 Mark A. Zaydman
Mark A. Zaydman Jianmin Cui
Jianmin Cui
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 27 May 2014
Sec. Membrane Physiology and Membrane Biophysics
Volume 5 - 2014 | https://doi.org/10.3389/fphys.2014.00195
This article is part of the Research Topic Ligand recognition and regulation of ion channel proteins View all 13 articles
Voltage-gated potassium (Kv) channels contain voltage-sensing (VSD) and pore-gate (PGD) structural domains. During voltage-dependent gating, conformational changes in the two domains are coupled giving rise to voltage-dependent opening of the channel. In addition to membrane voltage, KCNQ (Kv7) channel opening requires the membrane lipid phosphatidylinositol 4,5-bisphosphate (PIP2). Recent studies suggest that PIP2 serves as a cofactor to mediate VSD-PGD coupling in KCNQ1 channels. In this review, we put these findings in the context of the current understanding of voltage-dependent gating, lipid modulation of Kv channel activation, and PIP2-regulation of KCNQ channels. We suggest that lipid-mediated coupling of functional domains is a common mechanism among KCNQ channels that may be applicable to other Kv channels and membrane proteins.
Lipids define the physical and chemical environment of voltage-gated ion channels, yet most of the literature in the ion channel field considers only the channel protein and its proteinacious interacting partners. This protein-centric view is a natural consequence of our powerful genetic tools for manipulating the primary sequence of ion channels and heterologously expressing mutants for functional study. In comparison, we have a limited set of tools for manipulating the lipid environment of ion channels in heterologous or native cells. Despite this limitation, the importance of non-specific (bulk) and specific (cofactor) lipid interactions to ion channel function is a subject of continuing interest. Phosphatidylinositol 4,5-bisphosphate (PIP2) is an anionic lipid found in the inner leaflet of the surface membrane where it makes up a small fraction (<1%) of the total pool of phospholipids (McLaughlin et al., 2002; Rusten and Stenmark, 2006). PIP2 is known to directly bind to and regulate a diverse set of ion channels (Hilgemann and Ball, 1996; Hilgemann et al., 2001; Suh and Hille, 2005, 2008). PIP2 regulation is an excellent model for studying the cofactor-lipid regulation of ion channels, as we have increasingly more tools to manipulate the abundance of PIP2 in heterologous cells (Suh et al., 2006; Suh and Hille, 2007a; Okamura et al., 2009). Additionally, changes in the abundance of PIP2 that are sufficient to alter channel activity are not likely to change the bulk membrane properties.
Voltage-gated ion channels are transmembrane proteins that sense membrane voltage and respond by opening or closing an ion conductive pathway across the membrane. The ionic currents from voltage-gated ion channels generate action potentials in excitable tissues; thereby, they control muscle contraction, neuronal signaling, immune activation, and neurohormonal secretion. Variations in the abundance of membrane PIP2 modulate the function of several families of voltage-gated ion channels, including K+ channels (Kv) (Bian et al., 2001; Loussouarn et al., 2003; Zhang et al., 2003; Oliver et al., 2004; Abderemane-Ali et al., 2012; Rodriguez-Menchaca et al., 2012), Ca2+ channels (Cav) (Wu et al., 2002; Gamper et al., 2004; Suh et al., 2010), hyperpolarization and cyclic nucleotide activated channels (HCN) (Pian et al., 2006; Zolles et al., 2006), and voltage and Ca2+-activated K+ channels (BK) (Vaithianathan et al., 2008). It should be noted that the physiological relevance of the PIP2-sensitivities of some of these channels remains controversial (Hilgemann, 2012; Kruse et al., 2012). Among these voltage- and PIP2-sensitive channels, the KCNQ (herein we use KCNQ to refer gene and protein product—i.e., Kv7) family is unique in that its members absolutely require PIP2 in order to conduct current (as discussed below) making them a relatively straight-forward model to study PIP2 regulation of voltage-dependent gating. The KCNQ family includes five voltage-gated potassium-selective channels (KCNQ1-5) that are known to play important roles in regulating cardiac action potential duration (Barhanin et al., 1996; Sanguinetti et al., 1996; Wang et al., 1996), modulating neuroexcitability (Wang et al., 1998), and maintaining endolymph K+ homeostasis in the inner ear (Neyroud et al., 1997). The physiological importance of PIP2 regulation of KCNQ channels is well established in neurons where silencing of KCNQ channel activity by PIP2 hydrolysis downstream of G-protein Gq signaling increases neuroexcitability (Delmas and Brown, 2005; Brown et al., 2007). The role of PIP2 modulation of native cardiac KCNQ1 channels is less well established. However, in the heart, α1 adrenergic receptors (α1 AR) activate Gq signaling pathways (Jensen et al., 2011) which has been shown in heterologous expression systems to modulate IKs channels (formed by KCNQ1 and the auxiliary subunit KCNE1) in through a combination of PIP2 hydrolysis and PKC phosphorylation (Matavel and Lopes, 2009). Furthermore, KCNQ1 channel mutations associated with cardiac arrythmias in patients have been shown to affect PIP2-dependent activation, suggesting that native IKs channels are sensitive to PIP2 binding (Park et al., 2005; Li et al., 2011; Zaydman et al., 2013). A detailed understanding of the biophysical and molecular mechanisms by which PIP2 potentiates the function of KCNQ channels is an important step to understanding the physiology and pathophysiology of the cardiac, nervous, and auditory systems, and to developing effective new therapeutics for their diseases. In the following pages, we will review and attempt to synthesize the current body of work studying these mechanisms. Although the gating properties of KCNQ channels can be very different and accessory subunits lead to further variation, we will consider evidence regarding different KCNQ family members as well as KCNQ1 channels associated with KCNE1 accessory subunits. As described below, all these channels are voltage-gated, they all require PIP2 to open, and it appears that they all share a conserved PIP2 binding site at the VSD-PGD interface (see below). Therefore, we believe that considering these studies together may provide additional insights and may lead the reader to consider shared fundamental mechanisms that can tested and validated experimentally.
Voltage-gated cation channels share a common core structure consisting of four voltage-sensing domains (VSDs) surrounding a central pore-gate domain (PGD). In voltage-gated potassium (Kv) channels, including the members of the KCNQ (Kv7) family, tetrameric assembly of Kv-α subunits yields this channel structure (Figure 1). Each Kv-α subunit contains six transmembrane segments, of which S1-S4 from each Kv-α subunit form a VSD, while the S5-S6s from all four Kv-α subunits form the PGD. In general terms, voltage-dependent gating involves three different processes: VSD activation, PGD opening, and VSD-PGD coupling. In VSD activation, a conformational change is directly driven by transmembrane voltage. The voltage-dependence of this transition arises from forces that the transmembrane electric field exerts on conserved basic residues in S4 (Bezanilla, 2008). Depolarization yields outward force and promotes displacement of S4 from the resting to the activated state. The PGD forms a gated pathway through the membrane for selected ions to flow down their electrochemical gradient. The PGD opens by a dilation of the pathway at the crossing of the C-terminal portions of the S6 segments (the S6 gate) (Yellen, 1998). Several lines of evidence show that the VSD and the PGD are modular units. First, crystal structures of voltage-dependent cation channels reveal a surprisingly limited protein-protein contact surface at the VSD-PGD interface (Long, 2005; Payandeh et al., 2011). Second, voltage-independent channels consisting of the PGD alone without a VSD are found in nature, and they likely share a common, PGD-only ancestor with voltage-gated channels (Nayak et al., 2009). Likewise, voltage-sensor only proteins (VSOP) containing a functioning VSD without a PGD have been identified (Murata et al., 2005; Ramsey et al., 2006; Sasaki, 2006). Third, channels consisting of an artificially isolated PGD from voltage-gated potassium (Santos et al., 2008, 2012) or sodium (Shaya et al., 2011) channels have been shown to fold and function. Likewise, the artificially isolated VSD from KvAP adopts a similar conformation as in various crystal structures of full-length Kv channels (Butterwick and MacKinnon, 2010). Finally, functional, voltage-gated ion channels have been engineered by fusing together VSDs and PGDs from different sources (Lu et al., 2001; Arrigoni et al., 2013).
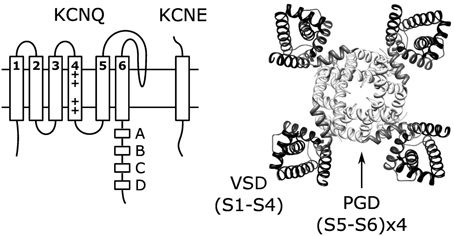
Figure 1. Topology of single KCNQ subunit and the structure (right) of KCNQ channels formed by coassembly of four KCNQ subunits. The structure shown is the KCNQ1 homology model (Smith et al., 2007) based on the Kv1.2 crystal structure (Long et al., 2005).
