ca handling dysfunction, a generally acknowledged arrhythmogenic factor of generating DADs, could play an important role in failing or age-related enhancement of atrial arrhythmogenicity.
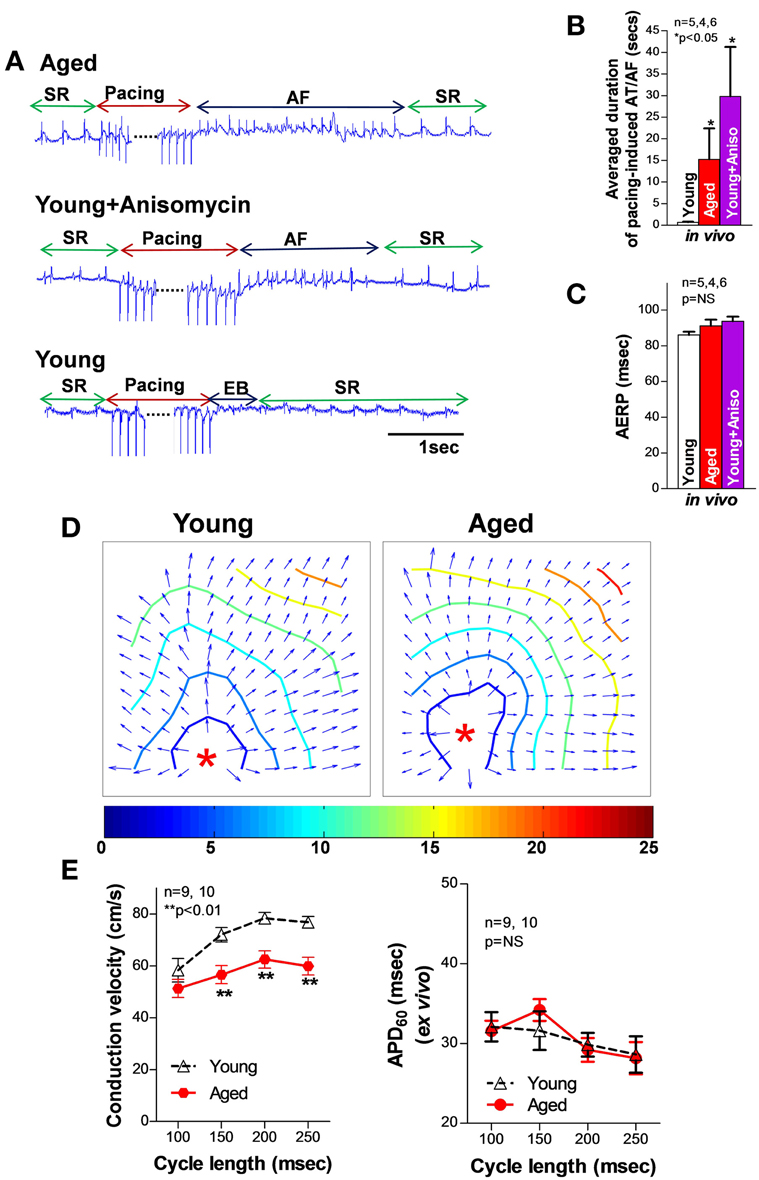
Figure 1. (A) Representative electrograms (EG) of burst pacing (for 30 s at 6× diastolic threshold, CL = ±5 ms of atrial effective refractory period (AERP) induced AF followed by self-reversion to sinus rhythm (SR) in open-chest aged (top row) and anisomycin-challenged young rabbit LA (middle row), and self-restored SR after burst pacing induced extra beats (EB) in sham control young rabbit LA (bottom row). (B–C) Summarized data of average duration of pacing-induced AT/AF as well as AERP in open-chest aged rabbit LA and young rabbit LA with or without anisomycin treatment (*p < 0.05 and p = NS, respectively). (D) Representative isochronal maps from young and aged rabbit hearts subjected to pacing at a CL of 200 ms (π indicates the pacing sites). (E) Summarized optical mapping CV data show that aged rabbits exhibited a CL-dependent, slower conduction but unchanged AERP vs. young controls. This figure is modified from Yan et al. (2013).
Although Ca handling in atrial myocytes is similar to that of ventricular myocytes, there are some important structural and cellular signal differences between atrial and ventricular myocytes. Atrial myocytes are thinner and longer, Walden et al. (2009) which may lead to a longer delay between APs and Ca transients at the center of the cells. This property of the atrial cell can increase the instability of Ca propagation, which is pro-arrhythmogenic. In addition, atrial myocytes exhibit a different Transverse tubules (T-tubules) structure compared to ventricular myocytes. T-tubules are an important sub-cellular network involved in SR Ca dynamics in ventricular myocytes (Wang et al., 2001; Brette and Orchard, 2003; Franzini-Armstrong et al., 2005; Ibrahim et al., 2010). T-tubules are located at the z-line of the myocyte and provide close coupling of L-type Ca channels to ryanodine receptors (RyRs) on the SR membrane. This structure allows rapid intracellular Ca triggered SR Ca release in response to electrical excitation (Franzini-Armstrong et al., 2005). Emerging evidence suggests that an atrial T-tubule network is present in large mammalian species including humans, sheep, dogs, cows, and horses (Dibb et al., 2009; Lenaerts et al., 2009; Wakili et al., 2010; Richards et al., 2011) although atrial T-tubular networks are less abundant and less organized compared to that in the ventricles. While it was previously believed that atrial T-tubules were virtually absent in the small rodents,(Forbes et al., 1990; Berlin, 1995) a recent report by Frisk et al. (2014) showed similar structural organization and density of the T-tubules in pig and rat atria. A disorganized T-tubule network has been found to contribute to SR Ca release dysfunction in failing ventricular myocytes from both human and HF animal models (Balijepalli et al., 2003; Louch et al., 2006; Heinzel et al., 2008; Lyon et al., 2009). In rapid pacing-induced failing dog atria, reduced T-tubular abundance was also found to be linked to altered subcellular Ca dynamics and AF development (Yeh et al., 2008; Dibb et al., 2009; Lenaerts et al., 2009). While accumulating evidence suggests that atrial T-tubular structure is present in most mammalian species, further investigations are clearly needed to understand whether there is remodeling in the failing and aged heart and its functional role in atrial SR Ca handling and AF development.
It is known that the cardiac Ca current during the normal AP contributes to the AP plateau and is involved in myocyte contraction. The voltage-gated L-type Ca channels (ICa) are activated by membrane depolarization that leads to a small amount of inward Ca flux (ICa) (Rougier et al., 1969). Ca entry via Ca current (ICa) along with a much smaller amount of Ca influx via Na-Ca exchange (NCX) activates large quantities of Ca release from SR via ryanodine receptor channels (RyR; also called Ca triggered SR Ca release channels). This Ca triggered SR Ca release involves a transient increase in intracellular Ca [Ca]i that initiates myocyte contraction as free Ca binds to the myofilaments (Bers, 2000). During the relaxation phase of the cells, intracellular free Ca ions will be removed from cytosol via: (1) pumping back to SR via a Ca pump SERCA2 (SR Ca-ATPase); (2) expulsion from the cell by NCXs; and (3) uptake by mitochondria via mitochondrial Ca uniporters (Bers, 2000).
