 Kurt A. Schalper
Kurt A. Schalper Daniel Carvajal-Hausdorf
Daniel Carvajal-Hausdorf Mauricio P. Oyarzo1
Mauricio P. Oyarzo1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Physiol. , 27 June 2014
Sec. Membrane Physiology and Membrane Biophysics
Volume 5 - 2014 | https://doi.org/10.3389/fphys.2014.00237
This article is part of the Research Topic Hemichannels; from the molecule to the function. View all 12 articles
In humans, connexins (Cxs) and pannexins (Panxs) are the building blocks of hemichannels. These proteins are frequently altered in neoplastic cells and have traditionally been considered as tumor suppressors. Alteration of Cxs and Panxs in cancer cells can be due to genetic, epigenetic and post-transcriptional/post-translational events. Activated hemichannels mediate the diffusional membrane transport of ions and small signaling molecules. In the last decade hemichannels have been shown to participate in diverse cell processes including the modulation of cell proliferation and survival. However, their possible role in tumor growth and expansion remains largely unexplored. Herein, we hypothesize about the possible role of hemichannels in carcinogenesis and tumor progression. To support this theory, we summarize the evidence regarding the involvement of hemichannels in cell proliferation and migration, as well as their possible role in the anti-tumor immune responses. In addition, we discuss the evidence linking hemichannels with cancer in diverse models and comment on the current technical limitations for their study.
Multicellular organisms have evolved with sophisticated routes for cell-to-cell communication. Such interactions are multidimensional (e.g., they occur between different types of cells, in diverse spatial orientation/direction, environmental conditions and time frames) and create signaling circuits within tissues, ultimately allowing for a more efficient use of resources and coordination of responses. In addition to a myriad of membrane receptors, channels and transporters, mammalian cells are equipped with relatively large, low-resistance proteinaceous conduits to transmit signaling molecules such as ions (e.g., Na+ and K+), second messengers (e.g., Ca2+ and phosphatidylinositol 1,4,5-trisphosphate [IP3]), nucleotides (e.g., cAMP, ADP, and ATP) and metabolites (e.g., glucose, adenosine, glutamate and glutathione) across electrochemical gradients. Such structures correspond to aqueous pores formed by hexamerization of subunits of two topologically related vertebrate protein families commonly referred to as “gap junction-type proteins”: connexins (Cxs) and pannexins (Panxs). In humans, there are 21 different Cxs genes with a wide genomic distribution and 3 genes encoding Panxs located in chromosomes 11 (Panx1 and 3) and 22 (Panx2) (Baranova et al., 2004; Söhl and Willecke, 2004). Cxs and Panxs show no sequence homology and there is no evidence supporting formation of mixed Cxs/Panxs based channels. Alternative splicing variants and pseudogenes for some of these transcripts have been described in humans, but their biological relevance is uncertain. Paradoxically (and for historical reasons), when such channels are recognized as single structures they are termed “hemichannels” or “connexons,” although they really are whole, functional cell membrane channels allowing the passage of relatively small solutes up to ~1.2 kDa. When two of such channels/connexons, each from one adjacent cell, converge to areas of intercellular membrane contact and serially dock allowing for continuous communication of cytosols, they are termed intercellular gap junction channels (Sáez et al., 2013). In a conservative view, hemichannels have been considered to remain preferentially closed in resting conditions and to serve mainly as structural precursors of gap junction channels through lateral diffusion and clustering after membrane insertion (Gaietta et al., 2002). However, independent site-specific and cytoskeleton-dependent membrane targeting routes for hemichannels, either to areas of cell-cell junctions or to unopposed membrane clusters, have been described (Shaw et al., 2007). The relevance of Cx-based hemichannels and intercellular channels in physiology has become evident by the demonstration of disease phenotypes associated with Cxs mutations affecting protein conformation, turnover and channels function (reviewed in Schalper et al., 2009; Pfenniger et al., 2011).
Diverse structural and functional factors limit the study Cx and Panx hemichannels and intercellular gap junction channels in biological systems. The first major challenge is clear discrimination between hemichannels and intercellular channels. Hemichannels and intercellular channels formed by the same Cx or Panx usually coexist in cells and show overlapping permeability to ions/molecules and a similar (not highly specific) pharmacological sensitivity (Retamal et al., 2007a; Giaume and Theis, 2010; Fiori et al., 2012). In addition, hemichannels and gap junction channels formed by most studied Cxs and Panxs share a common amino acid sequence, secondary structure and transcriptional/post-transcriptional regulation (Sáez et al., 2013; Riquelme et al., 2013a). Therefore, molecular methods or conventional antibody-based detection are usually limited to study the levels, location, stoichiometry and participation of each channel type in any given response. In addition, the difficulties to recapitulate in vitro the complex multi-cellular/multidimensional tissue conditions have limited a clear dissection of the relative contribution of each channel type to various physiological and pathological processes. To overcome some of the aforementioned limitations, mimetic peptides and antibodies targeting specific regions at the extracellular (docking) domains have been used to allow structure-specific recognition/blockade of hemichannels (recently reviewed in Riquelme et al., 2013a).
The second major problem is discriminating between the contribution of Cx and Panx-based channels to any given response. Channels and hemichannels formed by Cxs or Panxs have functional, pharmacological similarities and overlapping expression patterns. In particular, Panxs have been shown to have glycosylation sites on the extracellular loop and a high glycosylation level could preclude the serial docking of Panx hemichannels (Boassa et al., 2008; Peñuela et al., 2013). This led to the notion that Panxs form exclusively hemichannels and not intercellular gap junction channels (Sosinsky et al., 2011). However, recent studies confirmed the early findings by Bruzzone et al. (2003) showing that at least Panx1 and 3 can form functional intercellular gap junction channels with independent properties (Sahu et al., 2014). Future studies exploring diverse cell/tissues and various experimental conditions will be required to support and extend this concept. Further details on the transcriptional regulation of Cx and Panx genes, structural and functional characteristics of Cx- and Panx-based channels, post-translational modifications, pharmacological properties and methodological considerations are discussed in comprehensive reviews published elsewhere by our group and by others (Goodenough and Paul, 2003; Sáez et al., 2013, 2010; Baranova et al., 2004; Söhl and Willecke, 2004; Panchin, 2005; Schalper et al., 2008b; Giaume and Theis, 2010; Kar et al., 2012; D'Hondt et al., 2013; Peñuela et al., 2013).
The aforementioned methodological limitations for the study of hemichannels both in vitro and in vivo and the possible “contamination” of results by additional yet anonymous transmembrane routes have pointed out possible flaws in the interpretation of correlative dyes/molecules uptake or release and electrophysiological studies demonstrating hemichannel existence and functions (Spray et al., 2006). However, the evidence on intercellular gap junction channels also largely relies on comparable correlative expression/function studies using dye transfer and electrophysiological experiments combined with pharmacological blockade. Direct intercellular communication pathways different from gap junction channels termed intercellular nanotubes have recently been described (reviewed in Sherer, 2013) and should be considered in the interpretation of gap junction studies. In addition, the intercellular transfer of regulatory molecules in specialized small bi-layered membranous vesicles termed exosomes (or ectosomes) could also contribute to some of the responses attributed exclusively to gap junction channels, particularly in the central nervous system (Kalani et al., 2014), immune system (Hwang, 2013) and cancer cells (Azmi et al., 2013). Channel-independent functions of Cxs and Panxs have also been well described and add difficulty to the interpretation of results (Vinken et al., 2012). Most studies evaluating the functions and properties of intercellular channels in various conditions have not simultaneously addressed possible changes in hemichannel functions. Thus, a comparable degree of skepticism should exist on the notion of the exclusive involvement of intercellular channels in many studies correlating Cx and Panx expression with certain responses or phenotypes. Finally, visual localization of hemichannels and gap junction channels has been performed largely using antibodies, some of which have not been thoroughly validated regarding their specificity, optimal titration/dynamic range, reproducibility and stability over time. The lack of specificity and reproducibility of commercial and in-house established antibodies represents a common flaw in biomedical research (Bordeaux et al., 2010). In this regard, personal experience working in the gap junction field, as well as reports by other authors have highlighted possible limitations and conflicting results using presumably specific antibodies (Yahalom et al., 1991; Coppen et al., 2002; Shurman et al., 2005; Brisset et al., 2009; Cone et al., 2013). Moreover, the relative contribution of technical error and biological variations in antibody-based results (e.g., diverse protein folding, sample fixation, antigen retrieval, blocking solutions, alternative transcripts, and posttranslational modifications) are not easy to discern. Careful validation of reagents and, more important, systematic reporting of antibodies lacking specificity/reproducibility could help to overcome this complex scenario.
After the seminal work by Drs. Werner Loewenstein and Yoshinobu Kanno showing impaired intercellular electrical coupling in chemically-induced and xenografted rat hepatocarcinomas (Loewenstein and Kanno, 1966, 1967), the role of gap junctions in cancer cells has been a subject of interest for the last five decades. A considerable number of articles on this subject have been published and the literature has been periodically reviewed (see Loewenstein, 1980; Trosko et al., 1983; Kanno, 1985; Yamasaki, 1990; Mesnil and Yamasaki, 1993; Mesnil et al., 1995; Trosko and Ruch, 1998; Mesnil, 2002; Naus, 2002; Mesnil et al., 2005; Cronier et al., 2009; Kandouz and Batist, 2010; Naus and Laird, 2010). In general, cancer cells from various types and after diverse experimental challenges have been shown to have lower expression of their native gap junction proteins than non-tumor samples. Also, highly proliferative tumor cells show atypical (e.g., predominantly cytoplasmic) expression of these proteins and impaired gap junctional intercellular communication. Restoration of the gap junction-type proteins and/or intercellular communication is frequently associated with reduced cell proliferation, which led to the broad concept that Cxs and Panxs were tumor suppressors. However, emerging data on the role of these proteins in tumor cell migration and metastasis have challenged this paradigm and pointed to situations in which these proteins could actually favor cancer progression (Cronier et al., 2009; Kandouz and Batist, 2010; Naus and Laird, 2010). Unfortunately, little of the knowledge on this subject has been translated to cancer medicine and the possible role of hemichannels in carcinogenesis and tumor progression remains largely unexplored. Previous studies on hemichannels have used transformed cell models. However, key experiments addressing the functions and impact of hemichannels in cancer cells have not yet been communicated and include: (i) detailed characterization of the presence and relative abundance of hemichannels in cancer cells; (ii) evaluation of hemichannel-mediated molecule uptake/release in tumor cells as compared to non-tumor cells; (iii) functional consequences of hemichannels activation and blockade in tumor cells and neoplasms and (iv) prognostic and predictive value of hemichannels expression/activation in human malignancies.
In this article, we hypothesize that functional hemichannels can be present in tumor cells (at least of some tumor subtypes or tumor cell subpopulations) and they could alter tumor cell proliferation and disease progression through the transmembrane exchange of signaling solutes such as nucleotides and Ca2+. To test this theory, we review the literature regarding the involvement of hemichannels in cancer-related processes such as cell proliferation/death and cell migration; and revisit some of the gap junction/cancer studies from a “hemichannel-centric” perspective. We also speculate on the possible role of hemichannels in cancer progression and as a clinically useful biomarker.
Most tumor cells harbor genetic and epigenetic defects that lead to altered protein expression and signaling. The presence of gene amplification, rearrangements and acquisition of the so-called “driver” activating mutations are hallmarks of many clinically actionable oncogenes including HER2, EGFR, KRAS, c-KIT, and ALK. In addition, malignant cells frequently downregulate or silence transcripts associated with anti-tumor characteristics (e.g., tumor suppressor genes) such as P53, RB, and PTEN. Silencing of such alleles can occur by gene deletion, inactivating mutations or by epigenetic alterations (e.g., promoter hypermethylation and histone modifications). Finally, tumor cells can also alter the expression of transcripts post-transcriptionally by microRNA-mediated silencing or using the more recently described long non-coding RNAs (lncRNAs) system (Hauptman and Glavac, 2013).
Cxs and Panxs have been frequently shown to display altered expression in human tumors and cancer cell lines. Diverse studies have addressed the underlying mechanisms and they are summarized in Table 1. Of note, recent efforts in exome/genome-wide sequencing using massive parallel sequencing strategies of diverse human malignancies such as the TCGA (The Cancer Genome Atlas) have been completed and to date, mutations in Cxs and Panxs have not emerged as frequent events. Moreover, naturally occurring mutations have been reported in Cx genes and are frequently associated with complex disease phenotypes (Schalper et al., 2009). To our knowledge, no mutations in human Panx genes have been communicated. Taken together, this suggests that regulation of gap junction-type proteins in cancer cells could be more frequently associated with epigenetic and post-transcriptional events, as suggested in pioneering experiments characterizing mRNA transcripts using substractive hybridization methods in cultured normal and tumor breast epithelial cells (Lee et al., 1991).
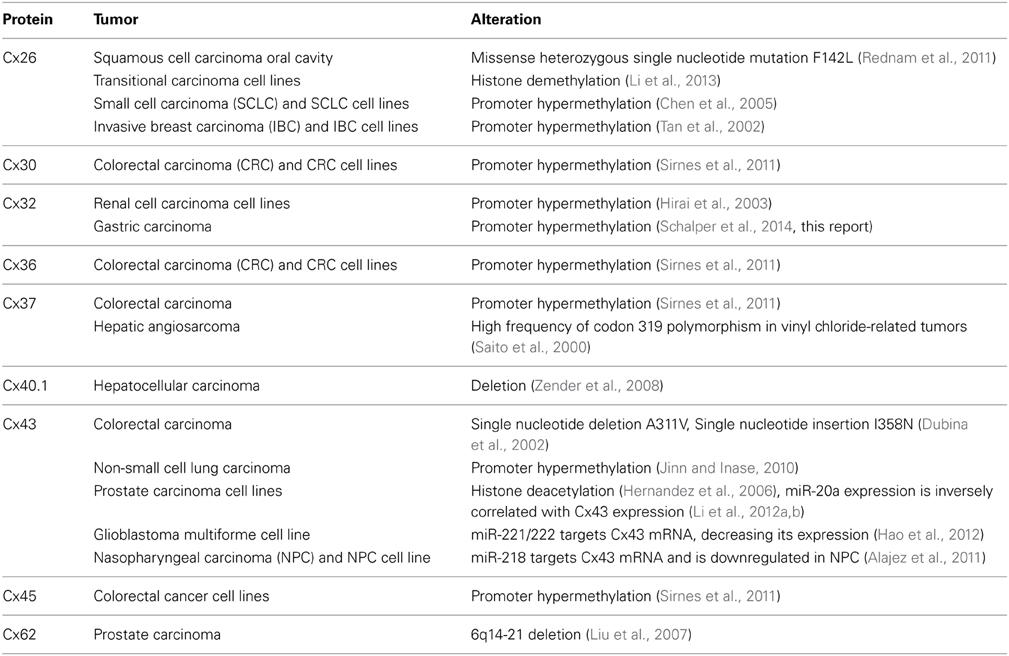
Table 1. Possible mechanisms responsible for reduced expression of connexins in human cancers and cell lines.
