
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 14 October 2014
Sec. Striated Muscle Physiology
Volume 5 - 2014 | https://doi.org/10.3389/fphys.2014.00393
This article is part of the Research Topic Myofilament Function In Health And Disease View all 15 articles
 Aziz Guellich1,2†Elisa Negroni3,4,5,6†
Aziz Guellich1,2†Elisa Negroni3,4,5,6† Valérie Decostre6Alexandre Demoule3,4,5,6,7
Valérie Decostre6Alexandre Demoule3,4,5,6,7 Catherine Coirault3,4,5,6*
Catherine Coirault3,4,5,6*Force and motion generated by skeletal muscle ultimately depends on the cyclical interaction of actin with myosin. This mechanical process is regulated by intracellular Ca2+ through the thin filament-associated regulatory proteins i.e.; troponins and tropomyosin. Muscular dystrophies are a group of heterogeneous genetic affections characterized by progressive degeneration and weakness of the skeletal muscle as a consequence of loss of muscle tissue which directly reduces the number of potential myosin cross-bridges involved in force production. Mutations in genes responsible for skeletal muscle dystrophies (MDs) have been shown to modify the function of contractile proteins and cross-bridge interactions. Altered gene expression or RNA splicing or post-translational modifications of contractile proteins such as those related to oxidative stress, may affect cross-bridge function by modifying key proteins of the excitation-contraction coupling. Micro-architectural change in myofilament is another mechanism of altered cross-bridge performance. In this review, we provide an overview about changes in cross-bridge performance in skeletal MDs and discuss their ultimate impacts on striated muscle function.
Muscular dystrophies (MDs) are a group of more than 30 clinical and molecular heterogeneous genetic disorders that cause progressive degeneration of the skeletal muscle fibers. They are characterized by severe muscle weakness that generally affects limb, axial, and/or facial muscles to a variable extent. The age of onset, severity and rate of progression greatly vary in the different forms of MD. The primary cause of various forms of MDs is an individual mutation in genes encoding a wide variety of proteins, including extracellular matrix (ECM) proteins, transmembrane, and membrane-associated proteins, cytoplasmic proteases and nuclear proteins. Detailed classification and list of causative genes in MD have been recently reviewed (Cohn and Campbell, 2000; Flanigan, 2012; Kaplan and Hamroun, 2012; Mercuri and Muntoni, 2013). Although still incompletely understood, considerable progress has been now made to reveal the pathophysiological mechanisms in MDs. It has been shown that most MDs share common pathologic features, such as altered Ca2+ homeostasis, infiltration of muscle tissue by inflammatory immune cells, accretion of proinflammatory and profibrotic cytokines, activation of proteolytic enzymes, metabolic/mitochondrial alterations, intracellular accumulation of reactive oxygen species (ROS) production, and/or defective autophagy, which can contribute to muscle wasting. At the earliest stages of the disease, reduced myofibrillar protein content can occur in apparent uninjured fibers, secondary to an imbalance between protein synthesis and proteolysis (McKeran et al., 1977; Warnes et al., 1981). Then, successive rounds of degeneration and regeneration lead to fibrosis and fatty replacement of muscle tissue and in turn reduce the number of potential cross-bridges (CBs) generating force (Figure 1). In addition, functional changes in the CB properties may contribute to muscle weakness in distinct types of muscle disease. It is well-established that shift in the relative myosin isoform expression is associated with modifications of the kinetics of actomyosin interactions (Bar and Pette, 1988; Schiaffino et al., 1989; Schiaffino and Reggiani, 1996). However, there are also increasing evidences that CB properties can change with no change in myosin isoform content (Coirault et al., 2002; Canepari et al., 2009), suggesting that post-translational modifications of contractile proteins have significant role in muscle weakness (Figure 1). The present review focuses on the changes in the function of contractile proteins and in CB performance that may occur in the context of skeletal muscle dystrophies (MDs) and their contribution to the pathophysiological mechanisms of these diseases.
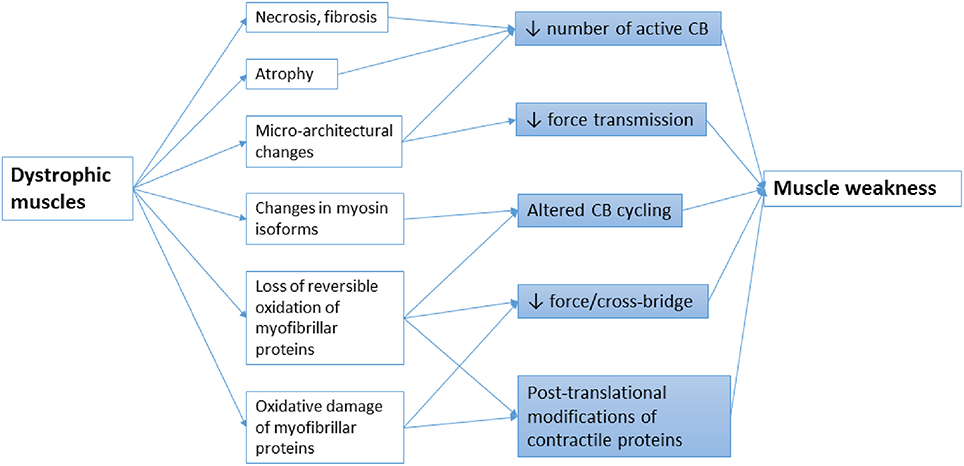
Figure 1. Schematic representation of the main mechanisms leading to muscle weakness in MDs. Altered CB cycle in muscular dystrophy can arise from multiples causes including progressive loss of muscle tissue due to necrosis, fibrosis, atrophy, or oxidative damage, micro-architectural alterations in myofilaments, altered redox regulation or pathological changes in contractile protein isoforms. These abnormalities in turn directly reduce the number, the force, the kinetics of active CBs or the transmission of force produced by the CBs.
Contractile force of striated muscle is produced within the half-sarcomere—the functional contractile unit—by the cyclical interactions of actin filament with myosin molecule, the muscle molecular motor. These cyclical interactions are controlled by membrane-located mechanisms that trigger intracellular raise of Ca2+and mechanisms inside the sarcomere itself. In skeletal muscle, sarcomeric mechanisms mainly involve the Ca2+-controlled conformational change of the regulatory proteins troponins and tropomyosin (Tm), the functional properties of myosin, and strong cooperative interaction between neighboring troponin-Tm unit along the actin filament (Gordon et al., 2000).
In resting muscle, intracellular Ca2+ concentration ([Ca2+]i) is low and the C-terminal domain of troponin I immobilizes the Tm in a position that prevents the binding of myosin to actin (Galinska-Rakoczy et al., 2008). Upon membrane depolarization, Ca2+ is released from the sarcoplasmic reticulum and binds to troponin C, initiating azimuthal movement of Tm around the actin-containing thin filament. This in turn exposes the sites on actin on which the myosin heads can attach (Kress et al., 1986; Geeves et al., 2005). In the conventional view, myosin binding is solely determined by the Ca2+ transient causing structural changes in the thin filament. However, numerous studies have reported Ca2+-induced structural change in the myosin-containing thick filaments before myosin binding to the thin filament (Huxley et al., 1982; Lowy and Poulsen, 1990; Yagi, 2003; Reconditi et al., 2011). This has led to the proposal of a modified model of muscle activation in which fast coordinated changes in the structures of both thick and thin filaments follow concomitantly upon the rise in intracellular free Ca2+ concentration (Reconditi et al., 2014).