Compared to ventricular myocytes, atrial myocytes have smaller Ca transient amplitude and a higher rate of intracellular Ca decay. This is due to an increased SERCA uptake and enhanced function of NCX to remove cytosolic Ca during the diastolic phase (Walden et al., 2009). The increased SERCA-dependent intracellular Ca removal is attributed to the greater amount of SERCA2 and less expression of SERCA inhibitory protein phospholamban (PLB) (Freestone et al., 2000; Walden et al., 2009). Another important feature of atrial myocytes is that atrial SR Ca content is greater than that of ventricular myocytes (Walden et al., 2009). With the greater atrial SR Ca content, atrial myocytes are prone to spontaneous diastolic SR Ca release when RyR channels are sensitized under pathological conditions (Venetucci et al., 2008; Bers, 2014).
We and others have previously discovered that increased diastolic SR Ca release causes abnormal ectopic activities, which lead to ventricular arrhythmogenesis in the failing heart (Ai et al., 2005; Yeh et al., 2008; Respress et al., 2012). During the diastolic phase, SR Ca release normally shuts off almost completely (~99%). However, increased diastolic RyR Ca release could be responsible for increased diastolic SR Ca leak and reduced systolic [Ca]FR for a given L-type voltage-gated Ca current (Ica) as the release trigger (Bassani et al., 1995; Shannon et al., 2000; Bers, 2014). The increased diastolic SR Ca leakage along with an impaired function of Ca uptake due to altered SERCA2 elevates the amount of [Ca]i and prolongs the [Ca]i decay phase in HF (Bers, 2000, 2014). Then, increased Na influx via NCX for [Ca]i removal can produce abnormal triggered activities (e.g., DADs) and initiate atrial arrhythmias (Bers, 2000, 2014). Studies suggest that alterations of Ca handling proteins including RyR2, PLB, and Cav1.2 contribute to changed intracellular Ca transients and diastolic SR Ca release (Schulman et al., 1992; DeSantiago et al., 2002; Wu et al., 1999). Others and we have previously demonstrated that activated CaMKII, a pro-arrhythmic signaling molecule, is critically involved in phosphorylation of RyR2-2815 and PLB-Thr17 (RyR2815-P, PLB17-P), which results in sensitized RyR channels that in turn leads to triggered activities and arrhythmia initiation due to diastolic SR Ca leak in pathologically altered ventricles (Hoch et al., 1999; Maier et al., 2003; Zhang et al., 2003; Ai et al., 2005; Yeh et al., 2008; Greiser et al., 2009; Sossalla et al., 2010; Respress et al., 2012). Recent studies indicate that alterations of CaMKII-dependent RyR phosphorylation are also exhibited in the atrium of chronic AF patients (Chelu et al., 2009; Neef et al., 2010). Results from several animal models have shown that these altered SR Ca handling proteins contribute to enhanced SR Ca leak and AF development (Chelu et al., 2009; Chiang et al., 2014). Although alteration of ICa could also contribute to abnormal SR Ca release, studies indicate that reduced ICa is a hallmark of AF induced electrical remodeling (Van Wagoner et al., 1999; Christ et al., 2004). CaMKII inhibition has been shown to improve the function of L-type Ca channel in mouse ventricular myocytes and cultured HL-1 atrial myocytes, which could be due to up-regulated expression of L-type Ca channel proteins (Zhang et al., 2005; Ronkainen et al., 2011). These results indicate that abnormal diastolic RyR Ca release could be the major cause of abnormal Ca handling in HF and chronic AF (Ai et al., 2005; Yeh et al., 2008 and Respress et al., 2012). However, other studies have reported inconsistent results of increased, reduced, or unchanged Ica preceding the onset of AF in postoperative patients compared to that of patients at low risk for AF (Van Wagoner et al., 1999; Christ et al., 2004; Dinanian et al., 2008; Workman et al., 2009). Thus, the underlying mechanisms of abnormal Ca handling in AF onset and maintenance in the pathologically altered heart require further investigation.
In addition to altered phosphorylation of Ca handling proteins regulated by kinases, some protein phosphatases (PP1, PP2A) have also been found to play roles in regulating the phosphorylation state of channel proteins in failing ventricular myocytes (Ai et al., 2005, 2011; Ai and Pogwizd, 2005). However, contradictory results of the expression and activity of protein phosphatases have been reported in humans and animal models with chronic AF or paroxysmal AF (Christ et al., 2004; Chelu et al., 2009; Heijman et al., 2013; Voigt et al., 2014). It is clear that the functional role of protein phosphatases in atrial Ca handling and AF genesis need to be further explored.
It has been shown that failing and aged hearts exhibit increased intrinsic stress and higher susceptibility to extrinsic stress stimuli (Belmin et al., 1995; Beckman and Ames, 1998; Juhaszova et al., 2005; Li et al., 2005a; Yang et al., 2005; Judge and Leeuwenburgh, 2007; Neuman et al., 2007; He et al., 2011; Ismahil et al., 2014). JNK, a family member of the MAPKs, was discovered by Davis in the early of 90s (Davis, 2000). And then JNK was found to be activated in response to stress challenges to regulate cell proliferation, differentiation, apoptosis, cell survival, cell mobility and cytokine production (Davis, 2000; Bogoyevitch and Kobe, 2006; Raman et al., 2007). It is known that the JNK signaling pathway is critical in the development of cancer, diabetes, and cardiovascular diseases (CVD; e.g., HF, myocardial infarction, atherosclerosis) (Davis, 2000; Karin and Gallagher, 2005; Rose et al., 2010). Emerging evidence suggests that enhanced JNK activation is also linked to significantly elevated intrinsic stress (e.g., oxidative stress or inflammatory stress) (Liu et al., 2014; Sun et al., 2014). Studies have shown that rapid transient JNK activation appears in cultured myocytes and animals that are subjected to exercise or severe pressure overload, Boluyt et al. (2003), Nadruz et al. (2004, 2005) and Pan et al. (2005) while 24 h mechanically stretched myocytes or exercise trained animals showed reduced or unchanged JNK activity (Boluyt et al., 2003; Miyamoto et al., 2004; Roussel et al., 2008). These results indicate that JNK activation could be a dynamic response to the stress stimuli. Our laboratory recently discovered and reported for the first time (Yan et al., 2013) that activated JNK plays an important role in reduced gap junction channels and slowed conduction (Figure 2) that is associated with markedly increased pacing-induced AF in vivo in aged rabbits. Young rabbits subjected to a JNK activator (anisomycin) (Hazzalin et al., 1998; Petrich et al., 2004) challenge in vivo also exhibited dramatically increased incidence and duration of pacing-induced AT/AF, which is comparable to that found in aged hearts (Figure 1). While a significantly increased propensity for AF in aged humans has been well-recorganized, Benjamin et al. (1994), Go et al. (2001) and Rich (2009) our recent observations (Wu et al., 2014) suggest an increase in activated JNK in aging human atrium from healthy donor hearts (which were rejected for heart transplant due to technical reasons). Moreover, we demonstrated that JNK-induced gap junction remodeling impairs atrial conduction and causes formation of reentrant circuits in cultured atrial myocytes (Figures 2C,D) (Yan et al., 2013). However, previous studies suggest that gap junction remodeling is most likely to contribute to stabilization and maintenance of AF (Elvan et al., 1997; van der Velden et al., 1998, 2000; Dupont et al., 2001; Polontchouk et al., 2001; Kostin et al., 2002; Nao et al., 2003; Kanagaratnam et al., 2004; Sakabe et al., 2004; Wetzel et al., 2005; Nattel et al., 2008). Therefore, other mechanisms such as SR Ca handling dysfunction could be responsible for the initiation of atrial arrhythmias in aged hearts. To date, the role of JNK in SR Ca handling and AF development in the failing and aged heart remains unknown. Our recent results suggest that activated JNK plays an important role in SR Ca leak and AF development in aged animals as well as young animals with manipulated JNK activity. A computer simulation study (Xie et al., 2010) suggested that generating an ectopic beat in heart tissue with poorly coupled neighboring myocytes (slowed AP conduction) requires much fewer EAD or DAD-producing myocytes than in normal tissue composed of well-coupled cells. In another words, impaired intercellular coupling could make cardiac tissue more vulnerable for generating ectopic triggers that may initiate arrhythmias. Therefore, JNK-induced slowed conduction in the aged atria may create a favorable environment for JNK-induced abnormal Ca activities to form ectopic beats and even to initiate AF. Many questions regarding the underlying mechanisms of JNK-induced AF genesis remain unanswered. Further investigations are clearly needed in this important research area.