In VSD-PGD coupling, the conformations of the two modules are linked so that VSD activation promotes PGD opening, yielding a voltage-dependent conductance. Previous studies have identified several sites at the VSD-PGD interface that are important for the coupling, including the interactions between the S4-S5 linker and S6 (Lu et al., 2001, 2002; Tristani-Firouzi et al., 2002; Long, 2005; Haddad and Blunck, 2011), S4 and S5 (Ledwell and Aldrich, 1999; Soler-Llavina et al., 2006; Grabe et al., 2007), and S1 and the outer vestibule (Lee et al., 2009). Relative to what is known about VSD activation and PGD opening, their coupling remains poorly understood and the literature is largely focused on the protein-protein interactions. As will described below, PIP2 was recently found as an absolutely required cofactor for coupling between the activated state of the VSD and the open state of the PGD in KCNQ1 channels (Zaydman et al., 2013), providing a new opportunity for studying this critical, yet mysterious, process. Interestingly, the Kv1.2-2.1 crystal structure contains several anionic lipids bound stably at the VSD-PGD interface (Long et al., 2007), suggesting that lipid involvement in VSD-PGD coupling may be a principle common to Kv channels.
Given that distinct VSD and PGD domains form Kv channels and that coupling is essential to voltage-dependent gating (Figure 2A), any model describing voltage-dependent gating should explicitly define coupling. Two mathematical models of voltage-dependent gating have found widespread application. A linear activation scheme (see Figure 2B for a simplified version) was first used to describe the gating of Shaker potassium channels (Schoppa et al., 1992; Zagotta et al., 1994). In this model, PGD opening occurs only after the independent activation of the four VSDs. In contrast, an allosteric scheme (see Figure 2C for a simplified version) was devised to describe the voltage-dependent gating (in the absence of calcium) of calcium-activated, large conductance potassium (BK) channels (Horrigan et al., 1999). In the allosteric model, the PGD can open or close when the VSDs are either activated or resting; however, the coupling interactions between the two domains make PGD opening relatively more likely when the VSDs are activated. The linear model is compact and intuitive, and fits data well in special cases where PGD opening when the VSDs are resting is extremely low, as shown in wt Shaker channels (Islas and Sigworth, 1999). In fact, under these specific conditions the allosteric model is well approximated by the linear scheme (for an excellent review of this point, see Chowdhury and Chanda, 2012). However, the linear scheme has often been applied to voltage-gated channels for which these special condition have not been verified, which can lead to possible errors in the interpretation and analysis of data. Given the evidence for the independent structures and functions of the VSD and PGD, the allosteric model better reflects the physical reality by allowing for resting-open (RO, Figure 2) and activated-closed (AC) conformations. Although it appears more complex, the allosteric model provides the conceptual framework required by allowing a mathematical definition of coupling (θ in Figure 2C). Even still, the scheme in Figure 2C is overly simple in that the θ parameter is a complex normalized term, it is influenced by the interactions between all states of the VSD and PGD. Furthermore, as a result of normalization, the gating parameters (Kv, Kg) are also a function of the domain-domain interactions and do not reflect the intrinsic gating of the two domains in isolation (See Chowdhury and Chanda, 2012 JGP for more details).
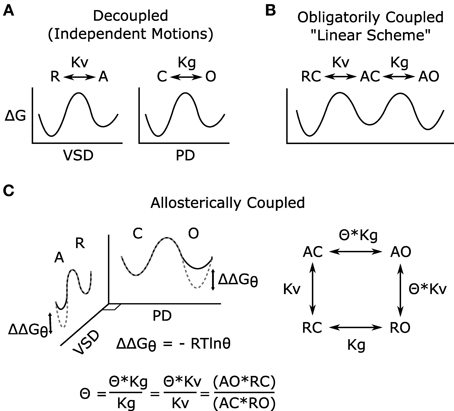
Figure 2. Models of voltage-dependent gating. (A) The VSD and PGD are independent structural and functional domains; each domain on its own may undergo conformational transitions that can be described by independent energy landscapes. These hypothetical reaction coordinates illustrate the free energy associated with the different states of the domains and the transitions among them. In the voltage-gated ion channel, the conformational changes of these domains are coupled, and such coupling has been modeled in two different ways (B,C). Here, for simplicity, all models are shown considering only one VSD, and both VSD activation and PGD opening are modeled as two-state processes, for full models see references (Zagotta et al., 1994; Horrigan and Aldrich, 1999). (B) Linear gating scheme in which VSD activation obligatorily precedes PGD opening, and coupling is not explicitly defined. (C) Allosteric gating scheme in which the distinctive nature of the two domains is represented, and the coupling is explicitly defined. In this simple model, the coupling factor (θ) has been normalized to represent the net coupling that is due to interactions among all states of the two domains and does not represent a single physical interaction (Chowdhury and Chanda, 2012). The effects of this coupling can be represented in the energy landscapes (C) where the difference between the dotted-gray and solid-black lines represent the free energy changes due to the VSD-PGD coupling. For example, within the PGD plane, solid-black and dotted-gray indicate the free energy landscape for PGD opening when the VSD is resting or activated, respectively. R, resting VSD; A, activated VSD; Kv, equilibrium constant for VSD activation; C, closed PGD; O, open PGD; Kg, equilibrium constant for PGD opening; Θ, allosteric coupling factor; ΔG, free energy of gating; ΔΔGθ, free energy of coupling. As illustrated (C), θ describes how much the open-close equilibrium is biased toward open when the VSD is activated. Equivalently, coupling can be quantified by measuring the effect of PGD opening on VSD activation, a measurement that is often more experimentally tractable (Arcisio-Miranda et al., 2010; Ryu and Yellen, 2012; Zaydman et al., 2013).
Several lines of experimental evidence strongly support allosteric coupling of KCNQ channels. First, even at low voltages, wt KCNQ1 channels display a small, but measurable, conductance (~5% of maximal conductance), which is sensitive to mutations of KCNQ1 and a KCNQ channel blocker, indicating that it is indeed a KCNQ1 conductance (Ma et al., 2011). Second, KCNQ1 channels display no separation between the voltage-dependence of VSD activation and conductance, indicating that KCNQ1 channel opening does not require prior activation of all four VSDs (Osteen et al., 2010). Studies in which WT and mutant KCNQ1 subunits are mixed provided additional evidence that channel opening does not require the prior activation of all four VSDs (Meisel et al., 2012; Osteen et al., 2012). Third, locking the KCNQ1 channel PGD in an open-state does not prevent the VSD from transitioning between resting and activated states (Zaydman et al., 2013), providing direct evidence against the obligatory coupling imposed by linear activation schemes (Figure 2B) These studies revealed allosteric coupling in KCNQ1. M-channels formed by KCNQ2 and KCNQ3 subunits may also have allosteric coupling as suggested by the report that these channels can open even when only the high affinity KCNQ3 subunits are bound to PIP2 (Telezhkin et al., 2012a), suggesting that opening is not concerted as in the linear scheme (Figure 2B). Interpretation of experimental data through the lenses of the linear vs. allosteric gating models can resolve some of the differences between the biophysical and molecular models that have been proposed for the regulation of KCNQ channels by PIP2, as described below.
How might specific lipid interactions affect voltage-dependent gating? Three possible mechanisms are suggested above: by affecting VSD activation, PGD opening, and/or VSD-PGD coupling. Foundational work establishing roles for lipids as cofactors in voltage-dependent gating came through the study of Kv channel interactions with phospholipids other than PIP2. These findings are presented here, briefly.
MacKinnon and colleagues showed that, to open in response to depolarization, KvAP channels reconstituted into a planar bilayer require phospholipids in the membrane. They further found that, in sustaining current, the lipid phosphate group is key, not the net headgroup charge, the acyl chain length, or the effects of the lipids on membrane curvature (Schmidt et al., 2006). These findings suggest that specific interactions between the lipid phosphodiesters and the channel protein are important for voltage-dependent gating. Along these lines, crystal structures and molecular dynamics simulations of Kv channels reveal probable interactions between the S4 arginines and the lipid phosphate groups, which may help to stabilize the activated-state of the VSD (Freites, 2005; Long, 2005; Treptow and Tarek, 2006; Delemotte et al., 2011; Jensen et al., 2012). Zheng et al. used an antibody to the S3b-S4 paddle motif to detect VSD activation in reconstituted KvAP channels. They found that phospholipids are required for the antibody to bind at 0 mV, indicating that phospholipids are required for VSD activation to occur at this voltage. However, these effects were not dependent on the charge at the first two-conserved arginine positions, arguing against a role for specific interaction between these gating charges and the lipid phosphodiesters (Zheng et al., 2011). While the structural and molecular dynamics studies support the possibility of a direct effect on VSD activation, the functional data do not rule out alternative mechanisms, such as indirect effects on VSD activation through direct modulation of PGD opening or VSD-PGD coupling, or through non-specific lipid effects.