Myosin head binding to actin occurs first in a low binding, pre-force-generating state (Figure 2, step a). At this state, the CB has already hydrolyzed the adenosine 5′-triphosphate (ATP), but the products adenosine 5′-diphosphate (ADP) and inorganic phosphate (Pi) are still bound to the myosin head (Lymn and Taylor, 1971; Pate and Cooke, 1989). Then, the CB goes through the power stroke (Figure 2, step b), during which the myosin head can generate a force of several piconewtons or an axial displacement of the actin filament toward the center of the sarcomere of 5–10 nm in vitro (Molloy et al., 1995; Veigel et al., 1998; Mehta et al., 1999; Reconditi et al., 2011) or 8–13 nm in situ (Reconditi et al., 2004). There are evidences that force generation precedes the Pi release (Dantzig et al., 1992; Caremani et al., 2008). Subsequent steps involve the release of ADP (Figure 2, step c), and the binding of ATP that rapidly dissociates the actomyosin complex (Figure 2, step d). When unbound from actin, myosin hydrolyzes ATP and reverses the power stroke, thus returning to its original position and allowing a new cycle to start (Eisenberg and Greene, 1980; Steffen and Sleep, 2004) (Figure 2, step e). Thus, at the molecular level, skeletal myosin is a molecular motor that transduces chemical energy produced by the hydrolysis of one ATP molecule into mechanical work (Huxley, 1957; Huxley and Simmons, 1971; Lymn and Taylor, 1971; Eisenberg et al., 1980; Pate and Cooke, 1989; Gordon et al., 2000). Skeletal MDs may reduce the number of active CBs producing force but may also potentially affect various steps of the cyclical interactions of actin with myosin, thus reducing the unitary force produced by each CB and/or modifying the CB kinetics.
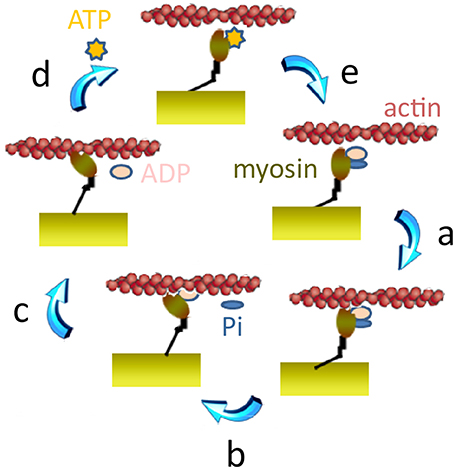
Figure 2. The CB cycle. Step a: attachment of myosin to actin. This step begins with the “weak binding” of actin (in magenta) to the myosin head (in brown). A stereospecific interaction between actin and myosin leads to strong binding of actin to myosin. Step b: power stroke. The loss of inorganic phosphate (Pi) is assumed to trigger the onset of the power stroke and a reversal of the previous conformational bent. Step c: ADP release step. The power stroke induces reopening of the nucleotide binding pocket and ADP release. Step d: ATP binding. ATP can then rapidly bind to myosin which dissociates from actin. Step e: ATP hydrolysis. ATP is then rapidly hydrolyzed to ADP and Pi.
The events in the CB cycle are essentially the same for all muscle myosins, but the kinetics of acto-myosin interaction varies widely among myosin heavy chain (MyHC) isoforms. Although the physiological differences between fast and slow skeletal muscle fibers depend on more than just differences in MyHC isoforms, it is well established that MyHC isoforms are a major determinant of the large variability in contractile and energetic properties of muscle fibers (Pette and Staron, 1990; Bottinelli et al., 1994a,b). The existence of several MyHC isoforms differentially distributed in various fibers makes MyHCs useful to study fiber heterogeneity and plasticity in mammalian muscles. In the adult skeletal muscle, four MyHC isoforms can be expressed, namely the slow MyHC -1 and the fast MyHC -2A, MyHC-2X, and MyHC-2B, coded respectively by the Myh7, Myh2, Myh1, and Myh4 genes (Bar and Pette, 1988; Schiaffino et al., 1989; Schiaffino and Reggiani, 1996).
Changes in the proportion of fast relative to slow fiber types are frequently observed in muscular dystrophy, and may result either from a reduced muscular activity in affected patients or from a physiopathological consequence of the gene mutation. The pattern of wasted muscle and affected fibers is highly variable among the types of MDs. Preferential involvement in fast type 2 muscle fibers is a common feature in Duchenne Muscular Dystrophy (DMD) (Webster et al., 1988), with early disappearance of muscle fibers expressing MyHC-2X transcripts (Pedemonte et al., 1999). Likewise, slow type 1 muscle fiber predominance has been reported in patients with facioscapulohumeral muscular dystrophy (FSHD) (Celegato et al., 2006), dysferlinopathy (Fanin and Angelini, 2002) and congenital muscular dystrophy type Ullrich (Schessl et al., 2008), suggesting a selective loss of fast-twitch/type 2 muscle fibers (D'Antona et al., 2007) or an active fiber typing conversion process (Schessl et al., 2008; De La Torre et al., 2009). This preferential involvement of the fastest fiber type leads to a fast to slow shift among fast MyHC isoforms in the dystrophic muscles (Stedman et al., 1991; Petrof et al., 1993; Coirault et al., 1999), which may represent an adaptive response that would tend to preserve the economy of force contraction. However, fiber-type composition has been shown to be differently affected in other MDs. Hypertrophy of type 2 fibers without type 1 fiber atrophy have been found in biopsies of patients with laminopathies (Kajino et al., 2014), whereas MyHC-2a fibers are markedly atrophied in affected cricopharyngeal muscle of oculopharyngeal muscular dystrophy patients (Gidaro et al., 2013). How mutations responsible for MDs differently affect myofiber-type specification and/or MyHC deserve further studies. In addition to adult myosin isoform remodeling, dystrophic fiber can express developmental embryonic and neonatal MyHCs, coded by Myh3 and Myh8, respectively (Wieczorek et al., 1985; Sartore et al., 1987). Such developmental or neonatal MyHC-expressing fibers can either be regenerating fibers or fibers that expressed inappropriate and immature MyHC, as has been observed in some congenital myopathies (Sewry, 1998).
Importantly, the different MyHC isoforms display large functional differences regarding the actin-activated ATPase activity of myosin, the rate of ADP release from acto-myosin during the time of attachment, and the velocity with which they can move actin. This results in large variability in contractile, thermodynamic, and kinetic coupling according to the fiber type (Barany, 1967). For instance, fibers containing MyHC-1 have nearly threefold slower ATPase and lower tension cost than fibers with MyHC-2X, while fibers with MyHC-2A are intermediate (Stienen et al., 1996). Therefore, an increased proportion of the slow MyHC isoforms would tend to improve muscle efficiency and may represent an adaptive response at least in some MDs. An opposite response, namely a slow-to-fast shift with preferential atrophy of slow fiber, accelerates the time cycle, reduces muscle efficiency (Coirault et al., 1999) and thus may contribute to the exercise intolerance in DMD patients. MyHC shift may also impact on CB recruitment, given that attachment of fast myosin involves a CB-mediated facilitation mechanism absent in slow MyHC isoforms (Galler et al., 2009). Indeed, fast myosin heads already attached to actin substantially accelerate the further attachment of neighboring myosin heads, thereby enabling a rapid change in the rate of force development at high levels of activation (Galler et al., 2009). In contrast, slow MyHC isoforms have moderate dependence on level of activation, enabling slow CB recruitment to become faster only gradually at higher levels of activation (Galler et al., 2009). Thus, pathologic changes in myosin isoforms modify specific steps of the CB cycling as well as the overall kinetics of the CB cycle, with functional consequences on muscle performance and energy cost of contraction.
