 Dominik J. Schaer
Dominik J. Schaer Francesca Vinchi
Francesca Vinchi Giada Ingoglia
Giada Ingoglia Emanuela Tolosano
Emanuela Tolosano Paul W. Buehler
Paul W. Buehler- 1Division of Internal Medicine, University of Zurich, Zurich, Switzerland
- 2Department of Molecular Biotechnology and Health Sciences, University of Torino, Torino, Italy
- 3Division of Hematology, Laboratory of Biochemistry and Vascular Biology, Center for Biologics Evaluation and Research, Food and Drug Administration, Bethesda, MD, USA
Hemolysis, which occurs in many disease states, can trigger a diverse pathophysiologic cascade that is related to the specific biochemical activities of free Hb and its porphyrin component heme. Normal erythropoiesis and concomitant removal of senescent red blood cells (RBC) from the circulation occurs at rates of approximately 2 × 106 RBCs/second. Within this physiologic range of RBC turnover, a small fraction of hemoglobin (Hb) is released into plasma as free extracellular Hb. In humans, there is an efficient multicomponent system of Hb sequestration, oxidative neutralization and clearance. Haptoglobin (Hp) is the primary Hb-binding protein in human plasma, which attenuates the adverse biochemical and physiologic effects of extracellular Hb. The cellular receptor target of Hp is the monocyte/macrophage scavenger receptor, CD163. Following Hb-Hp binding to CD163, cellular internalization of the complex leads to globin and heme metabolism, which is followed by adaptive changes in antioxidant and iron metabolism pathways and macrophage phenotype polarization. When Hb is released from RBCs within the physiologic range of Hp, the potential deleterious effects of Hb are prevented. However, during hyper-hemolytic conditions or with chronic hemolysis, Hp is depleted and Hb readily distributes to tissues where it might be exposed to oxidative conditions. In such conditions, heme can be released from ferric Hb. The free heme can then accelerate tissue damage by promoting peroxidative reactions and activation of inflammatory cascades. Hemopexin (Hx) is another plasma glycoprotein able to bind heme with high affinity. Hx sequesters heme in an inert, non-toxic form and transports it to the liver for catabolism and excretion. In the present review we discuss the components of physiologic Hb/heme detoxification and their potential therapeutic application in a wide range of hemolytic conditions.
Introduction
Hemolysis with release of free hemoglobin (Hb) and heme occurs in a wide range of disease states and clinical interventions, including genetic and acquired anemias such as sickle cell disease, burns, extracorporeal circulation and massive blood transfusion (Schaer et al., 2013c). Additionally, chemical or recombinant modified Hbs have been evaluated as oxygen therapeutics in several disease states or when blood is not available (Silverman and Weiskopf, 2009). The study of these therapeutic candidates has also provided relevant information regarding the concentration-dependent pathophysiology associated with extracellular Hb in circulation and tissue compartments. Critical reviews provide greater detail concerning their dose dependent toxicity (Buehler et al., 2010).
The primary pathophysiologic effects associated with free Hb/heme are acute hemodynamic instability and acute or chronic tissue injury (Schaer et al., 2013c). The underlying biochemistry of these adverse effects appears to be related to the nitric oxide and oxidant reactivity of free Hb and heme. The ability to counteract extracellular Hb resulting from normal red blood cell turnover and mild hemolysis is one function of haptoglobin (Hp), an α2-sialoglycoprotein, which is the primary Hb-binding protein in plasma. Hb-bound Hp targets a specific cellular pathway of clearance through the monocyte/macrophage surface receptor CD163 (Kristiansen et al., 2001). Depending on the extent and frequency of hemolysis, Hp may become depleted, rendering this pathway ineffective. Previous reports from patients with sickle cell disease, spherocytosis, autoimmune hemolytic anemia, erythropoietic protoporphyria and pyruvate kinase deficiency suggest that Hp depletion in plasma occurs prior to the decline of hemopexin (Hx) concentrations (Muller-Eberhard et al., 1968). The disease related consumption of the plasma Hp pool during hemolysis makes Hp a specific clinical marker for intravascular hemolysis (Kormoczi et al., 2006).
Heme released following oxidation of Hb to met-Hb or from heme saturated hepatocytes is bound by albumin and rapidly transferred to Hx, the plasma protein with the highest binding affinity for heme. Hx is another glycoprotein produced by the liver with a plasma concentration of 1–2 mg/ml (Muller-Eberhard et al., 1968). Hx prevents heme's pro-oxidant and pro-inflammatory effects and promotes its detoxification, particularly when Hp concentrations are low or depleted in cases of severe or prolonged hemolysis. Both Hp and Hx are acute-phase proteins, induced during infection and inflammatory states in order to minimize tissue injury and facilitate tissue repair.
The present review will describe the primary mechanisms by which Hp and Hx prevent Hb/heme toxicity prior to monocyte/macrophage-hepatocyte clearance, critically evaluate the difference in genetic phenotype function and describe the rationale for exogenous Hp and Hx as therapeutic proteins for use in hemolytic disease states.
Mechanisms by which Hp and Hx Protect against Hb/heme Toxicity
Mechanistic research on the in vitro and in vivo function of Hp and Hx has suggested well defined mechanisms of protection (Schaer et al., 2013c). The best-characterized function of Hp is intravascular sequestration of extracellular Hb following formation of large Hb-Hp protein complexes, a process that prevents extravasation of free Hb into tissues. This effect is particularly evident in the kidneys where oxidative reactions at the heme moiety of Hb lead to globin deposition (hyaline casts), iron overload, lipid peroxidation and renal tubular injury (Qian et al., 2010; Ballarin et al., 2011; Billings et al., 2011) (Figure 1). Similarly, by sequestering heme within a protein complex, Hx prevents heme's ability to intercalate into cell membranes and plasma lipoproteins, where it participates in reactions with organic peroxides, the result of which is the generation of free radicals in localized tissue spaces followed by lipid, protein and DNA oxidation. Additionally, Hx blocks heme activation of immune receptors and vascular inflammatory processes (Belcher et al., 2014). The combined antioxidant and Hb/heme binding characteristics of Hp and Hx allow Hb-Hp and heme-Hx complexes to circulate in the blood in a less toxic form until they can be cleared by their respective scavenger receptors. In the following sections, Hp and Hx will be discussed with an emphasis on tissue protection from the deleterious effects of extracellular Hb and heme.
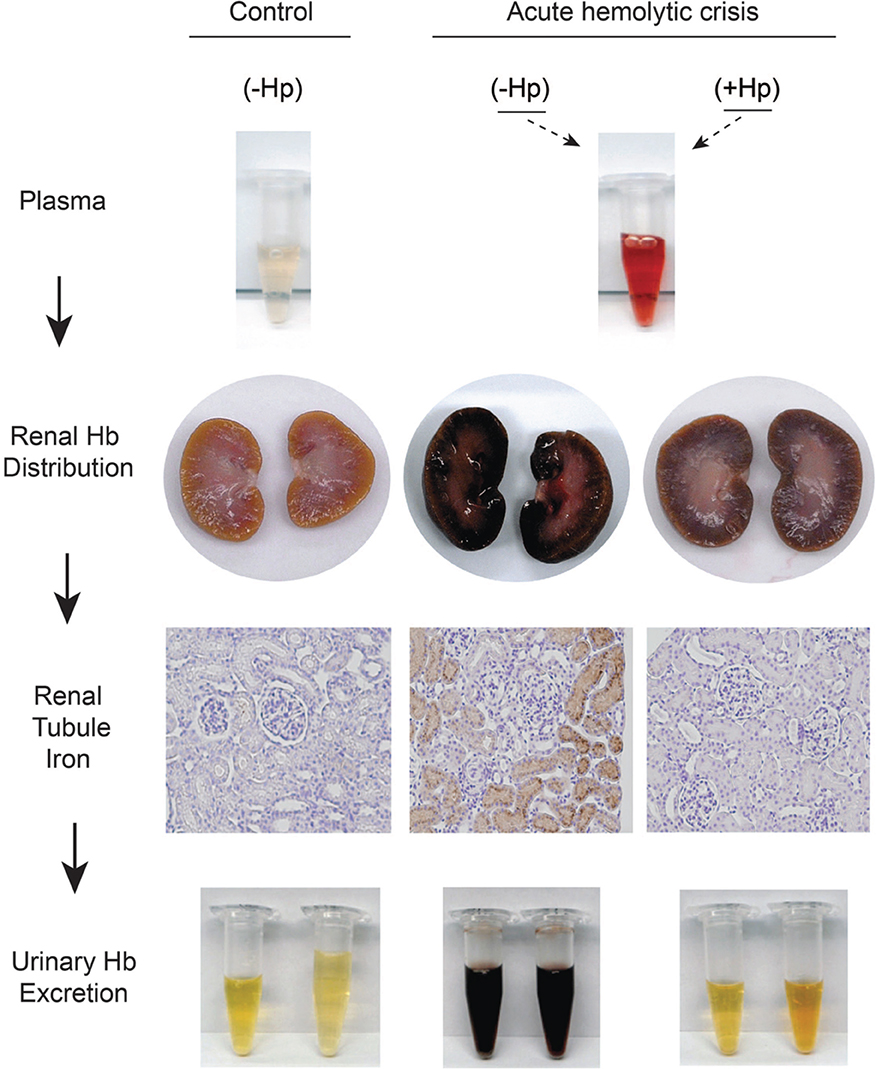
Figure 1. Sequence of renal plasma clearance of hemoglobin: The sequence represents sustained intravascular hemolysis in a guinea pig model. The red color of Hb in plasma during hemolysis suggests ferrous Hb accumulation. In the presence of haptoglobin free Hb is sequestrated within the Hb-Hp complex. Processes of Hb-Hp clearance are saturated during massive hemolysis and the complex remains in circulation for an extended period of time, leading to the brownish color or ferric heme in bound Hb dimers, which is a result of auto-oxidation or reaction with NO or other oxidants in plasma. Free hemoglobin readily dissociates into dimers in plasma, leading to extensive glomerular filtration, which is blocked by Hp. The filtration of Hb leads to renal tubule iron deposition (brown) and hemoglobinuria. This is not observed in control animals or when Hp is administered and Hb remains sequestered in the Hb-Hp complex.