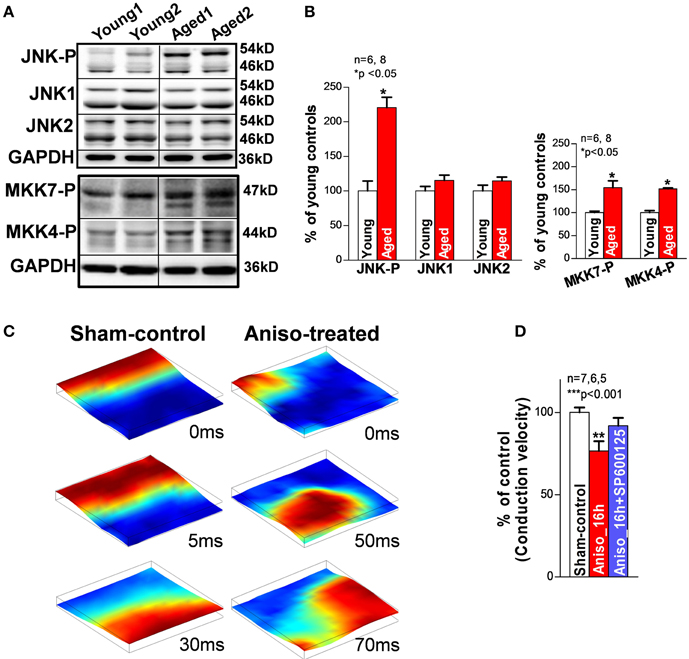
Figure 2. (A,B) Immunoblotting images and summarized data of phosphorylated JNK (JNK-P), JNK1, JNK2, and phosphorylated MKK7 and MKK4 (MKK7-P, MKK4-P) in young and aged rabbit LA. (C) Representative sequential images of uniformly propagated action potentials (CL = 200 ms) in sham-control HL-1 monolayers and broken and reentrant AP wave in an anisomycin-treated (24 h) monolayer. (D) Summarized data of conduction velocity between the three groups (**p < 0.01 vs. sham-control). This figure is modified from Yan et al. (2013).
ERKs and p38 MAPKs are the other two important stress-response signaling pathways in cellular biology (Ramos, 2008; Rose et al., 2010). At the cellular level, the two stress signaling pathways modulate cell proliferation and differentiation, cytokinesis, transcription, cell death, and cell adhesion. Like JNK, both ERK and p38 are involved in various pathologies such as cardiovascular diseases, diabetes, and cancers (Davis, 2000; Kyriakis and Avruch, 2001; Karin and Gallagher, 2005; Kyoi et al., 2006; Yoon and Seger, 2006; Rose et al., 2010). While enhanced activity of ERK or p38 alone may or may not be required or sufficient for facilitating cardiac hypertrophy, both ERK and p38 were found to be activated in HF and these activated stress kinases are involved in pathological remodeling and AF development in the failing heart (Zechner et al., 1997; Wang et al., 1998; Li et al., 2001, 2005b; Cardin et al., 2003; Nishida et al., 2004; Purcell et al., 2007). Studies suggest that hypertrophic stimuli lead to an increase in L-type Ca transients and down-regulation of SERCA2 expression via activated ERK (Takahashi et al., 2004; Hagiwara et al., 2007; Huang et al., 2014). Ras, a GTPase, is able to activate ERK through a Ras-Raf-MEK cascade (Avruch et al., 2001). Ras signaling activated ERK was found to contribute to down-regulation of L-type Ca channels and reduced channel activity along with reduced SERCA2 protein expression in cultured myocytes (Ho et al., 1998, 2001; Huang et al., 2014). It was also found that Ras-ERK-modulated molecular remodeling led to decreased intracellular Ca transients and impaired SR Ca uptake, which could lead to enhanced arrhythmogenicity (Zheng et al., 2004). Moreover, recent work reported by Scharf et al. (2013) suggests that p38 directly regulates SERCA2 mRNA and protein expression via transcription factors Egr-1 and SP1. Taken together, emerging evidence indicates that the stress-response MAP kinases signaling cascades could be involved in cardiac Ca handling and AF development (Figure 3). However, more work needs to be done to further understand the underlying molecular and electrophysiological mechanisms of altered stress signaling cascades and their crosstalking in AF development in the failing and aged heart.
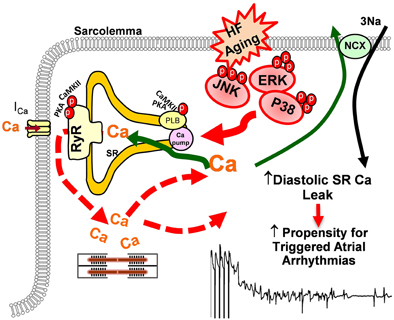
Figure 3. Schematic outline of the potential impact of stress-response MAP kinases on sarcolemma reticulum (SR) Ca handling that may increase AF propensity in failing and/or aged atria.
Accumulating evidence suggests that abnormal SR Ca handling is associated with the initiation and development of AF. However, much work still needs to be done to further uncover the underlying molecular and electrophysiological mechanisms of AF initiation and maintenance in diseased and aged hearts. To date, most of the mechanistic studies of SR Ca dynamics have been performed in isolated myocytes. However, isolated myocytes provide limited information regarding the spatial complexity of SR Ca kinetics in the 3-dimensional myocardial structure, which is completely disrupted by the enzymatic dissociation procedure of cell isolation. Thus, measuring Ca dynamics in intact atria using high-resolution Ca imaging should be considered in future studies to obtain important information about the relationship of SR Ca handling and APs, as well as their role in arrhythmogenesis. At present, emerging evidence indicates a link between altered stress signaling cascades and abnormal Ca handling in pathologically altered atrium. Further understanding of the underlying mechanisms of stress-induced AF development in the failing and/or aged heart could reveal potential effective therapeutic strategies for AF prevention and treatment.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by American Heart Association (10GRNT3770030 & 12GRNT12050478 to XA) and National Institutes of Health (HL113640 to XA).