Striated muscles are characterized by a highly ordered structure that is critical for normal function and muscle homeostasis. Micro-architectural changes in myofilaments are likely contributors to muscle weakness in MDs, either directly by affecting the function of critical structural proteins, or indirectly, by increasing the activity of Ca2+-dependent proteases.
In striated muscle, the dystrophin glycoprotein complex is preferentially located at the costamere (Ervasti and Campbell, 1991), a protein network connecting the outermost myofibrils to the sarcolemma at each Z-disc (Pardo et al., 1983; Ervasti and Campbell, 1991; Bloch and Gonzalez-Serratos, 2003; Ervasti, 2003; Michele and Campbell, 2003). These lateral linkages are critical in the maintenance of sarcomere stability and in the transmission of forces generated by the CBs (Figure 3) (Rybakova et al., 2000; Bloch and Gonzalez-Serratos, 2003; Ervasti, 2003). Indeed, while part of the forces generated in sarcomeres is longitudinally transmitted down myofibrils in muscle to the tendon, costameres are critical to transmit the forces laterally to the ECM and neighboring muscle fibers (Street, 1983; Ervasti, 2003; Bloch et al., 2004; Ramaswamy et al., 2011). Dystrophin deficiency reduces the lateral transfer of forces between activated fibers (Ramaswamy et al., 2011), thus compromising the homogeneity of sarcomere contraction between adjacent fibers and impairing the efficacy of force transmission. As a result, each fiber tends to act as an independent longitudinal force generator, thus increasing the susceptibility of muscle membrane to contraction-induced muscle damage.
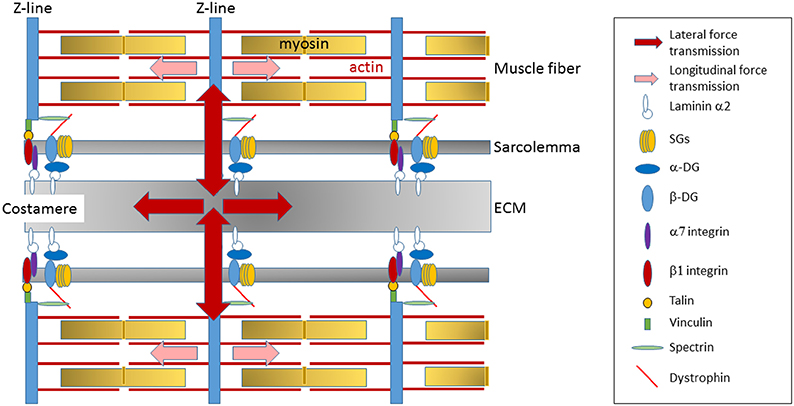
Figure 3. Transmission of the forces generated in sarcomere. The dystrophin-glycoprotein complex is preferentially located at the costamere, a protein network that reside at the sarcolemma membrane in register with the Z-lines of sarcomeres and that is critical in the lateral transmission of forces generated by the CB to the ECM and neighboring muscle fibers. Dystrophin deficiency at the lateral membrane alters costamere assembly and thus may impair the efficacy of lateral force transmission.
Skeletal muscle fiber contains the ubiquitously expressed and well-conserved family of Ca2+-dependent cysteine proteases calpains, μ-calpain, and m-calpain, as well as a muscle-specific calpain i.e., calpain-3 (Goll et al., 2003; Bartoli and Richard, 2005). Elevated calpain amount, primarily due to a significant increase in m-calpain concentration is observed in dystrophic muscles from the mdx mice (Spencer et al., 1995), a mouse model of DMD. Increased activity of calpains has been implicated in the progression of muscle wasting in dystrophic muscles (Spencer et al., 1995; Alderton and Steinhardt, 2000; Tidball and Spencer, 2000; Zhang et al., 2008). The two ubiquitous μ- and m-calpains both cleave the same substrates in vitro, including the troponin complex (Troponin C, Troponin I, and Troponin T), Tm, α-actinin, titin, desmin, the Z-disk protein fodrin and the sarcolemmal associated spectrin complex of proteins (Goll et al., 2003). Such substrates are consistent with a role of μ- and m-calpains in sarcomeric organization and/or dismantling. Interestingly, the two sarcomeric tropomodulin isoforms Tmod1 and Tmod4 have been recently identified as proteolytic targets of m-calpain in dystrophic muscle (Gokhin et al., 2014). Tropomodulins are dynamic actin filament pointed-end-capping proteins (Weber et al., 1994), that localize in sarcomere to each side of the M-line (Fowler et al., 1993). Absence of Tmod1 and its replacement by Tmod3 and Tmod4 in turn impair initial Tm movement over actin subunits during thin filament activation, thus reducing both the fraction of actomyosin CBs in the strongly bound state and fiber force-generating capacity (Ochala et al., 2014). Calpain-3 binds to the N2A region of titin (Sorimachi et al., 1995), but has also been identified in the nucleus of muscle cells (Baghdiguian et al., 1999). Interestingly, it has been shown that calpain-3 binding to the myofibrillar structure in vitro is modulated by the presence of CBs locked in rigor (Murphy et al., 2006; Murphy and Lamb, 2009), thus supporting the hypothesis that CB interactions modulate calpain-3 activity. This may explain why in vivo, eccentric exercise is the only physiological circumstance shown to result in the activation of calpain-3 (Murphy et al., 2007). A number of myofibrillar proteins have been identified as potential calpain-3 substrates in vitro, but none have been confirmed as in vivo targets. These include titin (at the PEVK region, adjacent to the N2A region of titin), and myosin light chain (Cohen et al., 2006). Future works are needed to determine the interrelation between calpain-3 and the CB interactions.
Oxidative stress has been reported as primum movens in both loss of cell viability and contractile dysfunction in MDs (Ragusa et al., 1997; Rando et al., 1998; Tidball and Wehling-Henricks, 2007; Menazza et al., 2010; Lawler, 2011). By definition, oxidative stress refers to a deregulation of the cellular redox-status due to an imbalance between reactive oxygen species (ROS) production and antioxidant capacities.
There are numerous potential intracellular sources of ROS in muscle tissue including the mitochondria (Murphy, 2009; Brand, 2010), the family of nicotinamide adenine dinucleotide phosphatate oxidases, now collectively known as NOX enzymes family, and a wide range of enzymes, such as xanthine oxidase, nitric oxide synthase, cyclo oxygenases, cytochrome P450 enzymes and lipoxygenases (Cheng et al., 2001). In addition, extracellular sources of ROS can arise from other non-muscle cell types, including activated neutrophils and macrophages (Moylan and Reid, 2007).
Because striated muscle cells have to adapt very rapidly and in a co-ordinated manner to changes in energy supply and oxygen flux, the control of ROS production and redox-status is tightly regulated during contraction. Importantly, the neuronal form of nitric oxide synthase (nNOS) is normally present in the sarcolemma of fast-twitch muscle fibers and associates with dystrophin (Brenman et al., 1995). Although the reaction between nitric oxide (NO) and the superoxide anion is known to form peroxynitrite, NO can also act as a protective molecule against the damaging actions of ROS (Wink et al., 1995). Accordingly, the absence of nNOS and NO in DMD muscle removes this protective action, increasing the susceptibility of DMD muscle to the damaging action of ROS (Brenman et al., 1995; Haycock et al., 1996; Disatnik et al., 1998).