Hp and Hx Sequester Extracellular Hb/heme within the Plasma Compartment Until Cellular Clearance
Within red blood cells (RBCs), Hb exists as a tetrameric heme containing protein with a molecular weight of 64 kD. Following release from RBCs during intravascular hemolysis, extracellular Hb is in a dynamic tetramer ↔ dimer equilibrium (Ackers and Halvorson, 1974). The dimer fraction of Hb is favored by low Hb concentration and an oxygenated Hb (Fe2+) state. The 32-kD Hb dimer readily passes through the glomerulus—if not bound by Hp—and is rapidly cleared by the kidneys (Murray et al., 1961; Andersen et al., 1966; Boretti et al., 2009). Renal filtration is therefore a primary route of clearance for extracellular Hb following Hp depletion. In guinea pigs and dogs, we have previously characterized plasma clearance of extracellular Hb with a circulatory half-life of approximately 10–60 min (Boretti et al., 2009, 2014). This is consistent with earlier findings using radio-labeled Hb in rabbit and dog (Bunn et al., 1969). Hb can also be filtered into the lymphatic system after intravascular injection, and translocates across intact endothelial monolayers both in vitro and in vivo (Nakai et al., 1998; Faivre-Fiorina et al., 1999; Matheson et al., 2000). The mechanisms underlying this “translocation/extravasation” phenomenon remain unclear, but may involve active transport mechanisms—for example, through the caveolar system, which is also responsible for the controlled transendothelial transport of other plasma proteins such as albumin (Faivre-Fiorina et al., 1999; Komarova and Malik, 2010).
Within the plasma compartment, the abundance of anti-oxidants (i.e., ascorbic acid and urate) stabilizes Hb in a reduced state (Butt et al., 2010). This physiologic function of plasma makes it less likely for Hb to participate in oxidative reactions that lead to rapid met-Hb formation and release of free heme. However, once dimerization of Hb occurs, autoxidation of dimers proceeds rapidly with a rate constant (kox = 0.24 h−1) 16-fold faster than tetrameric Hb (kox = 0.015 h−1) (Griffon et al., 1998). Furthermore, after translocation into the extravascular space, Hb may encounter much harsher oxidative conditions than in plasma. This may be particularly true in diseased and inflamed tissues or after an ischemic insult when oxidants accumulate and an acidic pH is created in local tissue sites. A disconnect of intravascular and extravascular Hb oxidation was observed in animal models of intravascular hemolysis or following infusion of ferrous (Fe2+) Hb. While oxidized ferric (Fe3+) Hb usually remains below the level of detection in plasma, a large fraction of excreted Hb ultimately appears in the urine in the oxidized ferric state (Boretti et al., 2009). This observation could be extrapolated to other tissues, such as the vascular wall, where Hb(Fe3+) may accumulate. The large Hb-Hp complex cannot be filtered by the kidney and in vitro translocation across endothelial monolayers is almost completely blocked (Lipiski et al., 2013). The most evident protective function of Hp is therefore the sequestration of extracellular Hb within the antioxidant rich plasma compartment (Figure 2). Following binding, Hp limits Hb's access to susceptible environments where heme release, oxidative processes and NO-consuming reactions are much more likely to occur and may finally lead to the sequelae of Hb toxicity, such as renal failure and vascular injury (Gladwin et al., 2012).
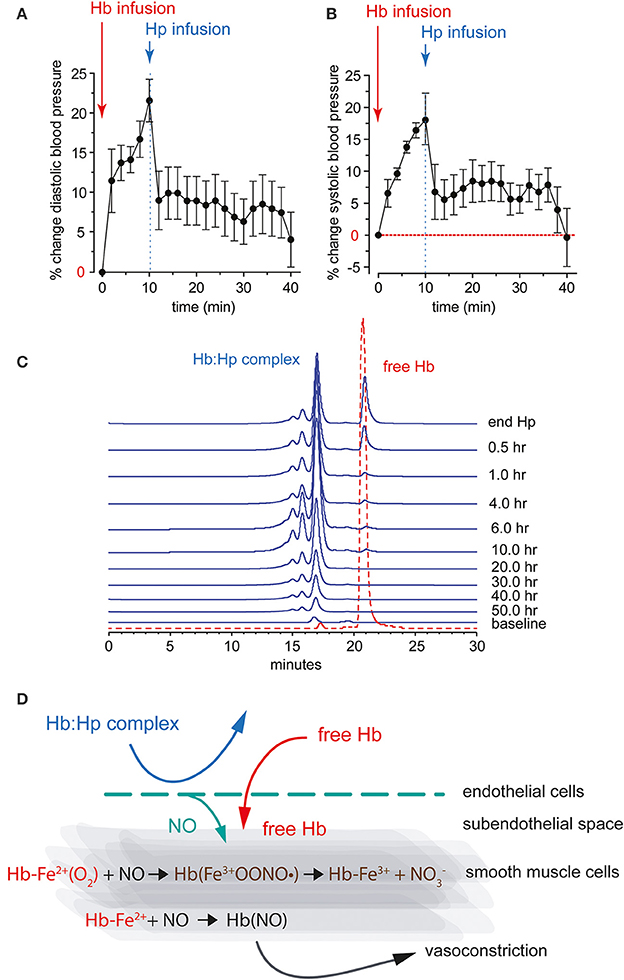
Figure 2. Diastolic (A) and systolic (B) systemic arterial blood pressure response after bolus infusion of free Hb followed by a bolus infusion of purified human Hp in guinea pigs. (C) HPLC analysis of guinea pig plasma after infusion of free Hb followed by Hp. The traces show free Hb co-eluting with an Hb standard (red). The Hb-Hp complex has a larger molecular weight, as indicated by the earlier elution time. The Hb-Hp complex has a long circulation half-life with detectable levels up to 50 h after infusion in this species. (D) NO reactions with free Hb in the vascular wall. Ferrous Hb (Fe2+) can react with the vasodilator NO via two reactions: (1) NO dioxygenation of oxy-Hb that generates nitrate (NO−3) and ferric Hb (Fe3+), and (2) iron nitrosylation of deoxy-Hb that occurs by direct iron binding of NO to non-liganded ferrous Hb (Fe2+). Both reactions lead to depletion of NO and explain the acute vasoactivity of extracellular Hb, which is attenuated by Hp.
A similar sequestering function is observed with Hx, which is capable of controlling the reactivity of free heme following orientation into Hx's “heme pockets.” By scavenging free heme, Hx prevents heme-mediated oxidative reactions with lipids, proteins, nucleic acids and other biological molecules. Along with Hx, several plasma proteins are able to bind heme, including albumin, lipoproteins, α1-microglobulin, but none of these proteins can efficiently prevent heme intercalation into lipid membranes or block heme's pro-oxidant and pro-inflammatory effects.
Hp and Hx Promote Free Hb and Heme Clearance via Receptor Targeted Pathways
Haptoglobin
Hb toxicity largely depends on the rate of hemolysis, tissue oxidant status and clearance capacity. In this network, Hp acts in concert with the plasma's small molecular reducing agents that maintain Hb in a reduced, less reactive ferrous (Fe2+) oxidation state (Buehler et al., 2007).
In addition to keeping Hb within the antioxidant environment of plasma, Hp plays an essential role in the clearance of Hb and metabolic detoxification of heme (Figure 3). In humans, the scavenger receptor CD163, previously assigned as the macrophage scavenger receptor M130 (Law et al., 1993), has been identified as an endocytosis receptor for Hb-Hp complexes (Kristiansen et al., 2001). CD163 is exclusively expressed by cells of the monocyte-macrophage lineage, with particularly high expression levels in red pulp macrophages of the spleen and liver Kupffer cells (Zwadlo et al., 1987; Bachli et al., 2006). Within the Hb-Hp complex, a high-affinity binding epitope has been identified within the interface region of the Hp β-chain and the Hb α-chain (Madsen et al., 2004; Andersen et al., 2012). Tetrameric Hb can also bind to and is internalized by CD163 in the absence of Hp (Schaer et al., 2006; Buehler et al., 2008). It is unknown whether this lower affinity interaction involves similar regions within Hb and CD163, or whether separate binding structures have independently evolved for Hb and the Hb-Hp complex, respectively (Buehler et al., 2008). Experimental evidence suggests that other Hb and Hb-Hp clearance pathways exist in addition to the CD163 scavenger receptor, and that efficient non-renal Hb clearance mechanisms may be active in the absence of Hp (Murray et al., 1961; Schaer et al., 2006; Etzerodt et al., 2013). However, the nature of these mechanisms requires further studies to be more thoroughly understood.
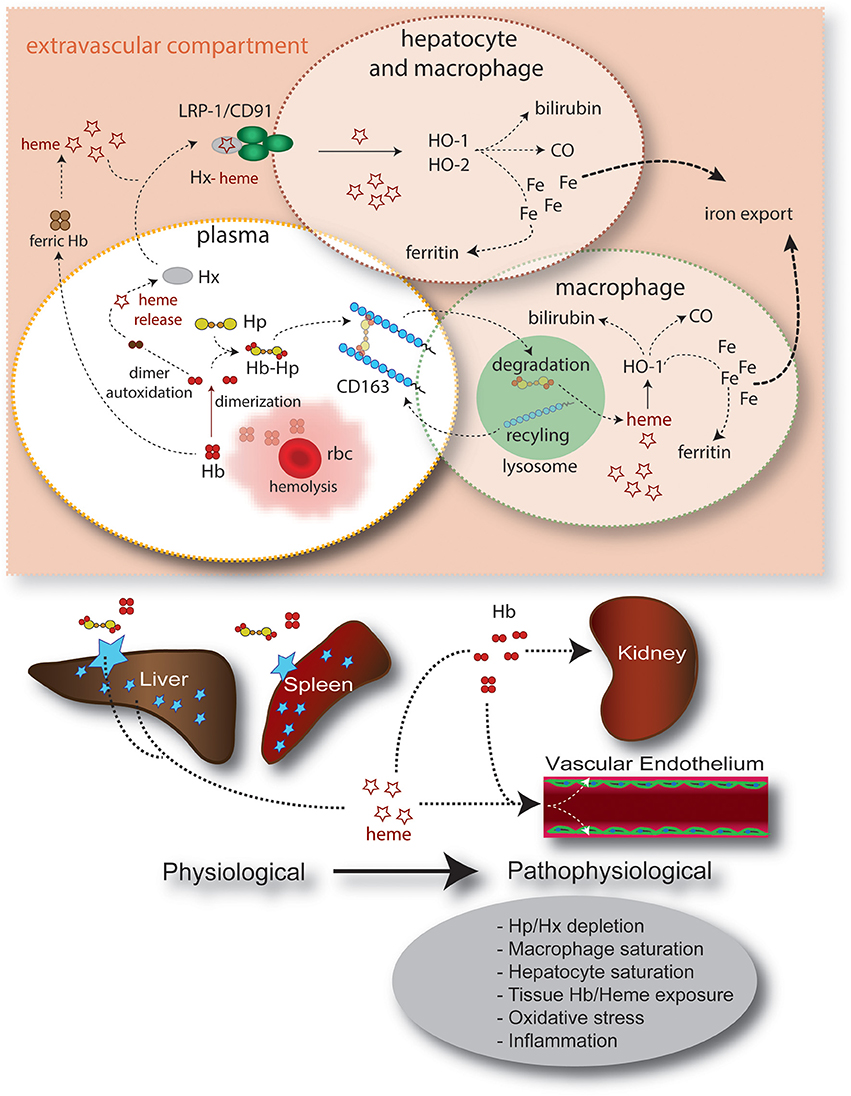
Figure 3. Summary of intravascular/extravascular Hb/heme clearance and cellular/tissue distribution. The main components of the Hb/heme detoxification system are haptoglobin (Hp), which sequesters Hb in the oxidatively protected Hb-Hp complex and hemopexin (Hx). The Hb-Hp complex is cleared and metabolized by CD163+ macrophages. Alternatively, during more severe hemolysis, free heme—after release from ferric Hb—can be detoxified by the hemopexin (Hx) rescue pathway. In all scenarios heme is finally detoxified by the heme oxygenases, which provide free iron for export by ferroportin or, alternatively, storage in the ferritin complex. The physiologic clearance and detoxification organs for Hb/heme are the liver and spleen, respectively, while the primary targets of Hb/heme toxic activities are the vascular endothelium and the kidney.