Another line of evidence for lipid modification of voltage-dependent gating comes from enzymatic modification of the headgroup of sphingomyelin, a zwitterionic lipid found primarily in the external leaflet of the cell membrane. Shingomyelinase D (Smase D) cleaves choline from the headgroup, leaving behind negatively charged ceramide-1-phosphate. Smase D treatment of oocytes expressing Kv channels was shown to left-shift the voltage dependence of PGD opening and VSD activation (Ramu et al., 2006). Again, these results alone do not provide exclusive evidence for a direct effect on VSD activation, as the voltage-dependence of VSD movement could be indirectly affected by alteration in the coupling or PGD opening (Colquhoun, 1998). However, Smase D was subsequently shown to left shift VSD activation in the voltage-sensitive enzyme CiVSP (Milescu et al., 2009). Provided that sphingomyelin acts by directly binding the channel, and not through aggregation of sphingomyelin-dependent lipid microdomains (Lingwood and Simons, 2009), this result provides direct evidence for an interaction between the sphongomyelin headgroup and the VSD, given that CiVSP contains only a VSD in its transmembrane domain (Murata et al., 2005). Sphingomyelinase C (Smase C), an enzyme that cleaves sphingomyelin after the phosphate, leaving behind a neutral headgroup, robustly inhibits the ionic current and gating charge movement in Kv channels. Unexpectedly, Smase C also inhibits currents from voltage-independent K+ channels (Xu et al., 2008) containing only a PGD in their transmembrane domain. This result suggests that sphingomyelin can act by directly affecting PGD opening, or through a non-specific mechanism.
Additional evidence for lipid interaction with the PGD comes from the presence of a lipid, phosphatidylglycerol (PG), in the crystal structure of the voltage-independent KCSA channel (Valiyaveetil et al., 2002), which is required for channel opening (Heginbotham, 1998; Valiyaveetil et al., 2002).
These studies have greatly enriched our understanding of the lipid regulation of voltage-dependent gating, but many questions remain unresolved. Particularly the role of lipids in VSD-PGD coupling was not explored. Previously, this coupling has been largely attributed to protein-protein interactions at the VSD-PGD interface. However, the presence of an anionic phospholipid bound at this interface in the Kv1.2/2.1 chimera channel crystal structure suggested that lipids may play a role as well (Long et al., 2007). Rodriguez-Menchaca et al. showed that mutations of the Kv1.2 S4-S5 linker, a structure that has been implicated in VSD-PGD coupling, increase the magnitude of GV shift and current inhibition caused by PIP2 depletion (Rodriguez-Menchaca et al., 2012). However, the effects of PIP2 on such Shaker-type channels are slight and complex (Abderemane-Ali et al., 2012; Rodriguez-Menchaca et al., 2012), and no biophysical mechanism has been established to explain these data. In contrast, PIP2 depletion completely inhibits the KCNQ currents, making KCNQ channels an excellent model system. We have recently developed an assay to directly detect the VSD-PGD coupling in KCNQ1 by using a mutation to lock the PGD in the open conformation and measuring the impact of PGD opening on VSD activation using voltage clamp fluorometry (VCF) (Zaydman et al., 2013). We found that the coupling between the activated-state of the VSD and the open-state of the PGD requires binding of PIP2 at the VSD-PGD interface (Figure 4), thereby providing functional evidence that lipids play a role in VSD-PGD coupling. The resemblance of the putative PIP2 binding site at the VSD-PGD interface of KCNQ1 to the anionic lipid-binding site in the Kv1.2/2.1 chimera channel structure (Long et al., 2007) suggests that lipid-mediated coupling may be a general property of voltage-gated channels. In the following pages we discuss these findings in the context of the large body of work on KCNQ-PIP2 interactions, generated by the efforts of many laboratories over the past decade.
All five members of the KCNQ family require PIP2 to conduct measurable current (Zhang et al., 2003). KCNQ currents are inhibited by various treatments that decrease the abundance of PIP2 in the membrane, including stimulating PIP2 hydrolysis through receptor-mediated activation of phospholipase C (Suh and Hille, 2002; Zhang et al., 2003), elevating the rate of PIP2 dephopsphorylation through activation of lipid phosphatases (Suh et al., 2006; Murata and Okamura, 2007; Li et al., 2011; Kruse et al., 2012; Zaydman et al., 2013), suppressing PIP2 generation by inhibiting PIP kinases (Zhang et al., 2003; Shen, 2005), or directly chelating free PIP2 by applying PIP2 antibodies or polycations (ex. Mg2+, polylysine) (Suh and Hille, 2007b; Piron et al., 2010). Additionally, KCNQ currents undergo a spontaneous PIP2-dependent rundown in excised patches, which can be prevented or reversed through application of exogenous PIP2 (Loussouarn et al., 2003; Zhang et al., 2003; Li et al., 2005, 2011; Telezhkin et al., 2012a,b, 2013; Zaydman et al., 2013). These results show that PIP2 is required to sustain KCNQ channel currents. As the magnitude of the macroscopic current is a function of multiple parameters, these results could indicate that PIP2 affects the number of channels in the membrane, the single channel conductance, the probability of opening (gating), or the driving force for selected ions (Zaydman et al., 2012). The fast kinetics of current inhibition upon PIP2 depletion (Zhang et al., 2003; Falkenburger et al., 2010) and the reversibility of PIP2 activation of KCNQ channels in excised patches (Loussouarn et al., 2003; Zhang et al., 2003; Li et al., 2005, 2011; Telezhkin et al., 2012a,b, 2013; Zaydman et al., 2013) suggest a mechanism that is independent of the number of channels in the membrane. In single channel recordings of KCNQ2, KCNQ3, KCNQ4, and KCNQ2/KCNQ3 channels, the open probability approaches zero when PIP2 is depleted, and it increases, in a dose-dependent manner, to approach unity at PIP2 saturation. On the other hand, the slope conductance was found to be insensitive to PIP2, indicating that PIP2 does not change the single channel conductance or ionic selectivity (Li et al., 2005). These experiments show that PIP2 is required for gating of KCNQ channels.
KCNQ channels are generally known to be voltage-gated, but the effects of PIP2 on their voltage-dependency may appear different among KCNQ channels. Following muscarinic stimulation to partially deplete PIP2, Shapiro et al. found that conductance-voltage (GV) relationship of KCNQ2+KCNQ3 channels was unaffected (Shapiro et al., 2000). Subsequently, Nakajo and Kubo observed a rightward shift in the GV curve following activation of muscarinic receptors for KCNQ2, KCNQ2+KCNQ3 or KCNQ4 channels. However, this shift was fully accounted for by the downstream activation of PKC, and application of Wortmannin, a relatively unspecific kinase inhibitor that is known to block the PI 4- kinase (Nakanishi et al., 1995), did not affect the GV curves of homomeric KCNQ1 or KCNQ2 channels (Nakajo and Kubo, 2005). Finally, Hille and colleagues reported that increasing membrane PIP2 using inducible kinases did not shift the GV relationship for KCNQ2+KCNQ3 channels (Suh et al., 2006). On the other hand, when Loussouarn et al. excised membrane patches from COS-7 cells expressing KCNQ1 subunits with KCNE1 accessory subunits into bath solution containing high concentrations of PIP2 (5 μ g/mL), they observed a small (ΔV1/2 ~12 mV) leftward shift in the GV relationship and a slowing of the deactivation kinetics (Loussouarn et al., 2003). They also observed a hastening of the deactivation kinetics during spontaneous rundown in PIP2 free solutions (Loussouarn et al., 2003). Similarly, Li et al. (2011) excised membrane patches from Xenopus oocytes expressing KCNQ1 and KCNE1 subunits into bath solutions containing high (300 μ M) PIP2 and observed a left shift in the GV (ΔV1/2 ~13 mV); however, no changes of deactivation kinetics were observed under these conditions or during spontaneous PIP2-dependent rundown. These seemingly contradictory kinetic results can be explained by different levels of expression of the KCNE1 subunit in these two studies. The currents recorded by Loussouarn et al. opened at hyperpolarized potentials (V1/2on cell ~−34 mV) and displayed a prominent tail hook indicative of inactivation. These are two hallmarks of homomeric KCNQ1 channels that are not usually present when KCNE1 is coexpressed (Barhanin et al., 1996; Sanguinetti et al., 1996; Pusch et al., 1998). As Loussouarn et al. measured deactivation at −40 mV, which is just below the half maximal activation, the effect of PIP2 on deactivation kinetics may be attributed to the shift in steady-state activation. Specifically, the deactivation slows at high PIP2 because −40 mV point moves higher up the steady-state activation curve, and vice versa. In contrast, the currents recorded by Li et al. manifested at more depolarized voltages (V1/2 on cell = +21.6 ± 3.2 mV) and did not display a tail hook, consistent with robust expression of KCNE1. Furthermore, Li et al. measured deactivation kinetics at −80 mV, at which point steady-state opening is minimal under conditions of both low and high PIP2. This result suggests, when isolated from activation, deactivation kinetics are insensitive to PIP2. While PIP2-dependent GV shifts are mild for wt KCNQ1 + wt KCNE1, the shift is greater (ΔV1/2 = −40 mV: on cell vs. 300 μM PIP2) when wt KCNQ1 is expressed with a mutant KCNE1 - K70N (Li et al., 2011). Several mutations of KCNQ1 or KCNE1, including K70N, right-shift the GV relationship of KCNQ1+KCNE1 channels, and they decrease the apparent affinity of KCNQ1+KCNE1 channels for exogenous PIP2 (Park et al., 2005; Li et al., 2011) or decrease the binding of C-terminal fragments of KCNQ1 to immobilized PIP2 (Thomas et al., 2011). Of these mutations, it has been shown for K70N that application of supranormal levels (300 μ M) of PIP2 reverses the shift, so that the GV relationship can be superimposed on that of wt channels under identical conditions (Li et al., 2011). These data suggest that the effects of K70N on PIP2 sensitivity can fully account for the shift in voltage-dependence. Altogether, these results provide evidence that PIP2 affects the voltage-dependent gating of KCNQ1+KCNE1 channels. It is important to bear in mind that the lack of a GV shift for other KCNQ channels, including homomeric KCNQ1, does not rule out an effect of PIP2 on their voltage-dependent gating (see below).