Ai, X., and Pogwizd, S. M. (2005). Connexin 43 downregulation and dephosphorylation in nonischemic heart failure is associated with enhanced colocalized protein phosphatase type 2a. Circ. Res. 96, 54–63. doi: 10.1161/01.RES.0000152325.07495.5a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum ca2+ leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.RES.0000194329.41863.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ai, X., Jiang, A., Ke, Y., Solaro, R. J., and Pogwizd, S. M. (2011). Enhanced activation of p21-activated kinase 1 in heart failure contributes to dephosphorylation of connexin 43. Cardiovasc. Res. 92, 106–114. doi: 10.1093/cvr/cvr163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Allessie, M. A., Bonke, F. I., and Schopman, F. J. (1976). Circus movement in rabbit atrial muscle as a mechanism of tachycardia. Ii. The role of nonuniform recovery of excitability in the occurrence of unidirectional block, as studied with multiple microelectrodes. Circ. Res. 39, 168–177. doi: 10.1161/01.RES.39.2.168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anyukhovsky, E. P., Sosunov, E. A., Chandra, P., Rosen, T. S., Boyden, P. A., Danilo, P. Jr., et al. (2005). Age-associated changes in electrophysiologic remodeling: a potential contributor to initiation of atrial fibrillation. Cardiovasc. Res. 66, 353–363. doi: 10.1016/j.cardiores.2004.10.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Avruch, J., Khokhlatchev, A., Kyriakis, J. M., Luo, Z., Tzivion, G., Vavvas, D., et al. (2001). Ras activation of the raf kinase: tyrosine kinase recruitment of the map kinase cascade. Recent Prog. Horm. Res. 56, 127–155. doi: 10.1210/rp.56.1.127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balijepalli, R. C., Lokuta, A. J., Maertz, N. A., Buck, J. M., Haworth, R. A., Valdivia, H. H., et al. (2003). Depletion of t-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovasc. Res. 59, 67–77. doi: 10.1016/S0008-6363(03)00325-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bassani, J. W., Yuan, W., and Bers, D. M. (1995). Fractional sr ca release is regulated by trigger ca and sr ca content in cardiac myocytes. Am. J. Physiol. 268, C1313–C1319.
Beckman, K. B., and Ames, B. N. (1998). The free radical theory of aging matures. Physiol. Rev. 78, 547–581.
Belmin, J., Bernard, C., Corman, B., Merval, R., Esposito, B., and Tedgui, A. (1995). Increased production of tumor necrosis factor and interleukin-6 by arterial wall of aged rats. Am. J. Physiol. 268, H2288–H2293.
Benjamin, E. J., Levy, D., Vaziri, S. M., D'Agostino, R. B., Belanger, A. J., and Wolf, P. A. (1994). Independent risk factors for atrial fibrillation in a population-based cohort. The framingham heart study. JAMA 271, 840–844.
Berlin, J. R. (1995). Spatiotemporal changes of ca2+ during electrically evoked contractions in atrial and ventricular cells. Am. J. Physiol. 269, H1165–H1170.
Bers, D. M. (2000). Calcium fluxes involved in control of cardiac myocyte contraction. Circ. Res. 87, 275–281. doi: 10.1161/01.RES.87.4.275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bers, D. M. (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127. doi: 10.1146/annurev-physiol-020911-153308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bogoyevitch, M. A., and Kobe, B. (2006). Uses for jnk: the many and varied substrates of the c-jun n-terminal kinases. Microbiol. Mol. Biol. Rev. 70, 1061–1095. doi: 10.1128/MMBR.00025-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boluyt, M. O., Loyd, A. M., Roth, M. H., Randall, M. J., and Song, E. Y. (2003). Activation of jnk in rat heart by exercise: effect of training. Am. J. Physiol. Heart Circ. Physiol. 285, H2639–H2647. doi: 10.1152/ajpheart.00596.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brette, F., and Orchard, C. (2003). T-tubule function in mammalian cardiac myocytes. Circ. Res. 92, 1182–1192. doi: 10.1161/01.RES.0000074908.17214.FD
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burashnikov, A., and Antzelevitch, C. (2003). Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation 107, 2355–2360. doi: 10.1161/01.CIR.0000065578.00869.7C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cardin, S., Li, D., Thorin-Trescases, N., Leung, T. K., Thorin, E., and Nattel, S. (2003). Evolution of the atrial fibrillation substrate in experimental congestive heart failure: angiotensin-dependent and -independent pathways. Cardiovasc. Res. 60, 315–325. doi: 10.1016/j.cardiores.2003.08.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chelu, M. G., Sarma, S., Sood, S., Wang, S., van Oort, R. J., Skapura, D. G., et al. (2009). Calmodulin kinase ii-mediated sarcoplasmic reticulum ca2+ leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951. doi: 10.1172/JCI37059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiang, D. Y., Kongchan, N., Beavers, D. L., Alsina, K. M., Voigt, N., Neilson, J. R., et al. (2014). Loss of microrna-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ. Arrhythm. Electrophysiol. 7, 1214–1222. doi: 10.1161/CIRCEP.114.001973
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christ, T., Boknik, P., Wohrl, S., Wettwer, E., Graf, E. M., Bosch, R. F., et al. (2004). L-type ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110, 2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cowie, M. R., Mosterd, A., Wood, D. A., Deckers, J. W., Poole-Wilson, P. A., Sutton, G. C., et al. (1997). The epidemiology of heart failure. Eur. Heart J. 18, 208–225. doi: 10.1093/oxfordjournals.eurheartj.a015223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Davis, R. J. (2000). Signal transduction by the jnk group of map kinases. Cell 103, 239–252. doi: 10.1016/S0092-8674(00)00116-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DeSantiago, J., Maier, L. S., and Bers, D. M. (2002). Frequency-dependent acceleration of relaxation in the heart depends on camkii, but not phospholamban. J. Mol. Cell. Cardiol. 34, 975–984. doi: 10.1006/jmcc.2002.2034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Di Lenarda, A., Scherillo, M., Maggioni, A. P., Acquarone, N., Ambrosio, G. B., Annicchiarico, M., et al. (2003). Current presentation and management of heart failure in cardiology and internal medicine hospital units: a tale of two worlds–the temistocle study. Am. Heart J. 146, E12. doi: 10.1016/S0002-8703(03)00315-6
Dibb, K. M., Clarke, J. D., Horn, M. A., Richards, M. A., Graham, H. K., Eisner, D. A., et al. (2009). Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ. Heart Fail. 2, 482–489. doi: 10.1161/CIRCHEARTFAILURE.109.852228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dinanian, S., Boixel, C., Juin, C., Hulot, J. S., Coulombe, A., Rucker-Martin, C., et al. (2008). Downregulation of the calcium current in human right atrial myocytes from patients in sinus rhythm but with a high risk of atrial fibrillation. Eur. Heart J. 29, 1190–1197. doi: 10.1093/eurheartj/ehn140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dupont, E., Ko, Y., Rothery, S., Coppen, S. R., Baghai, M., Haw, M., et al. (2001). The gap-junctional protein connexin40 is elevated in patients susceptible to postoperative atrial fibrillation. Circulation 103, 842–849. doi: 10.1161/01.CIR.103.6.842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ehrlich, J. R., Nattel, S., and Hohnloser, S. H. (2002). Atrial fibrillation and congestive heart failure: specific considerations at the intersection of two common and important cardiac disease sets. J. Cardiovasc. Electrophysiol. 13, 399–405. doi: 10.1046/j.1540-8167.2002.00399.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elvan, A., Huang, X. D., Pressler, M. L., and Zipes, D. P. (1997). Radiofrequency catheter ablation of the atria eliminates pacing-induced sustained atrial fibrillation and reduces connexin 43 in dogs. Circulation 96, 1675–1685. doi: 10.1161/01.CIR.96.5.1675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Forbes, M. S., Van Niel, E. E., and Purdy-Ramos, S. I. (1990). The atrial myocardial cells of mouse heart: a structural and stereological study. J. Struct. Biol. 103, 266–279. doi: 10.1016/1047-8477(90)90045-E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Franzini-Armstrong, C., Protasi, F., and Tijskens, P. (2005). The assembly of calcium release units in cardiac muscle. Ann. N. Y. Acad. Sci. 1047, 76–85. doi: 10.1196/annals.1341.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Freestone, N. S., Ribaric, S., Scheuermann, M., Mauser, U., Paul, M., and Vetter, R. (2000). Differential lusitropic responsiveness to beta-adrenergic stimulation in rat atrial and ventricular cardiac myocytes. Pflugers Arch. 441, 78–87. doi: 10.1007/s004240000397