Inflammatory and anti-inflammatory processes modulate the activity of the Hb-Hp scavenger pathway at multiple levels. Systemic inflammation, particularly if it involves the interleukin (IL)-6 effector pathway, increases expression of Hp in the liver and many parenchymal and non-parenchymal cells. In many species, Hp is one of the most extensively responding acute-phase plasma proteins. IL-6 has also been reported to enhance expression of CD163 on macrophages, suggesting that enhanced Hb sequestration and clearance capacity are general adaptive responses to infection and tissue injury (Buechler et al., 2000). Intriguingly, however, some inflammatory mediators, such as endotoxin and other Toll-like receptor (TLR) agonists or tumor necrosis factor-α (TNF-α) trigger protease-mediated shedding of CD163 from the cell surface of monocytes and macrophages (Droste et al., 1999; Hintz et al., 2002; Moller et al., 2002; Weaver et al., 2006; Etzerodt et al., 2010). This shedding acutely blocks clearance of Hb-Hp complexes by monocytes (Schaer et al., 2007). High levels of soluble CD163 are consequently found in patients with sepsis or more specific macrophage activation syndromes (Schaer et al., 2005; Moller et al., 2006). The physiologic role of this process is unknown, but it implies that complex regulatory mechanisms control Hb detoxification and cellular exposure. Additionally, sCD163 may itself contribute the oxidative detoxification of extracellular Hb and Fcγ-receptor-mediated endocytosis of sCD163-Hb-IgG complexes by monocytes/macrophages, and endothelial cells have been proposed to be an alternative route for Hb clearance (Subramanian et al., 2013).
Of special consideration is the regulation of the Hb clearance system by anti-inflammatory glucocorticoids. In global proteome and transcriptome exploration studies, CD163 was found to be one of the principal glucocorticoid-induced genes in human and mouse macrophages, and patients treated with high-dose pulse glucocorticoids had a significantly enhanced Hb-Hp clearance capacity of their peripheral blood monocytes (Schaer et al., 2001, 2002; Vallelian et al., 2010). Glucocorticoids also act as a transcriptional (co-)activator on Hp synthesis in many species, including human (Marinkovic and Baumann, 1990). In a dog model, a glucocorticoid-supported high Hp level was completely protective against hemoglobinuria and hypertension during an 8-h infusion of free Hb (Boretti et al., 2009). Cumulatively, inflammation and glucocorticoids appear to enhance tolerance against extracellular Hb. In contrast, other drugs such as the antimalarial chloroquine can negatively affect Hb/heme detoxification in macrophages and potentially enhance free Hb toxicity (Schaer et al., 2013b). The potential clinical relevance of these observations—for example, for the treatment of hemolytic anemias—remains to be resolved.
Hemopexin
The heme scavenging function of Hx primarily attenuates heme toxicity on the vascular endothelium (Vinchi et al., 2013). While heme easily enters into endothelial cells when bound to albumin, this translocation is completely blocked in the presence of Hx. Heme sequestration within the Hx complex further ensures protection against heme-driven oxidative processes in the extracellular space and it prevents heme-triggered inflammation and adhesion molecule expression.
In vivo, the heme-Hx complex is primarily cleared by hepatocytes via receptor-mediated endocytosis (Vinchi et al., 2013). To date, the only known heme-Hx complex receptor is the LDL receptor-related protein 1 (LRP 1), a multi-ligand scavenger receptor (Hvidberg et al., 2005). In addition to hepatocytes, LRP1 is expressed on several other cell types, including macrophages, neurons, and syncytiotrophoblasts. The role of these LRP1 expressing cell types and the possible contribution of alternative receptors in heme-Hx complex clearance remain to be studied.
Some studies have suggested that Hx could be recycled as an intact molecule to the extracellular milieu. However, Hvidberg et al., have shown that most Hx is degraded in lysosomes (Hvidberg et al., 2005). This agrees with the observation that plasma Hx level usually deplete in disorders associated with free heme exposures in both humans and mice.
Hp and Hx Support Local NO Homeostasis
Depletion of NO, which occurs when the gaseous vasodilator reacts with Hb, explains the acute vasoactivity of extracellular Hb, which is characterized by an acute systemic and, to some extent, pulmonary hypertensive response (Figure 2) (Doherty et al., 1998; Reiter et al., 2002; Gladwin et al., 2004; Olson et al., 2004; Minneci et al., 2005). In experimental animals, consistent acute hypertensive responses can be observed at extracellular Hb levels exceeding approximately 40 μM (heme). The observation that infusion of an Hb-Hp complex lacks the vasoactivity of an equimolar quantity of extracellular Hb has initially suggested that Hp may interfere with Hb's reactions with NO. However, when the NO reactivity of the Hb-Hp complex was measured in vitro or in animal plasma, no differences in NO-driven heme oxidation and/or NO consumption were found between free Hb and the Hb-Hp complex, respectively (Boretti et al., 2009). Additionally, Hb-triggered changes in NO metabolite plasma concentrations were not attenuated by Hp treatment at a dose that completely suppressed Hb's hypertensive response (Baek et al., 2012). Therefore, it was speculated that Hp supports local NO bioavailability by keeping the small Hb molecule away from sites of NO production or NO effector functions. Such vulnerable anatomic sites might be the endothelial caveolar system, which represents the primary localization of endothelial NO synthase, the subendothelial space, or the NO responsive contractile smooth muscle cell layers within the wall of arterial resistance vessels (Buehler et al., 2010).
Hx also appears to support vascular NO homeostasis as demonstrated by studies in heme-overloaded Hx-null mice and mouse models of hemolytic disorders (Chiabrando et al., 2014). It is also likely that the beneficial effect of Hx on NO bioavailability is due to heme sequestration away from sites of NO production. Several scenarios could contribute to heme-driven reduction of NO availability. The increased production of reactive oxygen species (ROS) induced by heme may promote NO consumption and formation of peroxynitrite (ONOO-) through the reaction of NO with superoxide (O·−2). Additionally, NO synthase (NOS), may be uncoupled due to oxidation of the essential cofactor BH4, thus leading to the generation of O·−2 in place of NO. Accumulation of ONOO- may further contribute to the reduction of NOS activity and the disruption of the NOS dimer.
In summary, although a sparing effect of Hp and Hx on local vascular NO homeostasis is strongly supported, an experimental proof of this concept is still warranted.
Hp Controls Oxidative Reactions of Hb
Increased tissue concentrations of peroxides [i.e., hydrogen peroxide (H2O2) or lipid peroxides] occur in many disease states, particularly within the context of inflammatory reactions and during cycles of ischemia followed by reperfusion (Niethammer et al., 2009). The biochemical reactions of Hb with peroxides have been extensively studied in vitro and can be summarized as follows: deoxy-Hb reacts with H2O2 via the following steps: (1) oxo-ferryl Hb [Hb(Fe4+ = O)] generation, (2) ferric Hb [Hb(Fe3+)] generation, and (3) protein radical generation [·Hb(Fe4+ = O)] (Boutaud et al., 2010).
The globin chain free radical shown in reaction 3 can participate in localized amino acid oxidations within Hb (“intrinsic reactions”) or it can transfer to susceptible external molecules such as lipoproteins (“extrinsic reactions”). During the “intrinsic” free radical reactions within Hb, a defined peptide hotspot that is located at the α-globin/β-globin interface becomes the primary site where amino acid oxidation occurs. Among these amino acids is the highly susceptible β-chain Cys93, which can be oxidized to cysteic acid, as well as the α-chain Tyr42, which appears to be an important radical hub within Hb (Jia et al., 2007; Reeder et al., 2008; Pimenova et al., 2010). The cumulative result of these oxidative processes becomes apparent as an unfolding, intermolecular crosslinking and progressive degradation of the Hb molecule (Jia et al., 2007; Vallelian et al., 2008). The oxidized protein can escape clearance by the Hp-CD163 scavenger pathway, and it appears that some Hb degradation products may eventually have direct pro-inflammatory and cytotoxic activities (Buehler et al., 2009; Schaer et al., 2013a). The result of the “extrinsic” radical reactions may be the generation of oxidized lipoproteins, damage to other (non-Hb) proteins and modification of cell membrane components (Miller et al., 1997; Schaer et al., 2013a). Hp binding does not reduce the primary reactivity of Hb with peroxides, a reaction that is generally measured as the initial change in the heme-iron oxidation states (i.e., oxidation of Fe2+ → Fe3+ → Fe4+) during the peroxidative reaction sequence. However, Hp binding limits secondary reactions of these compounds with internal (i.e., globin amino acids) and/or external substrates (Miller et al., 1997; Cooper et al., 2013).
