 Juan-Juan Sheng
Juan-Juan Sheng Jian-Ping Jin
Jian-Ping Jin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 30 April 2014
Sec. Striated Muscle Physiology
Volume 5 - 2014 | https://doi.org/10.3389/fphys.2014.00165
Troponin plays a central role in regulating the contraction and relaxation of vertebrate striated muscles. This review focuses on the isoform gene regulation, alternative RNA splicing, and posttranslational modifications of troponin subunits in cardiac development and adaptation. Transcriptional and posttranscriptional regulations such as phosphorylation and proteolysis modifications, and structure-function relationships of troponin subunit proteins are summarized. The physiological and pathophysiological significances are discussed for impacts on cardiac muscle contractility, heart function, and adaptations in health and diseases.
The primary contractile unit of striated muscles, e.g., the vertebrate cardiac and skeletal muscles, is the sarcomere. A sarcomere is comprised of overlapping myosin thick filaments and actin thin filaments. The interaction between myosin and actin activates myosin ATPase and powers myofilament sliding and muscle contraction. This process is regulated by the level of cytosolic Ca2+ through the thin filament-associated troponin-tropomyosin system (Gordon et al., 2000).
Troponin plays a central role in regulating the contraction and relaxation of striated muscles. The structure and function of troponin have been extensively investigated in the past four decades as comprehensively summarized in several recent review articles (Murphy, 2006; Jin et al., 2008; Wei and Jin, 2011). To provide an overview of the current understanding of the function and regulation of troponin in cardiac muscle, the present review focuses on the isoform genes, splice-forms and posttranslational modifications of troponin in cardiac function during postnatal development and physiological and pathophysiological adaptations.
The troponin complex is a heterotrimer consisting of three protein subunits. Named according to their functions, they are the Ca2+-binding subunit troponin C (TnC), the actomyosin ATPase inhibitory subunit troponin I (TnI), and the tropomyosin-binding subunit troponin T (TnT) (Greaser and Gergely, 1971) (Figure 1). Low-resolution X-ray crystallography (White et al., 1987) and electron microscopic (Flicker et al., 1982) studies demonstrated that the troponin complex may be divided into two structural domains: The TnT extension that binds tropomyosin and the core domain that bears most of the regulatory function of troponin. High-resolution crystallographic structure further revealed that the core domain of cardiac troponin contains two structurally distinct subdomains that are the regulatory head (amino acid residues 3–84 of TnC and amino acid residues 150–159 of TnI) and the I-T arm (residues 93–161 of TnC, residues 42–136 of TnI and residues 203–271 of TnT). They are dominated with α-helices connected by flexible linkers that make the molecule asymmetric and highly flexible, a crucial feature for the function of troponin in the regulation of muscle contraction and relaxation (Takeda et al., 2003; Vinogradova et al., 2005).
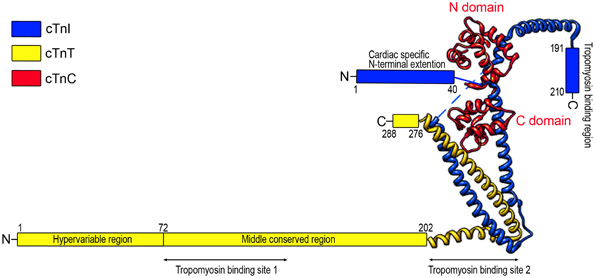
Figure 1. Schematic structure of cardiac troponin. Troponin is a protein complex consisting of three subunits: TnC, TnI, and TnT. The structure of troponin can be divided into two structural domains: the core domain and the TnT extension. High resolution crystallographic structure (Takeda et al., 2003) further revealed that the core domain contains two structural subdomains: the regulatory head (residues 3–84 of TnC and residues 150–159 of TnI) and the I-T arm (residues 93–161 of TnC, residues 42–136 of TnI, and residues 203–271 of TnT). The crystal structure portion of the illustration was redrawn from Takeda et al. (2003) using UCSF (Chimera software, alpha version 1.3).
The three troponin subunits are encoded by separate genes. Each of the genes had evolved into muscle type-specific isoform genes. Their expression is regulated during embryonic and postnatal development as well as physiological and pathological adaptations (Jin et al., 2008; Chong and Jin, 2009; Wei and Jin, 2011). TnI and TnT both have three muscle type-specific isoforms encoded by slow skeletal muscle TnI (TNNI1), fast skeletal muscle TnI (TNNI2), cardiac TnI (TNNI3), slow skeletal muscle TnT (TNNT1), fast skeletal muscle TnT (TNNT3), and cardiac TnT (TNNT2) genes. These TnI and TnT isoform genes are closely linked in three tandem pairs in the vertebrate genomes: Fast TnI-fast TnT (TNNI2-TNNT3), cardiac TnI-slow TnT (TNNI3-TNNT1) and slow TnI-cardiac TnT (TNNI1-TNNT2) (Jin et al., 2008; Chong and Jin, 2009; Feng et al., 2009b), supporting the hypothesis that TnI and TnT genes were duplicated from one common ancestral gene.
In contrast to the presence of three muscle type-specific TnI and TnT isoform genes, TnC is present in only two isoforms in the three striated muscle fiber types. Whereas fast skeletal muscle expresses fast TnC encoded by TNNC2, mature cardiac muscle and slow skeletal muscle share one isoform, the cardiac/slow skeletal muscle TnC encoded by TNNC1 (Schreier et al., 1990; Collins, 1991; Prigozy et al., 1997).
The diversity of isoform genes encoding the subunits of troponin endues the heart with adaptation during development. Discussed in more details in later sections, embryonic heart expresses solely slow TnI paired with cardiac TnT. An isoform transition from slow TnI to solely cardiac TnI in adult heart occurs during development. Cardiac TnI has a heart-specific N-terminal extension that is a regulatory structure specific to the adult cardiac muscle (Chong and Jin, 2009). On the other hand, slow TnI expression in embryonic hearts increases Ca2+ sensitivity of myofilament and the tolerance to acidosis, although it diminishes length dependence of Ca2+ activation (Arteaga et al., 2000). Cardiac TnT also has an N-terminal variable region that undergoes developmentally regulated alternative splicing (Jin and Lin, 1988, 1989) whereas no alternative RNA splicing is found for the transcripts of any of the three TnI isoform genes.
Troponin C belongs to the calmodulin super family of genes, containing four EF-hand helix coil-helix divalent metal ion-binding sites (Collins, 1991; Kawasaki et al., 1998). TnC is a dumbbell-shaped molecule with the N- and C-terminal globular domains connected by a nine turn α-helix (Herzberg and James, 1985). The C-terminal domain of TnC contains two high affinity Ca2+ or Mg2+ binding sites (Site III and Site IV), which are primarily occupied by Mg2+ in resting muscle cells and can become partially bound with Ca2+ during the activation of contraction (Robertson et al., 1981). The C-terminal domain of TnC plays a structural role of maintaining the anchoring affinity of the whole troponin complex to the thin filament (Zot and Potter, 1982).
The N-terminal domain of fast skeletal muscle TnC contains two low affinity metal ion-binding sites designated as Site I and Site II that are regulatory Ca2+-binding sites responsible for the regulation of muscle contraction (Sheng et al., 1990; Sweeney et al., 1990). The transient rise of cytosolic Ca2+ during the activation of contraction results in Ca2+ binding to the N-terminal domain of TnC and induces a cascade of conformational changes in the troponin complex and sarcomeric thin filament (Robertson et al., 1981; Collins, 1991; Gordon et al., 2000; Solaro, 2010). The conformational changes increase the binding affinity of TnC for TnI, promoting a detachment of TnI from actin, which releases the inhibition of actomyosin ATPase and activates myofilament sliding and shortening of the sarcomere (Grabarek et al., 1992).
Different from the fast skeletal muscle TnC, the N-terminal domain of cardiac/slow TnC contains only one active Ca2+ binding site (Site II), whereas Site I had lost the ability of binding Ca2+ (Van Eerd and Takahashi, 1976). Elimination of Ca2+ binding Site II in cardiac/slow TnC renders a cardiac fiber insensitive to Ca2+, whereas reengineering an active Ca2+-binding Site I does not compensate for the effect of Site II mutation. Therefore, Site II plays a critical role responsible for the regulatory function of cardiac/slow TnC (Sweeney et al., 1990). Nonetheless, engineered cardiac/slow TnC in which both Site I and Site II are actively binding to Ca2+ showed increased Ca2+ sensitivity than that of wild type cardiac TnC in which only Site II is active (Sweeney et al., 1990). The Ca2+ sensitivity of cardiac/slow TnC can also be regulated by other myofilament proteins, such as TnI, TnT, tropomyosin, actin, myosin binding protein-C, and myosin (Blumenschein et al., 2001; Burkart et al., 2003a). No alternative splicing or posttranslational regulation of TnC has been observed during development or pathological adaptations.