As described above, voltage-dependent gating involves three fundamental processes: VSD activation, VSD-PGD coupling, and PGD opening. Recently, we have investigated which step(s) requires PIP2 in KCNQ1 channels. To test if PIP2 affects VSD activation in KCNQ1 channels, VSD activation was detected by using voltage clamp fluorometry (VCF) (Mannuzzu et al., 1996; Osteen et al., 2010) while dynamically depleting the endogenous PIP2 by expressing and activating the lipid-phosphatase CiVSP (Murata et al., 2005; Murata and Okamura, 2007). PIP2 depletion did not prevent movement of the VSD in response to membrane voltage; meanwhile, the ionic currents were robustly inhibited (Zaydman et al., 2013). Furthermore, the fluorescence-voltage (FV) relationship, a measure of the steady-state voltage-dependence of VSD activation, was insensitive to PIP2 depletion suggesting that VSD movements are not directly affected by PIP2. As a complementary method to VCF, we detected VSD activation as a voltage-dependent conformational change altering the chemical accessibility of the residue I230C within S4 to extracellular MTSES. Modification of I230C is voltage-dependent in the presence of endogenous PIP2 and after PIP2 depletion by CiVSP, confirming that PIP2 is not required for VSD activation in KCNQ1 (Zaydman et al., 2013). These results suggest that PIP2 is required for either PGD opening or VSD-PGD coupling.
We then tested if PIP2 is required for coupling by measuring the impact of PGD opening on VSD activation both in the presence of endogenous PIP2 and after PIP2 depletion by CiVSP. In the presence of PIP2, VSD-PGD coupling caused a leftward shift in the FV relationship when the PGD was locked open by the mutation L353K. After depletion of PIP2, L353K KCNQ1 channels remained open, but the shift was eliminated and the FV relationships of L353K and wt could be superimposed (Zaydman et al., 2013). This result demonstrates that PIP2 is required for PGD opening to promote VSD activation, in addition to being required for VSD activation to promote PGD opening. These bidirectional effects are consistent with a model in which PIP2 is required for allosteric coupling between the activated-state of the VSD and the open-state of the PGD. A simple version of such a model globally fits the steady-state voltage- and PIP2-dependencies of PGD opening and VSD activation in KCNQ1 (Zaydman et al., 2013). This model demonstrates that the effect of PIP2 on coupling is sufficient to quantitatively model the gating behavior of KCNQ1 without any direct effects on PGD opening; however, these results do not rule out the possibility that PIP2 affects PGD opening as well.
Loussouarn et al. proposed a model for PIP2 regulation of KCNQ1+KCNE1 channels in which the role of PIP2 is to stabilize the PGD open-state (Loussouarn et al., 2003). This model recapitulated their observations that PIP2 left-shifts the GV relationship and slows current deactivation without changing the kinetics of current activation. This model is in agreement with the model of Zaydman et al. in the sense that PIP2 does not directly affect VSD activation; however, the linear architecture of this model does not allow one to identify effects on VSD-PGD coupling independently of PGD opening. Furthermore, it is debatable if deactivation kinetics are directly sensitive to PIP2 (see above). Loussouarn et al. draw an insightful analogy to voltage-independent Kir channels for which PIP2 binding at the C-terminus of the PGD has been suggested to stabilize the PGD open-state. However, recent crystallographic evidence suggests that PIP2 mediates coupling between the pore and cytosolic domains, not PGD opening, in Kir channels (Hansen et al., 2011; Whorton and MacKinnon, 2011). To the best of our knowledge, there is no firm evidence for or against a direct effect of PIP2 binding on the PGD opening in a KCNQ channel.
Identification of the binding site is critical to mechanistically understanding how PIP2 regulates KCNQ channels, and may hold the key to developing new therapeutics targeting PIP2-dependent activation. The search for putative PIP2 interacting partners has focused on basic residues that are exposed to the cytosol based on the rationale that PIP2 is restricted to the intracellular leaflet of the membrane and that crystalographically verified PIP2 binding sites in other proteins contain multiple basic residues for coordination of the negatively charged headgroup phosphates (McLaughlin et al., 2002; Rosenhouse-Dantsker and Logothetis, 2007; Suh and Hille, 2008; Hansen et al., 2011; Whorton and MacKinnon, 2011). The importance of electrostatic interactions on PIP2 activation of KCNQ has been verified by Brown and colleagues by studying the ability of various exogenous lipids to sustain the single channel open probability of KCNQ2+KCNQ3 channels at 0 mV. They found that a phosphate on the lipid headgroup is required, and the apparent affinity to different phosphoinositide species increases with headgroup charge; i.e., more phosphates yield higher apparent affinity (Telezhkin et al., 2012b). On the other hand, synthesized dic4-PIP2, dic8-PIP2, and long-chain PIP2 purified from bovine brain have similar effects on the activation of KCNQ1 channels (Li et al., 2011), suggesting that the acyl chains of PIP2 molecules do not affect the extent of channel activation. Implicit in the logic of studying basic residues is the idea that they are participating in a direct electrostatic interaction with the PIP2 headgroup. However, a high level of caution is required when putative binding residues are identified through functional studies because both binding and gating will determine the apparent affinity (for an excellent review of this topic, see Colquhoun, 1998). Many residues have been proposed to be important for the PIP2 dependent activation of KCNQ channels, which can be grouped into three sites: the VSD-PGD interface site, the helix A-B linker site, and the distal C-terminus site (Figure 3).
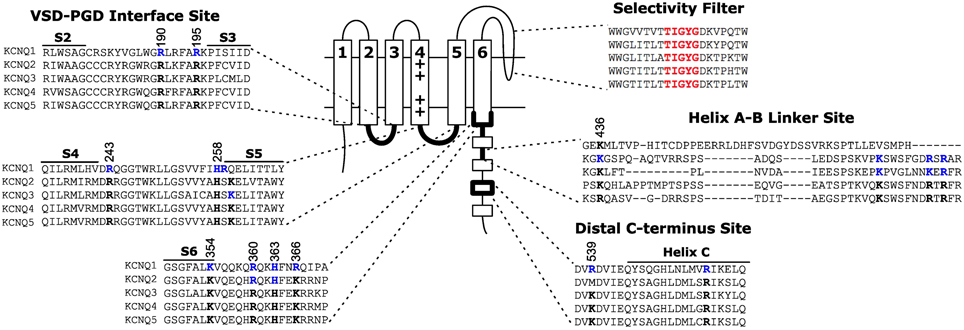
Figure 3. Location of three proposed PIP2 interacting sites on KCNQ. The VSD-PGD interface site includes contributions from the S2-S3 linker, S4-S5 linker, S6 and proximal C-terminus. Other sites have been proposed at the helix A-B linker or the distal C-terminus. Blue residues indicate residues for which mutations have been reported to affect PIP2 dependent activation. Black residues highlight the conservation of such residues among other members of the KCNQ family. Numbering indicates the positions of highlighted residues in the human KCNQ1 channel sequence.