Early efforts looking for Cx mutations in cancer specimens found that 4 of 7 primary human hepatic angiosarcomas carried a proline-to-serine substitution in codon 319 of the Cx37 gene (Saito et al., 2000). The same genetic variant was found in DNA extracts from adjacent non-tumor tissue, suggesting that it corresponded to a germline mutation or a single nucleotide polymorphism. The amino acid 319 is located in the cytoplasmic tail of the protein that contains diverse regulatory motifs. Of note, Cx37 has been shown to form functional hemichannels (Wong et al., 2006) and its expression reduced the proliferation of cultured rat insulinoma cells (Burt et al., 2008). In follow-up studies it was shown that introduction of Cx37 mutations that abrogate both the intercellular channel and hemichannel functions (T154A and C61A-C65A) failed to suppress the proliferation (Good et al., 2011, 2012). The inablity of the mutant protein to mimic the effect of the wild type Cx37 in cell proliferation indicates that the passage of molecules through Cx37-based channels is required to influence cell growth/survival. In addition, association between the C1019T polymorphism of the Cx37 gene, gastric adenocarcinomas and H. pylori infection was identified in a retrospective Chinese cohort (Jing et al., 2012).
Another study looking for Cx mutations in sporadic colorectal cancers found 2 frameshift Cx43 mutations in 3 of 6 studied tumors (a single nucleotide deletion A311V in 2 samples and a single nucleotide insertion I358N in 1 tumor) (Dubina et al., 2002). No alterations in Cx32 gene were identified. The Cx43 variants were mapped to the carboxy-terminal tail of the peptide and were not detected in DNA extracts from peripheral blood samples, confirming their somatic nature. However, such mutations have not been mechanistically characterized or further reported in larger tumor series.
Using a high-resolution array-based CGH platform to screen human hepatocellular carcinomas, deletions in the region encoding the Cx40.1 gene were identified (Zender et al., 2008). In support of a tumorigenic effect of this variant, an in vivo, xenograft-based RNAi screen showed that Cx40.1 downregulation using shRNA elicited a prominent acceleration of tumor growth. Little is known about the properties of human Cx40.1. For instance, it is expressed in the human heart (Söhl and Willecke, 2004), but its channel forming capacity remains to be proven.
Deletions of an 817 kb area at 6q14-21 containing the Cx62 gene were found in DNA extracts from 21 of 55 human prostatic adenocarcinomas using a high-resolution single nucleotide polymorphism array (Liu et al., 2007). However, the levels of Cx62 mRNA from the ONCOMINE database were comparable between tumor samples and normal controls, suggesting that another transcript located in this deleted segment such as MAP3K7 might have tumor suppressive actions.
Aggressive oral squamous cell carcinomas are predominantly associated with alcohol and tobacco use and are extremely rare in children. Interestingly, the case of a 6-year old girl was reported with psoriasiform dermatitis and sensorineural hearing loss that presented with a high grade, 3.3 cm palatal and nasal squamous cell carcinoma (Rednam et al., 2011). Due to the clinical findings the patient was screened and the single nucleotide Cx26 mutation F142L was found. Although there was no demonstration of causal relationship between the tumor and the Cx26 variant, other Cx26 mutations associated with excessive keratinocyte proliferation and syndromic hearing loss have been shown to produce hemichannels with increased function and altered permeability (Sánchez et al., 2010; Mese et al., 2011).
Epigenetic regulation of Cxs is a well-described process (reviewed in Vinken et al., 2009; Oyamada et al., 2013). Reduction in the expression of diverse Cxs with concomitant promoter hypermethylation has been shown in multiple human neoplasms and tumor cell lines. Downregulation of Cx32 gene expression by promoter hypermethylation was shown using bisulphite modified DNA-based PCR and a restriction enzyme-based assay in HK-2 and Caki-2 human renal cell carcinoma cells (Hirai et al., 2003). Treatment of cells with the demethylating agent 5-Aza-2′-Deoxycytidine restored Cx32 expression, confirming that Cx32 expression was suppressed. Similarly, downregulation of Cx43 in nasopharyngeal carcinoma CNE-1 cells was found to be associated with GJA1 (Cx43) gene hypermethylation (Yi et al., 2007). A high frequency of Cx30, Cx36, and Cx37 promoter hypermethylation was also detected in colorectal carcinoma samples, as well as in Cx30, Cx36, and Cx45 in colon cancer cell lines (Sirnes et al., 2011). However, correlation between reduced transcript levels and the presence of promoter methylation was found only for Cx45. A more recent study identified Cx45 promoter hypermethylation in 33% of 485 colorectal carcinomas and a positive association with the actionable oncogenic BRAF exon 15 mutation (e.g., V600E) was noted (Ahmed et al., 2011).
Low Cx43 protein levels in human non-small cell carcinomas were associated with Cx43 CpG island promoter methylation and with heavy tobacco use (Jinn and Inase, 2010). Strikingly, the presence of Cx43 promoter hypermethylation in the non-tumor peritumoral lung tissue, but not in the tumor tissue proper, was significantly associated with lymph node positivity in non-small cell cancer patients (Chen et al., 2003). The biological determinants of this association are not clear. Reduced Cx26 protein and mRNA levels in a variety of lung carcinomas (including small-cell neuroendocrine carcinomas) and cell lines was also correlated with promoter hypermethylation and reverted by 5-Aza-2′-Deoxycytidine (Chen et al., 2005). Cx26 promoter hypermethylation was also found in 11 of 20 human invasive breast carcinomas and in cell lines (Tan et al., 2002). However, the absence of Cx26 expression in human squamous esophageal carcinoma cells was not correlated with the presence of Cx26 promoter hypermethylation, suggesting the presence of a different silencing mechanism (Loncarek et al., 2003). Using methylation specific PCR, we found Cx32 promoter hypermethylation in 4 of 9 frozen samples from human gastric adenocarcinomas (Schalper et al., unpublished observation, Figure 1). The 246 bp methylation-specific band was detected exclusively in the tumor and not in non-tumor areas of the samples (Figure 1A). However, we found no clear relationship between the presence of Cx32 promoter hypermethylation and Cx32 protein levels as detected by immunohistochemistry (Figures 1B,C). Taken together, these findings suggest that although more frequent in cancer tissues, promoter hypermethylation in Cxs genes is not unequivocally related with reduced/absent protein.
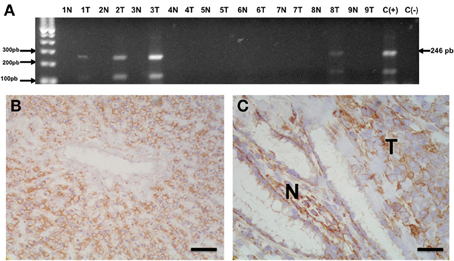
Figure 1. Methylation of Cx32 promoter region in human gastric adenocarcinomas. (A) Agarose gel showing the results from methylation specific PCR of sodium bisulphite-modified DNA extracted from 9 frozen gastric adenocarcinomas (1T-9T) and non-tumor tissue from the same subjects (1N-9N). In 4 cases, a 246 bp band corresponding to the methylated sequence in the Cx32 promoter region was identified. C(+) = Positive control sample of methylated DNA; C(−) = Negative control lacking template DNA. (B,C) Microphotographs showing Cx32 immunostaining in morphologically normal human liver (B) and non-tumor (N)/tumor (T) interface from one gastric adenocarcinoma sample (C). Liver was used as positive control and showed homogenous and intense membranous-like staining. Cx32 immunoreactivity was also detected in the normal foveolar gastric epithelium (N) and in malignant epithelial carcinoma cells (T). The previously reported (Sánchez et al., 2009) monoclonal anti-Cx32 antibody clone 72F (dilution 1:1500, overnight incubation) was used to stain frozen sections. Preparations were then counterstained with hematoxilin. Bar = 100 μm.
To our knowledge, regulation of Panx genes by epigenetic control has not been reported. Using the NIH PROSCAN webtool (http://www-bimas.cit.nih.gov/molbio/proscan/), we evaluated a 1000 bp region upstream of the initiation codon of the human Panx1 gene. We identified a 434 bp region with diverse transcription factor binding motifs and a significant probability of being a promoter region (Promoter score = 88.86; Promoter cutoff = 53.00). In the same genomic region and using the EMBL-EBI Cpgplot tool (https://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/) we detected a 554 bp region with >50% C/G nucleotides, consistent with a CpG island. The latter suggests that human Panx1 has a CpG nucleotide-rich promoter region and could be modulated by hypermethylation.
Few events associated with histone modifications have been shown to affect Cx26 and Cx43 genes in human neoplasms. Li and collaborators (Li et al., 2013) recently found an inverse relationship between Cx26 and JARID1B (also known as KDM5B) histone demethylase protein levels in transitional cancer cell lines and advanced human bladder tumors. Overexpression of JARID1B was associated with reduced Cx26 protein in HT1376 and T24 cells, indicating that this demethylase represses Cx26 expression.
In androgen dependent (LNCaP) and androgen-independent human prostate cancer cell lines (DU145 and PC3), treatment with the histone deacetylase inhibitor Trichostatin A prominently increased Cx43 mRNA and protein expression. The intercellular transfer of the fluorescent dye Lucifer yellow was also increased by Trichostatin A, indicating increased gap junction mediated intercellular communication (Hernandez et al., 2006). Similar findings were observed in cultured non-malignant human peritoneal mesothelial cells (Ogawa et al., 2005).
Post-transcriptional modulation by small non-coding RNAs of Cx genes has also been reported in human tumors. Li et al. (2012a,b) found an inverse relationship between miR-20a and Cx43 protein and mRNA expression in human prostate tumors and cell lines. In this model, downregulation of miR-20a caused a 4-fold increase in Cx43 expression and reduced the cell proliferation. Using a luciferase reporter assay, the miR-20a antagomir increased the luciferase activity using the wild type Cx43 sequence, but was ineffective with Cx43 mutated at the miR-20a binding site at the 3′UTR region. The latter demonstrates that Cx43 is a direct target of miR-20a. Using a similar approach, miR-221/222 was shown to bind Cx43 mRNA, reduce Cx43 expression and promote cell growth and invasion in the human glioblastoma multiforme cell line U251 (Hao et al., 2012). Finally, transfection of miR-218 in nasopharyngeal carcinoma cells reduced Cx43 expression and was associated with increased apoptosis. Moreover, miR-218 transfection reduced the tumor growth after intramuscular implantation of C666-1 cells in SCID mice (Alajez et al., 2011). Although likely to occur, regulation of Cxs expression by lncRNAs has not been reported yet. In addition and to our knowledge, modulation of Panxs by non-coding RNAs has not been communicated.
The cellular consequences of altered hemichannel functions are believed to be mediated mainly by defective transmembrane transport of signaling molecules passing through them, leading to altered activation of intracellular pathways and autocrine/paracrine signals. Key molecules associated with cell proliferation/survival and shown to permeate activated hemichannels include NAD+ (nicotinamide adenine dinucleotide), cADPR (cyclic ADP-ribose), ATP, Ca2+, IP3, glutathione, and prostaglandin E2 (Wang et al., 2013a,b). However, additional, yet unidentified, relatively small-sized solutes could also mediate autocrine/paracrine effect in cells. In particular, the nucleotides NAD+ and ATP have been directly involved in hemichannel mediated cell proliferation (of non-tumor cells) by increasing the intracellular Ca2+ levels. Therefore, the exchange of these nucleotides and Ca2+ through hemichannels will be the main focus of this section. Also, the transmembrane passage of nucleotides constitutes a unique feeder system for extracellular nucleotidases that have a prominent role in the anti-tumor immune responses (see below). In addition, the increasingly recognized paracrine signaling between tumor and stromal cells that may occur through hemichannels could also support their involvement in tumor growth and cancer progression. A summary of the possible functions of hemichannels in cancer biology is presented in Figure 2.
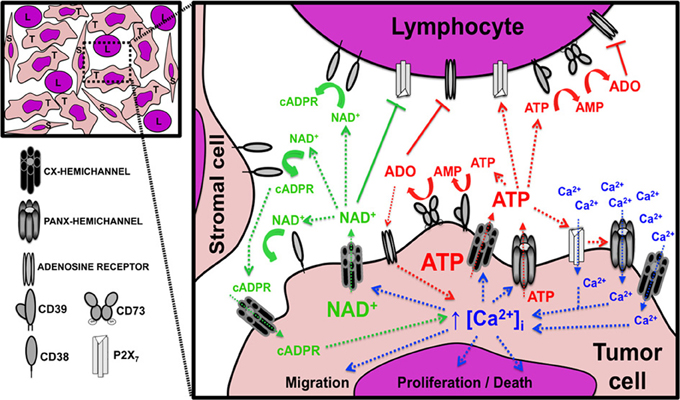
Figure 2. Possible role of hemichannels in tumor growth and progression. Diagram showing the paracrine interactions mediated by the passage of nucleotides and Ca2+ through hemichannels between tumor cells (T), lymphocytes (L), and stromal cells (S). The image in the upper left corner depicts the tumor microenvironment components with intimate relationship between the tumor and non-tumor cells. The expanded inset on the right represents a magnification of one tumor area and indicates the signaling circuits associated with NAD+ (green text and arrows), ATP (red text and arrows), and Ca2+ (blue text and arrows). The image also shows the presence and possible interactions between connexin (Cx) and pannexin (Panx) hemichannels, ectonucleotidases, purinergic receptors, and adenosine receptors. Arrows with dashed lines indicate a positive/stimulatory effect, arrows with continuous lines indicate a negative/inhibitory effect; and curved lines indicate (enzymatic) metabolic activity. ADO, adenosine; AMP, adenosine monophosphate; ATP, adenosine triphosphate; cADPR, cyclic adenosine diphosphate-ribose; [Ca2+]i, free intracellular Ca2+ concentration; NAD+, nicotinamide adenine dinucleotide; P2X7, P2X purinoreceptor 7. See text for detailed explanation of the biological consequences of each pathway.
The cellular NAD+ homeostasis is tightly regulated in all organisms and contributes to the maintenance of the energetic balance, intracellular redox potential and signal transduction. Tumor cells undergo metabolic adaptations to support growth and survival, including a pronounced shift from oxidative phosphorylation toward more NAD+ and lactate production through aerobic glycolysis, a phenomenon known as the Warburg effect (Chiarugi et al., 2012). The net cellular NAD+ content is the result from its synthesis by various enzymatic cascades involving niacin (vitamin B3), tryptophan, aspartic acid and reutilization of intracellular nicotinamide-related compounds, as well as NAD+ degradation through enzymatic hydrolysis, ribosylation and deacetylation. CD38 is the major mammalian NAD+ glycohydrolase and ADP-ribosyl cyclase, thus directly participating in NAD+ metabolism and production of second messengers (Chiarugi et al., 2012). However, the catalytic domain of CD38 is located in the extracellular domain, apart from its cognate intracellular substrate.