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frisk, M., Koivumaki, J. T., Norseng, P. A., Maleckar, M. M., Sejersted, O. M., and Louch, W. E. (2014). Variable t-tubule organization and ca2+ homeostasis across the atria. Am. J. Physiol. Heart Circ. Physiol. 307, H609–H620. doi: 10.1152/ajpheart.00295.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Go, A. S., Hylek, E. M., Phillips, K. A., Chang, Y., Henault, L. E., Selby, J. V., et al. (2001). Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the anticoagulation and risk factors in atrial fibrillation (atria) study. JAMA 285, 2370–2375. doi: 10.1001/jama.285.18.2370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greiser, M., Neuberger, H. R., Harks, E., El-Armouche, A., Boknik, P., de Haan, S., et al. (2009). Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J. Mol. Cell. Cardiol. 46, 385–394. doi: 10.1016/j.yjmcc.2008.11.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hagiwara, Y., Miyoshi, S., Fukuda, K., Nishiyama, N., Ikegami, Y., Tanimoto, K., et al. (2007). Shp2-mediated signaling cascade through gp130 is essential for lif-dependent i cal, [ca2+]i transient, and apd increase in cardiomyocytes. J. Mol. Cell. Cardiol. 43, 710–716. doi: 10.1016/j.yjmcc.2007.09.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hazzalin, C. A., Le Panse, R., Cano, E., and Mahadevan, L. C. (1998). Anisomycin selectively desensitizes signalling components involved in stress kinase activation and fos and jun induction. Mol. Cell. Biol. 18, 1844–1854.
He, B. J., Joiner, M. L., Singh, M. V., Luczak, E. D., Swaminathan, P. D., Koval, O. M., et al. (2011). Oxidation of camkii determines the cardiotoxic effects of aldosterone. Nat. Med. 17, 1610–1618. doi: 10.1038/nm.2506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heijman, J., Dewenter, M., El-Armouche, A., and Dobrev, D. (2013). Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J. Mol. Cell. Cardiol. 64, 90–98. doi: 10.1016/j.yjmcc.2013.09.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heinzel, F. R., Bito, V., Biesmans, L., Wu, M., Detre, E., von Wegner, F., et al. (2008). Remodeling of t-tubules and reduced synchrony of ca2+ release in myocytes from chronically ischemic myocardium. Circ. Res. 102, 338–346. doi: 10.1161/CIRCRESAHA.107.160085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hershberger, R. E., Nauman, D., Walker, T. L., Dutton, D., and Burgess, D. (2003). Care processes and clinical outcomes of continuous outpatient support with inotropes (cosi) in patients with refractory endstage heart failure. J. Card. Fail. 9, 180–187. doi: 10.1054/jcaf.2003.24
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ho, P. D., Fan, J. S., Hayes, N. L., Saada, N., Palade, P. T., Glembotski, C. C., et al. (2001). Ras reduces l-type calcium channel current in cardiac myocytes. Corrective effects of l-channels and serca2 on [ca(2+)](i) regulation and cell morphology. Circ. Res. 88, 63–69. doi: 10.1161/01.RES.88.1.63
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ho, P. D., Zechner, D. K., He, H., Dillmann, W. H., Glembotski, C. C., and McDonough, P. M. (1998). The raf-mek-erk cascade represents a common pathway for alteration of intracellular calcium by ras and protein kinase c in cardiac myocytes. J. Biol. Chem. 273, 21730–21735. doi: 10.1074/jbc.273.34.21730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoch, B., Meyer, R., Hetzer, R., Krause, E. G., and Karczewski, P. (1999). Identification and expression of delta-isoforms of the multifunctional ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ. Res. 84, 713–721. doi: 10.1161/01.RES.84.6.713
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, C., Ding, W., Li, L., and Zhao, D. (2006). Differences in the aging-associated trends of the monophasic action potential duration and effective refractory period of the right and left atria of the rat. Circ. J. 70, 352–357. doi: 10.1253/circj.70.352
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, H., Joseph, L. C., Gurin, M. I., Thorp, E. B., and Morrow, J. P. (2014). Extracellular signal-regulated kinase activation during cardiac hypertrophy reduces sarcoplasmic/endoplasmic reticulum calcium atpase 2 (serca2) transcription. J. Mol. Cell. Cardiol. 75, 58–63. doi: 10.1016/j.yjmcc.2014.06.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huang, J. L., Tai, C. T., Chen, J. T., Ting, C. T., Chen, Y. T., Chang, M. S., et al. (2003). Effect of atrial dilatation on electrophysiologic properties and inducibility of atrial fibrillation. Basic Res. Cardiol. 98, 16–24. doi: 10.1007/s00395-003-0385-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ibrahim, M., Al Masri, A., Navaratnarajah, M., Siedlecka, U., Soppa, G. K., Moshkov, A., et al. (2010). Prolonged mechanical unloading affects cardiomyocyte excitation-contraction coupling, transverse-tubule structure, and the cell surface. FASEB J. 24, 3321–3329. doi: 10.1096/fj.10-156638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ismahil, M. A., Hamid, T., Bansal, S. S., Patel, B., Kingery, J. R., and Prabhu, S. D. (2014). Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ. Res. 114, 266–282. doi: 10.1161/CIRCRESAHA.113.301720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Judge, S., and Leeuwenburgh, C. (2007). Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am. J. Physiol. Cell Physiol. 292, C1983–C1992. doi: 10.1152/ajpcell.00285.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Juhaszova, M., Rabuel, C., Zorov, D. B., Lakatta, E. G., and Sollott, S. J. (2005). Protection in the aged heart: preventing the heart-break of old age? Cardiovasc. Res. 66, 233–244. doi: 10.1016/j.cardiores.2004.12.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kanagaratnam, P., Cherian, A., Stanbridge, R. D., Glenville, B., Severs, N. J., and Peters, N. S. (2004). Relationship between connexins and atrial activation during human atrial fibrillation. J. Cardiovasc. Electrophysiol. 15, 206–216. doi: 10.1046/j.1540-8167.2004.03280.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kanagaratnam, P., Kojodjojo, P., and Peters, N. S. (2008). Electrophysiological abnormalities occur prior to the development of clinical episodes of atrial fibrillation: observations from human epicardial mapping. Pacing Clin. Electrophysiol. 31, 443–453. doi: 10.1111/j.1540-8159.2008.01014.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kannel, W. B., Wolf, P. A., Benjamin, E. J., and Levy, D. (1998). Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: population-based estimates. Am. J. Cardiol. 82, 2N–9N. doi: 10.1016/S0002-9149(98)00583-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karin, M., and Gallagher, E. (2005). From jnk to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 57, 283–295. doi: 10.1080/15216540500097111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kostin, S., Klein, G., Szalay, Z., Hein, S., Bauer, E. P., and Schaper, J. (2002). Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 54, 361–379. doi: 10.1016/S0008-6363(02)00273-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kyoi, S., Otani, H., Matsuhisa, S., Akita, Y., Tatsumi, K., Enoki, C., et al. (2006). Opposing effect of p38 map kinase and jnk inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc. Res. 69, 888–898. doi: 10.1016/j.cardiores.2005.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kyriakis, J. M., and Avruch, J. (2001). Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869.