Troponin I is the inhibitory subunit of the troponin complex. In the absence of Ca2+, its inhibitory region binds with actin, and thereby inhibits actomyosin ATPase (Farah et al., 1994). In the presence of Ca2+, the C-terminal domain of TnC interacts with TnI to induce conformational changes of TnI, releases the inhibitory effect, and initiates muscle contraction (Farah et al., 1994; Perry, 1999).
In vertebrate striated muscles, the three TnI isoform genes (Hastings, 1997; Perry, 1999; Chong and Jin, 2009) are differentially expressed under fiber type-specific and developmentally regulated transcriptional control. Fast skeletal muscle fibers only express fast TnI, and slow skeletal muscle fibers express only slow TnI. Accordingly, a slow to fast TnI (and TnT) isoform switching occurs during the slow to fast fiber type transition in muscle adaptation to unloading (Stevens et al., 2002; Yu et al., 2007).
As indicated above, cardiac muscle switches TnI isoforms during development (Saggin et al., 1989). The slow skeletal muscle TnI gene is expressed in the embryonic heart and switches off during development. Around birth, the expression of cardiac TnI gene up-regulates to completely replace slow TnI in adult cardiac muscle (Saggin et al., 1989; Sasse et al., 1993). The adult heart expresses cardiac TnI as the sole isoform and it does not change under pathological conditions such as ischemic heart disease, dilated cardiomyopathy, or end-stage heart failure (Sasse et al., 1993). This developmental TnI isoform transition may contribute to the differences in the Ca2+ sensitivity and pH responsiveness of force development of cardiomyocytes (Westfall et al., 1999). Over-expression of slow TnI in cardiac muscle of adult transgenic mice impaired cardiomyocyte relaxation and diastolic cardiac function due to increased Ca2+ sensitivity (Fentzke et al., 1999). On the other hand, slow TnI increased the tolerance of cardiomyocytes to acidosis-induced decrease in myofilament Ca2+ sensitivity (Westfall et al., 2000). These findings indicate that slow TnI produces a higher Ca2+ affinity of the troponin complex than that of cardiac TnI, which may maintain Ca2+ sensitivity of myofilament at the lower pH (6.5 vs. 7.0) in embryonic cardiomyocytes (Solaro et al., 1988).
Based on in vitro structure-function relationship studies, the structure of cardiac TnI can be divided into six functional segments (Li et al., 2004) (Figure 1): (a) cardiac-specific N-terminal extension (amino acids 1–30) that is not present in fast TnI and slow TnI; (b) an N-terminal region (amino acids 42–79) that binds the C domain of TnC; (c) a TnT-binding region (amino acids 80–136); (d) the inhibitory peptide (amino acids 128–147) that interacts with TnC and actin–tropomyosin; (e) the switch or triggering region (amino acids 148–163) that binds the N domain of TnC; and (f) the C-terminal region (amino acids 164–210) that binds actin–tropomyosin and is the most conserved segment highly similar among isoforms and across species (Jin et al., 2001, 2008). Recent studies demonstrated that the last 20 amino acids of the C-terminal end segment of TnI (amino acids 191–210), encoded by exon 8 is a Ca2+-modulated allosteric structure (Jin et al., 2001; Zhang et al., 2011a). Protein binding experiments showed that this segment functions through Ca2+-regulated conformational changes and interactions with tropomyosin (Solaro et al., 2008; Zhang et al., 2011a).
There is no alternative RNA splicing found for the transcripts of any TnI genes. In the meantime, posttranslational modifications have major roles in regulating the structure and function of cardiac TnI (Solaro et al., 2008). The mechanisms include amino acid side chain modifications and cleavages of the polypeptide chain, which induce conformational changes that modify the interaction with cardiac TnC and effects on cardiac muscle contractility (Pi et al., 2003; Layland et al., 2005; Westfall et al., 2005; Solaro and Van Der Velden, 2010; Akhter et al., 2012). Phosphorylation also regulates the degradation of cardiac TnI (Di Lisa et al., 1995).
It is well-accepted that phosphorylation of cardiac TnI at Ser23 and Ser24 in the adult heart-specific N-terminal extension regulates the diastolic function of cardiac muscle (Solaro and Kobayashi, 2011). Compiling evidences showed that Ser23 and Ser24 are sequentially phosphorylated by protein kinase A (PKA) under the regulation of adrenergic signaling cascades (Quirk et al., 1995; Solaro et al., 2008; Solaro and Kobayashi, 2011; Rao et al., 2012), reducing the Ca2+-binding affinity of the N domain regulatory site of cardiac TnC (Zhang et al., 1995b) and enhancing diastolic function of cardiac muscle (Zhang et al., 1995a; Stelzer et al., 2007; Li et al., 2010). Bisphosphorylation at Ser23 and Ser24 results in weakening interactions of cardiac TnI with the N-lobe of cardiac TnC and favoring the intra-molecular interaction between the N-terminal extension and the inhibitory region of cardiac TnI (Howarth et al., 2007). These two serine residues have also been reported to be phosphorylated in vitro by PKC-β, PKC-ε (Kobayashi et al., 2005), PKD (previously named PKCμ) (Haworth et al., 2004; Cuello et al., 2007; Bardswell et al., 2010) and PKG (Layland et al., 2002).
While PKA phosphorylation of cardiac TnI at Ser23/Ser24 increases myocardial relaxation, PKC phosphorylation of cardiac TnI exerts an antithetic role (Sakthivel et al., 2005; Kooij et al., 2011). PKC phosphorylates cardiac TnI at Ser43 and Ser45 (residue # in mouse sequence) in the region binding the C domain of TnC and Thr144 in the inhibitory region, slowing cardiac relaxation and increasing the duration of calcium transient and twitch contraction (Macgowan et al., 2001; Pi et al., 2002; Burkart et al., 2003b; Westfall et al., 2005). In mouse heart, phosphorylation of Thr144 of cardiac TnI by PKC-βII increased myofilament Ca2+ sensitivity (Wang et al., 2006). Substitution with Pro at Thr144 delayed relaxation, suggesting a role of Thr144 in accelerating relaxation in cardiomyocytes (Westfall et al., 2005). However, another study found that Thr144 phosphorylation did not modify the thin filament Ca2+ sensitivity, but depressed cooperative activation of thin filaments (Lu et al., 2010). The mechanism how phosphorylation of Thr144 regulates cardiac troponin requires further investigation.
It was reported that phosphorylation of cardiac TnI at Thr143 by PKC impaired the interaction between the inhibitory region and TnC, leading to depressed actomyosin ATPase activity and contractility (Lindhout et al., 2002; Li et al., 2003). PKC phosphorylation of cardiac TnI also inhibited ATPase activity (Noland and Kuo, 1991) and thin filament sliding velocity, which may protect the heart from ischemia-reperfusion injury (Macgowan et al., 2001).
Ser150 has also been found to be a phosphorylation site in cardiac TnI, which can be phosphorylated by P21-activated kinase (Pak) to increase the Ca2+ sensitivity of cardiac myofibrils (Buscemi et al., 2002; Ke et al., 2004). Recently, it was demonstrated that AMP-activated Protein Kinase (AMPK) phosphorylates cardiac TnI in vitro at Ser150 (Oliveira et al., 2012) adjacent to the inhibitory loop (Sancho Solis et al., 2011), which increased sensitivity of calcium-dependent force development (Nixon et al., 2012), prolonged relaxation (Oliveira et al., 2012), and increased the effect of adrenergic-induced myocardial hypertrophy (Taglieri et al., 2011). As AMPK is thought to act as a cellular energy sensor, phosphorylation of Ser150 may provide an adaptive mechanism in energy deprivation.
In vitro studies showed that human cardiac TnI was phosphorylated by mammalian sterile 20-like kinase 1 (Mst1) at Thr31, Thr51, Thr129, and Thr143, among which Thr31 is a preferential site (You et al., 2009). Several new phosphorylation sites in the N-terminal region (Ser5/Ser6/Tyr26) have also been identified with decreased phosphorylation in heart failure, whereas phosphorylation of Ser166/Thr181/Ser199 in the C-terminal region and Ser77/Thr78 at the TnI-TnT interface (I-T arm) was increased (Zhang et al., 2012).