There is increasing evidence that ROS reversibly modulate important intracellular pathways involved in muscle homeostasis (Droge, 2002; Smith and Reid, 2006; Jackson, 2008; Musaro et al., 2010). Small, compartmentalized, and transient increases in ROS regulate intracellular signals by reversible oxidation of specific protein residues (Meng et al., 2002; Ghezzi, 2005; Janssen-Heininger et al., 2008; Drazic et al., 2013). Hydrogen peroxide modifies protein function by oxidizing the thiol (-SH) groups of redox sensitive cysteine residues to form disulfide bonds with adjacent cysteine residues, glutathione (glutathionylation), or small protein thiols such as thioredoxin. Reduction of the disulfide bond can then be obtained through the action of enzymes such as glutaredoxin or the peroxiredoxins (Rhee, 2006; Hashemy et al., 2007). Accordingly, specific cysteine and methionine residues can function as redox-dependent switches, thereby modulating the function of myofibrillar proteins (Nogueira et al., 2009; Mollica et al., 2012; Gross and Lehman, 2013), phosphatase or transcriptional activities (Xanthoudakis et al., 1992; Ji et al., 2004; Powers et al., 2005, 2011; Jackson, 2008; Ugarte et al., 2010). Indeed, recent findings indicate that myosin, actin, troponin I and Tm from skeletal muscle have cysteines that are critical to the effects from oxidants (Nogueira et al., 2009; Mollica et al., 2012; Gross and Lehman, 2013), and that accessibility depends on the conformation of the CB (Gross and Lehman, 2013). In addition, the function of myosin (Nogueira et al., 2009) and of the ryanodine 1 receptor (Bellinger et al., 2009) can be reversibly modified by S-nitrosylation, a redox-related modification of cysteine thiol by NO. Reversible oxidation of contractile proteins likely regulates muscle force production and actomyosin interactions, thereby contributing to the physiological adaptation of muscle to mechanical challenge (Powers and Jackson, 2008). Disturbance or removal of this redox sensitive modulations may limit the adaptability of the CB kinetics, with deleterious effects on muscle function and homeostasis (Stone and Yang, 2006; Linnane et al., 2007). Apart cysteine, methionine residue can be oxidized to its sulfoxyde, generating a mixture of two diastereoimers, denoted S and R. The S and the R forms are reduced back to methionine by methionine sulfoxyde reductase A (MsrA) and B (MsrB), respectively (Stadtman et al., 2002; Petropoulos and Friguet, 2005; Ugarte et al., 2010). Interestingly, recent studies have highlighted a new role for these Msr enzymes, identifying them as actors controlling the assembly/disassembly of actin filaments (Hung et al., 2013; Lee et al., 2013). A specific oxidation-reduction (redox) enzyme, the Mical protein, selectively modifies two methionines on the conserved pointed-end in actin filaments. Oxidized methionines at these positions simultaneously disassemble and decrease the polymerization of actin filaments (Hung et al., 2013). Recently, Hung et al. and Lee et al. showed in Drosophila bristle processes model and in mouse macrophages that MsrB1 stereospecifically reverses the oxidative modification of the methionines introduced by Mical, indicating that Msr system antagonizes the Mical-actin depolymerization dependent process and reversibly control actin polymerization (Hung et al., 2013; Lee et al., 2013). These data indicate new post-translational regulatory mechanisms involving oxidoreductase systems, but also open up new paths of investigation in the field of muscle diseases.
In contrast to reversible modifications caused by low oxidative stress, chronic disruption of the oxidative balance and/or high levels of free ROS radicals cause potential biological damage, termed oxidative stress. This is a complex process that probably depends on the type of oxidant, on the site and intensity of its production, on the composition and activity of various antioxidants and on the ability of skeletal muscle to repair damaged fibers. Irreversible damage of myofibrillar proteins may affect the CB cycle, as observed after nitrosylation and carbonylation of myosin (Coirault et al., 2007; Guellich et al., 2007), and target them for catabolic proteolysis (Jung et al., 2007). Alternatively, oxidative stress can activate caspases that in turn cleave myofibrillar proteins and impair CB properties (Du et al., 2004).
The functional consequences of altered CB cycle in MD remain to be precisely determined. It has been reported that the reduced muscle strength per cross section in mdx diaphragm is associated with an accelerated cycle compared with control diaphragm muscle (Coirault et al., 1999). Assuming that one molecule of ATP is hydrolyzed per CB cycle (Huxley, 1957; Huxley and Simmons, 1971; Eisenberg et al., 1980), these data suggested that the overall cycle of ATP splitting takes place more rapidly in mdx than in control mouse diaphragm, thus reducing the efficiency of muscle contraction to sustain force. Overall, these data suggested that absence of the sarcolemmal dystrophin protein leads to damaged or dysfunctional myosin molecules that impair CB kinetics and may account for the contractile deficit in mdx diaphragm. However, while mechanical differences are observed between mdx and control diaphragms at the intact muscle strip level, these differences are not observed at the single permeabilized cell (Bates et al., 2013). In addition, it was reported that myosin extracted from bulk mdx mouse diaphragm muscle moves actin filaments in an “in vitro motility assay” at a lower velocity than myosin extracted from controls (Coirault et al., 2002). Consistent reduction in actin sliding velocity has been found in pure MyHC-2 isoform from mdx gastrocnemius muscle, but not in type 1 myosin from wild-type and mdx muscle (Canepari et al., 2009). Therefore, observed changes in myosin velocity have been related to a change in the intrinsic properties of the molecule, but was not attributed to a change in the proportion of different myosin isoforms in the sample. In addition, it has been shown that nebulin in skeletal muscle increases thin filament activation via increasing CB cycling kinetics leading to an increased force and efficiency of contraction (Chandra et al., 2009). Results from that study provide novel insights regarding nebulin-based nemaline myopathy. Thus, complex mechanisms other than translational changes in myosin isoform may contribute to CB modifications in muscular disorders. Future studies are needed to precisely determine the contribution of post-translational modifications of contractile proteins to muscle weakness in MDs.
The mechanisms of muscle weakness have been a fundamental question of the physiopathology of muscle dystrophy for more than 50 years. It is now clear that the molecular causes responsible for the impaired performance in MDs involve both structural and functional modifications of the acto-myosin interactions. The precise mechanisms are complex with probably some specificities related to each individual mutation responsible for the MD, the severity of the translational and post-translational alterations in myofibrillar proteins, and on the ability of skeletal muscle to adapt to these changes. Whereas the functional consequences of translational changes in contractile proteins have been extensively studied, the impact of post-translational changes on the function of various contractile proteins still remains to be precisely assessed in the context of MDs. This is a particularly exciting time to study this question as recent findings strongly suggest that the loss of transient structural modifications of the contractile proteins such as reversible oxidation of specific protein residues impair the ability of CBs to adapt very rapidly to changes in workload and energy supply. Future works will assess the impact of non-permanent structural modifications of actin, myosin, and regulatory proteins on muscle performance and determine their potential contribution to muscle weakness in MDs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ADP, adenosine 5′-diphosphate; ATP, adenosine 5′-triphosphate; CB, cross-bridge; DMD, Duchenne muscular dystrophy; ECM, extracellular matrix; FSHD, fascioscapulohumeral muscular dystrophy; MD, muscular dystrophy; MsrA and MrB, methionine sulfoxyde reductase A and B; MyHC, myosin heavy chain; nNOS, neuronal NO synthase; NO, nitric oxide; Pi, inorganic phosphate; ROS, reactive oxygen species; Tm, tropomyosin.