The structural basis of this protection is not yet understood. Hp complex formation may physically limit the transfer of radicals located on the surface of Hb toward external acceptor molecules. Additionally, Hp may limit direct heme pocket access for external substrates. Furthermore, Hb dimerization that is forced by Hb-Hp complex formation may interfere with intra-molecular free radical translocation pathways that normally exist across the Hb dimer interface. Lastly, it has been proposed that Hp may function as a suicidal free radical scavenger within the complex (Pimenova et al., 2010). In vitro, the primary result of this oxidative protection becomes apparent as a stabilization of Hb's protein structure (Pimenova et al., 2010).
Hp Prevents Heme Release from Hb
Heme is an extremely hydrophobic compound that cannot persist as a monomeric soluble molecule under physiologic pH conditions. The term “free heme” is therefore a misnomer, which in fact refers to heme that is not stably located within the heme pocket of a classic heme protein such as Hb or myoglobin. Instead, heme can readily be transferred from ferric (but not from ferrous) Hb to low-affinity heme acceptors (Bunn and Jandl, 1968; Gattoni et al., 1996). The most extensively studied low-affinity heme acceptors are lipoproteins and albumin; however, the group of low-affinity heme acceptors likely involves a very diverse group of soluble and membrane-based proteins and lipids. In the case of lipoproteins, the transferred heme can trigger a lipid-peroxidation cascade, which ultimately results in structural modification of the particle (Balla et al., 1991). Oxidized lipoproteins are cytotoxic and represent one of the principal mediators of Hb toxicity (Nagy et al., 2010; Schaer et al., 2013a). Less understood effects of heme transfer (or “free heme”) are its direct cytotoxic and inflammatory effects (Balla et al., 1993; Belcher et al., 2000, 2003; Wagener et al., 2001; Jeney et al., 2002; Larsen et al., 2010; Fortes et al., 2012). Proposed mechanisms involve stimulation of Toll-like receptors, such as the endotoxin receptor TLR-4 and inhibition of the proteasome, among others (Figueiredo et al., 2007; Lin et al., 2012a; Vallelian et al., 2014).
It is in this context that the fourth known protective activity of Hp appears: the prevention of heme release from ferric (Fe3+) Hb (Bunn and Jandl, 1968; Lipiski et al., 2013; Mollan et al., 2014). We have previously learned that ferric Hb (Fe3+)—the substrate for heme transfer—does not usually accumulate in plasma, even under severe hemolytic conditions (Baek et al., 2012). It is therefore unlikely that heme transfer from Hb is a relevant process that occurs during hemolysis within the plasma. However, unlike extracellular Hb, the Hb-Hp complex has a slower and saturable clearance, which allows ferric (Fe3+) Hb-Hp to accumulate and to reach significant plasma concentrations. In a hemolysis model in guinea pigs, we detected up to 50% of ferric Hb-Hp 24 h after infusion of an Hp bolus for treatment of severe blood transfusion-associated hemolysis. Conceptually, the Hb-Hp complex in plasma may therefore be better compared with a polymerized Hb such as a hemoglobin-based oxygen carrier (HBOC). The intentionally slow clearance rates of HBOCs also allow these substances for significant accumulation of ferric species in vivo. Following infusion of most HBOCs there is no barrier against heme release and the oxidative damage that is associated with heme loss may be a significant component of vascular toxicity that accompanies administration of these therapeutic candidates (Natanson et al., 2008). As an evolutionary hypothesis, it is possible that the protective strategy of Hb sequestration within the plasma compartment was only achievable in combination with the sophisticated heme retention and oxidation control capacities of Hp. Only these functions may allow the large Hb-Hp complex to circulate in a less toxic form until cleared. Heme retention and oxidative stability may therefore discriminate the Hb-Hp complex from the adverse event profile of some HBOCs with comparable molecular size and long intravascular retention times (Pimenova et al., 2009; Silverman et al., 2009).
Hx Blocks Interactions of Free Heme with Biological Molecules
Hx has the highest binding affinity for heme (Kd < 10−13 M). Hx sequesters heme in an oxidatively inert conformation in a complex with a 1:1 stoichiometry, until it is cleared by the liver. Heme transfer to more reactive biomolecules is therefore suppressed by Hx. Accordingly, heme-overloaded Hx-null mice show increased oxidative stress in blood vessels, endothelial activation, enhanced inflammation and decreased NO bioavailability (Vinchi et al., 2013). Moreover, Hx-null mice display defective heme accumulation and catabolism in hepatocytes and reduced heme excretion in the bile (Tolosano et al., 1999; Vinchi et al., 2008, 2013).
The work of Belcher et al. (2014) has provided additional insight into how heme sequestration within the heme-Hx complex could attenuate heme-triggered inflammation. Heme is a weak activator of TLR4 and in endothelial cells heme-mediated TLR4 activation promotes Weibel-Palade body (WPB) degranulation, enhanced expression of adhesion molecules and activation of NF-κ B (Belcher et al., 2014). The heme-triggered activation of the TLR4 pathway is blocked by Hx. By similar mechanisms, Hp and Hx may interrupt the synergistic pro-inflammatory and toxic activities of Hb that occur in synergy with HMGB1 and LPS, respectively (Liang et al., 2009; Baek et al., 2014). Other anti-inflammatory effects of Hx may also represent a direct macrophage suppressor effect, which is independent of Hb/heme-triggered signaling (Lin et al., 2012b).
Haptoglobin and Hemopexin in Pathology and Drug Development
Cardiovascular Diseases
Due to its unique anatomic location, the vascular wall appears to be the principal target of free Hb and heme exposure during hemolysis. The diverse Hb/heme-triggered disease processes (i.e., NO consumption, lipid peroxidative processes) are therefore likely to modulate pathologies of the cardiovascular system such as atherosclerosis or typical vascular complications of hemolytic anemias. Oxidized lipoproteins are an established promoter of atherosclerosis and both Hb and heme can promote LDL oxidation (Balla et al., 1991; Belcher et al., 2010; Nagy et al., 2010). Likewise, free heme appears to be an endogenous trigger of endothelium and monocyte/macrophage inflammation, which are both essential in the progression of atherosclerotic lesions (Belcher et al., 2014). Furthermore, heme that is released during microhemorrhage in the vascular wall appears to be a fundamental modulator of the resident macrophage phenotype in atherosclerotic plaques (Boyle et al., 2009, 2012; Kaempfer et al., 2011; Finn et al., 2012). However, despite all this biochemical and cell biologic evidence there is so far no direct experimental or epidemiologic proof that chronic hemolysis or microhemorrhage related Hb release into the vascular wall could directly aggravate atherosclerosis. Accordingly, we do not know whether substitution of Hp or Hx at supra-physiologic levels could attenuate this process. More evidence supports a direct contribution of Hb and heme-triggered reactions to the cardiovascular complication of sickle cell disease, primarily in relation to pulmonary hypertension and the acute chest syndrome. Nitric oxide consumption by free Hb and heme-triggered endothelial inflammatory activation are the principal pathophysiologic components (Rother et al., 2005; Ghosh et al., 2013; Belcher et al., 2014).
Hp Polymorphism and Cardiovascular Disease Associations
In humans, an Hp gene polymorphism exists that determines the three major phenotypes: 1-1, 2-1, and 2-2 (Levy et al., 2010). As a result of a partial intragenic duplication within the Hb α-chain coding region on chromosome 16 (16q22.2), Hp α-chain 2, as opposed to Hp α-chain 1, has two cysteines for disulfide bonding with two other α-chains. Therefore, Hp 2-2 is secreted as a heterogeneous mixture of Hp αβ-chain polymers, whereas the Hp 1-1 phenotype is produced as a homogeneous αβ-dimer. Phenotype 2-1 is a mixed phenotype composed of a range of dimers and polymers. The phenotype distribution varies according to ethnicity; however, in the Western world, it approaches 15% for Hp 1-1, 50% for Hp 2-1, and 35% for Hp 2-2 (Langlois and Delanghe, 1996). The crystal structure of the Hb-Hp complex confirmed earlier biochemical studies predicting that only the non-polymorphic β-chain subunit of Hp interacts with bound Hb αβ-dimer, although no functional role of the α-chain could be inferred from these studies (Andersen et al., 2012). Therefore, per milligram, Hp protein, Hb binding and the physiologic “neutralization” capacity of the three phenotypes should be comparable. The assumption of equal binding has also been confirmed by independent studies in human plasma and with purified Hb-Hp complexes (Delanghe et al., 2000; Lipiski et al., 2013).
In the past, multiple functions of Hp, such as its anti-oxidative and CD163 adaptor functions, have been reported to be phenotype dependent (Asleh et al., 2003, 2005; Levy et al., 2007). Cumulatively, these studies implied that the Hp 2-2 phenotype might be dysfunctional compared to Hp 1-1, with less protective capacity against Hb-triggered pathologies. However, more recently, larger quantities of more standardized and better-characterized Hp proteins became available for experimental studies, including in vivo administration studies. These experimental Hp products resulted from industrial efforts to develop phenotype-specific therapeutic Hp products from pooled human plasma. Comparative investigations of two Hp products with predominant Hp 1-1 or Hp 2-2 composition found no significant differences in the intravascular sequestration or renal and vascular short term protection provided by dimeric and multimeric Hp phenotypes in a therapeutic setting of acute intravascular Hb exposure in guinea pigs and dogs (Lipiski et al., 2013; Boretti et al., 2014). Additionally, a range of biochemical studies examining oxidative Hb reactions, prevention of low-density lipoprotein oxidation, and heme retention in the Hb-Hp complex provided evidence of equal protective functions of the dimeric and multimeric phenotypes (Lipiski et al., 2013). Also, NO consumption appears to be identical with the Hb:Hp 1-1 and 2-2 complexes (Azarov et al., 2008; Lipiski et al., 2013).
Epidemiologic studies have also found controversial associations of Hp genotypes with the prevalence and clinical sequelae of atherosclerosis in the general population and in specific patient populations, particularly those with diabetes mellitus. However, more stratified analysis of several large observational studies now convincingly revealed that the Hp 2-2 genotype/phenotype is associated with increased relative risk of coronary artery disease of up to 10-fold in diabetic patients with a glycosylated HbA1c > 6.5 (Cahill et al., 2013). Accompanying mechanistic studies have found higher tissue iron accumulation, additional signs of oxidative damage and a higher number of apoptotic macrophages in aortic atherosclerotic plaques from patients with the Hp 2-2 genotype (Moreno et al., 2008; Purushothaman et al., 2012).