In human end-stage dilated cardiomyopathy, baseline phosphorylation of cardiac TnI was diminished with increased myofilament Ca2+ affinity (Zakhary et al., 1999). In failing human heart, the PKA sites Ser23/Ser24 in cardiac TnI are dephosphorylated (Bodor et al., 1997) and the PKC site Ser43/Ser45/Thr144 are increasingly phosphorylated (Zhang et al., 2012), resulting in ventricular diastolic dysfunction. Cardiac TnI R21C mutation in transgenic mouse heart showed dephosphorylation of Ser23/Ser24 and developed cardiac hypertrophy and fibrosis (Wang et al., 2012b). In remodeling myocardium after myocardial infarction, expression of PKA was significantly down-regulated in cardiomyocytes and thus PKA-mediated phosphorylation of cardiac TnI was consequently decreased (Van Der Velden et al., 2004). Dephosphorylation of Ser23/Ser24 in cardiac TnI could also account for the contractile defect in end-stage heart failure (Messer et al., 2007), and the significantly reduced inotropic responsiveness to β-adrenergic stimulation in decompensated cardiac hypertrophy (McConnell et al., 1998).
The structural-functional domains of cardiac TnI, phosphorylation sites, and proteolytic modifications (see below) are summarized in Figure 2 (all residue #s reflected that in human cardiac TnI and included Met1).
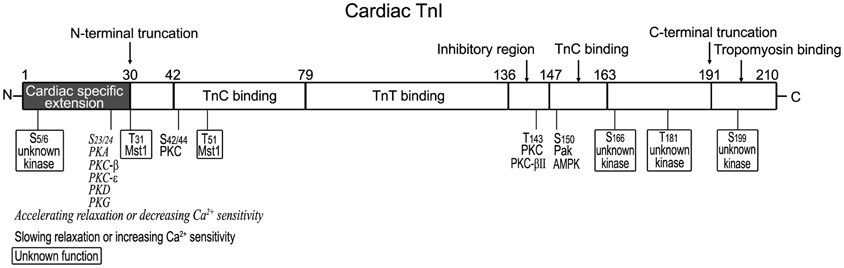
Figure 2. Structural and functional domains of cardiac TnI and posttranslational modifications. Indicated on this linear map of cardiac TnI polypeptide (residue # corresponds to that in human sequence including Met1), Ser23/Ser24 are phosphorylated by PKA (Solaro and Kobayashi, 2011), decreasing Ca2+ sensitivity and accelerating relaxation. They have also been reported to be phosphorylated by PKC-β, PKC-ε (Kobayashi et al., 2005), PKD (Haworth et al., 2004; Cuello et al., 2007; Bardswell et al., 2010) and PKG (Layland et al., 2002). Ser42/Ser44, Thr143, and Ser150 are phosphorylated by PKC, Pak or AMPK, decreasing Ca2+ sensitivity and slowing relaxation (Macgowan et al., 2001; Buscemi et al., 2002; Pi et al., 2002; Burkart et al., 2003b; Westfall et al., 2005). Thr31 and Thr51 are phosphorylated by Mst1 (You et al., 2009) with unknown function. New phosphorylation sites have been identified in the N-terminal region (Ser5/Ser6, with decreased levels in heart failure) and in the C-terminal region (Ser166/Thr181/Ser199, with unknown kinases and functions) (Zhang et al., 2012). A restrictive N-terminal truncation of cardiac TnI occurs in adaptation to hemodynamic stresses to selectively remove the adult heart-specific N-terminal extension with an effect on increasing myocardial relaxation, similar to the effect of PKA phosphorylation at Ser23/Ser24 (Barbato et al., 2005). A deletion of the C-terminal 19 amino acids was found in myocardial ischemia-reperfusion injury (McDonough et al., 1999) and myocardial stunning (McDonough et al., 2001), slowing down the rates of force development and relaxation (Narolska et al., 2006).
The half-life of cardiac TnI in adult cardiomyocytes is estimated to be ~3.2 days and there is a pool of unassembled cardiac TnI in the cytoplasm (Martin, 1981), indicating that cardiac TnI is synthesized in excess. Study on transgenic mouse hearts over-expressing modified cardiac TnI demonstrated that the stoichiometry of total TnI is determined by the incorporation into myofilaments (Feng et al., 2009a).
Cardiac TnI is also a substrate of intracellular modifying proteases, with a demonstrated sensitivity to μ-calpain (Di Lisa et al., 1995). Its degradation by μ-calpain was modulated by phosphorylation, in which phosphorylation by PKA reduced the sensitivity of cardiac TnI whereas phosphorylation by PKC increased the sensitivity of cardiac TnI to μ-calpain (Di Lisa et al., 1995).
The C-terminal end segment is the most conserved region of the TnI polypeptide (Jin et al., 2001). As an allosteric structure regulated by Ca2+ (Jin et al., 2001; Zhang et al., 2011a), it binds and stabilizes tropomyosin in the absence of Ca2+ (Galiñska et al., 2010; Zhang et al., 2011a). Mutations R193H and R205H in the C-terminal end segment altered conformation and function of the I-T interface and increased cardiac TnI binding affinity for TnT, indicating the regulatory role of the C-terminal end segment (Akhter et al., 2014).
A deletion of the C-terminal 19 amino acids was found during myocardial ischemia-reperfusion injury in Langendorff perfused rat hearts (McDonough et al., 1999). It was also seen in myocardial stunning in coronary bypass patients (McDonough et al., 2001). Over-expression of the C-terminal truncated cardiac TnI (cTnI1−192) in transgenic mouse heart resulted in a phenotype of myocardial stunning, and systolic and diastolic dysfunctions (Murphy et al., 2000). 50% replacement of intact cardiac TnI with cTnT1−192 in myofibrils in vitro and cardiomyocytes ex vivo did not affect maximal tension development but slowed down the rates of force redevelopment as well as relaxation (Narolska et al., 2006). cTnI1−192 significantly increased Ca2+-activated actomyosin ATPase and sliding velocity as compared with troponin containing intact cardiac TnI (Foster et al., 2003).
However, the pathological significance of the C-terminal truncation of cardiac TnI remains controversial. No C-terminal truncated cardiac TnI was found in swine hearts subjected to in vivo regional ischemia-reperfusion (Thomas et al., 1999). Another study suggested that the myocardial stunning in pigs induced by regional ischemia was due to dephosphorylation of phospholamban without degradation of cardiac TnI (Kim et al., 2001). No significant degradation of cardiac TnI was detected in the hearts of conscious dogs after reversible ischemia (Lüss et al., 2000; Sherman et al., 2000). A hypothesis is that the marked elevation of preload after global ischemia in Langendorff perfused heart (>30 mmHg) rather than ischemia per se activated μ-calpain and caused cardiac TnI proteolysis (Feng et al., 2001).
Different from the C-terminal truncation, a selective removal of the N-terminal extension of cardiac TnI has been found to be a regulatory mechanism in cardiac adaptation in physiological and pathological stress conditions. The N-terminal extension of approximately 30 amino acids is an adult heart-specific structure absent in fast and slow skeletal muscle TnI (Perry, 1999; Chong and Jin, 2009). The N-terminal extension contains the PKA phosphorylation sites and plays a role in modulating the overall molecular conformation and function of cardiac TnI (Akhter et al., 2012). A restrictive N-terminal truncation of cardiac TnI occurs at low levels in normal hearts of many species examined including human and significantly increases in adaptation to hemodynamic changes such as that in a tail suspension rat model of simulated microgravity (Yu et al., 2001) and Gsα deficiency-caused failing mouse hearts (Feng et al., 2008b).
Experimental evidence showed that the N-terminal extension truncated cardiac TnI (cTnI-ND) increased myocardial relaxation and improved ventricular filling, similar to the effect of PKA phosphorylation (Barbato et al., 2005). While expression of a similarly N-terminal truncated cardiac TnI did not cause functional defect in cardiomyocytes (Guo et al., 1994), over-expression of cTnI-ND improved the diastolic function of ex vivo working hearts of Gsα deficiency mice (Feng et al., 2008b) and cardiac function in vivo in aging mice (Biesiadecki et al., 2010). Co-expression of cTnI-ND corrected the diastolic dysfunction of restrictive cardiomyopathy hearts caused by cTnI193His mutation (Li et al., 2010). Isolated cardiomyocytes from cTnI-ND mouse hearts showed larger shortening amplitude and higher systolic and diastolic velocities (Wei and Jin, 2013). Whereas the N-terminal extension of cardiac TnI does not directly interact with other known proteins in the thin filament regulatory system, the molecular mechanism of cTnI-ND's function involves alterations of the conformation and function of the middle region of cardiac TnI (Akhter et al., 2012).