Alderton, J. M., and Steinhardt, R. A. (2000). Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J. Biol. Chem. 275, 9452–9460. doi: 10.1074/jbc.275.13.9452
Baghdiguian, S., Martin, M., Richard, I., Pons, F., Astier, C., Bourg, N., et al. (1999). Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IkappaB alpha/NF-kappaB pathway in limb-girdle muscular dystrophy type 2A. Nat. Med. 5, 503–511. doi: 10.1038/10579
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bar, A., and Pette, D. (1988). Three fast myosin heavy chains in adult rat skeletal muscle. FEBS Lett. 235, 153–155. doi: 10.1016/0014-5793(88)81253-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barany, M. (1967). ATPase activity of myosin correlated with speed of muscle shortening. J. Gen. Physiol. 50(Suppl.), 197–218. doi: 10.1085/jgp.50.6.197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bartoli, M., and Richard, I. (2005). Calpains in muscle wasting. Int. J. Biochem. Cell Biol. 37, 2115–2133. doi: 10.1016/j.biocel.2004.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bates, G., Sigurdardottir, S., Kachmar, L., Zitouni, N. B., Benedetti, A., Petrof, B. J., et al. (2013). Molecular, cellular, and muscle strip mechanics of the mdx mouse diaphragm. Am. J. Physiol. Cell Physiol. 304, C873–C880. doi: 10.1152/ajpcell.00220.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bellinger, A. M., Reiken, S., Carlson, C., Mongillo, M., Liu, X., Rothman, L., et al. (2009). Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 15, 325–330. doi: 10.1038/nm.1916
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bloch, R. J., and Gonzalez-Serratos, H. (2003). Lateral force transmission across costameres in skeletal muscle. Exerc. Sport Sci. Rev. 31, 73–78. doi: 10.1097/00003677-200304000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bloch, R. J., Reed, P., O'Neill, A., Strong, J., Williams, M., Porter, N., et al. (2004). Costameres mediate force transduction in healthy skeletal muscle and are altered in muscular dystrophies. J. Muscle Res. Cell Motil. 25, 590–592.
Bottinelli, R., Betto, R., Schiaffino, S., and Reggiani, C. (1994a). Maximum shortening velocity and coexistence of myosin heavy chain isoforms in single skinned fast fibres of rat skeletal muscle. J. Muscle Res. Cell Motil. 15, 413–419. doi: 10.1007/BF00122115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bottinelli, R., Canepari, M., Reggiani, C., and Stienen, G. J. (1994b). Myofibrillar ATPase activity during isometric contraction and isomyosin composition in rat single skinned muscle fibres. J. Physiol. 481(Pt 3), 663–675.
Brand, M. D. (2010). The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472. doi: 10.1016/j.exger.2010.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brenman, J. E., Chao, D. S., Xia, H., Aldape, K., and Bredt, D. S. (1995). Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82, 743–752. doi: 10.1016/0092-8674(95)90471-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Canepari, M., Rossi, R., Pansarasa, O., Maffei, M., and Bottinelli, R. (2009). Actin sliding velocity on pure myosin isoforms from dystrophic mouse muscles. Muscle Nerve 40, 249–256. doi: 10.1002/mus.21302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caremani, M., Dantzig, J., Goldman, Y. E., Lombardi, V., and Linari, M. (2008). Effect of inorganic phosphate on the force and number of myosin cross-bridges during the isometric contraction of permeabilized muscle fibers from rabbit psoas. Biophys. J. 95, 5798–5808. doi: 10.1529/biophysj.108.130435
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Celegato, B., Capitanio, D., Pescatori, M., Romualdi, C., Pacchioni, B., Cagnin, S., et al. (2006). Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics 6, 5303–5321. doi: 10.1002/pmic.200600056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chandra, M., Mamidi, R., Ford, S., Hidalgo, C., Witt, C., Ottenheijm, C., et al. (2009). Nebulin alters cross-bridge cycling kinetics and increases thin filament activation: a novel mechanism for increasing tension and reducing tension cost. J. Biol. Chem. 284, 30889–30896. doi: 10.1074/jbc.M109.049718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheng, G., Cao, Z., Xu, X., Van Meir, E. G., and Lambeth, J. D. (2001). Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 269, 131–140. doi: 10.1016/S0378-1119(01)00449-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cohen, N., Kudryashova, E., Kramerova, I., Anderson, L. V., Beckmann, J. S., Bushby, K., et al. (2006). Identification of putative in vivo substrates of calpain 3 by comparative proteomics of overexpressing transgenic and nontransgenic mice. Proteomics 6, 6075–6084. doi: 10.1002/pmic.200600199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cohn, R. D., and Campbell, K. P. (2000). Molecular basis of muscular dystrophies. Muscle Nerve 23, 1456–1471. doi: 10.1002/1097-4598(200010)23:10<1456::AID-MUS2>3.0.CO;2-T
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coirault, C., Guellich, A., Barbry, T., Samuel, J. L., Riou, B., and Lecarpentier, Y. (2007). Oxidative stress of myosin contributes to skeletal muscle dysfunction in rats with chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 292, H1009–H1017. doi: 10.1152/ajpheart.00438.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coirault, C., Lambert, F., Marchand-Adam, S., Attal, P., Chemla, D., and Lecarpentier, Y. (1999). Myosin molecular motor dysfunction in dystrophic mouse diaphragm. Am. J. Physiol. 277, C1170–C1176.
Coirault, C., Lambert, F., Pourny, J. C., and Lecarpentier, Y. (2002). Velocity of actomyosin sliding in vitro is reduced in dystrophic mouse diaphragm. Am. J. Respir. Crit. Care Med. 165, 250–253. doi: 10.1164/ajrccm.165.2.2105088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'Antona, G., Brocca, L., Pansarasa, O., Rinaldi, C., Tupler, R., and Bottinelli, R. (2007). Structural and functional alterations of muscle fibres in the novel mouse model of facioscapulohumeral muscular dystrophy. J. Physiol. 584, 997–1009. doi: 10.1113/jphysiol.2007.141481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dantzig, J. A., Goldman, Y. E., Millar, N. C., Lacktis, J., and Homsher, E. (1992). Reversal of the cross-bridge force-generating transition by photogeneration of phosphate in rabbit psoas muscle fibres. J. Physiol. 451, 247–278.