The first experimental evidence for the involvement of Cx43 hemichannels in transmembrane NAD+ transport came from studies using cultured NIH3T3 fibroblasts, HeLa cells and proteoliposomes (Bruzzone et al., 2001). In these models, NAD+ efflux required (and was paralleled by) Cx43 expression. Moreover, the NAD+ release was increased by lowering the extracellular Ca2+ concentration and prominently blocked by beta-glycyrrhetinic acid, La3+ and a Cx43 monoclonal antibody, thus implicating Cx43 hemichannels (Bruzzone et al., 2001). Shortly after, the same group convincingly demonstrated the involvement of Cx43 hemichannels in the [Ca2+]i-dependent, cADPR-induced cell proliferation in co-cultured mouse NIH/373 fibroblasts with or without CD38 expression (Franco et al., 2001; De Flora et al., 2004). In this model, the efflux of NAD+ through activated hemichannels allowed the interaction of this intracellularly produced dinucleotide with the extracellular catalytic segment of CD38, giving a mechanistic response to this topographical paradox. After its production by CD38-mediated NAD+ ribosylation, the ubiquitous second messenger cADPR can re-enter the cells to induce IP3-pathway independent-/ryanodine receptor induced Ca2+ transients and ultimately mediate cell cycling and proliferation. Later studies by another research group extended these observations and showed that cADPR re-uptake is mediated by Cx43 hemichannels through bidirectional NAD+ extrusion/cADPR import upon FCγ receptor stimulation in cultured murine J774A.1 macrophages (Song et al., 2011) (Figure 2, green colored arrows and text).
Although CD38 is expressed in various mammalian tissues including brain, prostate and muscle, it is most prominently found in lymphoid/blood cells (Deaglio et al., 2008). CD38 is frequently upregulated and has been indicated as a prominent oncogenic and adverse prognostic factor in hematologic malignancies including multiple myeloma and chronic lymphocytic leukemia. Moreover, humanized monoclonal antibodies targeting CD38 (e.g., daratumumab, MOR03087, and SAR650984) are under evaluation in early phase clinical trials to treat patients with advanced hematological B-cell malignancies (www.clinicaltrials.gov; trial identifiers NCT01615029, NCT00574288, NCT01749969, NCT01084252 and NCT01421186). As an integral component of the CD38-Cx43-cADPR axis and NAD+ degradation pathway, it is tempting to speculate that expression of Cx43 in neoplastic cells could serve as a prognostic/predictive biomarker for such compounds and modulation of Cx43 hemichannels may itself represent a novel anti-tumor therapeutic target, especially in CD38 expressing B cell malignancies.
Cx43 was shown to be expressed in isolated human B cells from tonsil and peripheral blood and Cx-based channels participate in immunoglobulin production and B cell maturation (Oviedo-Orta et al., 2000, 2001). In addition, Cx43 defective knockout mice have lower IgM-positive B cells than their wild type littermates and the defect is more pronounced in the homozygous than in the heterozygous animal, supporting a gene-dosage effect (Montecino-Rodriguez and Dorshkind, 2001). Although some authors have ascribed the functional effects of lymphocyte Cx43 to their participation in the pool of intercellular channels located at the cell-cell contacts between activated immune cells (e.g., “the immunological synapse”), the involvement of hemichannels in these processes has not been excluded. This is particularly relevant in immune cells that are expected to spend a considerable part of their lifespan detached from other cells. In this regard, the reduction in immunoglobulin levels observed by Oviedo-Orta and collaborators in primary human lymphocytes was triggered by traditional Cx-channel blockers and by a Cx43 mimetic peptide corresponding to the second extracellular loop (e.g., Gap27) (Oviedo-Orta et al., 2001), known to inhibit Cx43 hemichannels (Evans et al., 2006; Wang et al., 2012). Notably, additional short peptides corresponding to the intracellular domains of Cx43 (e.g., Gap18 and Gap20) were ineffective in this model.
More recently, Cx43 was shown to be required for activation and migration of cultured murine B cells (Machtaler et al., 2011). In this study, B cell responses were independent of intercellular gap junction channels and immunofluorescence experiments of Cx43 in the murine B lymphoma cell lines WEHI 231 and A20, and in primary murine B splenocytes, showed a predominant homogenous membranous distribution. The latter indicates a lack of preferential clustering/enrichment of Cx43 in areas of cell-cell contact. A similar diffuse membranous Cx43/CD38 co-localization staining pattern was observed in cultured J774A.1 macrophages (Song et al., 2011).
Surprisingly few studies have explored the expression of Cxs in native human lymphoid tissues and human lymphoid neoplasms. For instance, Cx43 was detected in germinal centers of fresh samples from human tonsil and spleen (Krenacs et al., 1997). Of note, Cx43 protein and mRNA were predominantly located in cells with irregular shape and co-expressing CD21 and CD35, consistent with follicular dendritic cells. Only scarce, more rounded cells with lymphocyte appearance and of undefined lineage were found to carry the Cx43 transcript in this study.
Aiming to characterize Cx43 expression in native human lymphoid tissues and lymphoid malignancies, preliminary experiments from our group using chromogenic immunohistochemistry detected low levels of Cx43 protein in formalin fixed-paraffin embedded samples from 4 non-tumor lymph nodes and absence of signal in a set of 22 malignant B cell lymphomas (13 Diffuse large B cell lymphomas [DLBCL] and 9 classical Hodgkin lymphomas [CHL]) (Figure 3). While distinctive Cx43-positive plaques were evident toward the intercalated discs of human myocardiocytes used as positive control (Figures 3A,B), only focal labeling of Cx43 in occasional germinal centers from reactive-type lymph nodes was seen in cells with morphology consistent with endothelial cells, macrophages and follicular dendritic cells (Figures 3C,D). The latter finding is somewhat consistent with previous observations by Krenacs and collaborators (Krenacs et al., 1997) and points to the possibility that Cx43 expression in lymphocytes might be too low to be detected by immunohistochemistry in unstimulated lymph nodes (e.g., lower than in myocardium). Alternatively, other Cxs known to be present in lymphocytes such as Cx40 (Oviedo-Orta et al., 2001) or Panxs could represent the main hemichannel forming pool in native human lymphoid tissues and lymphomas. Of note, Panx1 hemichannels have been previously shown to be expressed in T lymphocytes and to allow the passage of ATP that is structurally related with NAD+ and has a similar molecular mass (MW NAD+ = 663.42 vs. ATP = 551.14) (Woehrle et al., 2010). In addition, we found membranous-like Panx1 expression in samples from high grade, DLBCL that showed prominent co-localization with the pan-B cell marker CD20 in confocal stacks (Shoji et al., unpublished observation). Future studies using more sensitive methods and functional assays will be required to clarify this.
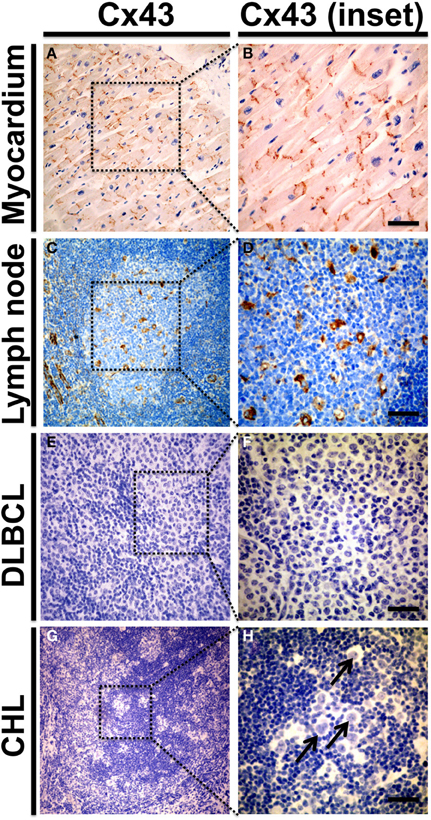
Figure 3. Cx43 protein expression in human reactive lymph nodes and lymphomas. Representative microphotographs from formalin fixed paraffin embedded samples of morphologically normal human myocardium (n = 3, A,B), reactive lymph node with follicular and parafollicular hyperplasia (n = 4, C,D), diffuse large B cell lymphomas (n = 13 DLBCL, E,F) and classical Hodgkin's lymphoma (n = 9 CHL, G,H). Samples were included in a tissue microarray and stained with chromogenic immunohistochemical method for Cx43 using a mouse monoclonal antibody at 1:100 dilution (Zymed Laboratories). Preparations were counterstained with hematoxylin to visualize nuclei. Right panels (B,D,F,H) show magnified insets from low power images on the left (A,C,E,G). (A,B) Depicts control Cx43 distribution in intercalated discs of human myocardiocytes. In non-tumor lymph nodes (C,D) Cx43 staining was predominantly detected in endothelial cells; and in cells with morphological features more consistent with macrophages and follicular dendritic cells. Cx43 was not detected in DLBCL and CHL samples (E–H). Arrow in (H) indicates Hodgkin/Reed-Stemberg cells. Bar = 50 μm.
The presence of hemichannels in tumor cells even without CD38 expression might confer on them growth/migration advantages through increased autocrine/paracrine NAD+ signaling and the possible interaction with CD38 present in the membrane of adjacent non-tumor cells (e.g., stromal cells). In addition, relatively low increases in the extracellular NAD+ concentration (in the micromolar range) through cell death/lysis or release through activated hemichannels can exert local immune suppressive effects through activation of ionotropic P2X7 purinergic receptors in immune cells, leading to effector cytotoxic T cell death and expansion of immune suppressive regulatory CD4+/CD25+/FoxP3+ T cells (Tregs) (Scheuplein et al., 2009; Hubert et al., 2010). The latter effect could favor tumor progression by shielding cancer cells from immune-mediated cell killing (Figure 2). Indeed, increasing studies have found elevated Cx43 and/or Cx26 to be associated with more aggressive (e.g., shorter survival) and disseminated disease in diverse human malignancies and in vitro cancer models (Kanczuga-Koda et al., 2006; Tate et al., 2006; Naoi et al., 2007; Kyo et al., 2008; Cronier et al., 2009; Bui et al., 2011; Lamiche et al., 2012; Stoletov et al., 2013; Tang et al., 2013; Ghosh et al., 2014). In addition, a recent study demonstrated that increased post-treatment Cx43 levels were significantly associated with response to neoadjuvant chemotherapy in human breast carcinomas (Teleki et al., 2013) and high tumor Cx43 expression by immunohistochemistry was predictive of response to platinum-based chemotherapy in non-small cell lung cancer (Du et al., 2013). Notably, many of these studies reported predominant cytoplasmic location of Cxs and failed to detect a corresponding increase in the intercellular gap junctional communication. Recently available antibodies capable of selectively detecting (and blocking) Cx hemichannels through recognition of extracellular loop sequences (Riquelme et al., 2013a) might shed light on the relative contribution of hemichannels and intercellular channels in these responses.
ATP is responsible for energy storage and transfer in the form of renewable high-energy phosphoryl bonds. In addition to its role in cell metabolism, intracellular ATP participates in the maintenance of electrochemical gradients through the fueling of pumps, active vesicular transport and also serves as a coenzyme and mediator for signal transduction and DNA synthesis. The cellular levels of ATP are determined by its constant production occurring mostly through oxidation of a variety of carbon-based substrates (e.g., aerobic cellular respiration); and degradation by hydrolysis into the lower level nucleotide phosphoforms ADP and AMP.
In addition to the prominent intracellular function, extracellular ATP serves as a pleiotropic intercellular messenger through its interaction with ionotropic (P2X1–P2X7) and metabotropic (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14) P2 purinergic membrane receptors (Falzoni et al., 2013). Examples of this include its function as neurotransmitter, proliferation stimulating agent, pro-inflammatory mediator and signaling factor between immune cells. In particular, extracellular ATP has been recognized to have a key role in the tumor-host interactions (reviewed in Di Virgilio, 2012). Despite its prominent extracellular role, the routes for cellular ATP release in both tumor and non-tumor cells are not well characterized. Diverse mechanisms have been proposed including passive transmembrane diffusion, vesicular release/exocytosis, cellular lysis and extrusion through specific chloride channels, ABC transporters, P2X7 receptors and Cx/Panx hemichannels (Di Virgilio, 2012; Baroja-Mazo et al., 2013; Falzoni et al., 2013; Orellana et al., 2013). The first direct indication of ATP release occurring through Cx hemichannels came from experiments showing that the propagation of Ca2+ waves in cultured astrocytes required the presence of extracellular ATP and Cxs expression, but not of functional intercellular channels (Cotrina et al., 1998). In addition, cultured C6, HeLa and U373 cells showed UTP-induced ATP release only after transfection with Cx26, 32, or 43 and the responses were mimicked by removal of extracellular Ca2+ (Cotrina et al., 1998). Later on, numerous studies confirmed the permeability of Cx hemichannels to ATP in diverse cell types and using an extensive repertoire of experimental conditions including the removal of extracellular divalents (Arcuino et al., 2002; Stout et al., 2002; Stout and Charles, 2003; Gomes et al., 2005; Pearson et al., 2005; Zhao et al., 2005; Bahima et al., 2006), mechanical stimulation (Arcuino et al., 2002; Stout et al., 2002; Gomes et al., 2005; Zhao et al., 2005; Richter et al., 2014), increased [Ca2+]i (Braet et al., 2003a,b; De Vuyst et al., 2009), hypoxia/ischemic like conditions (Faigle et al., 2008; Clarke et al., 2009), membrane depolarization (Kang et al., 2008; Nualart-Marti et al., 2013), application of bacterial lypopolysaccharide (De Vuyst et al., 2007), treatment with amyloid beta peptide (Orellana et al., 2011), exposure to hypotonic stress (Lu et al., 2012), after spinal cord injury (Huang et al., 2012), in activated polymorphonuclear granulocytes (Eltzschig et al., 2006), induced by gamma irradiation (Ohshima et al., 2012) and after air-stimulation of cultured keratinocytes (Barr et al., 2013). Hemichannels formed by Panx1 have also been consistently shown to allow the passage of ATP in diverse cell types and experimental models (Bao et al., 2004; Locovei et al., 2006; Huang et al., 2007; Reigada et al., 2008; Schenk et al., 2008; Buvinic et al., 2009; Iglesias et al., 2009; Qiu and Dahl, 2009; Ransford et al., 2009; Chekeni et al., 2010; Garré et al., 2010; Murata et al., 2010; Sridharan et al., 2010; Woehrle et al., 2010; Seminario-Vidal et al., 2011; Dolmatova et al., 2012; Iglesias and Spray, 2012; Li et al., 2012a,b; Lohman et al., 2012; Sandilos et al., 2012; Xia et al., 2012; Xiao et al., 2012; Pinheiro et al., 2013; Riquelme et al., 2013b). Moreover, Panx1 hemichannels are functionally coupled to P2X and P2Y receptors activation (Locovei et al., 2006; Pelegrin and Surprenant, 2006; Wang et al., 2013a,b).