A study on trout cardiac TnI that lacks the N-terminal extension showed that troponin complex containing trout cardiac TnI had a greater Ca2+ affinity than human troponin (Kirkpatrick et al., 2011). Although trout cardiac TnI lacks the two substrate residues of PKA phosphorylation, myofilament Ca2+ affinity decreased when treated with PKA, similar to the response of mammalian cardiac TnI with the N-terminal extension (Kirkpatrick et al., 2011). This apparently N-terminal extension-independent PKA regulation and enhancement of relaxation is worth further investigation.
Troponin T is a striated muscle-specific protein of ~250–305 amino acids with molecular weights ranging from 31-kDa to 36-kDa. Same as the differentiated TnI isoform genes, three muscle type-specific TnT isoform genes are present in vertebrates and expressed in fiber-specific and developmentally regulated manner (Jin et al., 2008; Wei and Jin, 2011). In addition to specific expression in cardiomyocytes, cardiac TnT also expresses at significant levels in embryonic skeletal muscle (Anderson et al., 1991; Jin, 1996) and myopathic skeletal muscle of patients and Duchenne muscular dystrophy (Ricchiuti and Apple, 1999), likely indicating active growth or regeneration.
Earlier studies had dissected the structure of TnT into two functional regions based on fragmentation using limited cleavages with chymotrypsin and CNBr, i.e., the T1 and T2 fragments (amino acids1–158 and 159–259, respectively, in rabbit skeletal muscle TnT) (Tanokura et al., 1983; Perry, 1998) (Figure 3). The T1 fragment binds the head-tail junction of tropomyosin mainly through a 39 amino acids segment in the N-terminal portion of the conserved middle region of TnT (Jin and Chong, 2010). The C-terminal T2 fragment contains binding sites for TnC, TnI, and F-actin (Heeley et al., 1987; Schaertl et al., 1995; Perry, 1998) as well as another tropomyosin-binding site in a segment of 25 amino acids near the beginning of the T2 region (Jin and Chong, 2010), which binds the central region of tropomyosin (Morris and Lehrer, 1984) (Figure 3). The current model states that TnT plays an anchoring role and transmits the signal from Ca2+-TnC binding to the thin filament regulatory system in striated muscles (Perry, 1998).
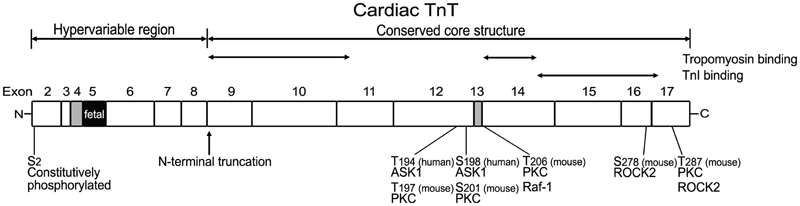
Figure 3. Structural and functional domains of cardiac TnT, alternative spliced exons, and posttranslational modifications. Outlined on this linear map of cardiac TnT polypeptide (residue #s are those published in the original papers, which used various isoforms from different species), the functional segments T1, T2, and the N-terminal hypervariable region as well as the alternatively spliced exons 4, 5, and 13 are indicated. Ser2 is a highly conserved residue constitutively and phosphorylated in cardiac TnT in vivo (Perry, 1998; Sancho Solis et al., 2008). In vitro studies demonstrated that cardiac TnT could be phosphorylated by PKC at Thr197, Ser201, Thr206, and Thr287 in the C-terminal region that contains binding sites for TnI, TnC, and tropomyosin (Jideama et al., 1996). Thr206 can also be phosphorylated by Raf-1 (Pfleiderer et al., 2009) and Ser278 and Thr287 by ROCK2, which inhibited tension development and ATPase activity in skinned fibers (Vahebi et al., 2005). Thr194 and Ser198 of cardiac TnT have been found to be phosphorylated by ASK1 with decreases in cardiomyocyte contractility (He et al., 2003).
The N-terminal region of TnT is a “hypervariable” region (Perry, 1998). This region has variable lengths and variable amino acid sequences. Cardiac TnT is of larger size than fast and slow skeletal muscle TnT, mainly due to a longer N-terminal variable region (Perry, 1998; Wei and Jin, 2011). The N-terminal variable region of TnT does not contain any binding sites for other known myofilament proteins (Perry, 1998; Jin et al., 2008; Wei and Jin, 2011).
Taking advantage of the presence of a cluster of transition metal ion-binding sites in the N-terminal variable region of fast skeletal muscle TnT of avian species in the orders of Galliformes and Craciformes (Jin and Smillie, 1994), antibody epitope analyses showed that Zn2+-binding to the N-terminal region of chicken breast muscle fast TnT altered the molecular conformation of epitopes outside of the N-terminal region, demonstrating a long-range modulatory effect (Wang and Jin, 1998). Fluorescence spectral analysis further showed that Cu2+ binding to the N-terminal region of chicken fast TnT induced changes in fluorescence intensity and anisotropy of Trp234, Trp236, and Trp285 or fluorescein-labeled Cys263 in the C-terminal region (Jin and Root, 2000). Protein-binding studies showed that the binding of Zn2+ to the N-terminal region of chicken fast TnT decreased the binding affinity for tropomyosin, TnI, and TnC (Ogut and Jin, 1996; Jin et al., 2000). These data indicated that the N-terminal variable region modulates the conformation and function of TnT core structure to fine-tune muscle contractility (Biesiadecki et al., 2007a).
We recently found that the heart of adult toads (Bufo) exclusively expresses slow skeletal muscle TnT instead of cardiac TnT while all other myofilament proteins, including cardiac TnI and cardiac myosin, remain to be the normal cardiac isoform (Feng et al., 2012). This unique biochemical content of toad cardiac muscle is correlated to a striking physiological feature of toads, i.e., it is highly adaptive to large changes in the volumes of body fluid and blood between rain and dry seasons (Boral and Deb, 1970) or under experimental conditions (Deb et al., 1974).
Functional studies demonstrated that toad hearts had faster contractile and relaxation velocities and a significantly higher tolerance to afterload (Feng et al., 2012). These findings demonstrate that the selective utilization of slow skeletal muscle TnT in toad heart was an adaptive change with significantly functional advantage and fitness value during evolutionary selection. This observation suggests that altering TnT function may be targeted for the improvement of systolic function and the treatment of congestive heart failure.
No expression of cardiac TnT was detected in either heart or skeletal muscle of the toad (Feng et al., 2012). Despite the unusual expression in the heart, slow skeletal muscle TnT is normally expressed specifically in toad slow twitch skeletal muscles (Feng et al., 2012). The mechanism of selectively activating the slow skeletal muscle TnT gene in toad heart and the inactivation of cardiac TnT gene remains to be further investigated.
Multiple alternative splice forms are expressed from each of the three TnT isoform genes to add structural and functional variations that fine-tune muscle contractility. Alternative splice forms of cardiac TnT are expressed in a regulated pattern during embryonic and postnatal heart development, and are found in diseased hearts (Jin and Lin, 1988, 1989; Townsend et al., 1995; Ricchiuti and Apple, 1999). In addition to physiological or pathophysiological adaptations, aberrant splicing has been found to cause dilated cardiomyopathy in turkeys and dogs (Biesiadecki and Jin, 2002; Biesiadecki et al., 2002).
Mammalian cardiac TnT gene contains 17 exons including 3 alternatively spliced exons (exon 4, exon 5, and exon 13) (Jin et al., 1992, 1996) (Figure 3). Exon 4 and exon 5 encode segments in the N-terminal variable region, and exon 13 encodes a variable segment between the T1 and T2 functional domains (Jin et al., 1996). Multiple splice forms of cardiac TnT are expressed in mammalian hearts (Jin and Lin, 1988, 1989; Anderson et al., 1991; Jin et al., 1992, 1996). The alternative splicing pattern of cardiac TnT is synchronized in developing cardiac and skeletal muscle independent of functional demands (Jin, 1996). It was further demonstrated that the abundant cardiomyocytes present in the walls of developing and adult rat and mouse thoracic veins exhibit patterns of cardiac TnT alternative splice forms identical to that in the heart (Kracklauer et al., 2013; Liu et al., 2014). These findings strongly support that the regulation of cardiac TnT alternative splicing during development and differentiation is under systemic control rather than directly responding to functional demands or adaptation.