Lenaerts, I., Bito, V., Heinzel, F. R., Driesen, R. B., Holemans, P., D'Hooge, J., et al. (2009). Ultrastructural and functional remodeling of the coupling between ca2+ influx and sarcoplasmic reticulum ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ. Res. 105, 876–885. doi: 10.1161/CIRCRESAHA.109.206276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, D., Shinagawa, K., Pang, L., Leung, T. K., Cardin, S., Wang, Z., et al. (2001). Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation 104, 2608–2614. doi: 10.1161/hc4601.099402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, M., Georgakopoulos, D., Lu, G., Hester, L., Kass, D. A., Hasday, J., et al. (2005b). P38 map kinase mediates inflammatory cytokine induction in cardiomyocytes and extracellular matrix remodeling in heart. Circulation 111, 2494–2502. doi: 10.1161/01.CIR.0000165117.71483.0C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, S. Y., Du, M., Dolence, E. K., Fang, C. X., Mayer, G. E., Ceylan-Isik, A. F., et al. (2005a). Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell 4, 57–64. doi: 10.1111/j.1474-9728.2005.00146.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Linne, A. B., Liedholm, H., Jendteg, S., and Israelsson, B. (2000). Health care costs of heart failure: results from a randomised study of patient education. Eur. J. Heart Fail. 2, 291–297. doi: 10.1016/S1388-9842(00)00089-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Y., Wang, J., Qi, S. Y., Ru, L. S., Ding, C., Wang, H. J., et al. (2014). Reduced endoplasmic reticulum stress might alter the course of heart failure via caspase-12 and jnk pathways. Can. J. Cardiol. 30, 368–375. doi: 10.1016/j.cjca.2013.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Louch, W. E., Mork, H. K., Sexton, J., Stromme, T. A., Laake, P., Sjaastad, I., et al. (2006). T-tubule disorganization and reduced synchrony of ca2+ release in murine cardiomyocytes following myocardial infarction. J. Physiol. 574, 519–533. doi: 10.1113/jphysiol.2006.107227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lyon, A. R., MacLeod, K. T., Zhang, Y., Garcia, E., Kanda, G. K., Lab, M. J., et al. (2009). Loss of t-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. U.S.A. 106, 6854–6859. doi: 10.1073/pnas.0809777106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maier, L. S., Zhang, T., Chen, L., DeSantiago, J., Brown, J. H., and Bers, D. M. (2003). Transgenic camkiideltac overexpression uniquely alters cardiac myocyte ca2+ handling: reduced sr ca2+ load and activated sr ca2+ release. Circ. Res. 92, 904–911. doi: 10.1161/01.RES.0000069685.20258.F1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mandapati, R., Skanes, A., Chen, J., Berenfeld, O., and Jalife, J. (2000). Stable microreentrant sources as a mechanism of atrial fibrillation in the isolated sheep heart. Circulation 101, 194–199. doi: 10.1161/01.CIR.101.2.194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Markides, V., and Peters, N. S. (2002). Mechanisms underlying the development of atrial arrhythmias in heart failure. Heart Fail. Rev. 7, 243–253. doi: 10.1023/A:1020077206796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). Pka phosphorylation dissociates fkbp12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/S0092-8674(00)80847-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyamoto, T., Takeishi, Y., Takahashi, H., Shishido, T., Arimoto, T., Tomoike, H., et al. (2004). Activation of distinct signal transduction pathways in hypertrophied hearts by pressure and volume overload. Basic Res. Cardiol. 99, 328–337. doi: 10.1007/s00395-004-0482-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyasaka, Y., Barnes, M. E., Gersh, B. J., Cha, S. S., Bailey, K. R., Abhayaratna, W. P., et al. (2006). Secular trends in incidence of atrial fibrillation in olmsted county, minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 114, 119–125. doi: 10.1161/CIRCULATIONAHA.105.595140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nadruz, W. Jr. Corat, M. A., Marin, T. M., Guimaraes Pereira, G. A., and Franchini, K. G. (2005). Focal adhesion kinase mediates mef2 and c-jun activation by stretch: role in the activation of the cardiac hypertrophic genetic program. Cardiovasc. Res. 68, 87–97. doi: 10.1016/j.cardiores.2005.05.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nadruz, W. Jr. Kobarg, C. B., Kobarg, J., and Franchini, K. G. (2004). C-jun is regulated by combination of enhanced expression and phosphorylation in acute-overloaded rat heart. Am. J. Physiol. Heart Circ. Physiol. 286, H760–H767. doi: 10.1152/ajpheart.00430.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nao, T., Ohkusa, T., Hisamatsu, Y., Inoue, N., Matsumoto, T., Yamada, J., et al. (2003). Comparison of expression of connexin in right atrial myocardium in patients with chronic atrial fibrillation versus those in sinus rhythm. Am. J. Cardiol. 91, 678–683. doi: 10.1016/S0002-9149(02)03403-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nattel, S., Burstein, B., and Dobrev, D. (2008). Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ. Arrhythm. Electrophysiol. 1, 62–73. doi: 10.1161/CIRCEP.107.754564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nattel, S., Maguy, A., Le Bouter, S., and Yeh, Y. H. (2007). Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 87, 425–456. doi: 10.1152/physrev.00014.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neef, S., Dybkova, N., Sossalla, S., Ort, K. R., Fluschnik, N., Neumann, K., et al. (2010). Camkii-dependent diastolic sr ca2+ leak and elevated diastolic ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 106, 1134–1144. doi: 10.1161/CIRCRESAHA.109.203836
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neuberger, H. R., Mewis, C., van Veldhuisen, D. J., Schotten, U., van Gelder, I. C., Allessie, M. A., et al. (2007). Management of atrial fibrillation in patients with heart failure. Eur. Heart J. 28, 2568–2577. doi: 10.1093/eurheartj/ehm341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Neuman, R. B., Bloom, H. L., Shukrullah, I., Darrow, L. A., Kleinbaum, D., Jones, D. P., et al. (2007). Oxidative stress markers are associated with persistent atrial fibrillation. Clin. Chem. 53, 1652–1657. doi: 10.1373/clinchem.2006.083923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishida, K., Yamaguchi, O., Hirotani, S., Hikoso, S., Higuchi, Y., Watanabe, T., et al. (2004). P38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 24, 10611–10620. doi: 10.1128/MCB.24.24.10611-10620.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oral, H., Ozaydin, M., Sticherling, C., Tada, H., Scharf, C., Chugh, A., et al. (2003). Effect of atrial fibrillation duration on probability of immediate recurrence after transthoracic cardioversion. J. Cardiovasc. Electrophysiol. 14, 182–185. doi: 10.1046/j.1540-8167.2003.02415.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pan, J., Singh, U. S., Takahashi, T., Oka, Y., Palm-Leis, A., Herbelin, B. S., et al. (2005). Pkc mediates cyclic stretch-induced cardiac hypertrophy through rho family gtpases and mitogen-activated protein kinases in cardiomyocytes. J. Cell. Physiol. 202, 536–553. doi: 10.1002/jcp.20151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patterson, E., Po, S. S., Scherlag, B. J., and Lazzara, R. (2005). Triggered firing in pulmonary veins initiated by in vitro autonomic nerve stimulation. Heart Rhythm 2, 624–631. doi: 10.1016/j.hrthm.2005.02.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Petrich, B. G., Eloff, B. C., Lerner, D. L., Kovacs, A., Saffitz, J. E., Rosenbaum, D. S., et al. (2004). Targeted activation of c-jun n-terminal kinase in vivo induces restrictive cardiomyopathy and conduction defects. J. Biol. Chem. 279, 15330–15338. doi: 10.1074/jbc.M314142200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Podrid, P. J. (1999). Atrial fibrillation in the elderly. Cardiol. Clin. 17, 173–188, ix-x. doi: 10.1016/S0733-8651(05)70063-1