The levels of extracellular ATP correspond to the result between cellular release through diverse routes including hemichannels and degradation by ectonucleotidases (e.g., CD39 and CD73). Such enzymes mediate the extracellular conversion of ATP to AMP (CD39) and to adenosine (CD73) that interacts with predominantly immune inhibitory adenosine receptors (e.g., A2A), thus driving the shift from ATP driven pro-inflammatory environment to an adenosine-driven antiinflammatory/immune suppressive state (Antonioli et al., 2013) (Figure 2, red arrows and text). The latter mechanism might represent a negative regulatory switch to limit the inflammatory response and control tissue damage. However, relatively high concentrations of ATP (milimolar range) acting on P2X and P2Y receptors can induce marked suppressive effects in lymphocytes (Trabanelli et al., 2012; Burnstock and Di Virgilio, 2013). Consequently, increased extracellular ATP can potentially favor or suppress the local anti-tumor immune response, depending on its concentration, the presence and relative abundance of adenosine and P2 receptors in tumor and inflammatory cells, and the rate of conversion to adenosine by nucleotidases (Di Virgilio, 2012). In general, tumor microenvironments are considered to be purine-rich and CD39 and CD73 are commonly upregulated by hypoxia in diverse cancers leading to a more immune-suppressive tumor niche (Di Virgilio, 2012; Ghiringhelli et al., 2012; Antonioli et al., 2013). Consistent with this notion, expression of CD39 and CD73 are associated with more aggressive and metastatic tumors. The recent emergence of immunostimulatory drugs blocking immune inhibitory checkpoint pathways such as CTLA-4 and the PD-1/PD-L1 axis have shown unprecedented prominent and lasting clinical responses in patients with advanced solid tumors (Quezada and Peggs, 2013). Therefore, it is not surprising that strategies to inhibit CD39 and CD73 in cancer patients are being actively pursued. Unfortunately, most available in vitro and animal models to study the role of hemichannels in cancer cells are not helpful in evaluating their possible effect in the anti-tumor immune response (e.g., monocultures and immune defficient animals). Future studies using reconstituted cell culture systems including tumor cells and lymphocytes/APCs or immunocompetent animal models will help clarifying this issue.
In addition to the inflammatory/immune effect, it has long been known that nucleotides have direct actions on tumor cells and most human neoplasms express a wide variety of purinergic receptors (Stagg and Smyth, 2010; Roger and Pelegrin, 2011; Burnstock and Di Virgilio, 2013). The effect of extracellular ATP in tumor cell proliferation/survival is complex and apparently depends on the ATP concentration, secretion pattern and the reportoir of purinergic receptors available in the target cell (Burnstock and Di Virgilio, 2013). Moreover, activation of the same receptor can induce apoptosis or even increase cell proliferation in different cell types. Extracellular nucleotides have been shown to induce anti-cancer effect in diverse tumor types and strategies to exploit this effect using ATP infusions or synthetic ATP analogs are ongoing. However, it is also clear that low doses of ATP as seen during spontaneous release from cells can have a pro-proliferative and growth promoting effect (Di Virgilio, 2012; Burnstock and Di Virgilio, 2013). In general, tumor cells have very high ATP content compared with non-tumor cells and strategies to reduce ATP have also been shown to enhance the activity of anti-cancer agents (Burnstock and Di Virgilio, 2013).
One of the most studied, yet still controversial P2 receptor in cancer cells is the so-called cytolytic P2X7 receptor. Although relatively high (millimolar range) concentrations of extracellular ATP can induce cell death through P2X7 receptor activation, more typically secreted (micromolar range) ATP concentrations are associated with promotion of cell survival/growth (Roger and Pelegrin, 2011; Di Virgilio, 2012). In support of this notion, expression of P2X7 receptor in some tumor models is associated with increased growth rate and metastases (Stagg and Smyth, 2010; Roger and Pelegrin, 2011; Di Virgilio, 2012). Also, signaling through some P2Y receptors such as P2Y2 has been associated with increased cell survival, proliferation and migration (White and Burnstock, 2006; Stagg and Smyth, 2010; Burnstock and Di Virgilio, 2013). In addition, it was recently shown that ATP released by non-tumor osteocytic cells treated with alendronate can either inhibit or stimulate the cell growth and migration of cultured MDA-MB231 breast cancer cell lines depending on the extracellular ATP concentration present (Zhou et al., 2014). While the inhibitory effect was seen with relatively low ATP concentrations and required P2X7 expression/activation; higher extracellular ATP levels were associated with increased tumor cell migration that was mediated by adenosine signaling through the A2A receptor. Similar observations were made in MDA-MB231 mouse xenographs, confirming the importance of this mechanism in vivo. Taken together, this indicates that both the release of ATP by adjecent non-tumor cells and the rate of conversion to adenosine participate in the ATP-mediated tumor cell proliferative responses. The biphasic effect of extracellular ATP could partially explain the dissimilar impact of ATP signaling in cell proliferation and survival seen in different models (Figure 2). Moreover, it suggests that treatment with non-hydrolizable forms of ATP could have prominent anti-cancer effects by limiting its conversion to pro-tumorigenic and immune supressive adenosine.
Notably, ATP release through activated Cx-hemichannels has been detected after stimulation with growth factors including nerve growth factor (NGF) (Belliveau et al., 2006); basic fibroblast growth factor (FGF-2) (De Vuyst et al., 2007) and acidic fibroblast growth factor (FGF-1) (Garré et al., 2010). NGF signaling has been implicated in diverse human malignancies (Molloy et al., 2011). In particular, NGF and its receptors are prominent oncogenic mediators in invasive human breast carcinomas and their expression in malignant cells is associated with adverse patient outcome (Dollé et al., 2004; Noh et al., 2013). Moreover, blockade of NGF signaling using monoclonal antibodies or reducing NGF expression with siRNA attenuates the proliferation and angiogenesis in MDA-MB-231 cells mouse xenografts (Adriaenssens et al., 2008). Aberrant FGFs signaling is also directly implicated in developmental tissue growth and in the progression of diverse human neoplasms and the genes encoding the four major FGF receptors are frequently amplified and/or mutated in human cancers (Dieci et al., 2013; Dienstmann et al., 2014). Research is ongoing to identify effective strategies to block FGF signaling using small molecule inhibitors and monoclonal antibodies to achieve anti-tumor effect with acceptable toxicity/safety profile. The aforementioned observations suggest that increased hemichannel activation possibly induced by excessive growth factors signaling could favor tumor cell proliferation and expansion. Supporting this possibility, Cx43 hemichannels were shown to mediate the proliferation of neural progenitor cells from the ventricular zone and the retinal epithelium through release of ATP, paracrine activation of purinergic P2Y receptors and subsequent [Ca2+]i transients (Weissman et al., 2004; Pearson et al., 2005). In addition, two naturally occurring mutations in the human Cx30 gene (G11R and A88V) associated with a hyperproliferative keratinocytic genodermatosis termed hidrotic ectodermal dysplasia (or Clouston syndrome) have been demonstrated to produce defective Cx30 hemichannels with increased ATP release (Essenfelder et al., 2004). Although not directly measuring ATP, blockade of Cx43 hemichannels using the Gap26 mimetic peptide or carbenoxolone reduced the neointimal formation after vascular injury through decreased proliferation of smooth muscle cells (Song et al., 2009) and opening of Cx43 hemichannels mediated the survival signals induced by alendronate in osteocytic/osteoblastic cells through a Src-MAP kinases transduction dependent mechanism (Plotkin et al., 2002). The possible involvement of increased extracellular adenosine signaling in these responses has not been studied.
Similar to Cx hemichannels, ATP release through Panx1 hemichannels has been associated with increased proliferation of stem cells, neural progenitors (Wicki-Stordeur et al., 2012) and primary human cultured fibroblasts stimulated with histamine (Pinheiro et al., 2013). In the latter model, P2Y1 receptor-dependent [Ca2+]i transients were triggered by Panx1-mediated ATP release. The release of ATP through activated Panx1 hemichannels was also shown to accelerate the assembly of multicellular C6 tumor cells aggregates in a 3D culture system (Bao et al., 2012). The latter effect was mediated by P2X7 receptors activation and remodeling of the F-actin cytoskeleton, and ultimately suggests that Panx1 could particiapte in tumor cell motility.
Consistent with a pro-tumorigenic effect, Panx1 (but not other Panxs) was found at relatively high levels in the melanoma cell lines B16 and was associated with membranous-like location and increased hemichannel function (Peñuela et al., 2012). In this model, down regulation of Panx1 using siRNAs significantly reduced the expression of the malignant melanoma markers vimentin, hsp70 and B-catenin, and diminished the cell density and motility, but increased melanin production suggesting acquisition of a more differentiated phenotype. Moreover, Panx1 silencing reduced the tumor size and the frequency of liver metastases in a chicken embryo tumor implantation model, suggesting that Panx1 hemichannels favor melanoma growth and progression (Peñuela et al., 2012). Similarly, silencing of Panx2 induced the acquisition of a more differentiated neuronal-like phenotype in N2A neuroblastoma cells (Swayne et al., 2010). Therefore, it would not be surprising to find specific tumor types that use hemichannels mediated mechanisms for their growth and dissemination. Also, the presence of subpopulations of cells with activated hemichannels, and clones with closed hemichannels or even lacking hemichannels in their membranes within a given tumor is possible. Dynamic adaptations of tumor cells to environmental pressure could also support the opportunistic transit between the functional states of hemichannels in tumor cells (e.g., proliferation vs. migration or invasion).
Although a good deal of evidence points to the possible pro-tumorigenic and proliferative effect of both Cx and Panx1 hemichannels activation, it is relevant to consider that association with reduced cell proliferation/survival is also apparent. Moreover, sustained or de-regulated opening of hemichannels can be deleterious for cells. In fact, activation of Cx43 or Panx1 hemichannels by noxious stimuli such as ischemic-like conditions or oxidative stress can accelerate self and paracrine cell death through massive ATP-release and [Ca2+]i-related mechanisms (Contreras et al., 2004; Retamal et al., 2007b; Decrock et al., 2011; Carette et al., 2014) (Figure 2). Consistent with an anti-tumor effect, exogenous Panx1 expression was associated with decreased proliferation, motility and anchorage-independent growth in cultured C6 glioma cells (Lai et al., 2007). Similar findings were obtained after expression of Panx2 (Lai et al., 2009). In addition, Panx1 and Panx2 expression were associated with reduced tumor volume in murine C6 tumor implants, which led to propose them as tumor suppressor genes. In line with this observation, forced expression of Panx1 and Panx3 increased the dye uptake and reduced the proliferation of cultured rat epidermal keratinocytes (Celetti et al., 2010). Data from our group revealed that exogenous Panx1 expression significantly reduced the cell density of cultured HeLa cells (Figure 4A). In addition and using a previously reported antibody (Buvinic et al., 2009), we found that Panx1 is present in morphologically normal human gallbladder epithelium and its levels are lower in gallbladder adenocarcinomas (Figure 4B). Also, in gallbladder tumors Panx1 signal is inversely related to the proliferative activity as determined by the nuclear Ki-67 positivity rate (Figure 4C). Of note, the rate of nuclear Ki-67 positive cells is frequently used as a metric of the tumor proliferation grade and has prognostic value in diverse human malignancies such as gliomas, breast carcinomas and melanomas (Weigel and Dowsett, 2010).

Figure 4. (A) Exogenous Panx1 expression reduces cell density in cultured HeLa cells. Microphotograph showing phase contrast (upper panel) and fluorescence images (lower panel) of cultured parental HeLa cells (left) and HeLa cells stably expressing Panx1-YFP (right, green channel 530 nm). Graph showing the cell density of parental (boxes) and Panx1-expressing HeLa cells (triangles) at different days of culture. Cells were exposed to 10% fetal bovine serum and counted each 24 h for 7 days. Mean ± s.e.m. of 6 independent experiments is shown. Bar = 30 μM. (B) Panx1 expression is lower in gallbladder adenocarcinomas. Upper-left panel: Immunohistochemical Panx1 expression using a previously reported antibody (Buvinic et al., 2009) in morphologically normal gallbladder showing positivity in epithelial cells (arrows) and mononuclear inflammatory cells from the lamina propria (arrowheads). Upper-right panel: Panx1 expression in gallbladder adenocarcinoma showing lower Panx1 expression in tumor cells. Lower panels: Panx1 expression (left) and signal intensity (right) in normal gallbladders, chronic cholecystitis, and gallbladder adenocarcinomas. The amount of positive cells is expressed as percentage ± s.e.m. Intensity is expressed as arbitrary levels (1: weak; 2: moderate; 3: strong). Experimental conditions remained the same in all experiments. The number of cases in each category is indicated within the bars. Bar = 25 μm (C) Panx1 expression is inversely related with proliferation in gallbladder carcinomas. Representative microphotographs showing immunohistochemical Panx1 signal (upper panels) and Ki67 nuclear staining (lower panels, arrows) in normal gallbladder (left) and in gallbladder adenocarcinoma (right). Graph showing the Ki67 index in normal samples (black boxes) and in carcinomas (triangles). Linear regression analysis showing inverse relationship between Panx1 and Ki67 positivity in gallbladder carcinomas. R2 = 0.48; *p < 0.05 and **p < 0.01. Bar = 400 μm.
The apparent discordances between the effects of Panx1 expression in proliferation and motility of melanoma, glioma and epithelial tumor cells could be due to tumor cell-specific and/or cell lineage-specific functions of hemichannels. Distinct alterations in oncogenic pathways between melanocytic, glial and epithelial tumors are well known.