Combinations of alternative splicing of exons 4 and 5 in the N-terminal variable region yield four cardiac TnT isoforms differing in size and charge: TnT1 (all exon present), TnT2 (splice out exon 4), TnT3 (splice out exon 5), and TnT4 (splice out exon 4 and 5), numbered in the order of decreasing molecular weight (Gomes et al., 2002). In vitro studies showed that both TnT1 and TnT2 reduced the ability of troponin to inhibit force development and ATPase activity, causing less relaxation of fibers (Gomes et al., 2002). Whether the expression of cardiac TnT splice forms is altered and plays a role in failing heart is controversial and an area of active investigation (Anderson et al., 1991, 1992; Mesnard et al., 1995).
Abnormal splice isoforms of cardiac TnT have been reported. Turkeys with inherited dilated cardiomyopathy and heart failure have an aberrant splice-out of the normally conserved exon 8-encoded segment in cardiac TnT (Biesiadecki and Jin, 2002). Similar abnormality (splice out of the equivalent exon 7) has been found in cardiac TnT of dog, pig and cat, which also have high incidence of dilated cardiomyopathies (Biesiadecki et al., 2002). In the heart of adult guinea pig, exon 6 that is significantly larger than exon 7 is spliced out (Biesiadecki et al., 2002). Overexpression of exon 7-deleted cardiac TnT in the heart of transgenic mice impaired systolic function (Wei et al., 2010).
In addition to the deletion of exon 7, embryonic exon 5 is abnormally included at significant levels in adult cardiac TnT in dilated cardiomyopathy dogs (Biesiadecki et al., 2002). Although the continuing expression of this embryonic specific exon in the N-terminal region of cardiac TnT in adult heart may have a value to compensate for the abnormality of exon 7 deletion, we have shown that the heterogeneity of TnT in adult ventricular muscle due to the co-presence of more TnT variants reduces cardiac efficiency by desynchronizing the Ca2+-activation of thin filaments (Feng and Jin, 2010) (discussed in more details below).
Aberrant splicing of cardiac TnT also occurs in chronic stress conditions. Splice out of the exon 4-encoded segment was increased in failing human heart (Anderson et al., 1991, 1995), diabetic rat heart (Akella et al., 1995), and familial hypertrophic cardiomyopathy human hearts (Thierfelder et al., 1994). In a rabbit model of mild cardiac hypertrophy, cardiac TnT splicing shifted toward the fetal pattern (Chen et al., 1997). Further investigations are needed to understand the function as well as regulatory mechanisms of such potentially adaptive alternative splicing of cardiac TnT under stress conditions.
The mechanism for the aberrantly spliced cardiac TnT to produce dilated cardiomyopathy has been investigated. Different alternative splice forms of cardiac TnT are of different functional properties (Gomes et al., 2002). As mentioned above, a hypothesis is that chronic co-existence of TnT variants in adult heart would produce split Ca2+ sensitivity among troponins in the thin filament, which will desynchronize activation of ventricular muscle and decrease the efficiency of cardiac pumping (Feng and Jin, 2010). Different from skeletal muscles that normally express multiple TnT isoforms and splice forms corresponding to the function of tetanic contractions, the ventricular muscle is electrically activated as a syncytium to produce uniform rhythmic contractions. Consistently, only adult isoform of cardiac TnT is present in adult heart after the developmental switch (Jin and Lin, 1988; Jin, 1996), corresponding to a uniform sensitivity to Ca2+ activation.
Studies on transgenic mice demonstrated that co-existence of a non-mutant fast TnT and the endogenous cardiac TnT in adult heart significantly impaired contractile functions (Huang et al., 2008; Yu et al., 2012). Therefore, the desynchronized troponin activity, other than a mutant structure in TnT, imposed negative impacts on myocardial function. Further studies on transgenic mice expressing one or more functionally distinct cardiac TnT variants in addition to the endogenous normal adult cardiac TnT produced lower left ventricular pressure development, slower contractile and relaxation velocities, and decreased stroke volume as compared with wild-type controls, further supporting the hypothesis that coexistence of functionally different cardiac TnT variants in adult ventricular muscle reduces cardiac efficiency due to desynchronized thin filament activation (Feng and Jin, 2010).
The alternative splice forms found in avian and mammalian cardiac TnT are summarized in Table 1. The molecular mechanism that regulates alternative splicing of TnT remains to be fully understood, in which both cis-regulatory elements (Biesiadecki and Jin, 2002) and trans-regulatory factors (Ward and Cooper, 2010) have been suggested for roles in regulating the alternative splicing of cardiac TnT.
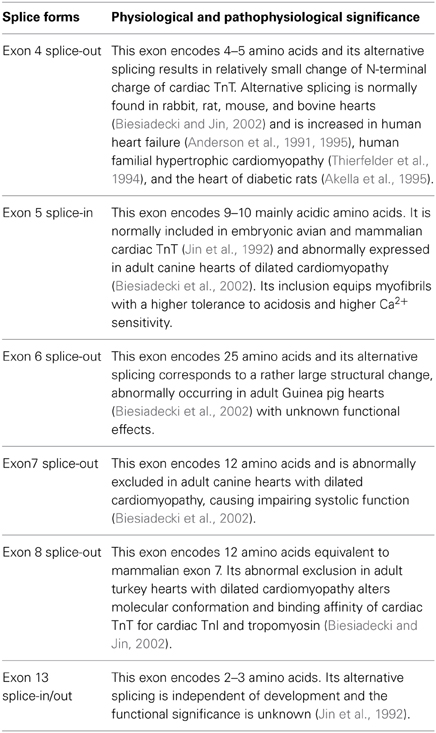
Table 1. Physiological and abnormal alternative splice forms of cardiac TnT.
Ser2 is a highly conserved residue in all three isoforms of avian and mammalian TnT (Jin et al., 2008) and is constitutively phosphorylated in cardiac TnT in vivo (Perry, 1998; Sancho Solis et al., 2008). Little is known regarding the kinase, regulation and functional significance of cardiac TnT Ser2 phosphorylation.
In vitro studies demonstrated that cardiac TnT could be phosphorylated by PKC at Thr197, Ser201, Thr206, and Thr287 (residue numbers in mouse cardiac TnT), which are located in the C-terminal region containing binding sites for TnI, TnC, and tropomyosin (Jideama et al., 1996). PKC-mediated phosphorylation of cardiac TnT has been shown to depress myofilament function, myocyte contractility, and ventricular pumping (Belin et al., 2007). PKC phosphorylation of cardiac TnT inhibited the Ca2+-stimulated Mg2+-ATPase activity without alteration of Ca2+-sensitivity (Noland and Kuo, 1993). When cardiac TnT was partially replaced with fast skeletal muscle TnT in transgenic mouse heart, which is not phosphorylated by PKC, the PKC-mediated depression of cardiac function was blunted (Montgomery et al., 2001). Studies with mutations at PKC phosphorylation sites supported the hypothesis that Thr206 is a regulatory site whose phosphorylation by PKCα or substitution with Glu to mimic phosphorylation significantly suppressed tension development, actomyosin Mg2+-ATPase activity, and myofilament Ca2+ sensitivity and cooperativity while Thr197, Ser201, and Thr287 had no significant effect (Sumandea et al., 2003).
Thr206 can also be phosphorylated by Raf-1, which links growth factor-dependent signaling to dynamic changes in cardiac contractile function (Pfleiderer et al., 2009). Ser278 and Thr287 of cardiac TnT were also found to be phosphorylated by Rho-dependent kinase (ROCK2), which inhibited tension development and ATPase activity in skinned fibers (Vahebi et al., 2005). Phosphorylation of cardiac TnT by ASK1 (a stress-activated kinase that has been implicated in TNFα and oxidant stress responses) at Thr194 and Ser198 has also been found with a decrease in cardiomyocyte contractility (He et al., 2003).
Protein phosphatase 1 (PP1) has been found to dephosphorylate cardiac TnT (Jideama et al., 2006). A coimmunoprecipitation study indicated that Pak1 (p21 activated kinase 1) is associated with cardiac TnT and regulates the phosphorylation level of cardiac TnT (Monasky et al., 2012). Hearts of Pak1 knockout mice showed a significant increase in TnT phosphorylation as compared with wild type controls (Ke et al., 2012). This modification may contribute to cardioprotection through Pak1 signaling and merits further investigation.