The VSD-PGD interface site includes contributions from the S2-S3 linker, S4-S5 linker and the proximal C-terminus (Figure 3). Logothetis and colleagues identified a mutation, H328C, in the proximal C-terminus of KCNQ2, that significantly decreased the apparent affinity of KCNQ2+KCNQ3 channels to exogenous PIP2 (Zhang et al., 2003). Subsequently, Loussouarn and colleagues demonstrated that the disease-associated mutation R243 in the S4-S5 linker decreases the apparent affinity of KCNQ1+KCNE1 channels for exogenous PIP2 (Park et al., 2005). Recently, Li and colleagues reported that K222A, in the S4-S5 linker, lowered the apparent PIP2 affinity of KCNQ3 channels (Zhou et al., 2013). In 2011, Tinker and colleagues found a cluster of basic residues (K354, K358, R360, K362) in the KCNQ1 proximal C-terminus is important for the binding of C-terminal fragments of KCNQ1 to immobilized phosphoinositides (Thomas et al., 2011). One concern with such an approach is that binding of a protein fragment may represent a non-specific electrostatic interaction that is not representative of PIP2 binding in the channel protein. Accordingly, these authors found that the identified mutations decreased the whole cell currents of full-length KCNQ1+KCNE1 channels, and a double mutation (K358A/R360A) had a blunted response to dialysis of 25 μ M diC8 PIP2. Studying the homologous residues in KCNQ2, Brown and colleagues found that the mutation R325A decreased the on-cell, single-channel open probability and the apparent affinity for diC8 PIP2 in excised patches; in contrast, the mutation K327A did not affect the dose response of KCNQ2 (Telezhkin et al., 2013). Lopes and colleagues found that the long QT syndrome-associated mutation R366Q increased the sensitivity of KCNQ1+KCNE1 channels to PIP2 hydrolysis (Matavel et al., 2010). We have recently shown that charge neutralizing and charge reversing mutations of basic residues at the VSD-PGD interface affect the whole cell current amplitude in a manner that is correlated with their effects on apparent PIP2 affinity and PIP2-dependent VSD-PGD coupling. Of particular interest, mutations of eight residues in KCNQ1 essentially eliminate VSD-PGD coupling and the whole cell current without preventing channel expression in the membrane or VSD activation (Zaydman et al., 2013). These residues are located in the S2-S3 linker (R190, R195), the S4-S5 linker (H258, R259) and the proximal C-terminus (K354, R360, H363, and R366). The residues at the end of S6 overlapped with some proposed earlier by other groups (Zhang et al., 2003; Matavel et al., 2010; Thomas et al., 2011; Telezhkin et al., 2013). Alignment of a KCNQ1 homology model (Smith et al., 2007) with the Kir-PIP2 structures (Hansen et al., 2011; Whorton and MacKinnon, 2011) revealed remarkable overlap of the VSD-PGD interface site in KCNQ1 the Kir PIP2 binding site (Zaydman et al., 2012). This evidence suggests that the VSD-PGD interface site constitutes a true PIP2 binding site that bridges basic residues in the VSD and the proximal C-terminus (including the S6 gate) (Figure 4). This molecular model is consistent with the biophysical model that PIP2 is required for coupling in KCNQ1. Furthermore, the basic residues within the VSD-PGD interface site are well conserved among all KCNQ channels (Figure 3) suggesting that PIP2 may be required for VSD-PGD coupling in KCNQ2-KCNQ5 as well. Along these lines, in a recent molecular dynamics simulation of KCNQ2, Yang and colleagues observed state-dependent interactions between PIP2 and K162 in the S2-S3 linker and K230 in the S4-S5 linker, and they observed, in experiments, that mutations of these residues cause a change in the apparent PIP2 affinity for PIP2 (Zhang et al., 2013).
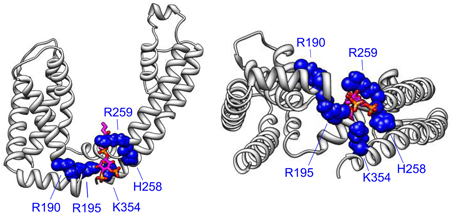
Figure 4. The VSD-PGD interface site. Side view (left) and bottom view (right) of a single KCNQ1 subunit from a homology model (Smith et al., 2007), which was built of the template of the Kv1.2 crystal structure (Long et al., 2005). PIP2 molecule (magenta, orange, and red) is positioned by aligning the Kir2.2-PIP2 crystal structure (Hansen et al., 2011) as done previously (Zaydman et al., 2013). Important residues for PIP2-dependent coupling are shown. Note that some additional residues reside in more C-terminal regions that are not represented in the homology model due to a lack of a structural template for the KCNQ C-terminus.
Shapiro and colleagues identified a possible interaction between the helix A-B linker of the C-terminus and PIP2 (Hernandez et al., 2008). They used a chimera approach to investigate the structural basis for the large difference in apparent PIP2 affinity between KCNQ3 and KCNQ4 channels. They found that chimeras that exchange portions of the C-terminus including the helix A-B linker yield chimeric channels with an apparent affinity that resembles the channel from which the A-B linker originates more than the background channel. For example, the KCNQ4 A-B linker confers wt KCNQ4-like sensitivity in the background of KCNQ3. The A-B linker contains several basic residues that are highly conserved among KCNQ2-KCNQ5 (Figure 3). Neutralizing and charge reversing mutations of these residues exhibit decreased on cell open probability, and a triple charge reversing mutation (K352E/R459E/R461E KCNQ2) decreases the apparent affinity to exogenous PIP2 (Hernandez et al., 2008) and the binding of KCNQ2 C-terminal fragments to immobilized phosphoinositides (Telezhkin et al., 2013). However, the critical A-B linker residues are not conserved in KCNQ1 (Figure 3), and chimeras replacing the KCNQ2 and KCNQ3 C-termini with that of KCNQ1 generates functional KCNQ2CTQ1/KCNQ3CTQ1 channels (Li et al., 2013). Furthermore, deletion of the A-B linker yields functional, PIP2-dependent KCNQ2 channels that are no more severely inhibited by PIP2 depletion than WT KCNQ2 (Aivar et al., 2012). These results suggest that the site at the A-B linker may not be the primary binding site for PIP2. Indeed, Brown and colleagues studied dic8-PIP2 dependence of the wild type KCNQ2/KCNQ3 and the wild type KCNQ2 coexpressed with the mutant KCNQ3 containing charge reversal of key residues in the A-B linker site, KCNQ2/KCNQ3(EEE), and suggested that the mutations in KCNQ3(EEE) do not change PIP2 binding affinity directly but reduce channel activation by markedly decreasing channel opening (Telezhkin et al., 2012a).
The residues in the distal C-terminus site (R539 and R555 in KCNQ1) were found by the Loussouarn group (Park et al., 2005). They studied the long QT syndrome-associated mutations R539W and R555C, located within helix C of the C-terminus, and found that these mutations decrease the apparent affinity KCNQ1+KCNE1 channels to exogenous PIP2. The distal position of R539 and R555 in the protein sequence makes it difficult to locate these residues with respect to the membrane because the structure of the large KCNQ C-terminus is unknown. There are several possible explanations for how these residues affect PIP2 dependent activation. First they could form an independent PIP2 binding site that affects channel gating. Second, the structure of the C-terminus could position these residues close to the VSD-PGD interface site to form a single PIP2 binding site. Third, the distal C-terminus site could be allosterically linked to a distant PIP2 binding site instead of directly coordinating PIP2. Further studies are required to address these possibilities.
The remarkable similarity in the location of the putative PIP2 binding site in KCNQ1 and the PIP2 binding site in the Kir crystal structure (Zaydman et al., 2013) suggests a conserved mechanism for PIP2 regulation of voltage-dependent and voltage independent K+ channels. Consistently, recent structural studies suggest that PIP2 is required to couple the Kir cytosolic domain, a sensor of intracellular factors, to the PGD. The Kir2.2 structures reveal that PIP2 binding pulls the cystosolic domain toward the PGD by roughly six angstroms and causes them to engage in a set of interactions that moves the PGD toward an open conformation (Hansen et al., 2011). The Kir3.1 structures suggest that PIP2 binding in between the cytosolic domain and the PGD is required to couple opening of the intracellular gate within the cytosolic domain to opening of the PGD (Whorton and MacKinnon, 2011). Thus the activation of Kir channels by PIP2, which has been attributed to open-state stabilization using linear activation schemes, is likely due to potentiation of coupling between the cytosolic domain and the PGD. Therefore, coupling between modular sensor domains and the PGD could be a conserved mechanism for PIP2 regulation of ion channels. Along these lines, Kohout et al. showed that PIP2 is required for mutations of the active site of the CiVSP phosphatase domain to shift the voltage-dependence of VSD activation, indicating that PIP2-dependent coupling may extend beyond PIP2-sensitive ion channels to PIP2 sensitive transmembrane proteins (Kohout et al., 2010). Even further yet, De Costa et al. demonstrated that anionic lipids (not PIP2) are required to couple the conformation of the extracellular ligand-sensing domain of the nicotinic acetylcholine receptor to the conformation of the transmembrane pore (daCosta and Baenziger, 2009; daCosta et al., 2009). From all these studies emerges a common theme, cofactor lipids modulate the function of transmembrane proteins by potentiating the coupling between modular sensor and effector domains. In fact this collection of work demonstrates this point in nearly every combination: transmembrane sensor to cytosolic effector (CiVSP), cytosolic sensor to transmembrane effector (Kir), extracellular sensor to transmembrane effector (nAChr), and transmembrane sensor to transmembrane effector (KCNQ1) (Figure 5).
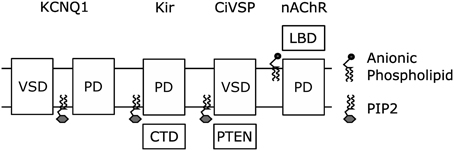
Figure 5. Coupling of modular sensor and effector domains as a general mechanism for the regulation of transmembrane proteins by PIP2 or other anionic phospholipids.