De La Torre, C., Illa, I., Faulkner, G., Soria, L., Robles-Cedeno, R., Dominguez-Perles, R., et al. (2009). Proteomics identification of differentially expressed proteins in the muscle of dysferlin myopathy patients. Proteomics Clin. Appl. 3, 486–497. doi: 10.1002/prca.200800087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Disatnik, M. H., Dhawan, J., Yu, Y., Beal, M. F., Whirl, M. M., Franco, A. A., et al. (1998). Evidence of oxidative stress in mdx mouse muscle: studies of the pre-necrotic state. J. Neurol. Sci. 161, 77–84. doi: 10.1016/S0022-510X(98)00258-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drazic, A., Miura, H., Peschek, J., Le, Y., Bach, N. C., Kriehuber, T., et al. (2013). Methionine oxidation activates a transcription factor in response to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 110, 9493–9498. doi: 10.1073/pnas.1300578110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Droge, W. (2002). Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95. doi: 10.1152/physrev.00018.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Du, J., Wang, X., Miereles, C., Bailey, J. L., Debigare, R., Zheng, B., et al. (2004). Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Invest. 113, 115–123. doi: 10.1172/JCI18330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eisenberg, E., and Greene, L. E. (1980). The relation of muscle biochemistry to muscle physiology. Annu. Rev. Physiol. 42, 293–309. doi: 10.1146/annurev.ph.42.030180.001453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eisenberg, E., Hill, T. L., and Chen, Y. (1980). Cross-bridge model of muscle contraction. Quantitative analysis. Biophys. J. 29, 195–227. doi: 10.1016/S0006-3495(80)85126-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ervasti, J. M. (2003). Costameres: the Achilles' heel of Herculean muscle. J. Biol. Chem. 278, 13591–13594. doi: 10.1074/jbc.R200021200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ervasti, J. M., and Campbell, K. P. (1991). Membrane organization of the dystrophin-glycoprotein complex. Cell 66, 1121–1131. doi: 10.1016/0092-8674(91)90035-W
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fanin, M., and Angelini, C. (2002). Muscle pathology in dysferlin deficiency. Neuropathol. Appl. Neurobiol. 28, 461–470. doi: 10.1046/j.1365-2990.2002.00417.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Flanigan, K. M. (2012). The muscular dystrophies. Semin. Neurol. 32, 255–263. doi: 10.1055/s-0032-1329199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fowler, V. M., Sussmann, M. A., Miller, P. G., Flucher, B. E., and Daniels, M. P. (1993). Tropomodulin is associated with the free (pointed) ends of the thin filaments in rat skeletal muscle. J. Cell Biol. 120, 411–420. doi: 10.1083/jcb.120.2.411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galinska-Rakoczy, A., Engel, P., Xu, C., Jung, H., Craig, R., Tobacman, L. S., et al. (2008). Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J. Mol. Biol. 379, 929–935. doi: 10.1016/j.jmb.2008.04.062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Galler, S., Andruchov, O., Stephenson, G. M., and Stephenson, D. G. (2009). Qualitatively different cross-bridge attachments in fast and slow muscle fiber types. Biochem. Biophys. Res. Commun. 385, 44–48. doi: 10.1016/j.bbrc.2009.05.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geeves, M. A., Fedorov, R., and Manstein, D. J. (2005). Molecular mechanism of actomyosin-based motility. Cell. Mol. Life Sci. 62, 1462–1477. doi: 10.1007/s00018-005-5015-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghezzi, P. (2005). Oxidoreduction of protein thiols in redox regulation. Biochem. Soc. Trans. 33, 1378–1381. doi: 10.1042/BST20051378
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gidaro, T., Negroni, E., Perie, S., Mirabella, M., Laine, J., Lacau St Guily, J., et al. (2013). Atrophy, fibrosis, and increased PAX7-positive cells in pharyngeal muscles of oculopharyngeal muscular dystrophy patients. J. Neuropathol. Exp. Neurol. 72, 234–243. doi: 10.1097/NEN.0b013e3182854c07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gokhin, D. S., Tierney, M. T., Sui, Z., Sacco, A., and Fowler, V. M. (2014). Calpain-mediated proteolysis of tropomodulin isoforms leads to thin filament elongation in dystrophic skeletal muscle. Mol. Biol. Cell 25, 852–865. doi: 10.1091/mbc.E13-10-0608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goll, D. E., Thompson, V. F., Li, H., Wei, W., and Cong, J. (2003). The calpain system. Physiol. Rev. 83, 731–801. doi: 10.1152/physrev.00029.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gordon, A. M., Homsher, E., and Regnier, M. (2000). Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924.
Gross, S. M., and Lehman, S. L. (2013). Accessibility of myofilament cysteines and effects on ATPase depend on the activation state during exposure to oxidants. PLoS ONE 8:e69110. doi: 10.1371/journal.pone.0069110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guellich, A., Damy, T., Lecarpentier, Y., Conti, M., Claes, V., Samuel, J. L., et al. (2007). Role of oxidative stress in cardiac dysfunction of PPARalpha-/- mice. Am. J. Physiol. Heart Circ. Physiol. 293, H93–H102. doi: 10.1152/ajpheart.00037.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hashemy, S. I., Johansson, C., Berndt, C., Lillig, C. H., and Holmgren, A. (2007). Oxidation and S-nitrosylation of cysteines in human cytosolic and mitochondrial glutaredoxins: effects on structure and activity. J. Biol. Chem. 282, 14428–14436. doi: 10.1074/jbc.M700927200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haycock, J. W., Jones, P., Harris, J. B., and Mantle, D. (1996). Differential susceptibility of human skeletal muscle proteins to free radical induced oxidative damage: a histochemical, immunocytochemical and electron microscopical study in vitro. Acta Neuropathol. 92, 331–340. doi: 10.1007/s004010050527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hung, R. J., Spaeth, C. S., Yesilyurt, H. G., and Terman, J. R. (2013). SelR reverses Mical-mediated oxidation of actin to regulate F-actin dynamics. Nat. Cell Biol. 15, 1445–1454. doi: 10.1038/ncb2871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huxley, A. F. (1957). Muscle structure and theories of contraction. Prog. Biophys. Biophys. Chem. 7, 255–318.
Huxley, A. F., and Simmons, R. M. (1971). Proposed mechanism of force generation in striated muscle. Nature 233, 533–538. doi: 10.1038/233533a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huxley, H. E., Faruqi, A. R., Kress, M., Bordas, J., and Koch, M. H. (1982). Time-resolved X-ray diffraction studies of the myosin layer-line reflections during muscle contraction. J. Mol. Biol. 158, 637–684. doi: 10.1016/0022-2836(82)90253-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jackson, M. J. (2008). Redox regulation of skeletal muscle. IUBMB Life 60, 497–501. doi: 10.1002/iub.72
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Janssen-Heininger, Y. M., Mossman, B. T., Heintz, N. H., Forman, H. J., Kalyanaraman, B., Finkel, T., et al. (2008). Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic. Biol. Med. 45, 1–17. doi: 10.1016/j.freeradbiomed.2008.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ji, L. L., Gomez-Cabrera, M. C., Steinhafel, N., and Vina, J. (2004). Acute exercise activates nuclear factor (NF)-kappaB signaling pathway in rat skeletal muscle. FASEB J. 18, 1499–1506. doi: 10.1096/fj.04-1846com
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jung, T., Bader, N., and Grune, T. (2007). Oxidized proteins: intracellular distribution and recognition by the proteasome. Arch. Biochem. Biophys. 462, 231–237. doi: 10.1016/j.abb.2007.01.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kajino, S., Ishihara, K., Goto, K., Ishigaki, K., Noguchi, S., Nonaka, I., et al. (2014). Congenital fiber type disproportion myopathy caused by LMNA mutations. J. Neurol. Sci. 340, 94–98. doi: 10.1016/j.jns.2014.02.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaplan, J. C., and Hamroun, D. (2012). The 2013 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 22, 1108–1135. doi: 10.1016/j.nmd.2012.10.021
Kress, M., Huxley, H. E., Faruqi, A. R., and Hendrix, J. (1986). Structural changes during activation of frog muscle studied by time-resolved X-ray diffraction. J. Mol. Biol. 188, 325–342. doi: 10.1016/0022-2836(86)90158-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lawler, J. M. (2011). Exacerbation of pathology by oxidative stress in respiratory and locomotor muscles with Duchenne muscular dystrophy. J. Physiol. 589, 2161–2170. doi: 10.1113/jphysiol.2011.207456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, B. C., Peterfi, Z., Hoffmann, F. W., Moore, R. E., Kaya, A., Avanesov, A., et al. (2013). MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol. Cell 51, 397–404. doi: 10.1016/j.molcel.2013.06.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Linnane, A. W., Kios, M., and Vitetta, L. (2007). The essential requirement for superoxide radical and nitric oxide formation for normal physiological function and healthy aging. Mitochondrion 7, 1–5. doi: 10.1016/j.mito.2006.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lowy, J., and Poulsen, F. R. (1990). Studies of the diffuse x-ray scattering from contracting frog skeletal muscles. Biophys. J. 57, 977–985. doi: 10.1016/S0006-3495(90)82617-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lymn, R. W., and Taylor, E. W. (1971). Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10, 4617–4624. doi: 10.1021/bi00801a004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McKeran, R. O., Halliday, D., and Purkiss, P. (1977). Increased myofibrillar protein catabolism in Duchenne muscular dystrophy measured by 3-methylhistidine excretion in the urine. J. Neurol. Neurosurg. Psychiatry 40, 979–981. doi: 10.1136/jnnp.40.10.979