Polontchouk, L., Haefliger, J. A., Ebelt, B., Schaefer, T., Stuhlmann, D., Mehlhorn, U., et al. (2001). Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J. Am. Coll. Cardiol. 38, 883–891. doi: 10.1016/S0735-1097(01)01443-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Psaty, B. M., Manolio, T. A., Kuller, L. H., Kronmal, R. A., Cushman, M., Fried, L. P., et al. (1997). Incidence of and risk factors for atrial fibrillation in older adults. Circulation 96, 2455–2461. doi: 10.1161/01.CIR.96.7.2455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purcell, N. H., Wilkins, B. J., York, A., Saba-El-Leil, M. K., Meloche, S., Robbins, J., et al. (2007). Genetic inhibition of cardiac erk1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 14074–14079. doi: 10.1073/pnas.0610906104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raman, M., Chen, W., and Cobb, M. H. (2007). Differential regulation and properties of mapks. Oncogene 26, 3100–3112. doi: 10.1038/sj.onc.1210392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramos, J. W. (2008). The regulation of extracellular signal-regulated kinase (erk) in mammalian cells. Int. J. Biochem. Cell Biol. 40, 2707–2719. doi: 10.1016/j.biocel.2008.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rensma, P. L., Allessie, M. A., Lammers, W. J., Bonke, F. I., and Schalij, M. J. (1988). Length of excitation wave and susceptibility to reentrant atrial arrhythmias in normal conscious dogs. Circ. Res. 62, 395–410. doi: 10.1161/01.RES.62.2.395
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Respress, J. L., van Oort, R. J., Li, N., Rolim, N., Dixit, S. S., deAlmeida, A., et al. (2012). Role of ryr2 phosphorylation at s2814 during heart failure progression. Circ. Res. 110, 1474–1483. doi: 10.1161/CIRCRESAHA.112.268094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rich, M. W. (2009). Epidemiology of atrial fibrillation. J. Interv. Card. Electrophysiol. 25, 3–8. doi: 10.1007/s10840-008-9337-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Richards, M. A., Clarke, J. D., Saravanan, P., Voigt, N., Dobrev, D., Eisner, D. A., et al. (2011). Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am. J. Physiol. Heart Circ. Physiol. 301, H1996–H2005. doi: 10.1152/ajpheart.00284.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ronkainen, J. J., Hanninen, S. L., Korhonen, T., Koivumaki, J. T., Skoumal, R., Rautio, S., et al. (2011). Ca2+-calmodulin-dependent protein kinase ii represses cardiac transcription of the l-type calcium channel alpha(1c)-subunit gene (cacna1c) by dream translocation. J. Physiol. 589, 2669–2686. doi: 10.1113/jphysiol.2010.201400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rose, B. A., Force, T., and Wang, Y. (2010). Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol. Rev. 90, 1507–1546. doi: 10.1152/physrev.00054.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rougier, O., Vassort, G., Garnier, D., Gargouil, Y. M., and Coraboeuf, E. (1969). Existence and role of a slow inward current during the frog atrial action potential. Pflugers Arch. 308, 91–110. doi: 10.1007/BF00587018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roussel, E., Gaudreau, M., Plante, E., Drolet, M. C., Breault, C., Couet, J., et al. (2008). Early responses of the left ventricle to pressure overload in wistar rats. Life Sci. 82, 265–272. doi: 10.1016/j.lfs.2007.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakabe, K., Fukuda, N., Nada, T., Shinohara, H., Tamura, Y., Wakatsuki, T., et al. (2003). Age-related changes in the electrophysiologic properties of the atrium in patients with no history of atrial fibrillation. Jpn. Heart J. 44, 385–393. doi: 10.1536/jhj.44.385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakabe, M., Fujiki, A., Nishida, K., Sugao, M., Nagasawa, H., Tsuneda, T., et al. (2004). Enalapril prevents perpetuation of atrial fibrillation by suppressing atrial fibrosis and over-expression of connexin43 in a canine model of atrial pacing-induced left ventricular dysfunction. J. Cardiovasc. Pharmacol. 43, 851–859. doi: 10.1097/00005344-200406000-00015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanders, P., Morton, J. B., Davidson, N. C., Spence, S. J., Vohra, J. K., Sparks, P. B., et al. (2003). Electrical remodeling of the atria in congestive heart failure: electrophysiological and electroanatomic mapping in humans. Circulation 108, 1461–1468. doi: 10.1161/01.CIR.0000090688.49283.67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scharf, M., Neef, S., Freund, R., Geers-Knorr, C., Franz-Wachtel, M., Brandis, A., et al. (2013). Mitogen-activated protein kinase-activated protein kinases 2 and 3 regulate serca2a expression and fiber type composition to modulate skeletal muscle and cardiomyocyte function. Mol. Cell. Biol. 33, 2586–2602. doi: 10.1128/MCB.01692-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schulman, H., Hanson, P. I., and Meyer, T. (1992). Decoding calcium signals by multifunctional cam kinase. Cell Calcium 13, 401–411. doi: 10.1016/0143-4160(92)90053-U
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shannon, T. R., Ginsburg, K. S., and Bers, D. M. (2000). Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys. J. 78, 334–343. doi: 10.1016/S0006-3495(00)76596-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sossalla, S., Fluschnik, N., Schotola, H., Ort, K. R., Neef, S., Schulte, T., et al. (2010). Inhibition of elevated ca2+/calmodulin-dependent protein kinase ii improves contractility in human failing myocardium. Circ. Res. 107, 1150–1161. doi: 10.1161/CIRCRESAHA.110.220418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sun, A., Zou, Y., Wang, P., Xu, D., Gong, H., Wang, S., et al. (2014). Mitochondrial aldehyde dehydrogenase 2 plays protective roles in heart failure after myocardial infarction via suppression of the cytosolic jnk/p53 pathway in mice. J. Am. Heart Assoc. 3:e000779. doi: 10.1161/JAHA.113.000779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takahashi, E., Fukuda, K., Miyoshi, S., Murata, M., Kato, T., Ita, M., et al. (2004). Leukemia inhibitory factor activates cardiac l-type ca2+ channels via phosphorylation of serine 1829 in the rabbit cav1.2 subunit. Circ. Res. 94, 1242–1248. doi: 10.1161/01.RES.0000126405.38858.BC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Timmermans, C., Rodriguez, L. M., Smeets, J. L., and Wellens, H. J. (1998). Immediate reinitiation of atrial fibrillation following internal atrial defibrillation. J. Cardiovasc. Electrophysiol. 9, 122–128. doi: 10.1111/j.1540-8167.1998.tb00893.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van der Velden, H. M., Ausma, J., Rook, M. B., Hellemons, A. J., van Veen, T. A., Allessie, M. A., et al. (2000). Gap junctional remodeling in relation to stabilization of atrial fibrillation in the goat. Cardiovasc. Res. 46, 476–486. doi: 10.1016/S0008-6363(00)00026-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