The phosphorylation of cardiac troponin could also be modulated by structure alterations. Deletion of Lys210 in cardiac TnT (ΔK210) decreased the phosphorylations of cardiac TnT by 30% and cardiac TnI by 46%, mainly at Ser23/24, in vivo as compared with wild-type controls (Sfichi-Duke et al., 2010). In vitro kinase assay indicated that ΔK210 increased phosphorylation propensity of Thr203 in cardiac TnT by three-fold, without changing Ser23/24 phosphorylation in cardiac TnI. Yeast two-hybrid studies indicated that cardiac TnT-ΔK210 bound stronger to cardiac TnI than that of wild type cardiac TnT (Sfichi-Duke et al., 2010), suggesting a possible explanation for cardiac TnT-ΔK210 mutation to correlate with dilated cardiomyopathy (Kamisago et al., 2000).
Cardiac TnT has a half-life of 3.5 days in vivo (Martin, 1981) and non-myofilament-associated TnT is rapidly degraded in cardiomyocytes (Wang et al., 2005; Jeong et al., 2009). The potent proteolysis capacity in cardiomyocytes may be critical to maintaining the integrity of myofilament contractile apparatus as well as to protecting cardiomyocytes from the cytotoxicity of TnT fragment (Jeong et al., 2009). In the absence of myofilaments, the C-terminal and middle fragments of TnT effectively induced cell apoptosis (Jeong et al., 2009). A hypothesis is that a peak release of cardiac TnT or cardiac TnT fragments from myofilaments exceeding the protective capacity of the proteolytic degradation would result in cytotoxicity and cause the death of cardiomyocytes in myocardial ischemia-reperfusion injury. No apoptosis-effect of N-terminal variable region was observed (Jeong et al., 2009). Along this line, an in vitro study showed that cardiac TnT was cleaved by activated caspase 3 to remove the N-terminal 92 amino acids and resulted in contractile dysfunction before cell death (Communal et al., 2002).
Restrictive deletion of the N-terminal 71 residues of mouse cardiac TnT was found in hearts after ischemia reperfusion (Zhang et al., 2006) and left ventricular pressure overload in vitro (Feng et al., 2008a). Amino acid sequencing and protein fragment reconstruction determined that this restrictive N-terminal proteolysis selectively removes the entire N-terminal variable region but preserves the conserved core structure of cardiac TnT intact (Zhang et al., 2006). Myofilament associated μ-calpain is found to contribute the restrictive N-terminal truncation of cardiac TnT (Zhang et al., 2006).
The selective removal of the N-terminal variable region had no significant effect on the binding affinities of cardiac TnT for TnI and tropomyosin. This observation demonstrates that the N-terminal variable region is not essential for the core function of TnT, and the restrictive N-terminal truncation of cardiac TnT may be a regulatory mechanism. In contrast, extended deletion to remove the N-terminal 91 residues of mouse cardiac TnT including a segment of the conserved middle region weakened the binding to tropomyosin (Biesiadecki et al., 2007a) as well as increased the Ca2+ sensitivity of troponin (Sumandea et al., 2009).
The restrictive cleavage of cardiac TnT can be induced with calcium overloading. The level of N-terminal truncated cardiac TnT (cTnT-ND) increased in primary cultures of adult mouse cardiomyocytes upon ouabain-produced Ca2+ overload (Zhang et al., 2011b). No degradation of cardiac TnI, a known substrate of μ-calpain, was detected and no significant alteration of phosphorylation was seen in cardiac TnT when Ca2+ overload produced cTnT-ND (Zhang et al., 2011b). These observations support a hypothesis that the induction of cTnT-ND in calcium overload is neither due to elevated overall activity of μ-calpain nor phosphorylation level of cardiac TnT. On the other hand, the structure of N-terminal region per se exhibited a role in determining the restrictive μ-calpain proteolysis. Deletion of exon 7-encoded segment made cardiac TnT more sensitive to μ-calpain modification (Zhang et al., 2011b).
Although the restrictive removal of the N-terminal variable region of cardiac TnT does not abolish the core function of troponin (Hinkle et al., 1999; Biesiadecki et al., 2007a), it results in conformational changes of cardiac TnT, modulates TnT's binding affinity for TnI, TnC, and tropomyosin, and alters Ca2+ activation of actomyosin ATPase (Wang and Jin, 1998; Jin and Root, 2000; Jin et al., 2000; Gomes et al., 2002). Using pyrene-labeled tropomyosin, studies demonstrated that N-terminal truncated cardiac TnT strengthened the interactions between cardiac TnT77−289 and tropomyosin and stabilized cardiac myofilaments in a sub-maximally activated state (Chandra et al., 1999).
Consistent with the notion that the N-terminal variable region of TnT is non-essential but a regulatory structure, overexpression of cTnT-ND in transgenic mouse hearts effectively replaced endogenous intact cardiac TnT and supported cardiac function. The hearts showed a slightly but statistically significant decrease in contractile velocity, which resulted in elongated time of left ventricular rapid ejection phase especially at high afterload (Feng et al., 2008a). This effect produced a significant increase in stroke volume and demonstrated that the restrictive N-terminal truncation of cardiac TnT is a mechanism to modulate thin filament function and alter myosin cross-bridge kinetics, suggesting a novel approach to compensating for cardiac output in energetic crisis (Feng et al., 2008a).
The structural-functional domains of cardiac TnT, alternative spliced exons, phosphorylation sites, and proteolytic modifications are summarized in Figure 3.
Numerous mutations in the genes encoding the three subunits of cardiac troponin have been found to cause cardiomyopathies. By increasing or decreasing Ca2+ sensitivity and force generation, troponin mutations contribute to the pathogeneses of inherited hypertrophic, restrictive and diastolic cardiomyopathies (Seidman and Seidman, 2001).
Mutations in cardiac/slow TnC account for approximately 0.4% of hypertrophic cardiomyopathy (Landstrom et al., 2008). L29Q mutation in TNNC1 was the first such mutation identified (Hoffmann et al., 2001). L29Q mutation in cardiac TnC hindered the PKA-dependent phosphorylation of cardiac TnI at Ser22/Ser23, and reduced Ca2+ sensitivity of myofilaments in ATPase assays using reconstituted skeletal muscle myofibrils containing cardiac troponin (Schmidtmann et al., 2005). However, the same mutation increased Ca2+ sensitivity of force development when it was used to replace endogenous TnC in skinned mouse cardiomyocytes (Liang et al., 2008). This difference may have resulted from the different experimental conditions or the intrinsic difference between cardiac and skeletal muscles.
More missense mutations, for example A8V, C84Y, E134D, and D145E, in TNNC1 have been reported in hypertrophic cardiomyopathies (Landstrom et al., 2008). Functional studies showed that A8V, C84Y, and D145E increased Ca2+ sensitivity of force development (Pinto et al., 2009). In addition, E59D, D75Y and G159D mutation in TNNC1 are found in dilated cardiomyopathy patients. E59D and D75Y localized in the regulatory Ca2+ binding site II decrease myofilament calcium responsiveness (Lim et al., 2008). G159D is localized in a metal ion-binding site and, therefore, alters the function of troponin complex in regulating cardiac muscle contractility (Mogensen et al., 2004).
Besides altering Ca2+-induced conformational changes, mutations in cardiac TnC may alter molecular conformations involved in Ca2+ affinity and binding to cardiac TnI. An example is that L48Q substitution in human cardiac TnC made the hydrophobic core more exposed to cardiac TnI, thus increased the binding affinity for TnI (Wang et al., 2012a). Mutation A31S in TNNC1 increases Ca2+ sensitivity, which may contribute to causing hypertrophic cardiomyopathy and arrhythmogenesis (Parvatiyar et al., 2012). Although G159D mutation in the C-lobe of cardiac TnC did not alter myofilament function, it blunted the myofilament desensitization induced by phosphorylation of cardiac TnI at Ser23/Ser24 (Finley et al., 1999; Biesiadecki et al., 2007b).
It is worth noting that mutations in the region of the inactive Ca2+-binding Site I of cardiac TnC are found at a much higher rate than that in the active Site II region (Hoffmann et al., 2001; Landstrom et al., 2008; Parvatiyar et al., 2012; Wang et al., 2012a). This observation suggests that most of the myopathic mutations in TnC fixed in the population are those causing relatively mild functional changes other than drastically destructive at critical sites of function, such as the sole regulatory site II of cardiac/slow TnC.
Cardiac TnI mutations account for approximately 5% of familial hypertrophic cardiomyopathy cases and at least 20 mutations of cardiac TnI have been reported to link to inherited restrictive cardiomyopathy with increased Ca2+ sensitivity and reduced ATPase activity and force development (Gomes and Potter, 2004; Gomes et al., 2005; Yumoto et al., 2005). These cardiac TnI mutations are mainly found in the inhibitory region and the C-terminal end segment, indicating functional relevance.