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mehta, A. D., Rief, M., Spudich, J. A., Smith, D. A., and Simmons, R. M. (1999). Single-molecule biomechanics with optical methods. Science 283, 1689–1695. doi: 10.1126/science.283.5408.1689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Menazza, S., Blaauw, B., Tiepolo, T., Toniolo, L., Braghetta, P., Spolaore, B., et al. (2010). Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum. Mol. Genet. 19, 4207–4215. doi: 10.1093/hmg/ddq339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meng, T. C., Fukada, T., and Tonks, N. K. (2002). Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 9, 387–399. doi: 10.1016/S1097-2765(02)00445-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mercuri, E., and Muntoni, F. (2013). Muscular dystrophies. Lancet 381, 845–860. doi: 10.1016/S0140-6736(12)61897-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michele, D. E., and Campbell, K. P. (2003). Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J. Biol. Chem. 278, 15457–15460. doi: 10.1074/jbc.R200031200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mollica, J. P., Dutka, T. L., Merry, T. L., Lamboley, C. R., McConell, G. K., McKenna, M. J., et al. (2012). S-glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast-twitch muscle fibres of rats and humans. J. Physiol. 590, 1443–1463. doi: 10.1113/jphysiol.2011.224535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Molloy, J. E., Burns, J. E., Kendrick-Jones, J., Tregear, R. T., and White, D. C. (1995). Movement and force produced by a single myosin head. Nature 378, 209–212. doi: 10.1038/378209a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moylan, J. S., and Reid, M. B. (2007). Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 35, 411–429. doi: 10.1002/mus.20743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, R. M., Goodman, C. A., McKenna, M. J., Bennie, J., Leikis, M., and Lamb, G. D. (2007). Calpain-3 is autolyzed and hence activated in human skeletal muscle 24h following a single bout of eccentric exercise. J. Appl. Physiol. (1985) 103, 926–931. doi: 10.1152/japplphysiol.01422.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, R. M., and Lamb, G. D. (2009). Endogenous calpain-3 activation is primarily governed by small increases in resting cytoplasmic [Ca2+] and is not dependent on stretch. J. Biol. Chem. 284, 7811–7819. doi: 10.1074/jbc.M808655200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Murphy, R. M., Verburg, E., and Lamb, G. D. (2006). Ca2+ activation of diffusible and bound pools of mu-calpain in rat skeletal muscle. J. Physiol. 576, 595–612. doi: 10.1113/jphysiol.2006.114090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Musaro, A., Fulle, S., and Fano, G. (2010). Oxidative stress and muscle homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 13, 236–242. doi: 10.1097/MCO.0b013e3283368188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nogueira, L., Figueiredo-Freitas, C., Casimiro-Lopes, G., Magdesian, M. H., Assreuy, J., and Sorenson, M. M. (2009). Myosin is reversibly inhibited by S-nitrosylation. Biochem. J. 424, 221–231. doi: 10.1042/BJ20091144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ochala, J., Gokhin, D. S., Iwamoto, H., and Fowler, V. M. (2014). Pointed-end capping by tropomodulin modulates actomyosin crossbridge formation in skeletal muscle fibers. FASEB J. 28, 408–415. doi: 10.1096/fj.13-239640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pardo, J. V., Siliciano, J. D., and Craig, S. W. (1983). A vinculin-containing cortical lattice in skeletal muscle: transverse lattice elements (“costameres”) mark sites of attachment between myofibrils and sarcolemma. Proc. Natl. Acad. Sci. U.S.A. 80, 1008–1012. doi: 10.1073/pnas.80.4.1008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pate, E., and Cooke, R. (1989). A model of crossbridge action: the effects of ATP, ADP and Pi. J. Muscle Res. Cell Motil. 10, 181–196. doi: 10.1007/BF01739809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pedemonte, M., Sandri, C., Schiaffino, S., and Minetti, C. (1999). Early decrease of IIx myosin heavy chain transcripts in Duchenne muscular dystrophy. Biochem. Biophys. Res. Commun. 255, 466–469. doi: 10.1006/bbrc.1999.0213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Petrof, B. J., Stedman, H. H., Shrager, J. B., Eby, J., Sweeney, H. L., and Kelly, A. M. (1993). Adaptations in myosin heavy chain expression and contractile function in dystrophic mouse diaphragm. Am. J. Physiol. 265, C834–C841.
Petropoulos, I., and Friguet, B. (2005). Protein maintenance in aging and replicative senescence: a role for the peptide methionine sulfoxide reductases. Biochim. Biophys. Acta 1703, 261–266. doi: 10.1016/j.bbapap.2004.08.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pette, D., and Staron, R. S. (1990). Cellular and molecular diversities of mammalian skeletal muscle fibers. Rev. Physiol. Biochem. Pharmacol. 116, 1–76.
Powers, S. K., and Jackson, M. J. (2008). Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol. Rev. 88, 1243–1276. doi: 10.1152/physrev.00031.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Powers, S. K., Ji, L. L., Kavazis, A. N., and Jackson, M. J. (2011). Reactive oxygen species: impact on skeletal muscle. Compr. Physiol. 1, 941–969. doi: 10.1002/cphy.c100054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Powers, S. K., Kavazis, A. N., and Deruisseau, K. C. (2005). Mechanisms of disuse muscle atrophy: role of oxidative stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R337–R344. doi: 10.1152/ajpregu.00469.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ragusa, R. J., Chow, C. K., and Porter, J. D. (1997). Oxidative stress as a potential pathogenic mechanism in an animal model of Duchenne muscular dystrophy. Neuromuscul. Disord. 7, 379–386. doi: 10.1016/S0960-8966(97)00096-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramaswamy, K. S., Palmer, M. L., van der Meulen, J. H., Renoux, A., Kostrominova, T. Y., Michele, D. E., et al. (2011). Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J. Physiol. 589, 1195–1208. doi: 10.1113/jphysiol.2010.201921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rando, T. A., Disatnik, M. H., Yu, Y., and Franco, A. (1998). Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul. Disord. 8, 14–21. doi: 10.1016/S0960-8966(97)00124-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reconditi, M., Brunello, E., Fusi, L., Linari, M., Martinez, M. F., Lombardi, V., et al. (2014). Sarcomere-length dependence of myosin filament structure in skeletal muscle fibres of the frog. J. Physiol. 592, 1119–1137. doi: 10.1113/jphysiol.2013.267849
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reconditi, M., Brunello, E., Linari, M., Bianco, P., Narayanan, T., Panine, P., et al. (2011). Motion of myosin head domains during activation and force development in skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 108, 7236–7240. doi: 10.1073/pnas.1018330108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reconditi, M., Linari, M., Lucii, L., Stewart, A., Sun, Y. B., Boesecke, P., et al. (2004). The myosin motor in muscle generates a smaller and slower working stroke at higher load. Nature 428, 578–581. doi: 10.1038/nature02380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rhee, S. G. (2006). Cell signaling. H2O2, a necessary evil for cell signaling. Science 312, 1882–1883. doi: 10.1126/science.1130481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rybakova, I. N., Patel, J. R., and Ervasti, J. M. (2000). The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 150, 1209–1214. doi: 10.1083/jcb.150.5.1209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sartore, S., Mascarello, F., Rowlerson, A., Gorza, L., Ausoni, S., Vianello, M., et al. (1987). Fibre types in extraocular muscles: a new myosin isoform in the fast fibres. J. Muscle Res. Cell Motil. 8, 161–172. doi: 10.1007/BF01753992
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schessl, J., Goemans, N. M., Magold, A. I., Zou, Y., Hu, Y., Kirschner, J., et al. (2008). Predominant fiber atrophy and fiber type disproportion in early ullrich disease. Muscle Nerve 38, 1184–1191. doi: 10.1002/mus.21088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schiaffino, S., Gorza, L., Sartore, S., Saggin, L., Ausoni, S., Vianello, M., et al. (1989). Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J. Muscle Res. Cell Motil. 10, 197–205. doi: 10.1007/BF01739810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schiaffino, S., and Reggiani, C. (1996). Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol. Rev. 76, 371–423.