Thus, far, the pathophysiology underlying these associations is unknown. According to studies discussed above, the primary Hb detoxifying functions of Hp 1-1 and Hp 2-2 are comparable and can therefore not easily explain the epidemiologic differences. However, the biologic functions of Hp that have been investigated so far are more representative of the short term protective functions of Hp during acute hemolysis and may not fully reflect longer term antioxidant or immune modulatory functions of the protein that might be more relevant for chronic vascular disease development.
Most epidemiologic studies of phenotype association with cardiovascular disease have been controlled for overall population heterogeneity and for established cardiovascular risk factors; however, these studies were not systematically controlled for differences in Hp plasma concentrations (Cahill et al., 2013). Generally, individuals with the Hp 2-2 phenotype have lower plasma Hp concentrations. Therefore, besides the postulated phenotype dependent molecular functions of Hp, lower Hp concentrations may have an additional impact on vascular disease development in individuals with the Hp 2-2 phenotype (Cid et al., 1993; Shen et al., 2012).
The controversial findings related to Hp phenotype-specific functions and associated disease state risks should certainly stimulate more extensive investigations. However, it now appears that, for therapeutic considerations, the Hp phenotype selection of a product will likely not be a primary determinant of clinical success (Lipiski et al., 2013).
Disease Risk and Hx
A genetic Hx polymorphism has been reported in populations of African ancestry (Kamboh et al., 1993). The biological significance of this polymorphism in hemolytic disorders is merely speculative.
The only available data suggesting a strong association between plasma Hx level and disease risk come from studies evaluating the mouse model of Hx deficiency (Tolosano et al., 1999). Several studies reported a higher susceptibility of Hx-null mice to hemolysis triggered organ damage and heme-driven vascular injury, as discussed below. In other diseases, the impact of Hx has not been directly related to its function as a heme scavenger, likely due to the technical difficulties in quantifying small amounts of “free” heme or possibly due to heme-independent functions of Hx. One such example is related to a regulatory function of Hx in neuroinflammation. Hx deficient mice are more susceptible to the development of experimental autoimmune encephalomyelitis (EAE), the murine model of multiple sclerosis (Rolla et al., 2013). In mice, Hx deficiency favors the differentiation of naïve CD4+ T cells toward Th17 lineage and enhances the stabilization and expansion of memory Th17 cells by IL-23. This is due to a higher disposition of naïve T cells to differentiate toward the Th17 lineage and to a higher production of Th17 differentiating cytokines IL-6 and IL-23 by APCs. These data indicate that Hx may have a negative regulatory role in Th17-mediated inflammation. Interestingly, Hp-null mice subjected to EAE also develop an exacerbated disease and display an increased expression of inflammatory cytokines in the central nervous system, suggesting that Hp and Hx share some anti-inflammatory effects (Galicia et al., 2009). The potential role of the common Hb-heme axis remains to be established.
Pre-Clinical Studies of Scavenger Protein Therapeutics
Mouse models of sickle cell disease have a phenotype of chronic heme-driven endothelial activation and dysfunction that could be recovered by repeated Hx administration (Vinchi et al., 2013). These findings highlight a causative role of the free Hb-heme axis in SCD associated vasculopathy. Acute increases in plasma Hb or extracellular heme concentrations can trigger acute vasco-occlusion as well as the acute chest syndrome in SCD mice. Two independent studies have shown that both vaso-occlusion and the acute chest syndrome can be prevented by the infusion of Hp or Hx, respectively (Ghosh et al., 2013; Belcher et al., 2014).
Hemolysis has also been recognized to exacerbate some renal and vascular complications related to the transfusion of stored red blood cells. Retrospective clinical observation studies suggested a link between the storage duration of red blood cells and the incidence of cardiovascular complications such as myocardial infarction, stroke and death. One component of the so-called red blood cell storage lesion is an increased RBC fragility, which may lead to hemolysis post-transfusion. In a guinea pig transfusion model, increased fragility of older RBC could be linked to increased hemolysis in the post-transfusion period, appearance of free Hb in the circulation and Hb-triggered injury in the kidney and vasculature. All these pathologic changes could be prevented by Hp treatment at the time of old blood transfusion (Baek et al., 2012; Lipiski et al., 2013).
Hemolysis with increased free Hb/heme levels and accompanying Hp/Hx depletion can be observed in some patients with severe sepsis. In a mouse sepsis model, Hx administration significantly reduced organ injury and mortality (Larsen et al., 2010), presumably by blocking free heme-mediated oxidative processes and inflammation. In contrast, other mouse studies provided evidence that Hx could even accelerate uncontrolled infection by a poorly defined activity that impairs leukocyte recruitment to the focus of primary infection (Spiller et al., 2011). Observational studies in human patients provided evidence for a positive association of mortality with free Hb release in adults with sepsis (Janz et al., 2013). In this patient cohort, preserved plasma Hp concentrations were significantly correlated with reduced mortality. Whether these data could point to a causative role of free Hb/heme in the progression of human sepsis remains speculative and further laboratory research and better-controlled clinical studies are needed to resolve the role of the Hb/heme-Hp/Hx axis in the control of severe infection and sepsis.
Potential Clinical uses of Plasma-Derived Human Hp and Hx
The rationale for the use of Hp and Hx as therapeutic agents is based on the idea that they act by scavenging circulating Hb and heme, with particular relevance to pathological conditions associated with hemolysis. Central to this concept is the notion that acute and chronic hemolytic conditions are characterized by depletion of the Hb and heme scavengers. Pre-clinical proof-of-concept animal models have demonstrated that Hp and Hx effectively attenuate Hb- and heme-induced vascular and renal pathologies. Based on these preliminary observations, it would appear rational to develop Hp and Hx for therapeutic use (Boretti et al., 2009; Dalton and Podmore, 2011). Plasma-purified Hp has been marketed in Japan since 1985 with primary indications for use in conjunction with extracorporeal circulation, massive transfusion and thermal injury (Schaer et al., 2013c). The primary therapeutic effect in these disease states is protection of the kidneys from Hb-induced toxicity (Hashimoto et al., 1993). Dosing approaches to minimize overall renal exposure to Hb in medically compromised patients are described in a few case reports (Homann et al., 1977; Tanaka et al., 1991; Imaizumi et al., 1994). However, the primary dosing strategy employed in these studies is consistent with a dose-to-effect approach, whereby the Hp dose is increased until amelioration of hemoglobinuria occurs. The Japanese medical communities' use of Hp encompasses a wide range of hemolytic events in approved and off-label disease states in which hemolysis occurs. A new therapeutic protein being considered for approval in the United States and European markets would require safety and efficacy trials in a specific disease state with measurable efficacy end points. For example, in the circumstance of treating a chronic hemolytic anemia, such as sickle cell disease, with repeated Hp and/or Hx doses, a measurable outcome such as significant reduction in vascular complications, reduced hospitalization or duration of hospitalization may be required. As a result, selection of the appropriate disease state(s) to demonstrate measurable improvements in morbidity remains a critical and challenging decision for the drug development process. Pre-clinical studies are still ongoing with the aim of better defining the common and protein-specific functions of Hp and Hx, their disease specific modulatory functions and potential off-target effects. This research may further support well-designed clinical trials translating the protective effects of heme scavengers into clinical progress.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Swiss National Science Foundation (grants 310030/120658 and 31003A/138500), University of Zurich Research Priority Program “Integrative Human Physiology,” Swiss Federal Commission for Technology and Innovation (CTI), FDA Internal Funding and the Telethon Grant GGP12082.
References
Ackers, G. K., and Halvorson, H. R. (1974). The linkage between oxygenation and subunit dissociation in human hemoglobin. Proc. Natl. Acad. Sci. U.S.A. 71, 4312–4316. doi: 10.1073/pnas.71.11.4312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andersen, C. B., Torvund-Jensen, M., Nielsen, M. J., De Oliveira, C. L., Hersleth, H. P., Andersen, N. H., et al. (2012). Structure of the haptoglobin-haemoglobin complex. Nature 489, 456–459. doi: 10.1038/nature11369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Andersen, M. N., Mouritzen, C. V., and Gabrielli, E. R. (1966). Mechanisms of plasma hemoglobin clearance after acute hemolysis in dogs: serum haptoglobin levels and selective deposition in liver and kidney. Ann. Surg. 164, 905–912. doi: 10.1097/00000658-196611000-00019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Asleh, R., Guetta, J., Kalet-Litman, S., Miller-Lotan, R., and Levy, A. P. (2005). Haptoglobin genotype- and diabetes-dependent differences in iron-mediated oxidative stress in vitro and in vivo. Circ. Res. 96, 435–441. doi: 10.1161/01.RES.0000156653.05853.b9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Asleh, R., Marsh, S., Shilkrut, M., Binah, O., Guetta, J., Lejbkowicz, F., et al. (2003). Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ. Res. 92, 1193–1200. doi: 10.1161/01.RES.0000076889.23082.F1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Azarov, I., He, X., Jeffers, A., Basu, S., Ucer, B., Hantgan, R. R., et al. (2008). Rate of nitric oxide scavenging by hemoglobin bound to haptoglobin. Nitric Oxide 18, 296–302. doi: 10.1016/j.niox.2008.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bachli, E. B., Schaer, D. J., Walter, R. B., Fehr, J., and Schoedon, G. (2006). Functional expression of the CD163 scavenger receptor on acute myeloid leukemia cells of monocytic lineage. J. Leukoc. Biol. 79, 312–318. doi: 10.1189/jlb.0605309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baek, J. H., D'agnillo, F., Vallelian, F., Pereira, C. P., Williams, M. C., Jia, Y., et al. (2012). Hemoglobin-driven pathophysiology is an in vivo consequence of the red blood cell storage lesion that can be attenuated in guinea pigs by haptoglobin therapy. J. Clin. Invest. 122, 1444–1458. doi: 10.1172/JCI59770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baek, J. H., Zhang, X., Williams, M. C., Schaer, D. J., Buehler, P. W., and D'agnillo, F. (2014). Extracellular Hb enhances cardiac toxicity in endotoxemic guinea pigs: protective role of haptoglobin. Toxins (Basel). 6, 1244–1259. doi: 10.3390/toxins6041244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balla, G., Jacob, H. S., Eaton, J. W., Belcher, J. D., and Vercellotti, G. M. (1991). Hemin: a possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. 11, 1700–1711. doi: 10.1161/01.ATV.11.6.1700
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balla, J., Jacob, H. S., Balla, G., Nath, K., Eaton, J. W., and Vercellotti, G. M. (1993). Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. U.S.A. 90, 9285–9289. doi: 10.1073/pnas.90.20.9285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ballarin, J., Arce, Y., Torra Balcells, R., Diaz Encarnacion, M., Manzarbeitia, F., Ortiz, A., et al. (2011). Acute renal failure associated to paroxysmal nocturnal haemoglobinuria leads to intratubular haemosiderin accumulation and CD163 expression. Nephrol. Dial. Transplant. 26, 3408–3411. doi: 10.1093/ndt/gfr391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belcher, J. D., Beckman, J. D., Balla, G., Balla, J., and Vercellotti, G. (2010). Heme degradation and vascular injury. Antioxid. Redox Signal. 12, 233–248. doi: 10.1089/ars.2009.2822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belcher, J. D., Bryant, C. J., Nguyen, J., Bowlin, P. R., Kielbik, M. C., Bischof, J. C., et al. (2003). Transgenic sickle mice have vascular inflammation. Blood 101, 3953–3959. doi: 10.1182/blood-2002-10-3313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belcher, J. D., Chen, C., Nguyen, J., Milbauer, L., Abdulla, F., Alayash, A. I., et al. (2014). Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123, 377–390. doi: 10.1182/blood-2013-04-495887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belcher, J. D., Marker, P. H., Weber, J. P., Hebbel, R. P., and Vercellotti, G. M. (2000). Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood 96, 2451–2459.