Voltage-gated ion channels are an appealing drug target (Kaczorowski et al., 2008). They are expressed in the surface membrane of many cells, directly determine cellular excitability, and couple to intracellular signaling pathways. Furthermore, a diverse set of diseases arise from inherited or acquired disturbances in ion channel function, and PIP2 sensitive channels have been implicated in many of these channelopathies (Logothetis et al., 2010). The predominant strategies for modulating voltage-gated ion channel function with small molecules have been targeted at VSD activation, PGD opening, or permeation, while VSD-PGD coupling has been relatively ignored. An argument can be made that VSD-PGD coupling is superior drug target. Theoretically, modulating coupling can increase or decrease the magnitude of the current without significantly changing the critical properties of voltage-dependence and time-dependence, as we have seen in the case of KCNQ1 (Zaydman et al., 2013). This is significant because voltage-gated ion channels often work in concert, and the voltage- and time- dependence of the current are critical to efficient and orderly excitation waveforms. With the discovery of PIP2 mediated VSD-PGD coupling in KCNQ1 and the characterization of the binding pocket for coupling (Zaydman et al., 2013) we now have a structural target for small molecules to modulate VSD-PGD coupling. Of course, many issues regarding safety, efficacy and specificity must be addressed before such a therapeutic can be brought to market. However, it is exciting that the compound zinc pyrithione has recently been shown to rescue KCNQ channel current following PIP2 depletion, compete with PIP2 for activation of KCNQ, and to lose activity when mutations are made to the putative PIP2 binding residues (Zhou et al., 2013). These results suggest that zinc pyrithione targets the VSD-PGD coupling that is typically mediated by PIP2 binding, in which case it would represent the first in its class.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abderemane-Ali, F., Es-Salah-Lamoureux, Z., Delemotte, L., Kasimova, M. A., Labro, A. J., Snyders, D. J., et al. (2012). Dual effect of PIP2 on Shaker potassium channels. J. Biol. Chem. 287, 36158–36167. doi: 10.1074/jbc.M112.382085
Aivar, P., Fernández-Orth, J., Gomis-Perez, C., Alberdi, A., Alaimo, A., Rodríguez, M. S., et al. (2012). Surface expression and subunit specific control of steady protein levels by the Kv7.2 helix A-B linker. PLoS ONE 7:e47263. doi: 10.1371/journal.pone.0047263
Arcisio-Miranda, M., Muroi, Y., Chowdhury, S., and Chanda, B. (2010). Molecular mechanism of allosteric modification of voltage-dependent sodium channels by local anesthetics. J. Gen. Physiol. 136, 541–554. doi: 10.1085/jgp.201010438
Arrigoni, C., Schroeder, I., Romani, G., Van Etten, J. L., Thiel, G., and Moroni, A. (2013). The voltage-sensing domain of a phosphatase gates the pore of a potassium channel. J. Gen. Physiol. 141, 389–395. doi: 10.1085/jgp.201210940
Barhanin, J., Lesage, F., Guillemare, E., Fink, M., Lazdunski, M., and Romey, G. (1996). K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80. doi: 10.1038/384078a0
Bezanilla, F. (2008). How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol. 9, 323–332. doi: 10.1038/nrm2376
Bian, J., Cui, J., and McDonald, T. V. (2001). HERG K(+) channel activity is regulated by changes in phosphatidyl inositol 4,5-bisphosphate. Circ. Res. 89, 1168–1176. doi: 10.1161/hh2401.101375
Brown, D. A., Hughes, S. A., Marsh, S. J., and Tinker, A. (2007). Regulation of M(Kv7.2/7.3) channels in neurons by PIP2 and products Of PIP2 hydrolysis: significance for receptor-mediated inhibition. J. Physiol. 582(Pt 3), 917–925. doi: 10.1113/jphysiol.2007.132498
Butterwick, J. A., and MacKinnon, R. (2010). Solution structure and phospholipid interactions of the isolated voltage-sensor domain from KvAP. J. Mol. Biol. 403, 591–606. doi: 10.1016/j.jmb.2010.09.012
Chowdhury, S., and Chanda, B. (2012). Thermodynamics of electromechanical coupling in voltage-gated ion channels. J. Gen. Physiol. 140, 10. doi: 10.1085/jgp.201210840
Colquhoun, D. (1998). Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br. J. Pharmacol. 125, 923–947. doi: 10.1038/sj.bjp.0702164
daCosta, C. J. B., and Baenziger, J. E. (2009). A lipid-dependent uncoupled conformation of the acetylcholine receptor. J. Biol. Chem. 284, 17819–17825. doi: 10.1074/jbc.M900030200
daCosta, C. J. B., Medaglia, S. A., Lavigne, N., Wang, S., Carswell, C. L., and Baenziger, J. E. (2009). Anionic lipids allosterically modulate multiple nicotinic acetylcholine receptor conformational equilibria. J. Biol. Chem. 284, 33841–33849. doi: 10.1074/jbc.M109.048280
Delemotte, L., Tarek, M., Klein, M. L., Amaral, C., and Treptow, W. (2011). Intermediate states of the Kv1.2 voltage sensor from atomistic molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 108, 6109. doi: 10.1073/pnas.1102724108
Delmas, P., and Brown, D. A. (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850–862. doi: 10.1038/nrn1785
Falkenburger, B. H., Jensen, J. B., and Hille, B. (2010). Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J. Gen. Physiol. 135, 99–114. doi: 10.1085/jgp.200910345
Freites, J. A. (2005). From the cover: interface connections of a transmembrane voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 102, 15059–15064. doi: 10.1073/pnas.0507618102
Gamper, N., Reznikov, V., Yamada, Y., Yang, J., and Shapiro, M. S. (2004). Phosphotidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-Type Ca2+ channels. J. Neurosci. 24, 10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004
Grabe, M., Lai, H. C., Jain, M., Nung Jan, Y., and Yeh Jan, L. (2007). Structure prediction for the down state of a potassium channel voltage sensor. Nature 445, 550–553. doi: 10.1038/nature05494
Haddad, G. A., and Blunck, R. (2011). Mode shift of the voltage sensors in Shaker K+ channels is caused by energetic coupling to the pore domain. J. Gen. Physiol. 137, 455–472. doi: 10.1085/jgp.201010573
Hansen, S. B., Tao, X., and MacKinnon, R. (2011). Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature 477, 495–498. doi: 10.1038/nature10370
Heginbotham, L. (1998). Functional reconstitution of a prokaryotic K+ channel. J. Gen. Physiol. 111, 741–749. doi: 10.1085/jgp.111.6.741
Hernandez, C. C., Zaika, O., and Shapiro, M. S. (2008). A carboxy-terminal inter-helix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J. Gen. Physiol. 132, 361. doi: 10.1085/jgp.200810007
Hilgemann, D. W. (2012). Fitting KV potassium channels into the PIP2 puzzle: Hille group connects dots between illustrious HH groups. J. Gen. Physiol. 140, 245–248. doi: 10.1085/jgp.201210874
Hilgemann, D. W., and Ball, R. (1996). Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 273, 956–959. doi: 10.1126/science.273.5277.956
Hilgemann, D. W., Feng, S., and Nasuhoglu, C. (2001). The complex and intriguing lives of PIP2 with Ion channels and transporters. Sci. STKE 2001, re19. doi: 10.1126/stke.2001.111.re19
Horrigan, F. T., and Aldrich, R. W. (1999). Allosteric voltage gating of potassium channels II. Mslo channel gating charge movement in the absence of Ca(2+). J. Gen. Physiol. 114, 305–336. doi: 10.1085/jgp.114.2.305
Horrigan, F. T., Cui, J., and Aldrich, R. W. (1999). Allosteric voltage gating of potassium channels I. Mslo ionic currents in the absence of Ca(2+). J. Gen. Physiol. 114, 277–304. doi: 10.1085/jgp.114.2.277
Islas, L. D., and Sigworth, F. J. (1999). Voltage sensitivity and gating charge in shaker and shab family potassium channels. J. Gen. Physiol. 114, 723. doi: 10.1085/jgp.114.5.723
Jensen, B. C., O'Connell, T. D., and Simpson, P. C. (2011). Alpha-1 adrenergic receptors: targets for agonist drugs to treat heart failure. J. Mol. Cell. Cardiol. 51, 229–233. doi: 10.1016/j.yjmcc.2010.11.014
Jensen, M. O., Jogini, V., Borhani, D. W., Leffler, A. E., Dror, R. O., and Shaw, D. E. (2012). Mechanism of voltage gating in potassium channels. Science 336, 229–233. doi: 10.1126/science.1216533
Kaczorowski, G. J., McManus, O. B., Priest, B. T., and Garcia, M. L. (2008). Ion channels as drug targets: the next GPSCRs. J. Gen. Physiol. 131, 399–405. doi: 10.1085/jgp.200709946
Kohout, S. C., Bell, S. C., Liu, L., Xu, Q., Minor, D. L., and Isacoff, E. Y. (2010). Electrochemical coupling in the voltage-dependent phosphatase Ci-VSP. Nat. Chem. Biol. 6, 369–375. doi: 10.1038/nchembio.349
Kruse, M., Hammond, G. R. V., and Hille, B. (2012). Regulation of voltage-gated potassium channels by PI(4,5)P2. J. Gen. Physiol. 140, 189–205. doi: 10.1085/jgp.201210806
Ledwell, J. L., and Aldrich, R. W. (1999). Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 113, 389–414. doi: 10.1085/jgp.113.3.389