van der Velden, H. M., van Kempen, M. J., Wijffels, M. C., van Zijverden, M., Groenewegen, W. A., Allessie, M. A., et al. (1998). Altered pattern of connexin40 distribution in persistent atrial fibrillation in the goat. J. Cardiovasc. Electrophysiol. 9, 596–607. doi: 10.1111/j.1540-8167.1998.tb00940.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Wagoner, D. R., Pond, A. L., Lamorgese, M., Rossie, S. S., McCarthy, P. M., and Nerbonne, J. M. (1999). Atrial l-type ca2+ currents and human atrial fibrillation. Circ. Res. 85, 428–436. doi: 10.1161/01.RES.85.5.428
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Venetucci, L. A., Trafford, A. W., O'Neill, S. C., and Eisner, D. A. (2008). The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 77, 285–292. doi: 10.1093/cvr/cvm009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Voigt, N., Heijman, J., Wang, Q., Chiang, D. Y., Li, N., Karck, M., et al. (2014). Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 129, 145–156. doi: 10.1161/CIRCULATIONAHA.113.006641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wakili, R., Yeh, Y. H., Yan Qi, X., Greiser, M., Chartier, D., Nishida, K., et al. (2010). Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ. Arrhythm. Electrophysiol. 3, 530–541. doi: 10.1161/CIRCEP.109.933036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walden, A. P., Dibb, K. M., and Trafford, A. W. (2009). Differences in intracellular calcium homeostasis between atrial and ventricular myocytes. J. Mol. Cell. Cardiol. 46, 463–473. doi: 10.1016/j.yjmcc.2008.11.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, S. Q., Song, L. S., Lakatta, E. G., and Cheng, H. (2001). Ca2+ signalling between single l-type ca2+ channels and ryanodine receptors in heart cells. Nature 410, 592–596. doi: 10.1038/35069083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, T. J., Larson, M. G., Levy, D., Vasan, R. S., Leip, E. P., Wolf, P. A., et al. (2003). Temporal relations of atrial fibrillation and congestive heart failure and their joint influence on mortality: the framingham heart study. Circulation 107, 2920–2925. doi: 10.1161/01.CIR.0000072767.89944.6E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Huang, S., Sah, V. P., Ross, J. Jr. Brown, J. H., Han, J., et al. (1998). Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J. Biol. Chem. 273, 2161–2168. doi: 10.1074/jbc.273.10.5423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wetzel, U., Boldt, A., Lauschke, J., Weigl, J., Schirdewahn, P., Dorszewski, A., et al. (2005). Expression of connexins 40 and 43 in human left atrium in atrial fibrillation of different aetiologies. Heart 91, 166–170. doi: 10.1136/hrt.2003.024216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Workman, A. J., Pau, D., Redpath, C. J., Marshall, G. E., Russell, J. A., Norrie, J., et al. (2009). Atrial cellular electrophysiological changes in patients with ventricular dysfunction may predispose to af. Heart Rhythm 6, 445–451. doi: 10.1016/j.hrthm.2008.12.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, X., Zhao, W., Corrillo, E., Chen, W., Yan, J., Bers, D. M., et al. (2014). “Novel stress signaling jnk regulates camkiiδ activity and expression in aged human atrium,” in AHA Annual Meeting (Chicago).
Wu, Y., MacMillan, L. B., McNeill, R. B., Colbran, R. J., and Anderson, M. E. (1999). Cam kinase augments cardiac l-type ca2+ current: a cellular mechanism for long q-t arrhythmias. Am. J. Physiol. 276, H2168–H2178.
Xie, Y., Sato, D., Garfinkel, A., Qu, Z., and Weiss, J. N. (2010). So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys. J. 99, 1408–1415. doi: 10.1016/j.bpj.2010.06.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yan, J., Kong, W., Zhang, Q., Beyer, E. C., Walcott, G., Fast, V. G., et al. (2013). C-jun n-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc. Res. 97, 589–597. doi: 10.1093/cvr/cvs366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, Z., Shen, W., Rottman, J. N., Wikswo, J. P., and Murray, K. T. (2005). Rapid stimulation causes electrical remodeling in cultured atrial myocytes. J. Mol. Cell. Cardiol. 38, 299–308. doi: 10.1016/j.yjmcc.2004.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yeh, Y. H., Wakili, R., Qi, X. Y., Chartier, D., Boknik, P., Kaab, S., et al. (2008). Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ. Arrhythm. Electrophysiol. 1, 93–102. doi: 10.1161/CIRCEP.107.754788
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoon, S., and Seger, R. (2006). The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44. doi: 10.1080/02699050500284218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zechner, D., Thuerauf, D. J., Hanford, D. S., McDonough, P. M., and Glembotski, C. C. (1997). A role for the p38 mitogen-activated protein kinase pathway in myocardial cell growth, sarcomeric organization, and cardiac-specific gene expression. J. Cell Biol. 139, 115–127. doi: 10.1083/jcb.139.1.115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, R., Dzhura, I., Grueter, C. E., Thiel, W., Colbran, R. J., and Anderson, M. E. (2005). A dynamic alpha-beta inter-subunit agonist signaling complex is a novel feedback mechanism for regulating l-type ca2+ channel opening. FASEB J. 19, 1573–1575. doi: 10.1096/fj.04-3283fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, T., Maier, L. S., Dalton, N. D., Miyamoto, S., Ross, J. Jr. Bers, D. M., et al. (2003). The deltac isoform of camkii is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92, 912–919. doi: 10.1161/01.RES.0000069686.31472.C5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, M., Dilly, K., Dos Santos Cruz, J., Li, M., Gu, Y., Ursitti, J. A., et al. (2004). Sarcoplasmic reticulum calcium defect in ras-induced hypertrophic cardiomyopathy heart. Am. J. Physiol. Heart Circ. Physiol. 286, H424–H433. doi: 10.1152/ajpheart.00110.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: atrial fibrillation, calcium handling, arrhythmogenesis, stress-response kinases, heart failure, aging
Citation: Ai X (2015) SR calcium handling dysfunction, stress-response signaling pathways, and atrial fibrillation. Front. Physiol. 6:46. doi: 10.3389/fphys.2015.00046
Received: 02 December 2014; Accepted: 30 January 2015;
Published online: 19 February 2015.
Edited by:
Ming Lei, University of Oxford, UKReviewed by:
Andrew F. James, University of Bristol, UKCopyright © 2015 Ai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xun Ai, Department of Cell and Molecular Physiology, Loyola University Chicago, 2160 South First Avenue, Maywood, IL 60153, USA e-mail:eGFpMUBsdWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.