Billings, F. T. T., Ball, S. K., Roberts, L. J. 2nd., and Pretorius, M. (2011). Postoperative acute kidney injury is associated with hemoglobinemia and an enhanced oxidative stress response. Free Radic. Biol. Med. 50, 1480–1487. doi: 10.1016/j.freeradbiomed.2011.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boretti, F. S., Baek, J. H., Palmer, A. F., Schaer, D. J., and Buehler, P. W. (2014). Modeling hemoglobin and hemoglobin:haptoglobin complex clearance in a non-rodent species-pharmacokinetic and therapeutic implications. Front. Physiol. 5:385. doi: 10.3389/fphys.2014.00385
Boretti, F. S., Buehler, P. W., D'agnillo, F., Kluge, K., Glaus, T., Butt, O. I., et al. (2009). Sequestration of extracellular hemoglobin within a haptoglobin complex decreases its hypertensive and oxidative effects in dogs and guinea pigs. J. Clin. Invest. 119, 2271–2280. doi: 10.1172/JCI39115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boutaud, O., Moore, K. P., Reeder, B. J., Harry, D., Howie, A. J., Wang, S., et al. (2010). Acetaminophen inhibits hemoprotein-catalyzed lipid peroxidation and attenuates rhabdomyolysis-induced renal failure. Proc. Natl. Acad. Sci. U.S.A. 107, 2699–2704. doi: 10.1073/pnas.0910174107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boyle, J. J., Harrington, H. A., Piper, E., Elderfield, K., Stark, J., Landis, R. C., et al. (2009). Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 174, 1097–1108. doi: 10.2353/ajpath.2009.080431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boyle, J. J., Johns, M., Kampfer, T., Nguyen, A. T., Game, L., Schaer, D. J., et al. (2012). Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ. Res. 110, 20–33. doi: 10.1161/CIRCRESAHA.111.247577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buechler, C., Ritter, M., Orso, E., Langmann, T., Klucken, J., and Schmitz, G. (2000). Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J. Leukoc. Biol. 67, 97–103.
Buehler, P. W., Abraham, B., Vallelian, F., Linnemayr, C., Pereira, C. P., Cipollo, J. F., et al. (2009). Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood 113, 2578–2586. doi: 10.1182/blood-2008-08-174466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buehler, P. W., D'agnillo, F., Hoffman, V., and Alayash, A. I. (2007). Effects of endogenous ascorbate on oxidation, oxygenation, and toxicokinetics of cell-free modified hemoglobin after exchange transfusion in rat and guinea pig. J. Pharmacol. Exp. Ther. 323, 49–60. doi: 10.1124/jpet.107.126409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buehler, P. W., D'agnillo, F., and Schaer, D. J. (2010). Hemoglobin-based oxygen carriers: from mechanisms of toxicity and clearance to rational drug design. Trends Mol. Med. 16, 447–457. doi: 10.1016/j.molmed.2010.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buehler, P. W., Vallelian, F., Mikolajczyk, M. G., Schoedon, G., Schweizer, T., Alayash, A. I., et al. (2008). Structural stabilization in tetrameric or polymeric hemoglobin determines its interaction with endogenous antioxidant scavenger pathways. Antioxid. Redox Signal. 10, 1449–1462. doi: 10.1089/ars.2008.2028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bunn, H. F., Esham, W. T., and Bull, R. W. (1969). The renal handling of hemoglobin. I. Glomerular filtration. J. Exp. Med. 129, 909–923. doi: 10.1084/jem.129.5.909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bunn, H. F., and Jandl, J. H. (1968). Exchange of heme among hemoglobins and between hemoglobin and albumin. J. Biol. Chem. 243, 465–475.
Butt, O. I., Buehler, P. W., and D'agnillo, F. (2010). Differential induction of renal heme oxygenase and ferritin in ascorbate and nonascorbate producing species transfused with modified cell-free hemoglobin. Antioxid. Redox Signal. 12, 199–208. doi: 10.1089/ars.2009.2798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cahill, L. E., Levy, A. P., Chiuve, S. E., Jensen, M. K., Wang, H., Shara, N. M., et al. (2013). Haptoglobin genotype is a consistent marker of coronary heart disease risk among individuals with elevated glycosylated hemoglobin. J. Am. Coll. Cardiol. 61, 728–737. doi: 10.1016/j.jacc.2012.09.063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chiabrando, D., Vinchi, F., Fiorito, V., Mercurio, S., and Tolosano, E. (2014). Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 5:61. doi: 10.3389/fphar.2014.00061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cid, M. C., Grant, D. S., Hoffman, G. S., Auerbach, R., Fauci, A. S., and Kleinman, H. K. (1993). Identification of haptoglobin as an angiogenic factor in sera from patients with systemic vasculitis. J. Clin. Invest. 91, 977–985. doi: 10.1172/JCI116319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cooper, C. E., Schaer, D. J., Buehler, P. W., Wilson, M. T., Reeder, B. J., Silkstone, G., et al. (2013). Haptoglobin binding stabilizes hemoglobin ferryl iron and the globin radical on tyrosine beta145. Antioxid. Redox Signal. 18, 2264–2273. doi: 10.1089/ars.2012.4547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalton, J., and Podmore, A. (2011). Enriched Haptoglobin Polymers for the Treatment of Disease. US Patent Application 20110021418.
Delanghe, J., Allcock, K., Langlois, M., Claeys, L., and De Buyzere, M. (2000). Fast determination of haptoglobin phenotype and calculation of hemoglobin binding capacity using high pressure gel permeation chromatography. Clin. Chim. Acta 291, 43–51. doi: 10.1016/S0009-8981(99)00194-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doherty, D. H., Doyle, M. P., Curry, S. R., Vali, R. J., Fattor, T. J., Olson, J. S., et al. (1998). Rate of reaction with nitric oxide determines the hypertensive effect of cell-free hemoglobin. Nat. Biotechnol. 16, 672–676. doi: 10.1038/nbt0798-672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Droste, A., Sorg, C., and Hogger, P. (1999). Shedding of CD163, a novel regulatory mechanism for a member of the scavenger receptor cysteine-rich family. Biochem. Biophys. Res. Commun. 256, 110–113. doi: 10.1006/bbrc.1999.0294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Etzerodt, A., Kjolby, M., Nielsen, M. J., Maniecki, M., Svendsen, P., and Moestrup, S. K. (2013). Plasma clearance of hemoglobin and haptoglobin in mice and effect of CD163 gene targeting disruption. Antioxid. Redox Signal. 18, 2254–2263. doi: 10.1089/ars.2012.4605
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Etzerodt, A., Maniecki, M. B., Moller, K., Moller, H. J., and Moestrup, S. K. (2010). Tumor necrosis factor alpha-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J. Leukoc. Biol. 88, 1201–1205. doi: 10.1189/jlb.0410235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Faivre-Fiorina, B., Caron, A., Fassot, C., Fries, I., Menu, P., Labrude, P., et al. (1999). Presence of hemoglobin inside aortic endothelial cells after cell-free hemoglobin administration in guinea pigs. Am. J. Physiol. 276, H766–H770.
Figueiredo, R. T., Fernandez, P. L., Mourao-Sa, D. S., Porto, B. N., Dutra, F. F., Alves, L. S., et al. (2007). Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 282, 20221–20229. doi: 10.1074/jbc.M610737200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Finn, A. V., Nakano, M., Polavarapu, R., Karmali, V., Saeed, O., Zhao, X., et al. (2012). Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 59, 166–177. doi: 10.1016/j.jacc.2011.10.852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fortes, G. B., Alves, L. S., De Oliveira, R., Dutra, F. F., Rodrigues, D., Fernandez, P. L., et al. (2012). Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 119, 2368–2375. doi: 10.1182/blood-2011-08-375303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galicia, G., Maes, W., Verbinnen, B., Kasran, A., Bullens, D., Arredouani, M., et al. (2009). Haptoglobin deficiency facilitates the development of autoimmune inflammation. Eur. J. Immunol. 39, 3404–3412. doi: 10.1002/eji.200939291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gattoni, M., Boffi, A., Sarti, P., and Chiancone, E. (1996). Stability of the heme-globin linkage in alphabeta dimers and isolated chains of human hemoglobin. A study of the heme transfer reaction from the immobilized proteins to albumin. J. Biol. Chem. 271, 10130–10136. doi: 10.1074/jbc.271.17.10130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghosh, S., Adisa, O. A., Chappa, P., Tan, F., Jackson, K. A., Archer, D. R., et al. (2013). Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J. Clin. Invest. 123, 4809–4820. doi: 10.1172/JCI64578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gladwin, M. T., Kanias, T., and Kim-Shapiro, D. B. (2012). Hemolysis and cell-free hemoglobin drive an intrinsic mechanism for human disease. J. Clin. Invest. 122, 1205–1208. doi: 10.1172/JCI62972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gladwin, M. T., Sachdev, V., Jison, M. L., Shizukuda, Y., Plehn, J. F., Minter, K., et al. (2004). Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N. Engl. J. Med. 350, 886–895. doi: 10.1056/NEJMoa035477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Griffon, N., Baudin, V., Dieryck, W., Dumoulin, A., Pagnier, J., Poyart, C., et al. (1998). Tetramer-dimer equilibrium of oxyhemoglobin mutants determined from auto-oxidation rates. Protein Sci. 7, 673–680. doi: 10.1002/pro.5560070316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hashimoto, K., Nomura, K., Nakano, M., Sasaki, T., and Kurosawa, H. (1993). Pharmacological intervention for renal protection during cardiopulmonary bypass. Heart Vessels 8, 203–210. doi: 10.1007/BF01744743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hintz, K. A., Rassias, A. J., Wardwell, K., Moss, M. L., Morganelli, P. M., Pioli, P. A., et al. (2002). Endotoxin induces rapid metalloproteinase-mediated shedding followed by up-regulation of the monocyte hemoglobin scavenger receptor CD163. J. Leukoc. Biol. 72, 711–717.