Lee, S.-Y., Banerjee, A., and MacKinnon, R. (2009). Two separate interfaces between the voltage sensor and pore are required for the function of voltage-dependent K(+) channels. PLoS Biol. 7:e47. doi: 10.1371/journal.pbio.1000047
Li, Y., Gamper, N., Hilgemann, D. W., and Shapiro, M. S. (2005). Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25, 9825–9835. doi: 10.1523/JNEUROSCI.2597-05.2005
Li, Y., Gao, J., Lu, Z., McFarland, K., Shi, J., Bock, K., et al. (2013). Intracellular ATP binding is required to activate the slowly activating K+ channel I(Ks). Proc. Natl. Acad. Sci. U.S.A. 110, 18922–18927. doi: 10.1073/pnas.1315649110
Li, Y., Zaydman, M. A., Wu, D., Shi, J., Guan, M., Virgin-Downey, B., et al. (2011). KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc. Natl. Acad. Sci. U.S.A. 108, 9095–9100. doi: 10.1073/pnas.1100872108
Lingwood, D., and Simons, K. (2009). Lipid rafts as a membrane-organizing principle. Science 327, 46–50. doi: 10.1126/science.1174621
Logothetis, D. E., Petrou, V. I., Adney, S. K., and Mahajan, R. (2010). Channelopathies linked to plasma membrane phosphoinositides. Pflügers Arch. 460, 321–341. doi: 10.1007/s00424-010-0828-y
Long, S. B. (2005). Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309, 903–908. doi: 10.1126/science.1116270
Long, S. B., Campbell, E. B., and MacKinnon, R. (2005). Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903. doi: 10.1126/science.1116269
Long, S. B., Tao, X., Campbell, E. B., and MacKinnon, R. (2007). Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. doi: 10.1038/nature06265
Loussouarn, G., Park, K.-H., Bellocq, C., Baró, I., Charpentier, F., and Escande, D. (2003). Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 22, 5412–5421. doi: 10.1093/emboj/cdg526
Lu, Z., Klem, A. M., and Ramu, Y. (2001). Ion conduction pore is conserved among potassium channels. Nature 413, 809–813. doi: 10.1038/35101535
Lu, Z., Klem, A. M., and Ramu, Y. (2002). Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J. Gen. Physiol. 120, 663–676. doi: 10.1085/jgp.20028696
Ma, L.-J., Ohmert, I., and Vardanyan, V. (2011). Allosteric features of KCNQ1 gating revealed by alanine scanning mutagenesis. Biophys. J. 100, 885–894. doi: 10.1016/j.bpj.2010.12.3726
Mannuzzu, L. M., Moronne, M. M., and Isacoff, E. Y. (1996). Direct physical measure of conformational rearrangement underlying potassium channel gating. Science 271, 213–216. doi: 10.1126/science.271.5246.213
Matavel, A., and Lopes, C. M. B. (2009). PKC activation and PIP2 depletion underlie biphasic regulation of IKs by Gq-coupled receptors. J. Mol. Cell. Cardiol. 46, 704–712. doi: 10.1016/j.yjmcc.2009.02.006
Matavel, A., Medei, E., and Lopes, C. M. B. (2010). PKA and PKC partially rescue Long QT type 1 phenotype by restoring channel-PIP2 interactions. Channels 4, 3–11. doi: 10.4161/chan.4.1.10227
McLaughlin, S., Wang, J., Gambhir, A., and Murray, D. (2002). PIP(2) and proteins: interactions, organization, and information flow. Annu. Rev. Biophys. Biomol. Struct. 31, 151–175. doi: 10.1146/annurev.biophys.31.082901.134259
Meisel, E., Meidan, D., Haitin, Y., Giladi, M., Peretz, A., and Attali, B. (2012). KCNQ1 channels do not undergo concerted but sequential gating transitions in both the absence and the presence of KCNE1 protein. J. Biol. Chem. 287, 34212–34224. doi: 10.1074/jbc.M112.364901
Milescu, M., Bosmans, F., Lee, S., Alabi, A. A., Kim, J. I., and Swartz, K. J. (2009). Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 16, 1080–1085. doi: 10.1038/nsmb.1679
Murata, Y., Iwasaki, H., Sasaki, M., Inaba, K., and Okamura, Y. (2005). Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243. doi: 10.1038/nature03650
Murata, Y., and Okamura, Y. (2007). Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J. Physiol. 583, 875–889. doi: 10.1113/jphysiol.2007.134775
Nakajo, K., and Kubo, Y. (2005). Protein kinase C shifts the voltage dependence of KCNQ/M channels expressed in Xenopus oocytes. J. Physiol. 569, 59–74. doi: 10.1113/jphysiol.2005.094995
Nakanishi, S., Catt, K. J., and Balla, T. (1995). A wortmannin-sensitive phosphatidylinositol 4-kinase that regulates hormone-sensitive pools of inositolphospholipids. Proc. Natl. Acad. Sci. U.S.A. 92, 5317. doi: 10.1073/pnas.92.12.5317
Nayak, S., Batalov, S., Jegla, T., and Zmasek, C. (2009). Evolution of the human ion channel set. Comb. Chem. High Throughput Screen. 12, 2–23. doi: 10.2174/138620709787047957
Neyroud, N., Tesson, F., Denjoy, I., Leibovici, M., Donger, C., Barhanin, J., et al. (1997). A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet. 15, 186–189. doi: 10.1038/ng0297-186
Okamura, Y., Murata, Y., and Iwasaki, H. (2009). Voltage-sensing phosphatase: actions and potentials. J. Physiol. 587, 513–520. doi: 10.1113/jphysiol.2008.163097
Oliver, D., Lien, C.-C., Soom, M., Baukrowitz, T., Jonas, P., and Fakler, B. (2004). Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science 304, 265–270. doi: 10.1126/science.1094113
Osteen, J. D., Barro-Soria, R., Robey, S., Sampson, K. J., Kass, R. S., and Larsson, H. P. (2012). Allosteric gating mechanism underlies the flexible gating of KCNQ1 potassium channels. Proc. Natl. Acad. Sci. U.S.A. 109, 7103–7108. doi: 10.1073/pnas.1201582109
Osteen, J. D., Gonzalez, C., Sampson, K. J., Iyer, V., Rebolledo, S., Larsson, H. P., et al. (2010). KCNE1 alters the voltage sensor movements necessary to open the KCNQ1 channel gate. Proc. Natl. Acad. Sci. U.S.A. 107, 22710–22715. doi: 10.1073/pnas.1016300108
Park, K.-H., Piron, J., Dahimene, S., Merot, J., Baro, I., Escande, D., et al. (2005). Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ. Res. 96, 730–739. doi: 10.1161/01.RES.0000161451.04649.a8
Payandeh, J., Scheuer, T., Zheng, N., and Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. doi: 10.1038/nature10238
Pian, P., Bucchi, A., Robinson, R. B., and Siegelbaum, S. A. (2006). Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J. Gen. Physiol. 128, 593–604. doi: 10.1085/jgp.200609648
Piron, J., Choveau, F. S., Amarouch, M. Y., Rodriguez, N., Charpentier, F., Merot, J., et al. (2010). KCNE1-KCNQ1 osmoregulation by interaction of phosphatidylinositol-4,5-bisphosphate with Mg2+and polyamines. J. Physiol. 588, 3471–3483. doi: 10.1113/jphysiol.2010.195313
Pusch, M., Magrassi, R., Wollnik, B., and Conti, F. (1998). Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75, 785–792. doi: 10.1016/S0006-3495(98)77568-X
Ramsey, I. S., Moran, M. M., Chong, J. A., and Clapham, D. E. (2006). A voltage-gated proton-selective channel lacking the pore domain. Nature 440, 1213–1216. doi: 10.1038/nature04700
Ramu, Y., Xu, Y., and Lu, Z. (2006). Enzymatic activation of voltage-gated potassium channels. Nature 442, 696–699. doi: 10.1038/nature04880
Rodriguez-Menchaca, A. A., Adney, S. K., Tang, Q.-Y., Meng, X.-Y., Rosenhouse-Dantsker, A., Cui, M., et al. (2012). PIP2 controls voltage-sensor movement and pore opening of Kv channels through the S4-S5 linker. Proc. Natl. Acad. Sci. U.S.A. 109, E2399–E2408. doi: 10.1073/pnas.1207901109
Rosenhouse-Dantsker, A., and Logothetis, D. E. (2007). Molecular characteristics of phosphoinositide binding. Pflugers Arch. 455, 45–53. doi: 10.1007/s00424-007-0291-6
Rusten, T. E., and Stenmark, H. (2006). Analyzing phosphoinositides and their interacting proteins. Nat. Methods 3, 251–258. doi: 10.1038/nmeth867
Ryu, S., and Yellen, G. (2012). Charge movement in gating-locked HCN channels reveals weak coupling of voltage sensors and gate. J. Gen. Physiol. 140, 469–479. doi: 10.1085/jgp.201210850
Sanguinetti, M. C., Curran, M. E., Zou, A., Shen, J., Spector, P. S., Atkinson, D. L., et al. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83. doi: 10.1038/384080a0
Santos, J. S., Asmar-Rovira, G. A., Han, G. W., Liu, W., Syeda, R., Cherezov, V., et al. (2012). Crystal structure of a voltage-gated K+ channel pore module in a closed state in lipid membranes. J. Biol. Chem. 287, 43063–43070. doi: 10.1074/jbc.M112.415091
Santos, J. S., Grigoriev, S. M., and Montal, M. (2008). Molecular template for a voltage sensor in a novel K+ channel. III. Functional reconstitution of a sensorless pore module from a prokaryotic Kv channel. J. Gen. Physiol. 132, 651–666. doi: 10.1085/jgp.200810077
Sasaki, M. (2006). A voltage sensor-domain protein is a voltage-gated proton channel. Science 312, 589–592. doi: 10.1126/science.1122352
Schmidt, D., Jiang, Q.-X., and MacKinnon, R. (2006). Phospholipids and the origin of cationic gating charges in voltage sensors. Nature 444, 775–779. doi: 10.1038/nature05416
Schoppa, N. E., McCormack, K., Tanouye, M. A., and Sigworth, F. J. (1992). The size of gating charge in wild-type and mutant shaker potassium channels. Science 255, 1712–1715. doi: 10.1126/science.1553560
Shapiro, M. S., Roche, J. P., Kaftan, E. J., Cruzblanca, H., Mackie, K., and Hille, B. (2000). Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J. Neurosci. 20, 1710–1721.