Sewry, C. A. (1998). The role of immunocytochemistry in congenital myopathies. Neuromuscul. Disord. 8, 394–400. doi: 10.1016/S0960-8966(98)00053-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, M. A., and Reid, M. B. (2006). Redox modulation of contractile function in respiratory and limb skeletal muscle. Respir. Physiol. Neurobiol. 151, 229–241. doi: 10.1016/j.resp.2005.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sorimachi, H., Kinbara, K., Kimura, S., Takahashi, M., Ishiura, S., Sasagawa, N., et al. (1995). Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. J. Biol. Chem. 270, 31158–31162. doi: 10.1074/jbc.270.52.31158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Spencer, M. J., Croall, D. E., and Tidball, J. G. (1995). Calpains are activated in necrotic fibers from mdx dystrophic mice. J. Biol. Chem. 270, 10909–10914. doi: 10.1074/jbc.270.18.10909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stadtman, E. R., Moskovitz, J., Berlett, B. S., and Levine, R. L. (2002). Cyclic oxidation and reduction of protein methionine residues is an important antioxidant mechanism. Mol. Cell Biochem. 234–235, 3–9. doi: 10.1023/A:1015916831583
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stedman, H. H., Sweeney, H. L., Shrager, J. B., Maguire, H. C., Panettieri, R. A., Petrof, B., et al. (1991). The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352, 536–539. doi: 10.1038/352536a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steffen, W., and Sleep, J. (2004). Repriming the actomyosin crossbridge cycle. Proc. Natl. Acad. Sci. U.S.A. 101, 12904–12909. doi: 10.1073/pnas.0400227101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stienen, G. J., Kiers, J. L., Bottinelli, R., and Reggiani, C. (1996). Myofibrillar ATPase activity in skinned human skeletal muscle fibres: fibre type and temperature dependence. J. Physiol. 493(Pt 2), 299–307.
Stone, J. R., and Yang, S. (2006). Hydrogen peroxide: a signaling messenger. Antioxid. Redox Signal. 8, 243–270. doi: 10.1089/ars.2006.8.243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Street, S. F. (1983). Lateral transmission of tension in frog myofibers: a myofibrillar network and transverse cytoskeletal connections are possible transmitters. J. Cell. Physiol. 114, 346–364. doi: 10.1002/jcp.1041140314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tidball, J. G., and Spencer, M. J. (2000). Calpains and muscular dystrophies. Int. J. Biochem. Cell Biol. 32, 1–5. doi: 10.1016/S1357-2725(99)00095-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tidball, J. G., and Wehling-Henricks, M. (2007). The role of free radicals in the pathophysiology of muscular dystrophy. J. Appl. Physiol. (1985) 102, 1677–1686. doi: 10.1152/japplphysiol.01145.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ugarte, N., Petropoulos, I., and Friguet, B. (2010). Oxidized mitochondrial protein degradation and repair in aging and oxidative stress. Antioxid. Redox Signal. 13, 539–549. doi: 10.1089/ars.2009.2998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veigel, C., Bartoo, M. L., White, D. C., Sparrow, J. C., and Molloy, J. E. (1998). The stiffness of rabbit skeletal actomyosin cross-bridges determined with an optical tweezers transducer. Biophys. J. 75, 1424–1438. doi: 10.1016/S0006-3495(98)74061-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Warnes, D. M., Tomas, F. M., and Ballard, F. J. (1981). Increased rates of myofibrillar protein breakdown in muscle-wasting diseases. Muscle Nerve 4, 62–66. doi: 10.1002/mus.880040111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weber, A., Pennise, C. R., Babcock, G. G., and Fowler, V. M. (1994). Tropomodulin caps the pointed ends of actin filaments. J. Cell Biol. 127, 1627–1635. doi: 10.1083/jcb.127.6.1627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Webster, C., Silberstein, L., Hays, A. P., and Blau, H. M. (1988). Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 52, 503–513. doi: 10.1016/0092-8674(88)90463-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wieczorek, D. F., Periasamy, M., Butler-Browne, G. S., Whalen, R. G., and Nadal-Ginard, B. (1985). Co-expression of multiple myosin heavy chain genes, in addition to a tissue-specific one, in extraocular musculature. J. Cell Biol. 101, 618–629. doi: 10.1083/jcb.101.2.618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wink, D. A., Cook, J. A., Pacelli, R., Liebmann, J., Krishna, M. C., and Mitchell, J. B. (1995). Nitric oxide (NO) protects against cellular damage by reactive oxygen species. Toxicol. Lett. 82–83, 221–226. doi: 10.1016/0378-4274(95)03557-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xanthoudakis, S., Miao, G., Wang, F., Pan, Y. C., and Curran, T. (1992). Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 11, 3323–3335.
Yagi, N. (2003). An x-ray diffraction study on early structural changes in skeletal muscle contraction. Biophys. J. 84, 1093–1102. doi: 10.1016/S0006-3495(03)74925-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, B. T., Yeung, S. S., Allen, D. G., Qin, L., and Yeung, E. W. (2008). Role of the calcium-calpain pathway in cytoskeletal damage after eccentric contractions. J. Appl. Physiol. (1985) 105, 352–357. doi: 10.1152/japplphysiol.90320.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: myosin, cross-bridge kinetics, skeletal muscle, muscle dystrophy, myopathies
Citation: Guellich A, Negroni E, Decostre V, Demoule A and Coirault C (2014) Altered cross-bridge properties in skeletal muscle dystrophies. Front. Physiol. 5:393. doi: 10.3389/fphys.2014.00393
Received: 22 August 2014; Accepted: 23 September 2014;
Published online: 14 October 2014.
Edited by:
Julien Ochala, King's College London, UKReviewed by:
Ranganath Mamidi, Case Western Reserve University, USACopyright © 2014 Guellich, Negroni, Decostre, Demoule and Coirault. This is an open-access article distributed under the terms of the This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine Coirault, Institut National de la Santé et de la Recherche Médicale U974, Bâtiment Babinski, 47 Bd de l'Hôpital, 75013 Paris, France e-mail:Y2F0aGVyaW5lLmNvaXJhdWx0QGluc2VybS5mcg==
†Co-first authors.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.