Cardiac TnI mutation R21C in the N-terminal extension associated with hypertrophic cardiomyopathy abolishes in vivo phosphorylation of Ser23/Ser24 (Wang et al., 2012a). The phenotype of this mutation supports the regulatory role of the N-terminal extension of cardiac TnI in diastolic function of the heart. Cardiac TnI mutation R145G found in familial hypertrophic cardiomyopathy is within the inhibitory region and alters the interaction of cardiac TnI with cardiac TnC. This mutation reduces the inhibition of actomyosin ATPase and thus increases Ca2+ sensitivity (Kimura et al., 1997; Lindhout et al., 2002). Lys184 deletion in the C-terminal region of cardiac TnI impairs relaxation kinetics and results in hypercontractility when overexpressed in mouse cardiomyocytes (Iorga et al., 2008). Similarly, transgenic mice over-expressing cardiac TnI with R193H mutation demonstrated impaired relaxation similar to that in human restrictive cardiomyopathy patient (Du et al., 2008). The negative impact of this cardiac TnI mutation on heart function showed a dose dependence (Li et al., 2013). These findings indicate the critical role of the C-terminal domain of TnI in muscle relaxation and diastolic function of the heart (Davis et al., 2007).
It is intriguing that over 94% of known disease-causing single nucleotide polymorphisms (SNPs) in cardiac TnI are located in the C-terminal half of the polypeptide chain (residues 128–210) (Palpant et al., 2010). This observation may indicate more stringent structure-function relationships in this region. Alternatively, this pattern may reflect that this region of TnI has a tolerance to structural modifications, allowing more mutations fixed in the population without reproductive lethality. These hypotheses require further investigation.
Mutations in cardiac TnT account for approximately 15% of familial hypertrophic cardiomyopathy cases. These mutations are characterized by severe myocardial disarray, relatively mild and often subclinical hypertrophy, and a high incidence of sudden cardiac death (Thierfelder et al., 1994; Watkins et al., 1995; Maron et al., 1996; Tardiff et al., 1998; Varnava et al., 2001; Sehnert et al., 2002). Together with the aberrant splicing of cardiac TnT found in turkey and dog cardiomyopathies, at least 52 point mutations of cardiac TnT have been reported to cause human heart diseases, including 50 missense mutations, one deletion and one splicing donor site mutation (Willott et al., 2010).
Mutations in different regions of cardiac TnT may contribute to the pathogenesis of cardiomyopathies via different mechanisms, including increasing the Ca2+-sensitivity of troponin complex, changing the binding affinity of cardiac TnT for cardiac TnI and the affinity of cardiac TnI for cardiac TnC, and perturbing the proper response of myocardial contraction to changes in pH (Harada and Potter, 2004).
Dilated cardiomyopathy is a major cause of heart failure, and genetic defects are a significant contributor to the disease (Lakdawala et al., 2012). Up to date, at least five cardiac TnT mutants: R131W (Mogensen et al., 2004), R141W (Li et al., 2001), R205L (Mogensen et al., 2004), Lys210 deletion (Kamisago et al., 2000), and D270N (Mogensen et al., 2004), all in the conserved core structure of cardiac TnT, are found to reduce Ca2+ sensitivity and produce phenotypes of dilated cardiomyopathy (Mirza et al., 2005).
It is worth noting that some troponin mutations have been reported with clinical phenotypes of more than one types of cardiomyopathy. A possible explanation is that while hypertrophic or restrictive cardiomyopathies may be the primary disease, dilated cardiomyopathy can develop in the later stages as the pathology progresses into congestive heart failure. It is also interesting to note that no clinical case of human cardiomyopathy mutation has been found in the N-terminal domain of cardiac TnT corresponding to the hypervariable region of TnT, which is naturally tolerant to structural variations (Jin et al., 2008; Wei and Jin, 2011).
As the mutations of different troponin subunits have different functional impacts, their combined phenotypes may indicate structural and functional relationships among the troponin subunits. For example, a mutation R111C is found in cardiac TnI of wild turkeys co-existing with the dilated cardiomyopathy-related aberrantly splice-out of exon 8 in cardiac TnT (Biesiadecki et al., 2004). By lowering the binding affinity of cardiac TnI for the mutant cardiac TnT that showed increased affinity for TnI, mutually compensatory effects were observed. While the mouse counterparts, cardiac TnI-K118C mutation and exon 7-deleted cardiac TnT, each alone has dominant negative phenotypes in transgenic mice, double transgenic mouse hearts co-expressing cardiac TnI-K118C mutation and exon 7-deleted cardiac TnT showed that the systolic abnormality of cardiac TnT exon 7 deletion and the diastolic abnormality of cardiac TnI-K118C mutation mutually canceled each other (Wei et al., 2010).
Another example is that the S69D and D73N mutations of cardiac TnC corrected the abnormal Ca2+ sensitivity increased by cardiac TnI-R192H mutation or ischemia-induced C-terminal truncation (cTnI1−192) of cardiac TnI (Liu et al., 2012).
Figure 4 summarizes most of the characterized human cardiomyopathic mutations found in cardiac TnC, cardiac TnI and cardiac TnT.
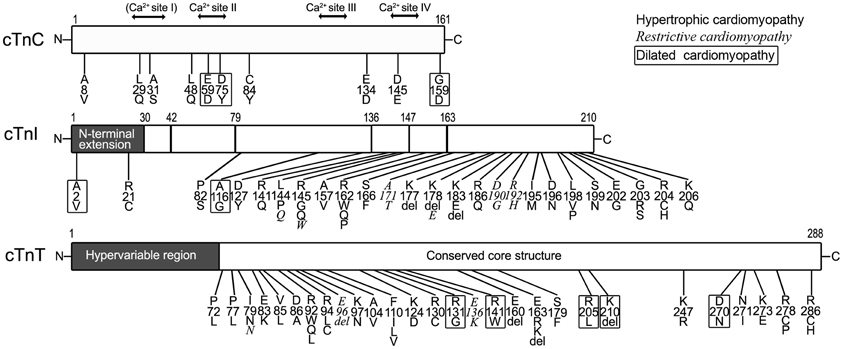
Figure 4. Myopathic mutations in troponin subunits. Amino acid substitutions and deletions found in human cardiomyopathies are indicated on the linear maps of cardiac TnC (cTnC, NP_003271.1), cardiac TnI (cTnI, NP_000354.4), and cardiac TnT (cTnT, NP_001263276.1). Different fonts were used to indicate that each of the mutations causes hypertrophic, restrictive (Italic), and dilated (boxed) cardiomyopathies. It is worth noting that no myopathic mutation was found in the N-terminal hypervariable region of cardiac TnT. The residue #s are counted including Met1.
The TnC, TnI, and TnT subunits of cardiac troponin function interactively as regulators of myofilament activation and force generation. Based on biochemical, biophysical, physiological and pathophysiological studies, mounting evidence for the molecular evolution, gene regulation, alternative splicing, and posttranslational modifications of cardiac troponin subunits has laid a solid foundation for understanding their structural diversity, structure-function relationships, adaptive regulations, and pathogenic mutations. For troponin's central role in muscle thin filament regulation and contractility, further elucidation of troponin structure and function will powerfully forward the prevention, diagnosis, and treatment of heart diseases.
In order to advance troponin research and translate the knowledge into clinical applications, there are several important questions remain to be answered. With modern molecular engineering methodology, it is important and feasible to fine map the interaction sites between troponin subunits and the allosteric and conformational relationships that are essential in regulation of cardiac muscle contraction. Alternative splicing is an important regulatory pathway of cardiac TnT and aberrant splicing of it has close relation with cardiomyopathy. However, the mechanism of cardiac TnT expression via alternative splicing is not well understood. The cell signaling pathway that controls RNA splicing and the production of cardiac TnT variants remains to be investigated. Restrictive N-terminal truncations of cardiac TnI and cardiac TnT are novel posttranslational regulatory mechanisms that have potent roles in cardiac adaptation to stress conditions. However, the cellular mechanisms that induce restrictive N-terminal truncations of cardiac TnI and cardiac TnT have not been established. It is worth noting that no single stress condition has been found to be able to produce restrictive N-terminal truncations of both cardiac TnI and cardiac TnT, indicating distinct mechanisms in the posttranslational regulation of the two troponin subunits that are structurally and functionally closely related. It is also an intriguing observation that the N-terminal truncation of cardiac TnI selectively enhances diastolic function whereas the N-terminal truncation of cardiac TnT selectively reduces systolic velocity of the hearts. To understand how structural modifications of the two subunits of troponin regulate muscle contraction and relaxation in a highly selective manner would lead to development of new therapeutic approaches for the treatments systolic and diastolic heart failures.