Homann, B., Kult, J., and Weis, K. H. (1977). [On the use of concentrated haptoglobin in the treatment of a haemolytic transfusion accident of the ABO-system (author's transl)]. Anaesthesist 26, 485–488.
Hvidberg, V., Maniecki, M. B., Jacobsen, C., Hojrup, P., Moller, H. J., and Moestrup, S. K. (2005). Identification of the receptor scavenging hemopexin-heme complexes. Blood 106, 2572–2579. doi: 10.1182/blood-2005-03-1185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imaizumi, H., Tsunoda, K., Ichimiya, N., Okamoto, T., and Namiki, A. (1994). Repeated large-dose haptoglobin therapy in an extensively burned patient: case report. J. Emerg. Med. 12, 33–37. doi: 10.1016/0736-4679(94)90009-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Janz, D. R., Bastarache, J. A., Sills, G., Wickersham, N., May, A. K., Bernard, G. R., et al. (2013). Association between haptoglobin, hemopexin and mortality in adults with sepsis. Crit. Care 17, R272. doi: 10.1186/cc13108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeney, V., Balla, J., Yachie, A., Varga, Z., Vercellotti, G. M., Eaton, J. W., et al. (2002). Pro-oxidant and cytotoxic effects of circulating heme. Blood 100, 879–887. doi: 10.1182/blood.V100.3.879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jia, Y., Buehler, P. W., Boykins, R. A., Venable, R. M., and Alayash, A. I. (2007). Structural basis of peroxide-mediated changes in human hemoglobin: a novel oxidative pathway. J. Biol. Chem. 282, 4894–4907. doi: 10.1074/jbc.M609955200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaempfer, T., Duerst, E., Gehrig, P., Roschitzki, B., Rutishauser, D., Grossmann, J., et al. (2011). Extracellular hemoglobin polarizes the macrophage proteome toward Hb-clearance, enhanced antioxidant capacity and suppressed HLA class 2 expression. J. Proteome Res. 10, 2397–2408. doi: 10.1021/pr101230y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kamboh, M. I., Bunker, C. H., Nwankwo, M. U., and Ferrell, R. E. (1993). Hemopexin: a unique genetic polymorphism in populations of African ancestry. Hum. Biol. 65, 655–660.
Komarova, Y., and Malik, A. B. (2010). Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 72, 463–493. doi: 10.1146/annurev-physiol-021909-135833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kormoczi, G. F., Saemann, M. D., Buchta, C., Peck-Radosavljevic, M., Mayr, W. R., Schwartz, D. W., et al. (2006). Influence of clinical factors on the haemolysis marker haptoglobin. Eur. J. Clin. Invest. 36, 202–209. doi: 10.1111/j.1365-2362.2006.01617.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kristiansen, M., Graversen, J. H., Jacobsen, C., Sonne, O., Hoffman, H. J., Law, S. K., et al. (2001). Identification of the haemoglobin scavenger receptor. Nature 409, 198–201. doi: 10.1038/35051594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Langlois, M. R., and Delanghe, J. R. (1996). Biological and clinical significance of haptoglobin polymorphism in humans. Clin. Chem. 42, 1589–1600.
Larsen, R., Gozzelino, R., Jeney, V., Tokaji, L., Bozza, F. A., Japiassu, A. M., et al. (2010). A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2, 51ra71. doi: 10.1126/scitranslmed.3001118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Law, S. K., Micklem, K. J., Shaw, J. M., Zhang, X. P., Dong, Y., Willis, A. C., et al. (1993). A new macrophage differentiation antigen which is a member of the scavenger receptor superfamily. Eur. J. Immunol. 23, 2320–2325. doi: 10.1002/eji.1830230940
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levy, A. P., Asleh, R., Blum, S., Levy, N. S., Miller-Lotan, R., Kalet-Litman, S., et al. (2010). Haptoglobin: basic and clinical aspects. Antioxid. Redox Signal. 12, 293–304. doi: 10.1089/ars.2009.2793
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levy, A. P., Purushothaman, K. R., Levy, N. S., Purushothaman, M., Strauss, M., Asleh, R., et al. (2007). Downregulation of the hemoglobin scavenger receptor in individuals with diabetes and the Hp 2-2 genotype: implications for the response to intraplaque hemorrhage and plaque vulnerability. Circ. Res. 101, 106–110. doi: 10.1161/CIRCRESAHA.107.149435
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liang, X., Lin, T., Sun, G., Beasley-Topliffe, L., Cavaillon, J. M., and Warren, H. S. (2009). Hemopexin down-regulates LPS-induced proinflammatory cytokines from macrophages. J. Leukoc. Biol. 86, 229–235. doi: 10.1189/jlb.1208742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, S., Yin, Q., Zhong, Q., Lv, F. L., Zhou, Y., Li, J. Q., et al. (2012a). Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflammation 9, 46. doi: 10.1186/1742-2094-9-46
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, T., Sammy, F., Yang, H., Thundivalappil, S., Hellman, J., Tracey, K. J., et al. (2012b). Identification of hemopexin as an anti-inflammatory factor that inhibits synergy of hemoglobin with HMGB1 in sterile and infectious inflammation. J. Immunol. 189, 2017–2022. doi: 10.4049/jimmunol.1103623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lipiski, M., Deuel, J. W., Baek, J. H., Engelsberger, W. R., Buehler, P. W., and Schaer, D. J. (2013). Human Hp1-1 and Hp2-2 phenotype-specific haptoglobin therapeutics are both effective in vitro and in guinea pigs to attenuate hemoglobin toxicity. Antioxid. Redox Signal. 19, 1619–1633. doi: 10.1089/ars.2012.5089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Madsen, M., Moller, H. J., Nielsen, M. J., Jacobsen, C., Graversen, J. H., Van Den Berg, T., et al. (2004). Molecular characterization of the haptoglobin.hemoglobin receptor CD163. Ligand binding properties of the scavenger receptor cysteine-rich domain region. J. Biol. Chem. 279, 51561–51567. doi: 10.1074/jbc.M409629200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marinkovic, S., and Baumann, H. (1990). Structure, hormonal regulation, and identification of the interleukin-6- and dexamethasone-responsive element of the rat haptoglobin gene. Mol. Cell. Biol. 10, 1573–1583.
Matheson, B., Razynska, A., Kwansa, H., and Bucci, E. (2000). Appearance of dissociable and cross-linked hemoglobins in the renal hilar lymph. J. Lab. Clin. Med. 135, 459–464. doi: 10.1067/mlc.2000.106458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, Y. I., Altamentova, S. M., and Shaklai, N. (1997). Oxidation of low-density lipoprotein by hemoglobin stems from a heme-initiated globin radical: antioxidant role of haptoglobin. Biochemistry 36, 12189–12198. doi: 10.1021/bi970258a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Minneci, P. C., Deans, K. J., Zhi, H., Yuen, P. S., Star, R. A., Banks, S. M., et al. (2005). Hemolysis-associated endothelial dysfunction mediated by accelerated NO inactivation by decompartmentalized oxyhemoglobin. J. Clin. Invest. 115, 3409–3417. doi: 10.1172/JCI25040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mollan, T. L., Jia, Y., Banerjee, S., Wu, G., Kreulen, R. T., Tsai, A. L., et al. (2014). Redox properties of human hemoglobin in complex with fractionated dimeric and polymeric human haptoglobin. Free Radic. Biol. Med. 69, 265–277. doi: 10.1016/j.freeradbiomed.2014.01.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moller, H. J., Moestrup, S. K., Weis, N., Wejse, C., Nielsen, H., Pedersen, S. S., et al. (2006). Macrophage serum markers in pneumococcal bacteremia: prediction of survival by soluble CD163. Crit. Care Med. 34, 2561–2566. doi: 10.1097/01.CCM.0000239120.32490.AB
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moller, H. J., Peterslund, N. A., Graversen, J. H., and Moestrup, S. K. (2002). Identification of the hemoglobin scavenger receptor/CD163 as a natural soluble protein in plasma. Blood 99, 378–380. doi: 10.1182/blood.V99.1.378
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moreno, P. R., Purushothaman, K. R., Purushothaman, M., Muntner, P., Levy, N. S., Fuster, V., et al. (2008). Haptoglobin genotype is a major determinant of the amount of iron in the human atherosclerotic plaque. J. Am. Coll. Cardiol. 52, 1049–1051. doi: 10.1016/j.jacc.2008.06.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muller-Eberhard, U., Javid, J., Liem, H. H., Hanstein, A., and Hanna, M. (1968). Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood 32, 811–815.
Murray, R. K., Connell, G. E., and Pert, J. H. (1961). The role of haptoglobin in the clearance and distribution of extracorpuscular hemoglobin. Blood 17, 45–53.