Intracellular Ca2+ is recognized as key second messenger and master regulator of cell fate. Altered cytosolic Ca2+ can be associated with cell death and cell proliferation, depending on the cell type, concentration and biological context. The intracellular Ca2+ balance is tightly regulated and active mechanisms have evolved to keep its levels in the low nanomolar range under resting conditions. The extracellular levels of Ca2+ are several orders of magnitude higher (e.g., milimolar range), creating a prominent inward concentration gradient. A complex signaling system administrates dynamic intracellular Ca2+ reservoirs within organelles such as the endoplasmic reticulum, mainly through the function of endomembranous ion channels, Ca2+ ATPases and binding to Ca2+ rich proteins (Berridge et al., 2003). Also, this system allows the functional coupling between intracellular Ca2+ stores and extracellular signals through activation of plasma membrane receptors. Such surface receptors include the dimeric tyrosine kinase growth factor-type receptors (e.g., EGFR, FGFR, PDGFR, etc.) and a wide array of seven-transmembrane domain G-protein coupled receptors (e.g., P2Y purinergic receptors, peptide hormone receptors, chemokine receptors, neurotransmitters, etc.). In addition, the influx of Ca2+ through ligand-activated channels also participates in the intracellular Ca2+ homeostasis. Intracellular Ca2+ signaling and the generation of intercellular Ca2+ waves have been involved in key cancer-related process including escape from apoptosis, perpetuation of growth, cell migration, metastasis and promotion of angiogenesis (Monteith et al., 2007; Parkash and Asotra, 2010; Monteith et al., 2012). In particular, Ca2+ increases have been shown to favor cell proliferation through altered transcription and enhance cell motility through remodeling of actin cytoskeleton and focal adhesions (Lee et al., 2011; Monteith et al., 2012). Moreover, remodeling of Ca2+ signaling is a feature of diverse cancers and consists frequently in altered expression and function of Ca2+ channels and pumps (Roderick and Cook, 2008; Monteith et al., 2012). For instance, purinergic receptors, TRP channels, store operated Ca2+ channels (e.g., ORAI and STIM) and Ca2+ pumps are deregulated in diverse human tumors (Lee et al., 2011; Chen et al., 2013). Intracellular effectors commonly involved in the pro-oncogenic effects of Ca2+ include protein kinases (e.g., PKC), phosphatases (e.g., calcineurin), transcription factors (e.g., NFAT) and Ca2+-sensitive signaling proteins (e.g., calmodulin).
As discussed, the activation of hemichannels is linked to the intracellular Ca2+ balance by mediating the autocrine/paracrine signaling through transmembrane exchange of nucleotides and Ca2+-modifying second messengers (reviewed in Wang et al., 2013a,b) (Figure 2, blue arrows and text). In addition, early observations suggested that hemichannels activated by metabolic inhibition induced massive Na+ and Ca2+ entry in cultured rabbit myocardiocytes (Li et al., 2001). In support of this notion, recent data using reconstituted systems support that hemichannels formed by Cx26 (Fiori et al., 2012), Cx32 (Sánchez et al., 2009) and Cx43 (Schalper et al., 2010) allow Ca2+ influx (reviewed in Orellana et al., 2012). Notably, increasing the [Ca2+]i with endogenous ligands (e.g., FGFs) or a Ca2+ ionophore enhances the activation of hemichannels formed by Cx32 and Cx43 (De Vuyst et al., 2006, 2009; Schalper et al., 2008a; Garré et al., 2010). Also, autocrine/paracrine signaling by additional (non-nucleotide) hemichannels permeants such as IP3, glutamate and prostaglandins involve increased intracellular Ca2+ (Schwartz and Alford, 2000; Meves, 2006; Gossman and Zhao, 2008; Traynelis et al., 2010), suggesting the possibility of hemichannels-mediated Ca2+ influx/Ca2+-increase/Ca2+ influx loops (see Figure 2). To our knowledge, Panx hemichannels have not yet been proven to be Ca2+ permeable. However, they can also be activated by increased [Ca2+]i and the permeability properties of Panx1 hemichannels suggest that they are likely to allow the passage of ions. Taken together, these data suggest that under specific pro-tumorigenic circumstances (e.g., altered signal transduction and tissue microenvironment) activated hemichannels could favor cancer progression by directly increasing intracellular Ca2+-dependent tumor cell proliferation and motility.
Cxs and Panxs are frequently altered in human tumors and cell lines. Emerging data has challenged the notion that these proteins are tumor suppressors and suggest that they could favor tumor growth and dissemination in some circumstances. Although diverse technical aspects limit the study of hemichannels, current data support their involvement in the modulation of cell proliferation and migration. The presence, regulation and characteristics of hemichannel functions in cancer cells remain largely unexplored, but their permeability to nucleotides and Ca2+ suggest a possible role in favoring tumor growth and disease progression. Future efforts to characterize the expression and specific functions of hemichannels in diverse human malignancies will support their use as prognostic/predictive markers and to design novel anti-cancer therapeutic strategies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Juan Carlos Sáez (P. Universidad Católica de Chile) for providing research supplies and technical support and Dr. Christian Naus (University of British Columbia) for critical review of the manuscript and helpful suggestions. This work was partially funded by Departamento Cientifico Docente, Clinica Alemana de Santiago, Chile; P. Universidad Católica de Chile, School of Medicine Graduate Training Grant PG-24/09 (awarded to Kurt A. Schalper); and FONDEF grant DO7I1086 (awarded to Kurt A. Schalper).
Adriaenssens, E., Vanhecke, E., Saule, P., Mougel, A., Page, A., Romon, R., et al. (2008). Nerve growth factor is a potential therapeutic target in breast cancer. Cancer Res. 68, 346–351. doi: 10.1158/0008-5472.CAN-07-1183
Ahmed, D., Lothe, R. A., Rivedal, E., and Lind, G. E. (2011). Quantitative validation of GJC1 promoter hypermethylation in benign and malignant colorectal tumors. Endocr. Relat. Cancer 18, C31–C34. doi: 10.1530/ERC-11-0204
Alajez, N. M., Lenarduzzi, M., Ito, E., Hui, A. B., Shi, W., Bruce, J., et al. (2011). MiR-218 suppresses nasopharyngeal cancer progression through downregulation of survivin and the SLIT2-ROBO1 pathway. Cancer Res. 71, 2381–2391. doi: 10.1158/0008-5472.CAN-10-2754
Antonioli, L., Pacher, P., Vizi, E. S., and Haskó, G. (2013). CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 19, 355–367. doi: 10.1016/j.molmed.2013.03.005
Arcuino, G., Lin, J. H., Takano, T., Liu, C., Jiang, L., Gao, Q., et al. (2002). Intercellular calcium signaling mediated by point-source burst release of ATP. Proc. Natl. Acad. Sci. U.S.A. 99, 9840–9845. doi: 10.1073/pnas.152588599
Azmi, A. S., Bao, B., and Sarkar, F. H. (2013). Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 32, 623–642. doi: 10.1007/s10555-013-9441-9
Bahima, L., Aleu, J., Elias, M., Martín-Satué, M., Muhaisen, A., Blasi, J., et al. (2006). Endogenous hemichannels play a role in the release of ATP from Xenopus oocytes. J. Cell. Physiol. 206, 95–102. doi: 10.1002/jcp.20440
Bao, B. A., Lai, C. P., Naus, C. C., and Morgan, J. R. (2012). Pannexin1 drives multicellular aggregate compaction via a signaling cascade that remodels the actin cytoskeleton. J. Biol. Chem. 287, 8407–8016. doi: 10.1074/jbc.M111.306522
Bao, L., Locovei, S., and Dahl, G. (2004). Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 572, 65–68. doi: 10.1016/j.febslet.2004.07.009
Baranova, A., Ivanov, D., Petrash, N., Pestova, A., Skoblov, M., Kelmanson, I., et al. (2004). The mammalian pannexin family is homologous to the invertebrate innexin gap junction proteins. Genomics 83, 706–716. doi: 10.1016/j.ygeno.2003.09.025
Baroja-Mazo, A., Barberà-Cremades, M., and Pelegrín, P. (2013). The participation of plasma membrane hemichannels to purinergic signaling. Biochim. Biophys. Acta 1828, 79–93. doi: 10.1016/j.bbamem.2012.01.002
Barr, T. P., Albrecht, P. J., Hou, Q., Mongin, A. A., Strichartz, G. R., and Rice, F. L. (2013). Air-stimulated ATP release from keratinocytes occurs through connexin hemichannels. PLoS ONE 8:e56744. doi: 10.1371/journal.pone.0056744
Belliveau, D. J., Bani-Yaghoub, M., McGirr, B., Naus, C. C., and Rushlow, W. J. (2006). Enhanced neurite outgrowth in PC12 cells mediated by connexin hemichannels and ATP. J. Biol. Chem. 281, 20920–20931. doi: 10.1074/jbc.M600026200
Berridge, M. J., Bootman, M. D., and Roderick, H. L. (2003). Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529. doi: 10.1038/nrm1155
Boassa, D., Qiu, F., Dahl, G., and Sosinsky, G. (2008). Trafficking dynamics of glycosylated pannexin 1 proteins. Cell Commun. Adhes. 15, 119–132. doi: 10.1080/15419060802013885
Bordeaux, J., Welsh, A., Agarwal, S., Killiam, E., Baquero, M., Hanna, J., et al. (2010). Antibody validation. Biotechniques 48, 197–209. doi: 10.2144/000113382
Braet, K., Aspeslagh, S., Vandamme, W., Willecke, K., Martin, P. E., Evans, W. H., et al. (2003a). Pharmacological sensitivity of ATP release triggered by photoliberation of inositol-1,4,5-trisphosphate and zero extracellular calcium in brain endothelial cells. J. Cell. Physiol. 197, 205–213. doi: 10.1002/jcp.10365
Braet, K., Vandamme, W., Martin, P. E., Evans, W. H., and Leybaert, L. (2003b). Photoliberating inositol-1,4,5-trisphosphate triggers ATP release that is blocked by the connexin mimetic peptide gap 26. Cell Calcium 33, 37–48. doi: 10.1016/S0143-4160(02)00180-X
Brisset, A. C., Isakson, B. E., and Kwak, B. R. (2009). Connexins in vascular physiology and pathology. Antioxid. Redox Signal. 11, 267–282. doi: 10.1089/ars.2008.2115
Bruzzone, R., Hormuzdi, S. G., Barbe, M. T., Herb, A., and Monyer, H. (2003). Pannexins, a family of gap junction proteins expressed in brain. Proc. Natl. Acad. Sci. U.S.A. 100, 13644–13649. doi: 10.1073/pnas.2233464100
Bruzzone, S., Guida, L., Zocchi, E., Franco, L., and de Flora, A. (2001). Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 15, 10–12. doi: 10.1096/fj.00-0566fje
Bui, M. M., Han, G., Acs, G., Reed, D., Gonzalez, R. J., Pasha, T. L., et al. (2011). Connexin 43 is a potential prognostic biomarker for ewing sarcoma/primitive neuroectodermal tumor. Sarcoma 2011:971050. doi: 10.1155/2011/971050
Burnstock, G., and Di Virgilio, F. (2013). Purinergic signalling and cancer. Purinergic Signal. 9, 491–540. doi: 10.1007/s11302-013-9372-5
Burt, J. M., Nelson, T. K., Simon, A. M., and Fang, J. S. (2008). Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am. J. Physiol. Cell Physiol. 295, C1103–C1112. doi: 10.1152/ajpcell.299.2008
Buvinic, S., Almarza, G., Bustamante, M., Casas, M., López, J., Riquelme, M., et al. (2009). ATP released by electrical stimuli elicits calcium transients and gene expression in skeletal muscle. J. Biol. Chem. 284, 34490–34505. doi: 10.1074/jbc.M109.057315
Carette, D., Gilleron, J., Chevallier, D., Segretain, D., and Pointis, G. (2014). Connexin a check-point component of cell apoptosis in normal and physiopathological conditions. Biochimie 101, 1–9. doi: 10.1016/j.biochi.2013.11.015
Celetti, S. J., Cowan, K. N., Penuela, S., Shao, Q., Churko, J., and Laird, D. W. (2010). Implications of pannexin 1 and pannexin 3 for keratinocyte differentiation. J. Cell Sci. 123(Pt 8), 1363–1372. doi: 10.1242/jcs.056093
Chekeni, F. B., Elliott, M. R., Sandilos, J. K., Walk, S. F., Kinchen, J. M., Lazarowski, E. R., et al. (2010). Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature 467, 863–867. doi: 10.1038/nature09413
Chen, J. T., Cheng, Y. W., Chou, M. C., Sen-Lin, T., Lai, W. W., Ho, W. L., et al. (2003). The correlation between aberrant connexin 43 mRNA expression induced by promoter methylation and nodal micrometastasis in non-small cell lung cancer. Clin. Cancer Res. 9, 4200–4204.