Shaya, D., Kreir, M., Robbins, R. A., Wong, S., Hammon, J., Brüggemann, A., et al. (2011). Voltage-gated sodium channel (NaV) protein dissection creates a set of functional pore-only proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 12313–12318. doi: 10.1073/pnas.1106811108
Shen, W. (2005). Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J. Neurosci. 25, 7449–7458. doi: 10.1523/JNEUROSCI.1381-05.2005
Smith, J., Vanoye, C. G., George, A. L. Jr., Meiler, J., and Sanders, C. R. (2007). Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry 46, 14141–14152. doi: 10.1021/bi701597s
Soler-Llavina, G. J., Chang, T.-H., and Swartz, K. J. (2006). Functional interactions at the interface between voltage-sensing and pore domains in the Shaker K(v) channel. Neuron 52, 623–634. doi: 10.1016/j.neuron.2006.10.005
Suh, B.-C., and Hille, B. (2002). Recovery from muscarinic modulation of M current chanels requires phosphatidyinositol 4,5-bisphosphate. Neuron 35, 507–520. doi: 10.1016/S0896-6273(02)00790-0
Suh, B.-C., and Hille, B. (2005). Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol. 15, 370–378. doi: 10.1016/j.conb.2005.05.005
Suh, B.-C., and Hille, B. (2007a). Regulation of KCNQ channels by manipulation of phosphoinositides. J. Physiol. 582(Pt 3), 911–916. doi: 10.1113/jphysiol.2007.132647
Suh, B.-C., and Hille, B. (2007b). Electrostatic interaction of internal Mg2+ with membrane PIP2 Seen with KCNQ K+ channels. J. Gen. Physiol. 130, 241–256. doi: 10.1085/jgp.200709821
Suh, B.-C., and Hille, B. (2008). PIP2 is a necessary cofactor for ion channel function: how and why? Annu. Rev. Biophys. 37, 175–195. doi: 10.1146/annurev.biophys.37.032807.125859
Suh, B.-C., Inoue, T., Meyer, T., and Hille, B. (2006). Rapid chemically induced changes of ptdins(4,5)P2 gate KCNQ ion channels. Sci. Signal. 314, 1454. doi: 10.1126/science.1131163
Suh, B.-C., Leal, K., and Hille, B. (2010). Modulation of high-voltage activated Ca(2+) channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron 67, 224–238. doi: 10.1016/j.neuron.2010.07.001
Telezhkin, V., Brown, D. A., and Gibb, A. J. (2012a). Distinct subunit contributions to the activation of M-type potassium channels by PI(4,5)P2. J. Gen. Physiol. 140, 41–53. doi: 10.1085/jgp.201210796
Telezhkin, V., Reilly, J. M., Thomas, A. M., Tinker, A., and Brown, D. A. (2012b). Structural requirements of membrane phospholipids for M-type potassium channel activation and binding. J. Biol. Chem. 287, 10001–10012. doi: 10.1074/jbc.M111.322552
Telezhkin, V., Thomas, A. M., Harmer, S. C., Tinker, A., and Brown, D. A. (2013). A basic residue in the proximal C-terminus is necessary for efficient activation of the M-channel subunit Kv7.2 by PI(4,5)P2. Pflgers Arch. 465, 945–953. doi: 10.1007/s00424-012-1199-3
Thomas, A. M., Harmer, S. C., Khambra, T., and Tinker, A. (2011). Characterization of a binding site for anionic phospholipids on KCNQ1. J. Biol. Chem. 286, 2088–2100. doi: 10.1074/jbc.M110.153551
Treptow, W., and Tarek, M. (2006). Environment of the gating charges in the Kv1.2 Shaker potassium channel. Biophys. J. 90, L64–L66. doi: 10.1529/biophysj.106.080754
Tristani-Firouzi, M., Chen, J., and Sanguinetti, M. C. (2002). Interactions between S4-S5 linker and S6 transmembrane domain modulate gating of HERG K+ channels. J. Biol. Chem. 277, 18994–19000. doi: 10.1074/jbc.M200410200
Vaithianathan, T., Bukiya, A., Liu, J., Liu, P., Asuncion-Chin, M., Fan, Z., et al. (2008). Direct regulation of BK channels by phosphatidylinositol 4,5-bisphosphate as a novel signaling pathway. J. Gen. Physiol. 132, 13–28. doi: 10.1085/jgp.200709913
Valiyaveetil, F., Zhou, Y., and MacKinnon, R. (2002). Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry 41, 10771–10777. doi: 10.1021/bi026215y
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893. doi: 10.1126/science.282.5395.1890
Wang, Q., Curran, M. E., Splawski, I., Burn, T. C., Millholland, J. M., VanRaay, T. J., et al. (1996). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 12, 17–23. doi: 10.1038/ng0196-17
Whorton, M. R., and MacKinnon, R. (2011). Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell 147, 199–208. doi: 10.1016/j.cell.2011.07.046
Wu, L., Bauer, C. S., Zhen, X.-G., Xie, C., and Yang, J. (2002). Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature 419, 947–952. doi: 10.1038/nature01118
Xu, Y., Ramu, Y., and Lu, Z. (2008). Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature 451, 826–830. doi: 10.1038/nature06618
Yellen, G. (1998). The moving parts of voltage-gated ion channels. Q. Rev. Biophys. 31, 239–295. doi: 10.1017/S0033583598003448
Zagotta, W. N., Hoshi, T., and Aldrich, R. W. (1994). Shaker potassium channel gating. III: evaluation of kinetic models for activation. J. Gen. Physiol. 103, 321–362. doi: 10.1085/jgp.103.2.321
Zaydman, M. A., Silva, J. R., and Cui, J. (2012). Ion channel associated diseases: overview of molecular mechanisms. Chem. Rev. 112, 6319–6333. doi: 10.1021/cr300360k
Zaydman, M. A., Silva, J. R., Delaloye, K., Li, Y., Liang, H., Larsson, H. P., et al. (2013). Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci. U.S.A. 110, 13180–13185. doi: 10.1073/pnas.1305167110
Zhang, H., Craciun, L., Mirshahi, T., Rohacs, T., Lopes, C., Jin, T., et al. (2003). PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975. doi: 10.1016/S0896-6273(03)00125-9
Zhang, Q., Zhou, P., Chen, Z., Li, M., Jiang, H., Gao, Z., et al. (2013). Dynamic PIP2 interactions with voltage sensor elements contribute to KCNQ2 channel gating. Proc. Natl. Acad. Sci. U.S.A. 110, 20093–20098. doi: 10.1073/pnas.1312483110
Zheng, H., Liu, W., Anderson, L. Y., and Jiang, Q.-X. (2011). Lipid-dependent gating of a voltage-gated potassium channel. Nat. Commun. 2, 250. doi: 10.1038/ncomms1254
Zhou, P., Yu, H., Gu, M., Nan, F. J., Gao, Z., and Li, M. (2013). Phosphatidylinositol 4,5-bisphosphate alters pharmacological selectivity for epilepsy-causing KCNQ potassium channels. Proc. Natl. Acad. Sci. U.S.A. 110, 8725–8731. doi: 10.1073/pnas.1302167110
Keywords: PIP2, voltage-gating, lipid modulations, ion channel, KCNQ
Citation: Zaydman MA and Cui J (2014) PIP2 regulation of KCNQ channels: biophysical and molecular mechanisms for lipid modulation of voltage-dependent gating. Front. Physiol. 5:195. doi: 10.3389/fphys.2014.00195
Received: 15 February 2014; Accepted: 08 May 2014;
Published online: 27 May 2014.
Edited by:
Harley Takatsuna Kurata, University of British Columbia, CanadaReviewed by:
Álvaro Villarroel, Euskal Herriko Unibertsitatea, SpainCopyright © 2014 Zaydman and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianmin Cui, Department of Biomedical Engineering, Washington University in St. Louis, Whitaker Rm 290C, One Brookings Drive, St. Louis, MO 63130, USA e-mail:amN1aUB3dXN0bC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.