Nagy, E., Eaton, J. W., Jeney, V., Soares, M. P., Varga, Z., Galajda, Z., et al. (2010). Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 30, 1347–1353. doi: 10.1161/ATVBAHA.110.206433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nakai, K., Sakuma, I., Ohta, T., Ando, J., Kitabatake, A., Nakazato, Y., et al. (1998). Permeability characteristics of hemoglobin derivatives across cultured endothelial cell monolayers. J. Lab. Clin. Med. 132, 313–319. doi: 10.1016/S0022-2143(98)90045-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Natanson, C., Kern, S. J., Lurie, P., Banks, S. M., and Wolfe, S. M. (2008). Cell-free hemoglobin-based blood substitutes and risk of myocardial infarction and death: a meta-analysis. JAMA 299, 2304–2312. doi: 10.1001/jama.299.19.jrv80007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niethammer, P., Grabher, C., Look, A. T., and Mitchison, T. J. (2009). A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 459, 996–999. doi: 10.1038/nature08119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Olson, J. S., Foley, E. W., Rogge, C., Tsai, A. L., Doyle, M. P., and Lemon, D. D. (2004). NO scavenging and the hypertensive effect of hemoglobin-based blood substitutes. Free Radic. Biol. Med. 36, 685–697. doi: 10.1016/j.freeradbiomed.2003.11.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pimenova, T., Pereira, C. P., Gehrig, P., Buehler, P. W., Schaer, D. J., and Zenobi, R. (2010). Quantitative mass spectrometry defines an oxidative hotspot in hemoglobin that is specifically protected by haptoglobin. J. Proteome Res. 9, 4061–4070. doi: 10.1021/pr100252e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pimenova, T., Pereira, C. P., Schaer, D. J., and Zenobi, R. (2009). Characterization of high molecular weight multimeric states of human haptoglobin and hemoglobin-based oxygen carriers by high-mass MALDI MS. J. Sep. Sci. 32, 1224–1230. doi: 10.1002/jssc.200800625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Purushothaman, K. R., Purushothaman, M., Levy, A. P., Lento, P. A., Evrard, S., Kovacic, J. C., et al. (2012). Increased expression of oxidation-specific epitopes and apoptosis are associated with haptoglobin genotype: possible implications for plaque progression in human atherosclerosis. J. Am. Coll. Cardiol. 60, 112–119. doi: 10.1016/j.jacc.2012.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qian, Q., Nath, K. A., Wu, Y., Daoud, T. M., and Sethi, S. (2010). Hemolysis and acute kidney failure. Am. J. Kidney Dis. 56, 780–784. doi: 10.1053/j.ajkd.2010.03.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reeder, B. J., Grey, M., Silaghi-Dumitrescu, R. L., Svistunenko, D. A., Bulow, L., Cooper, C. E., et al. (2008). Tyrosine residues as redox cofactors in human hemoglobin: implications for engineering nontoxic blood substitutes. J. Biol. Chem. 283, 30780–30787. doi: 10.1074/jbc.M804709200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reiter, C. D., Wang, X., Tanus-Santos, J. E., Hogg, N., Cannon, R. O. 3rd., Schechter, A. N., et al. (2002). Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 8, 1383–1389. doi: 10.1038/nm1202-799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rolla, S., Ingoglia, G., Bardina, V., Silengo, L., Altruda, F., Novelli, F., et al. (2013). Acute-phase protein hemopexin is a negative regulator of Th17 response and experimental autoimmune encephalomyelitis development. J. Immunol. 191, 5451–5459. doi: 10.4049/jimmunol.1203076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rother, R. P., Bell, L., Hillmen, P., and Gladwin, M. T. (2005). The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA 293, 1653–1662. doi: 10.1001/jama.293.13.1653
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, C. A., Deuel, J. W., Bittermann, A. G., Rubio, I. G., Schoedon, G., Spahn, D. R., et al. (2013a). Mechanisms of haptoglobin protection against hemoglobin peroxidation triggered endothelial damage. Cell Death Differ. 20, 1569–1579. doi: 10.1038/cdd.2013.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, C. A., Laczko, E., Schoedon, G., Schaer, D. J., and Vallelian, F. (2013b). Chloroquine interference with hemoglobin endocytic trafficking suppresses adaptive heme and iron homeostasis in macrophages: the paradox of an antimalarial agent. Oxid. Med. Cell. Longev. 2013, 870472. doi: 10.1155/2013/870472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, C. A., Vallelian, F., Imhof, A., Schoedon, G., and Schaer, D. J. (2007). CD163-expressing monocytes constitute an endotoxin-sensitive Hb clearance compartment within the vascular system. J. Leukoc. Biol. 82, 106–110. doi: 10.1189/jlb.0706453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, D. J., Boretti, F. S., Hongegger, A., Poehler, D., Linnscheid, P., Staege, H., et al. (2001). Molecular cloning and characterization of the mouse CD163 homologue, a highly glucocorticoid-inducible member of the scavenger receptor cysteine-rich family. Immunogenetics 53, 170–177. doi: 10.1007/s002510100304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, D. J., Boretti, F. S., Schoedon, G., and Schaffner, A. (2002). Induction of the CD163-dependent haemoglobin uptake by macrophages as a novel anti-inflammatory action of glucocorticoids. Br. J. Haematol. 119, 239–243. doi: 10.1046/j.1365-2141.2002.03790.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, D. J., Buehler, P. W., Alayash, A. I., Belcher, J. D., and Vercellotti, G. M. (2013c). Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121, 1276–1284. doi: 10.1182/blood-2012-11-451229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, D. J., Schaer, C. A., Buehler, P. W., Boykins, R. A., Schoedon, G., Alayash, A. I., et al. (2006). CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood 107, 373–380. doi: 10.1182/blood-2005-03-1014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaer, D. J., Schleiffenbaum, B., Kurrer, M., Imhof, A., Bachli, E., Fehr, J., et al. (2005). Soluble hemoglobin-haptoglobin scavenger receptor CD163 as a lineage-specific marker in the reactive hemophagocytic syndrome. Eur. J. Haematol. 74, 6–10. doi: 10.1111/j.1600-0609.2004.00318.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shen, H., Song, Y., Colangelo, C. M., Wu, T., Bruce, C., Scabia, G., et al. (2012). Haptoglobin activates innate immunity to enhance acute transplant rejection in mice. J. Clin. Invest. 122, 383–387. doi: 10.1172/JCI58344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Silverman, T. A., and Weiskopf, R. B. (2009). Hemoglobin-based oxygen carriers: current status and future directions. Transfusion 49, 2495–2515. doi: 10.1111/j.1537-2995.2009.02356.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Silverman, T. A., Weiskopf, R. B., Planning, C., and The, S. (2009). Hemoglobin-based oxygen carriers: current status and future directions. Anesthesiology 111, 946–963. doi: 10.1097/ALN.0b013e3181ba3c2c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spiller, F., Costa, C., Souto, F. O., Vinchi, F., Mestriner, F. L., Laure, H. J., et al. (2011). Inhibition of neutrophil migration by hemopexin leads to increased mortality due to sepsis in mice. Am. J. Respir. Crit. Care Med. 183, 922–931. doi: 10.1164/rccm.201002-0223OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Subramanian, K., Du, R., Tan, N. S., Ho, B., and Ding, J. L. (2013). CD163 and IgG codefend against cytotoxic hemoglobin via autocrine and paracrine mechanisms. J. Immunol. 190, 5267–5278. doi: 10.4049/jimmunol.1202648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tanaka, K., Kanamori, Y., Sato, T., Kondo, C., Katayama, Y., Yada, I., et al. (1991). Administration of haptoglobin during cardiopulmonary bypass surgery. ASAIO Trans. 37, M482–M483.
Tolosano, E., Hirsch, E., Patrucco, E., Camaschella, C., Navone, R., Silengo, L., et al. (1999). Defective recovery and severe renal damage after acute hemolysis in hemopexin-deficient mice. Blood 94, 3906–3914.
Vallelian, F., Deuel, J. W., Opitz, L., Schaer, C. A., Puglia, M., Lönn, M., et al. (2014). Proteasome inhibition and oxidative reactions disrupt cellular homeostasis during heme stress. Cell Death Differ. doi: 10.1038/cdd.2014.154. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vallelian, F., Pimenova, T., Pereira, C. P., Abraham, B., Mikolajczyk, M. G., Schoedon, G., et al. (2008). The reaction of hydrogen peroxide with hemoglobin induces extensive alpha-globin crosslinking and impairs the interaction of hemoglobin with endogenous scavenger pathways. Free Radic. Biol. Med. 45, 1150–1158. doi: 10.1016/j.freeradbiomed.2008.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vallelian, F., Schaer, C. A., Kaempfer, T., Gehrig, P., Duerst, E., Schoedon, G., et al. (2010). Glucocorticoid treatment skews human monocyte differentiation into a hemoglobin-clearance phenotype with enhanced heme-iron recycling and antioxidant capacity. Blood 116, 5347–5356. doi: 10.1182/blood-2010-04-277319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vinchi, F., De Franceschi, L., Ghigo, A., Townes, T., Cimino, J., Silengo, L., et al. (2013). Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 127, 1317–1329. doi: 10.1161/CIRCULATIONAHA.112.130179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vinchi, F., Gastaldi, S., Silengo, L., Altruda, F., and Tolosano, E. (2008). Hemopexin prevents endothelial damage and liver congestion in a mouse model of heme overload. Am. J. Pathol. 173, 289–299. doi: 10.2353/ajpath.2008.071130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wagener, F. A., Eggert, A., Boerman, O. C., Oyen, W. J., Verhofstad, A., Abraham, N. G., et al. (2001). Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 98, 1802–1811. doi: 10.1182/blood.V98.6.1802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weaver, L. K., Hintz-Goldstein, K. A., Pioli, P. A., Wardwell, K., Qureshi, N., Vogel, S. N., et al. (2006). Pivotal advance: activation of cell surface Toll-like receptors causes shedding of the hemoglobin scavenger receptor CD163. J. Leukoc. Biol. 80, 26–35. doi: 10.1189/jlb.1205756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: hemolysis, haptoglobin, hemopexin, CD163, vascular diseases, transfusion, sickle cell disease
Citation: Schaer DJ, Vinchi F, Ingoglia G, Tolosano E and Buehler PW (2014) Haptoglobin, hemopexin, and related defense pathways—basic science, clinical perspectives, and drug development. Front. Physiol. 5:415. doi: 10.3389/fphys.2014.00415
Received: 28 June 2014; Accepted: 08 October 2014;
Published online: 28 October 2014.
Edited by:
Magnus Gram, Lund University, SwedenReviewed by:
Sruti Shiva, University of Pittsburgh, USAJeak Ling Ding, National University of Singapore, Singapore
Joseph M. Rifkind, National Institute on Aging, USA
Soren Kragh Moestrup, Aarhus University, Denmark
Copyright © 2014 Schaer, Vinchi, Ingoglia, Tolosano and Buehler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominik J. Schaer, Division of Internal Medicine, University Hospital, Ramistrasse 100, Zurich CH-8091, Switzerland e-mail:ZG9taW5pay5zY2hhZXJAdXN6LmNo
†The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.