Chen, Y., Hühn, D., Knösel, T., Pacyna-Gengelbach, M., Deutschmann, N., and Petersen, I. (2005). Downregulation of connexin 26 in human lung cancer is related to promoter methylation. Int. J. Cancer 113, 14–21. doi: 10.1002/ijc.20498
Chen, Y. F., Chen, Y. T., Chiu, W. T., and Shen, M. R. (2013). Remodeling of calcium signaling in tumor progression. J. Biomed. Sci. 20:23. doi: 10.1186/1423-0127-20-23
Chiarugi, A., Dölle, C., Felici, R., and Ziegler, M. (2012). The NAD metabolome–a key determinant of cancer cell biology. Nat. Rev. Cancer 12, 741–752. doi: 10.1038/nrc3340
Clarke, T. C., Williams, O. J., Martin, P. E., and Evans, W. H. (2009). ATP release by cardiac myocytes in a simulated ischaemia model: inhibition by a connexin mimetic and enhancement by an antiarrhythmic peptide. Eur. J. Pharmacol. 605, 9–14. doi: 10.1016/j.ejphar.2008.12.005
Cone, A. C., Ambrosi, C., Scemes, E., Martone, M. E., and Sosinsky, G. E. (2013). A comparative antibody analysis of pannexin1 expression in four rat brain regions reveals varying subcellular localizations. Front. Pharmacol. 4:6. doi: 10.3389/fphar.2013.00006
Contreras, J. E., Sánchez, H. A., Véliz, L. P., Bukauskas, F. F., Bennett, M. V., and Sáez, J. C. (2004). Role of connexin-based gap junction channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res. Brain Res. Rev. 47, 290–303. doi: 10.1016/j.brainresrev.2004.08.002
Coppen, S. R., Dupont, E., and Severs, N. J. (2002). Re: the sinoatrial node, connexin distribution patterns and specific immunodetection of connexin45. Cardiovasc. Res. 53, 1043–1045; author reply 1046. doi: 10.1016/S0008-6363(01)00564-8
Cotrina, M. L., Lin, J. H., Alves-Rodrigues, A., Liu, S., Li, J., Azmi-Ghadimi, H., et al. (1998). Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. U.S.A. 95, 15735–15740. doi: 10.1073/pnas.95.26.15735
Cronier, L., Crespin, S., Strale, P. O., Defamie, N., and Mesnil, M. (2009). Gap junctions and cancer: new functions for an old story. Antioxid. Redox Signal. 11, 323–338. doi: 10.1089/ars.2008.2153
Deaglio, S., Aydin, S., Vaisitti, T., Bergui, L., and Malavasi, F. (2008). CD38 at the junction between prognostic marker and therapeutic target. Trends Mol. Med. 14, 210–218. doi: 10.1016/j.molmed.2008.02.005
Decrock, E., Vinken, M., Bol, M., D'Herde, K., Rogiers, V., Vandenabeele, P., et al. (2011). Calcium and connexin-based intercellular communication, a deadly catch? Cell Calcium 50, 310–321. doi: 10.1016/j.ceca.2011.05.007
De Flora, A., Zocchi, E., Guida, L., Franco, L., and Bruzzone, S. (2004). Autocrine and paracrine calcium signaling by the CD38/NAD+/cyclic ADP-ribose system. Ann. N.Y. Acad. Sci. 1028, 176–191. doi: 10.1196/annals.1322.021
De Vuyst, E., Decrock, E., Cabooter, L., Dubyak, G. R., Naus, C. C., Evans, W. H., et al. (2006). Intracellular calcium changes trigger connexin 32 hemichannel opening. EMBO J. 25, 34–44. doi: 10.1038/sj.emboj.7600908
De Vuyst, E., Decrock, E., De Bock, M., Yamasaki, H., Naus, C. C., Evans, W. H., et al. (2007). Connexin hemichannels and gap junction channels are differentially influenced by lipopolysaccharide and basic fibroblast growth factor. Mol. Biol. Cell. 18, 34–46. doi: 10.1091/mbc.E06-03-0182
De Vuyst, E., Wang, N., Decrock, E., De Bock, M., Vinken, M., Van Moorhem, M., et al. (2009). Ca(2+) regulation of connexin 43 hemichannels in C6 glioma and glial cells. Cell Calcium 46, 176–187. doi: 10.1016/j.ceca.2009.07.002
D'Hondt, C., Iyyathurai, J., Vinken, M., Rogiers, V., Leybaert, L., Himpens, B., et al. (2013). Regulation of connexin- and pannexin-based channels by post-translational modifications. Biol. Cell 105, 373–398. doi: 10.1111/boc.201200096
Dieci, M. V., Arnedos, M., Andre, F., and Soria, J. C. (2013). Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov. 3, 264–279. doi: 10.1158/2159-8290.CD-12-0362
Dienstmann, R., Rodon, J., Prat, A., Perez-Garcia, J., Adamo, B., Felip, E., et al. (2014). Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann. Oncol. 25, 552–563. doi: 10.1093/annonc/mdt419
Di Virgilio, F. (2012). Purines, purinergic receptors, and cancer. Cancer Res. 72, 5441–5447. doi: 10.1158/0008-5472.CAN-12-1600
Dollé, L., Adriaenssens, E., El Yazidi-Belkoura, I., Le Bourhis, X., Nurcombe, V., and Hondermarck, H. (2004). Nerve growth factor receptors and signaling in breast cancer. Curr. Cancer Drug Targets 4, 463–470. doi: 10.2174/1568009043332853
Dolmatova, E., Spagnol, G., Boassa, D., Baum, J. R., Keith, K., Ambrosi, C., et al. (2012). Cardiomyocyte ATP release through pannexin 1 aids in early fibroblast activation. Am. J. Physiol. Heart Circ. Physiol. 303, H1208–H1218. doi: 10.1152/ajpheart.00251.2012
Du, G., Yang, Y., Zhang, Y., Sun, T., Liu, W., Wang, Y., et al. (2013). Thrombocytosis and immunohistochemical expression of connexin 43 at diagnosis predict survival in advanced non-small-cell lung cancer treated with cisplatin-based chemotherapy. Cancer Chemother. Pharmacol. 71, 893–904. doi: 10.1007/s00280-013-2080-6
Dubina, M. V., Iatckii, N. A., Popov, D. E., Vasil'ev, S. V., and Krutovskikh, V. A. (2002). Connexin 43, but not connexin 32, is mutated at advanced stages of human sporadic colon cancer. Oncogene 21, 4992–4996. doi: 10.1038/sj.onc.1205630
Eltzschig, H. K., Eckle, T., Mager, A., Küper, N., Karcher, C., Weissmüller, T., et al. (2006). ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 99, 1100–1108. doi: 10.1161/01.RES.0000250174.31269.70
Essenfelder, G. M., Bruzzone, R., Lamartine, J., Charollais, A., Blanchet-Bardon, C., Barbe, M. T., et al. (2004). Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum. Mol. Genet. 13, 1703–1714. doi: 10.1093/hmg/ddh191
Evans, W. H., De Vuyst, E., and Leybaert, L. (2006). The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J. 397, 1–14. doi: 10.1042/BJ20060175
Faigle, M., Seessle, J., Zug, S., El Kasmi, K. C., and Eltzschig, H. K. (2008). ATP release from vascular endothelia occurs across Cx43 hemichannels and is attenuated during hypoxia. PLoS ONE 3:e2801. doi: 10.1371/journal.pone.0002801
Falzoni, S., Donvito, G., and Di Virgilio, F. (2013). Detecting adenosine triphosphate in the pericellular space. Interface Focus 3:20120101. doi: 10.1098/rsfs.2012.0101
Fiori, M. C., Figueroa, V., Zoghbi, M. E., Saéz, J. C., Reuss, L., and Altenberg, G. A. (2012). Permeation of calcium through purified connexin 26 hemichannels. J. Biol. Chem. 287, 40826–40834. doi: 10.1074/jbc.M112.383281
Franco, L., Zocchi, E., Usai, C., Guida, L., Bruzzone, S., Costa, A., et al. (2001). Paracrine roles of NAD+ and cyclic ADP-ribose in increasing intracellular calcium and enhancing cell proliferation of 3T3 fibroblasts. J. Biol. Chem. 276, 21642–21648. doi: 10.1074/jbc.M010536200
Gaietta, G., Deerinck, T. J., Adams, S. R., Bouwer, J., Tour, O., Laird, D. W., et al. (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296, 503–507. doi: 10.1126/science.1068793
Garré, J. M., Retamal, M. A., Cassina, P., Barbeito, L., Bukauskas, F. F., Sáez, J. C., et al. (2010). FGF-1 induces ATP release from spinal astrocytes in culture and opens pannexin and connexin hemichannels. Proc. Natl. Acad. Sci. U.S.A. 107, 22659–22664. doi: 10.1073/pnas.1013793107
Ghiringhelli, F., Bruchard, M., Chalmin, F., and Rébé, C. (2012). Production of adenosine by ectonucleotidases: a key factor in tumor immunoescape. J. Biomed. Biotechnol. 2012:473712. doi: 10.1155/2012/473712
Ghosh, S., Kumar, A., Tripathi, R. P., and Chandna, S. (2014). Connexin-43 regulates p38-mediated cell migration and invasion induced selectively in tumour cells by low doses of γ-radiation in an ERK-1/2-independent manner. Carcinogenesis 35, 383–395. doi: 10.1093/carcin/bgt303
Giaume, C., and Theis, M. (2010). Pharmacological and genetic approaches to study connexin-mediated channels in glial cells of the central nervous system. Brain Res. Rev. 63, 160–176. doi: 10.1016/j.brainresrev.2009.11.005
Gomes, P., Srinivas, S. P., Van Driessche, W., Vereecke, J., and Himpens, B. (2005). ATP release through connexin hemichannels in corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 46, 1208–1218. doi: 10.1167/iovs.04-1181
Good, M. E., Ek-Vitorín, J. F., and Burt, J. M. (2012). Extracellular loop cysteine mutant of cx37 fails to suppress proliferation of rat insulinoma cells. J. Membr. Biol. 245, 369–380. doi: 10.1007/s00232-012-9459-x
Good, M. E., Nelson, T. K., Simon, A. M., and Burt, J. M. (2011). A functional channel is necessary for growth suppression by Cx37. J. Cell Sci. 124(Pt 14), 2448–2456. doi: 10.1242/jcs.081695
Goodenough, D. A., and Paul, D. L. (2003). Beyond the gap: functions of unpaired connexon channels. Nat. Rev. Mol. Cell Biol. 4, 285–294. doi: 10.1038/nrm1072
Gossman, D. G., and Zhao, H. B. (2008). Hemichannel-mediated inositol 1,4,5-trisphosphate (IP3) release in the cochlea: a novel mechanism of IP3 intercellular signaling. Cell Commun. Adhes. 15, 305–315. doi: 10.1080/15419060802357217
Hao, J., Zhang, C., Zhang, A., Wang, K., Jia, Z., Wang, G., et al. (2012). miR-221/222 is the regulator of Cx43 expression in human glioblastoma cells. Oncol. Rep. 27, 1504–1510. doi: 10.3892/or.2012.1652
Hauptman, N., and Glavac, D. (2013). MicroRNAs and long non-coding RNAs: prospects in diagnostics and therapy of cancer. Radiol. Oncol. 47, 311–318. doi: 10.2478/raon-2013-0062
Hernandez, M., Shao, Q., Yang, X. J., Luh, S. P., Kandouz, M., Batist, G., et al. (2006). A histone deacetylation-dependent mechanism for transcriptional repression of the gap junction gene cx43 in prostate cancer cells. Prostate 66, 1151–1161. doi: 10.1002/pros.20451
Hirai, A., Yano, T., Nishikawa, K., Suzuki, K., Asano, R., Satoh, H., et al. (2003). Down-regulation of connexin 32 gene expression through DNA methylation in a human renal cell carcinoma cell. Am. J. Nephrol. 23, 172–177. doi: 10.1159/000070653
Huang, C., Han, X., Li, X., Lam, E., Peng, W., Lou, N., et al. (2012). Critical role of connexin 43 in secondary expansion of traumatic spinal cord injury. J. Neurosci. 32, 3333–3338. doi: 10.1523/JNEUROSCI.1216-11.2012
Huang, Y. J., Maruyama, Y., Dvoryanchikov, G., Pereira, E., Chaudhari, N., and Roper, S. D. (2007). The role of pannexin 1 hemichannels in ATP release and cell-cell communication in mouse taste buds. Proc. Natl. Acad. Sci. U.S A. 104, 6436–6441. doi: 10.1073/pnas.0611280104
Hubert, S., Rissiek, B., Klages, K., Huehn, J., Sparwasser, T., Haag, F., et al. (2010). Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J. Exp. Med. 207, 2561–2568. doi: 10.1084/jem.20091154
Hwang, I. (2013). Cell-cell communication via extracellular membrane vesicles and its role in the immune response. Mol. Cells 36, 105–111. doi: 10.1007/s10059-013-0154-2
Iglesias, R., Dahl, G., Qiu, F., Spray, D. C., Scemes, E., Iglesias, et al. (2009). Pannexin 1: the molecular substrate of astrocyte hemichannels. J. Neurosci. 29, 7092–7097. doi: 10.1523/JNEUROSCI.6062-08.2009
Iglesias, R. M., and Spray, D. C. (2012). Pannexin1-mediated ATP release provides signal transmission between Neuro2A cells. Neurochem. Res. 37, 1355–1363. doi: 10.1007/s11064-012-0720-6
Jinn, Y., and Inase, N. (2010). Connexin 43, E-cadherin, beta-catenin and ZO-1 expression, and aberrant methylation of the connexin 43 gene in NSCLC. Anticancer Res. 30, 2271–2278.
Jing, Y. M., Guo, S. X., Zhang, X. P., Sun, A. J., Tao, F., and Qian, H. X. (2012). Association between C1019T polymorphism in the connexin 37 gene and Helicobacter pylori infection in patients with gastric cancer. Asian Pac. J. Cancer Prev. 13, 2363–2367. doi: 10.7314/APJCP.2012.13.5.2363
Kalani, A., Tyagi, A., and Tyagi, N. (2014). Exosomes: mediators of neurodegeneration, neuroprotection and therapeutics. Mol. Neurobiol. 49, 590–600. doi: 10.1007/s12035-013-8544-1
Kanczuga-Koda, L., Sulkowski, S., Lenczewski, A., Koda, M., Wincewicz, A., Baltaziak, M., et al. (2006). Increased expression of connexins 26 and 43 in lymph node metastases of breast cancer. J. Clin. Pathol. 59, 429–433. doi: 10.1136/jcp.2005.029272
Kandouz, M., and Batist, G. (2010). Gap junctions and connexins as therapeutic targets in cancer. Expert Opin. Ther. Targets 14, 681–692. doi: 10.1517/14728222.2010.487866
Kang, J., Kang, N., Lovatt, D., Torres, A., Zhao, Z., Lin, J., et al. (2008). Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 28, 4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008
Kanno, Y. (1985). Modulation of cell communication and carcinogenesis. Jpn. J. Physiol. 35, 693–707. doi: 10.2170/jjphysiol.35.693
Kar, R., Batra, N., Riquelme, M. A., and Jiang, J. X. (2012). Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 524, 2–15. doi: 10.1016/j.abb.2012.03.008
Krenacs, T., van Dartel, M., Lindhout, E., and Rosendaal, M. (1997). Direct cell/cell communication in the lymphoid germinal center: connexin43 gap junctions functionally couple follicular dendritic cells to each other and to B lymphocytes. Eur. J. Immunol. 27, 1489–1497. doi: 10.1002/eji.1830270627
Kyo, N., Yamamoto, H., Takeda, Y., Ezumi, K., Ngan, C. Y., Terayama, M., et al. (2008). Overexpression of connexin 26 in carcinoma of the pancreas. Oncol. Rep. 19, 627–631. doi: 10.3892/or.19.3.627
Lai, C. P., Bechberger, J. F., and Naus, C. C. (2009). Pannexin2 as a novel growth regulator in C6 glioma cells. Oncogene 28, 4402–4408. doi: 10.1038/onc.2009.283
Lai, C. P., Bechberger, J. F., Thompson, R. J., MacVicar, B. A., Bruzzone, R., and Naus, C. C. (2007). Tumor-suppressive effects of pannexin 1 in C6 glioma cells. Cancer Res. 67, 1545–1554. doi: 10.1158/0008-5472.CAN-06-1396
Lamiche, C., Clarhaut, J., Strale, P. O., Crespin, S., Pedretti, N., Bernard, F. X., et al. (2012). The gap junction protein Cx43 is involved in the bone-targeted metastatic behaviour of human prostate cancer cells. Clin. Exp. Metastas. 29, 111–122. doi: 10.1007/s10585-011-9434-4
Lee, J. M., Davis, F. M., Roberts-Thomson, S. J., and Monteith, G. R. (2011). Ion channels and transporters in cancer. 4. Remodeling of Ca(2+) signaling in tumorigenesis: role of Ca(2+) transport. Am. J. Physiol. Cell Physiol. 301, C969–C976. doi: 10.1152/ajpcell.00136.2011
Lee, S. W., Tomasetto, C., and Sager, R. (1991). Positive selection of candidate tumor-suppressor genes by subtractive hybridization. Proc. Natl. Acad. Sci. U.S.A. 88, 2825–2829. doi: 10.1073/pnas.88.7.2825
Li, A., Leung, C. T., Peterson-Yantorno, K., Stamer, W. D., Mitchell, C. H., and Civan, M. M. (2012a). Mechanisms of ATP release by human trabecular meshwork cells, the enabling step in purinergic regulation of aqueous humor outflow. J. Cell Physiol. 227, 172–182. doi: 10.1002/jcp.22715
Li, F., Sugishita, K., Su, Z., Ueda, I., and Barry, W. H. (2001). Activation of connexin-43 hemichannels can elevate [Ca(2+)]i and [Na(+)]i in rabbit ventricular myocytes during metabolic inhibition. J. Mol. Cell Cardiol. 33, 2145–2155. doi: 10.1006/jmcc.2001.1477
Li, X., Pan, J. H., Song, B., Xiong, E. Q., Chen, Z. W., Zhou, Z. S., et al. (2012b). Suppression of CX43 expression by miR-20a in the progression of human prostate cancer. Cancer Biol. Ther. 13, 890–898. doi: 10.4161/cbt.20841
Li, X., Su, Y., Pan, J., Zhou, Z., Song, B., Xiong, E., et al. (2013). Connexin 26 is down-regulated by KDM5B in the progression of bladder cancer. Int. J. Mol. Sci. 14, 7866–7879. doi: 10.3390/ijms14047866
Liu, W., Chang, B. L., Cramer, S., Koty, P. P., Li, T., Sun, J., et al. (2007). Deletion of a small consensus region at 6q15, including the MAP3K7 gene, is significantly associated with high-grade prostate cancers. Clin. Cancer Res. 13, 5028–5033. doi: 10.1158/1078-0432.CCR-07-0300
Locovei, S., Bao, L., and Dahl, G. (2006). Pannexin 1 in erythrocytes: function without a gap. Proc. Natl. Acad. Sci. U.S.A. 103, 7655–7659. doi: 10.1073/pnas.0601037103
Loewenstein, W. R. (1980). Junctional cell-to-cell communication and growth control. Ann. N.Y. Acad. Sci. 339, 39–45. doi: 10.1111/j.1749-6632.1980.tb15966.x
Loewenstein, W. R., and Kanno, Y. (1966). Intercellular communication and the control of tissue growth: lack of communication between cancer cells. Nature 209, 1248–1249. doi: 10.1038/2091248a0
Loewenstein, W. R., and Kanno, Y. (1967). Intercellular communication and tissue growth. I. Cancerous growth. J. Cell Biol. 33, 225–234. doi: 10.1083/jcb.33.2.225
Lohman, A. W., Weaver, J. L., Billaud, M., Sandilos, J. K., Griffiths, R., Straub, A. C., et al. (2012). S-nitrosylation inhibits pannexin 1 channel function. J. Biol. Chem. 287, 39602–39612. doi: 10.1074/jbc.M112.397976
Loncarek, J., Yamasaki, H., Levillain, P., Milinkevitch, S., and Mesnil, M. (2003). The expression of the tumor suppressor gene connexin 26 is not mediated by methylation in human esophageal cancer cells. Mol. Carcinog. 36, 74–81. doi: 10.1002/mc.10102
Lu, D., Soleymani, S., Madakshire, R., and Insel, P. A. (2012). ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. FASEB J. 26, 2580–2591. doi: 10.1096/fj.12-204677
Machtaler, S., Dang-Lawson, M., Choi, K., Jang, C., Naus, C. C., and Matsuuchi, L. (2011). The gap junction protein Cx43 regulates B-lymphocyte spreading and adhesion. J. Cell Sci. 124(Pt 15), 2611–2621. doi: 10.1242/jcs.089532
Mese, G., Sellitto, C., Li, L., Wang, H. Z., Valiunas, V., Richard, G., et al. (2011). The Cx26-G45E mutation displays increased hemichannel activity in a mouse model of the lethal form of keratitis-ichthyosis-deafness syndrome. Mol. Biol. Cell. 22, 4776–4786. doi: 10.1091/mbc.E11-09-0778
Mesnil, M., Crespin, S., Avanzo, J. L., and Zaidan-Dagli, M. L. (2005). Defective gap junctional intercellular communication in the carcinogenic process. Biochim. Biophys. Acta 1719, 125–145. doi: 10.1016/j.bbamem.2005.11.004
Mesnil, M., Krutovskikh, V., Omori, Y., and Yamasaki, H. (1995). Role of blocked gap junctional intercellular communication in non-genotoxic carcinogenesis. Toxicol. Lett. 82–83, 701–706. doi: 10.1016/0378-4274(95)03588-5
Mesnil, M., and Yamasaki, H. (1993). Cell-cell communication and growth control of normal and cancer cells: evidence and hypothesis. Mol. Carcinog. 7, 14–17. doi: 10.1002/mc.2940070103
Meves, H. (2006). The action of prostaglandins on ion channels. Curr. Neuropharmacol. 4, 41–57. doi: 10.2174/157015906775203048
Molloy, N. H., Read, D. E., and Gorman, A. M. (2011). Nerve growth factor in cancer cell death and survival. Cancers 3, 510–530. doi: 10.3390/cancers3010510
Montecino-Rodriguez, E., and Dorshkind, K. (2001). Regulation of hematopoiesis by gap junction-mediated intercellular communication. J. Leukoc. Biol. 70, 341–347.
Monteith, G. R., Davis, F. M., and Roberts-Thomson, S. J. (2012). Calcium channels and pumps in cancer: changes and consequences. J. Biol. Chem. 287, 31666–31673. doi: 10.1074/jbc.R112.343061
Monteith, G. R., McAndrew, D., Faddy, H. M., and Roberts-Thomson, S. J. (2007). Calcium and cancer: targeting Ca2+ transport. Nat. Rev. Cancer 7, 519–530. doi: 10.1038/nrc2171
Murata, Y., Yasuo, T., Yoshida, R., Obata, K., Yanagawa, Y., Margolskee, R. F., et al. (2010). Action potential-enhanced ATP release from taste cells through hemichannels. J. Neurophysiol. 104, 896–901. doi: 10.1152/jn.00414.2010
Naoi, Y., Miyoshi, Y., Taguchi, T., Kim, S. J., Arai, T., Tamaki, Y., et al. (2007). Connexin26 expression is associated with lymphatic vessel invasion and poor prognosis in human breast cancer. Breast Cancer Res. Treat. 106, 11–17. doi: 10.1007/s10549-006-9465-8
Naus, C. C. (2002). Gap junctions and tumour progression. Can. J. Physiol. Pharmacol. 80, 136–141. doi: 10.1139/y02-009
Naus, C. C., and Laird, D. W. (2010). Implications and challenges of connexin connections to cancer. Nat. Rev. Cancer 10, 435–441. doi: 10.1038/nrc2841
Noh, S. J., Bae, J. S., Jamiyandorj, U., Park, H. S., Kwon, K. S., Jung, S. H., et al. (2013). Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer 13:516. doi: 10.1186/1471-2407-13-516
Nualart-Marti, A., del Molino, E. M., Grandes, X., Bahima, L., Martin-Satué, M., Puchal, R., et al. (2013). Role of connexin 32 hemichannels in the release of ATP from peripheral nerves. Glia 61, 1976–1989. doi: 10.1002/glia.22568
Ogawa, T., Hayashi, T., Tokunou, M., Nakachi, K., Trosko, J. E., Chang, C. C., et al. (2005). Suberoylanilide hydroxamic acid enhances gap junctional intercellular communication via acetylation of histone containing connexin 43 gene locus. Cancer Res. 65, 9771–9778. doi: 10.1158/0008-5472.CAN-05-0227
Ohshima, Y., Tsukimoto, M., Harada, H., and Kojima, S. (2012). Involvement of connexin43 hemichannel in ATP release after γ-irradiation. J. Radiat. Res. 53, 551–557. doi: 10.1093/jrr/rrs014
Orellana, J. A., Martinez, A. D., and Retamal, M. A. (2013). Gap junction channels and hemichannels in the CNS: regulation by signaling molecules. Neuropharmacology 75, 567–582. doi: 10.1016/j.neuropharm.2013.02.020
Orellana, J. A., Sánchez, H. A., Schalper, K. A., Figueroa, V., and Sáez, J. C. (2012). Regulation of intercellular calcium signaling through calcium interactions with connexin-based channels. Adv. Exp. Med. Biol. 740, 777–794. doi: 10.1007/978-94-007-2888-2_34
Orellana, J. A., Shoji, K. F., Abudara, V., Ezan, P., Amigou, E., Sáez, P. J., et al. (2011). Amyloid β-induced death in neurons involves glial and neuronal hemichannels. J. Neurosci. 31, 4962–4977. doi: 10.1523/JNEUROSCI.6417-10.2011
Oviedo-Orta, E., Gasque, P., and Evans, W. H. (2001). Immunoglobulin and cytokine expression in mixed lymphocyte cultures is reduced by disruption of gap junction intercellular communication. FASEB J. 15, 768–774. doi: 10.1096/fj.00-0288com
Oviedo-Orta, E., Hoy, T., and Evans, W. H. (2000). Intercellular communication in the immune system: differential expression of connexin40 and 43, and perturbation of gap junction channel functions in peripheral blood and tonsil human lymphocyte subpopulations. Immunology 99, 578–590. doi: 10.1046/j.1365-2567.2000.00991.x
Oyamada, M., Takebe, K., and Oyamada, Y. (2013). Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta 1828, 118–133. doi: 10.1016/j.bbamem.2011.12.031
Panchin, Y. V. (2005). Evolution of gap junction proteins–the pannexin alternative. J. Exp. Biol. 208(Pt 8), 1415–1419. doi: 10.1242/jeb.01547
Parkash, J., and Asotra, K. (2010). Calcium wave signaling in cancer cells. Life Sci. 87, 587–595. doi: 10.1016/j.lfs.2010.09.013
Pearson, R. A., Dale, N., Llaudet, E., and Mobbs, P. (2005). ATP released via gap junction hemichannels from the pigment epithelium regulates neural retinal progenitor proliferation. Neuron 46, 731–744. doi: 10.1016/j.neuron.2005.04.024
Pelegrin, P., and Surprenant, A. (2006). Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 25, 5071–5082. doi: 10.1038/sj.emboj.7601378
Peñuela, S., Gehi, R., and Laird, D. W. (2013). The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 1828, 15–22. doi: 10.1016/j.bbamem.2012.01.017
Peñuela, S., Gyenis, L., Ablack, A., Churko, J. M., Berger, A. C., Litchfield, D. W., et al. (2012). Loss of pannexin 1 attenuates melanoma progression by reversion to a melanocytic phenotype. J. Biol. Chem. 287, 29184–29193. doi: 10.1074/jbc.M112.377176
Pfenniger, A., Wohlwend, A., and Kwak, B. R. (2011). Mutations in connexin genes and disease. Eur. J. Clin. Invest. 41, 103–116. doi: 10.1111/j.1365-2362.2010.02378.x
Pinheiro, A. R., Paramos-de-Carvalho, D., Certal, M., Costa, M. A., Costa, C., Magalhães-Cardoso, M. T., et al. (2013). Histamine induces ATP release from human subcutaneous fibroblasts, via pannexin-1 hemichannels, leading to Ca2+ mobilization and cell proliferation. J. Biol. Chem. 288, 27571–27583. doi: 10.1074/jbc.M113.460865
Plotkin, L. I., Manolagas, S. C., and Bellido, T. (2002). Transduction of cell survival signals by connexin-43 hemichannels. J. Biol. Chem. 277, 8648–8657. doi: 10.1074/jbc.M108625200
Qiu, F., and Dahl, G. (2009). A permeant regulating its permeation pore: inhibition of pannexin 1 channels by ATP. Am. J. Physiol. Cell Physiol. 296, C250–C255. doi: 10.1152/ajpcell.00433.2008
Quezada, S. A., and Peggs, K. S. (2013). Exploiting CTLA-4, PD-1 and PD-L1 to reactivate the host immune response against cancer. Br. J. Cancer 108, 1560–1565. doi: 10.1038/bjc.2013.117
Ransford, G. A., Fregien, N., Qiu, F., Dahl, G., Conner, G. E., and Salathe, M. (2009). Pannexin 1 contributes to ATP release in airway epithelia. Am. J. Respir. Cell Mol. Biol. 41, 525–534. doi: 10.1165/rcmb.2008-0367OC
Rednam, S., Hicks, J., Levy, M. L., and Pappo, A. S. (2011). Metastatic squamous cell carcinoma of the oropharynx in a child with a mutation in the Connexin 26 gene. J. Pediatr. Hematol. Oncol. 33, 387–389. doi: 10.1097/MPH.0b013e3181e65c1c
Reigada, D., Lu, W., Zhang, M., and Mitchell, C. H. (2008). Elevated pressure triggers a physiological release of ATP from the retina: possible role for pannexin hemichannels. Neuroscience 157, 396–404. doi: 10.1016/j.neuroscience.2008.08.036
Retamal, M. A., Froger, N., Palacios-Prado, N., Ezan, P., Sáez, P. J., Sáez, J. C., et al. (2007a). Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J. Neurosci. 27, 13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007
Retamal, M. A., Schalper, K. A., Shoji, K. F., Orellana, J. A., Bennett, M. V., and Sáez, JC. (2007b). Possible involvement of different connexin43 domains in plasma membrane permeabilization induced by ischemia-reperfusion. J. Membr. Biol. 218, 49–63. doi: 10.1007/s00232-007-9043-y
Richter, K., Kiefer, K. P., Grzesik, B. A., Clauss, W. G., and Fronius, M. (2014). Hydrostatic pressure activates ATP-sensitive K+ channels in lung epithelium by ATP release through pannexin and connexin hemichannels. FASEB J. 28, 45–55. doi: 10.1096/fj.13-229252
Riquelme, M. A., Cea, L. A., Vega, J. L., Boric, M. P., Monyer, H., Bennett, M. V., et al. (2013b). The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology 75, 594–603. doi: 10.1016/j.neuropharm.2013.03.022
Riquelme, M. A., Kar, R., Gu, S., and Jiang, J. X. (2013a). Antibodies targeting extracellular domain of connexins for studies of hemichannels. Neuropharmacology 75, 525–532. doi: 10.1016/j.neuropharm.2013.02.021
Roderick, H. L., and Cook, S. J. (2008). Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 8, 361–375. doi: 10.1038/nrc2374
Roger, S., and Pelegrin, P. (2011). P2X7 receptor antagonism in the treatment of cancers. Expert Opin. Invest. Drugs 20, 875–880. doi: 10.1517/13543784.2011.583918
Sáez, J. C., Berthoud, V. M., Brañes, M. C., Martinez, A. D., and Beyer, E. C. (2003). Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83, 1359–1400. doi: 10.1152/physrev.00007.2003
Sáez, J. C., Schalper, K. A., Retamal, M. A., Orellana, J. A., Shoji, K. F., and Bennett, M. V. (2010). Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp. Cell Res. 316, 2377–2389. doi: 10.1016/j.yexcr.2010.05.026
Sahu, G., Sukumaran, S., and Bera, A. K. (2014). Pannexins form gap junctions with electrophysiological and pharmacological properties distinct from connexins. Sci. Rep. 4:4955. doi: 10.1038/srep04955
Saito, T., Krutovskikh, V., Marion, M. J., Ishak, K. G., Bennett, W. P., and Yamasaki, H. (2000). Human hemangiosarcomas have a common polymorphism but no mutations in the connexin37 gene. Int. J. Cancer 86, 67–70. doi: 10.1002/(SICI)1097-0215(20000401)86:1%3C67::AID-IJC10%3E3.0.CO;2-1
Sánchez, H. A., Mese, G., Srinivas, M., White, T. W., and Verselis, V. K. (2010). Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J. Gen. Physiol. 136, 47–62. doi: 10.1085/jgp.201010433
Sánchez, H. A., Orellana, J. A., Verselis, V. K., and Sáez, J. C. (2009). Metabolic inhibition increases activity of connexin-32 hemichannels permeable to Ca2+ in transfected HeLa cells. Am. J. Physiol. Cell Physiol. 297, C665–C678. doi: 10.1152/ajpcell.00200.2009
Sandilos, J. K., Chiu, Y. H., Chekeni, F. B., Armstrong, A. J., Walk, S. F., Ravichandran, K. S., et al. (2012). Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J. Biol. Chem. 287, 11303–11311. doi: 10.1074/jbc.M111.323378
Schalper, K. A., Orellana, J. A., Berthoud, V. M., and Sáez, J. C. (2009). Dysfunctions of the diffusional membrane pathways mediated by hemichannels in inherited and acquired human diseases. Curr. Vasc. Pharmacol. 7, 486–505. doi: 10.2174/157016109789043937
Schalper, K. A., Palacios-Prado, N., Orellana, J. A., and Sáez, J. C. (2008b). Currently used methods for identification and characterization of hemichannels. Cell Commun. Adhes. 15, 207–218. doi: 10.1080/15419060802014198
Schalper, K. A., Palacios-Prado, N., Retamal, M. A., Shoji, K. F., Martínez, A. D., and Sáez, J. C. (2008a). Connexin hemichannel composition determines the FGF-1-induced membrane permeability and free [Ca2+]i responses. Mol. Biol. Cell 19, 3501–3513. doi: 10.1091/mbc.E07-12-1240
Schalper, K. A., Riquelme, M. A., Brañes, M. C., Martínez, A. D., Vega, J. L., Berthoud, V. M., et al. (2014). Modulation of gap junction channels and hemichannels by growth factors. Mol. Biosyst. 8, 685–698. doi: 10.1039/c1mb05294b
Schalper, K. A., Sánchez, H. A., Lee, S. C., Altenberg, G. A., Nathanson, M. H., and Sáez, J. C. (2010). Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 299, C1504–C1515. doi: 10.1152/ajpcell.00015.2010
Schenk, U., Westendorf, A. M., Radaelli, E., Casati, A., Ferro, M., Fumagalli, M., et al. (2008). Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci. Signal. 1:ra6. doi: 10.1126/scisignal.1160583
Scheuplein, F., Schwarz, N., Adriouch, S., Krebs, C., Bannas, P., Rissiek, B., et al. (2009). NAD+ and ATP released from injured cells induce P2X7-dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. J. Immunol. 182, 2898–2908. doi: 10.4049/jimmunol.0801711
Schwartz, N. E., and Alford, S. (2000). Physiological activation of presynaptic metabotropic glutamate receptors increases intracellular calcium and glutamate release. J. Neurophysiol. 84, 415–427.
Seminario-Vidal, L., Okada, S. F., Sesma, J. I., Kreda, S. M., van Heusden, C. A., Zhu, Y., et al. (2011). Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J. Biol. Chem. 286, 26277–26286. doi: 10.1074/jbc.M111.260562
Shaw, R. M., Fay, A. J., Puthenveedu, M. A., von Zastrow, M., Jan, Y. N., and Jan, L. Y. (2007). Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128, 547–560. doi: 10.1016/j.cell.2006.12.037
Sherer, N. M. (2013). Long-distance relationships: do membrane nanotubes regulate cell-cell communication and disease progression? Mol. Biol. Cell 24, 1095–1098. doi: 10.1091/mbc.E12-08-0622
Shurman, D. L., Glazewski, L., Gumpert, A., Zieske, J. D., and Richard, G. (2005). In vivo and in vitro expression of connexins in the human corneal epithelium. Invest. Ophthalmol. Vis. Sci. 46, 1957–1965. doi: 10.1167/iovs.04-1364
Sirnes, S., Honne, H., Ahmed, D., Danielsen, S. A., Rognum, T. O., Meling, G. I., et al. (2011). DNA methylation analyses of the connexin gene family reveal silencing of GJC1 (Connexin45) by promoter hypermethylation in colorectal cancer. Epigenetics 6, 602–609. doi: 10.4161/epi.6.5.15237
Söhl, G., and Willecke, K. (2004). Gap junctions and the connexin protein family. Cardiovasc. Res. 62, 228–232. doi: 10.1016/j.cardiores.2003.11.013
Song, E. K., Rah, S. Y., Lee, Y. R., Yoo, C. H., Kim, Y. R., Yeom, J. H., et al. (2011). Connexin-43 hemichannels mediate cyclic ADP-ribose generation and its Ca2+-mobilizing activity by NAD+/cyclic ADP-ribose transport. J. Biol. Chem. 286, 44480–44490. doi: 10.1074/jbc.M111.307645
Song, M., Yu, X., Cui, X., Zhu, G., Zhao, G., Chen, J., et al. (2009). Blockade of connexin 43 hemichannels reduces neointima formation after vascular injury by inhibiting proliferation and phenotypic modulation of smooth muscle cells. Exp. Biol. Med. (Maywood) 234, 1192–1200. doi: 10.3181/0902-RM-80
Sosinsky, G. E., Boassa, D., Dermietzel, R., Duffy, H. S., Laird, D. W., MacVicar, B., et al. (2011). Pannexin channels are not gap junction hemichannels. Channels (Austin) 5, 193–197. doi: 10.4161/chan.5.3.15765
Spray, D. C., Ye, Z. C., and Ransom, B. R. (2006). Functional connexin hemichannels: a critical appraisal. Glia 54, 758–773. doi: 10.1002/glia.20429
Sridharan, M., Adderley, S. P., Bowles, E. A., Egan, T. M., Stephenson, A. H., Ellsworth, M. L., et al. (2010). Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am. J. Physiol. Heart Circ. Physiol. 299, H1146–H1152. doi: 10.1152/ajpheart.00301.2010
Stagg, J., and Smyth, M. J. (2010). Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 29, 5346–5358. doi: 10.1038/onc.2010.292
Stoletov, K., Strnadel, J., Zardouzian, E., Momiyama, M., Park, F. D., Kelber, J. A., et al. (2013). Role of connexins in metastatic breast cancer and melanoma brain colonization. J. Cell Sci. 126(Pt 4), 904–913. doi: 10.1242/jcs.112748
Stout, C., and Charles, A. (2003). Modulation of intercellular calcium signaling in astrocytes by extracellular calcium and magnesium. Glia 43, 265–273. doi: 10.1002/glia.10257
Stout, C. E., Costantin, J. L., Naus, C. C., and Charles, A. C. (2002). Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 277, 10482–10488. doi: 10.1074/jbc.M109902200
Swayne, L. A., Sorbara, C. D., and Bennett, S. A. (2010). Pannexin 2 is expressed by postnatal hippocampal neural progenitors and modulates neuronal commitment. J. Biol. Chem. 285, 24977–24986. doi: 10.1074/jbc.M110.130054
Tan, L. W., Bianco, T., and Dobrovic, A. (2002). Variable promoter region CpG island methylation of the putative tumor suppressor gene Connexin 26 in breast cancer. Carcinogenesis 23, 231–236. doi: 10.1093/carcin/23.2.231
Tang, B., Peng, Z. H., Yu, P. W., Yu, G., Qian, F., Zeng, D. Z., et al. (2013). Aberrant expression of cx43 is associated with the peritoneal metastasis of gastric cancer and cx43-mediated gap junction enhances gastric cancer cell diapedesis from peritoneal mesothelium. PLoS ONE 8:e74527. doi: 10.1371/journal.pone.0074527
Tate, A. W., Lung, T., Radhakrishnan, A., Lim, S. D., Lin, X., and Edlund, M. (2006). Changes in gap junctional connexin isoforms during prostate cancer progression. Prostate 66, 19–31. doi: 10.1002/pros.20317
Teleki, I., Krenacs, T., Szasz, M. A., Kulka, J., Wichmann, B., Leo, C., et al. (2013). The potential prognostic value of connexin 26 and 46 expression in neoadjuvant-treated breast cancer. BMC Cancer 13:50. doi: 10.1186/1471-2407-13-50
Trabanelli, S., Ocadlíková, D., Gulinelli, S., Curti, A., Salvestrini, V., Vieira, R. P., et al. (2012). Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J. Immunol. 189, 1303–1310. doi: 10.4049/jimmunol.1103800
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496. doi: 10.1124/pr.109.002451
Trosko, J. E., Chang, C. C., and Medcalf, A. (1983). Mechanisms of tumor promotion: potential role of intercellular communication. Cancer Invest. 1, 511–526. doi: 10.3109/07357908309020276
Trosko, J. E., and Ruch, R. J. (1998). Cell-cell communication in carcinogenesis. Front. Biosci. 3, d208–d236.
Vinken, M., Decrock, E., Leybaert, L., Bultynck, G., Himpens, B., Vanhaecke, T., et al. (2012). Non-channel functions of connexins in cell growth and cell death. Biochim. Biophys. Acta 1818, 2002–2008. doi: 10.1016/j.bbamem.2011.06.011
Vinken, M., De Rop, E., Decrock, E., De Vuyst, E., Leybaert, L., Vanhaecke, T., et al. (2009). Epigenetic regulation of gap junctional intercellular communication: more than a way to keep cells quiet? Biochim. Biophys. Acta 1795, 53–61. doi: 10.1016/j.bbcan.2008.08.002
Wang, J., Jackson, D. G., and Dahl, G. (2013b). The food dye FD&C Blue No. 1 is a selective inhibitor of the ATP release channel Panx1. J. Gen. Physiol. 141, 649–656. doi: 10.1085/jgp.201310966
Wang, N., De Bock, M., Antoons, G., Gadicherla, A. K., Bol, M., Decrock, E., et al. (2012). Connexin mimetic peptides inhibit Cx43 hemichannel opening triggered by voltage and intracellular Ca2+ elevation. Basic Res. Cardiol. 107:304. doi: 10.1007/s00395-012-0304-2
Wang, N., De Bock, M., Decrock, E., Bol, M., Gadicherla, A., Bultynck, G., et al. (2013a). Connexin targeting peptides as inhibitors of voltage- and intracellular Ca(2+)-triggered Cx43 hemichannel opening. Neuropharmacology. 75, 506–516. doi: 10.1016/j.neuropharm.2013.08.021
Weigel, M. T., and Dowsett, M. (2010). Current and emerging biomarkers in breast cancer: prognosis and prediction. Endocr. Relat. Cancer 17, R245–R262. doi: 10.1677/ERC-10-0136
Weissman, T. A., Riquelme, P. A., Ivic, L., Flint, A. C., and Kriegstein, A. R. (2004). Calcium waves propagate through radial glial cells and modulate proliferation in the developing neocortex. Neuron 43, 647–661. doi: 10.1016/j.neuron.2004.08.015
White, N., and Burnstock, G. (2006). P2 receptors and cancer. Trends Pharmacol. Sci. 27, 211–217. doi: 10.1016/j.tips.2006.02.004
Wicki-Stordeur, L. E., Dzugalo, A. D., Swansburg, R. M., Suits, J. M., and Swayne, L. A. (2012). Pannexin 1 regulates postnatal neural stem and progenitor cell proliferation. Neural Dev. 7:11. doi: 10.1186/1749-8104-7-11
Woehrle, T., Yip, L., Elkhal, A., Sumi, Y., Chen, Y., Yao, Y., et al. (2010). Pannexin-1 hemichannel-mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood 116, 3475–3484. doi: 10.1182/blood-2010-04-277707
Wong, C. W., Christen, T., Roth, I., Chadjichristos, C. E., Derouette, J. P., Foglia, B. F., et al. (2006). Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 12, 950–954. doi: 10.1038/nm1441
Xia, J., Lim, J. C., Lu, W., Beckel, J. M., Macarak, E. J., Laties, A. M., et al. (2012). Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J. Physiol. 590(Pt 10), 2285–2304. doi: 10.1113/jphysiol.2012.227983
Xiao, F., Waldrop, S. L., Khimji, A. K., and Kilic, G. (2012). Pannexin1 contributes to pathophysiological ATP release in lipoapoptosis induced by saturated free fatty acids in liver cells. Am. J. Physiol. Cell Physiol. 303, C1034–C1044. doi: 10.1152/ajpcell.00175.2012
Yahalom, A., Warmbrodt, R. D., Laird, D. W., Traub, O., Revel, J. P., Willecke, K., et al. (1991). Maize mesocotyl plasmodesmata proteins cross-react with connexin gap junction protein antibodies. Plant Cell 3, 407–417. doi: 10.1105/tpc.3.4.407
Yamasaki, H. (1990). Gap junctional intercellular communication and carcinogenesis. Carcinogenesis 11, 1051–1058. doi: 10.1093/carcin/11.7.1051
Yi, Z. C., Wang, H., Zhang, G. Y., and Xia, B. (2007). Downregulation of connexin 43 in nasopharyngeal carcinoma cells is related to promoter methylation. Oral. Oncol. 43, 898–904. doi: 10.1016/j.oraloncology.2006.11.004
Zender, L., Xue, W., Zuber, J., Semighini, C. P., Krasnitz, A., Ma, B., et al. (2008). An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 135, 852–864. doi: 10.1016/j.cell.2008.09.061
Zhao, H. B., Yu, N., and Fleming, C. R. (2005). Gap junctional hemichannel-mediated ATP release and hearing controls in the inner ear. Proc. Natl. Acad. Sci. U.S.A. 102, 18724–18729. doi: 10.1073/pnas.0506481102
Keywords: hemichannels, connexins, pannexins, cancer
Citation: Schalper KA, Carvajal-Hausdorf D and Oyarzo MP (2014) Possible role of hemichannels in cancer. Front. Physiol. 5:237. doi: 10.3389/fphys.2014.00237
Received: 09 January 2014; Accepted: 09 June 2014;
Published online: 27 June 2014.
Edited by:
Mauricio Antonio Retamal, Universidad del Desarrollo, ChileReviewed by:
Núria Comes, University of Barcelona, SpainCopyright © 2014 Schalper, Carvajal-Hausdorf and Oyarzo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kurt A. Schalper, Servicio Anatomía Patológica, Clinica Alemana de Santiago, Av. Manquehue 1410, Segundo Subterráneo, Santiago 7630000, Chile e-mail:a3VydC5zY2hhbHBlckB5YWxlLmVkdQ==;a3NjaGFscGVyQGFsZW1hbmEuY2w=
†Present address: Kurt A. Schalper, Department of Pathology, Yale School of Medicine, New Haven, USA
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.