Continued in depth research is required to answer these and new emerging questions toward the goal of fully understanding the function of troponin in cardiac muscle contraction in order to improve the treatment and prevention of myocardial diseases and heart failure.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the current and past members of our laboratory who have made invaluable contributions to our troponin research discussed in this review. This work was supported by grants from the National Institutes of Health (AR048816 and HL098945) and a Multidisciplinary Incubator Grant from the Office of Vice President for Research Wayne State University to Jian-Ping Jin. Juan-Juan Sheng is recipient of a graduate fellowship from the China Scholarship Council.
Akella, A. B., Ding, X. L., Cheng, R., and Gulati, J. (1995). Diminished Ca2+ sensitivity of skinned cardiac muscle contractility coincident with troponin T-band shifts in the diabetic rat. Circ. Res. 76, 600–606. doi: 10.1161/01.RES.76.4.600
Akhter, S., Bueltmann, K. Jr., Huang, X., and Jin, J. P. (2014). Restrictive cardiomyopathy mutations demonstrate functions of the C-terminal end-segment of troponin I. Arch. Biochem. Biophys. doi: 10.1016/j.abb.2013.12.001. [Epub ahead of print].
Akhter, S., Zhang, Z., Jin, J. P. (2012). The heart-specific NH2-terminal extension regulates the molecular conformation and function of cardiac troponin I. Am. J. Physiol. Heart Circ. Physiol. 302, H923–H933. doi: 10.1152/ajpheart.00637.2011
Anderson, P. A., Greig, A., Mark, T. M., Malouf, N. N., Oakeley, A. E., Ungerleider, R. M., et al. (1995). Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circ. Res. 76, 681–686. doi: 10.1161/01.RES.76.4.681
Anderson, P. A., Malouf, N. N., Oakeley, A. E., Pagani, E. D., and Allen, P. D. (1991). Troponin T isoform expression in humans. A comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circ. Res. 69, 1226–1233. doi: 10.1161/01.RES.69.5.1226
Anderson, P. A., Malouf, N. N., Oakeley, A. E., Pagani, E. D., and Allen, P. D. (1992). Troponin T isoform expression in the normal and failing human left ventricle: a correlation with myofibrillar ATPase activity. Basic Res. Cardiol. 87(Suppl. 1), 117–127.
Arteaga, G. M., Palmiter, K. A., Leiden, J. M., and Solaro, R. J. (2000). Attenuation of length dependence of calcium activation in myofilaments of transgenic mouse hearts expressing slow skeletal troponin I. J. Physiol. 526(Pt 3), 541–549. doi: 10.1111/j.1469-7793.2000.t01-1-00541.x
Barbato, J. C., Huang, Q. Q., Hossain, M. M., Bond, M., and Jin, J. P. (2005). Proteolytic N-terminal truncation of cardiac troponin I enhances ventricular diastolic function. J. Biol. Chem. 280, 6602–6609. doi: 10.1074/jbc.M408525200
Bardswell, S. C., Cuello, F., Rowland, A. J., Sadayappan, S., Robbins, J., Gautel, M., et al. (2010). Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J. Biol. Chem. 285, 5674–5682. doi: 10.1074/jbc.M109.066456
Belin, R. J., Sumandea, M. P., Allen, E. J., Schoenfelt, K., Wang, H., Solaro, R. J., et al. (2007). Augmented protein kinase C-α–induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ. Res. 101, 195–204. doi: 10.1161/CIRCRESAHA.107.148288
Biesiadecki, B. J., Chong, S. M., Nosek, T. M., and Jin, J. P. (2007a). Troponin T core structure and the regulatory NH2-terminal variable region. Biochemistry 46, 1368–1379. doi: 10.1021/bi061949m
Biesiadecki, B. J., Elder, B. D., Yu, Z. B., and Jin, J. P. (2002). Cardiac troponin T variants produced by aberrant splicing of multiple exons in animals with high instances of dilated cardiomyopathy. J. Biol. Chem. 277, 50275–50285. doi: 10.1074/jbc.M206369200
Biesiadecki, B. J., and Jin, J. P. (2002). Exon skipping in cardiac troponin T of turkeys with inherited dilated cardiomyopathy. J. Biol. Chem. 277, 18459–18468. doi: 10.1074/jbc.M200788200
Biesiadecki, B. J., Kobayashi, T., Walker, J. S., John Solaro, R., and De Tombe, P. P. (2007b). The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ. Res. 100, 1486–1493. doi: 10.1161/01.RES.0000267744.92677.7f
Biesiadecki, B. J., Schneider, K. L., Yu, Z. B., Chong, S. M., and Jin, J. P. (2004). An R111C polymorphism in wild turkey cardiac troponin I accompanying the dilated cardiomyopathy-related abnormal splicing variant of cardiac troponin T with potentially compensatory effects. J. Biol. Chem. 279, 13825–13832. doi: 10.1074/jbc.M314225200
Biesiadecki, B. J., Tachampa, K., Yuan, C., Jin, J. P., De Tombe, P. P., and Solaro, R. J. (2010). Removal of the cardiac troponin I N-terminal extension improves cardiac function in aged mice. J. Biol. Chem. 285, 19688–19698. doi: 10.1074/jbc.M109.086892
Blumenschein, T. M. A., Tripet, B. P., Hodges, R. S., and Sykes, B. D. (2001). Mapping the interacting regions between troponins T and C: binding of TnT and TnI peptides to TnC and NMR mapping of the TnT-binding site on TnC. J. Biol. Chem. 276, 36606–36612. doi: 10.1074/jbc.M105130200
Bodor, G. S., Oakeley, A. E., Allen, P. D., Crimmins, D. L., Ladenson, J. H., and Anderson, P. A. W. (1997). Troponin I phosphorylation in the normal and failing adult human heart. Circulation 96, 1495–1500. doi: 10.1161/01.CIR.96.5.1495
Boral, M. C., and Deb, C. (1970). Seasonal changes in body fluids and haematology in toad Bufo melanostictus a poikilothermic cold torpor. Proc. Ind. Natl. Sci. Acad. 36, 369–379.
Burkart, E. M., Arteaga, G. M., Sumandea, M. P., Prabhakar, R., Wieczorek, D. F., and Solaro, R. J. (2003a). Altered signaling surrounding the C-lobe of cardiac troponin C in myofilaments containing an alpha-tropomyosin mutation linked to familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 35, 1285. doi: 10.1016/S0022-2828(03)00240-2
Burkart, E. M., Sumandea, M. P., Kobayashi, T., Nili, M., Martin, A. F., Homsher, E., et al. (2003b). Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J. Biol. Chem. 278, 11265–11272. doi: 10.1074/jbc.M210712200
Buscemi, N., Foster, D. B., Neverova, I., and Van Eyk, J. E. (2002). p21-activated kinase increases the calcium sensitivity of rat triton-skinned cardiac muscle fiber bundles via a mechanism potentially involving novel phosphorylation of troponin I. Circ. Res. 91, 509–516. doi: 10.1161/01.RES.0000035246.27856.53
Chandra, M., Montgomery, D. E., Kim, J. J., and Solaro, R. J. (1999). The N-terminal region of troponin T is essential for the maximal activation of rat cardiac myofilaments. J. Mol. Cell. Cardiol. 31, 867–880. doi: 10.1006/jmcc.1999.0928
Chen, Z., Higashiyama, A., Yaku, H., Bell, S., Fabian, J., Watkins, M. W., et al. (1997). Altered expression of troponin T isoforms in mild left ventricular hypertrophy in the rabbit. J. Mol. Cell. Cardiol. 29, 2345. doi: 10.1006/jmcc.1997.0468
Chong, S. M., and Jin, J. P. (2009). To investigate protein evolution by detecting suppressed epitope structures. J. Mol. Evol. 68, 448–460. doi: 10.1007/s00239-009-9202-0
Collins, J. H. (1991). Myosin light chains and troponin C: structural and evolutionary relationships revealed by amino acid sequence comparisons. J. Muscle Res. Cell Motil. 12, 3–25. doi: 10.1007/BF01781170
Communal, C., Sumandea, M., De Tombe, P., Narula, J., Solaro, R. J., and Hajjar, R. J. (2002). Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 99, 6252–6256. doi: 10.1073/pnas.092022999
Cuello, F., Bardswell, S. C., Haworth, R. S., Yin, X., Lutz, S., Wieland, T., et al. (2007). Protein kinase D selectively targets cardiac troponin I and regulates myofilament Ca2+ sensitivity in ventricular myocytes. Circ. Res. 100, 864–873. doi: 10.1161/01.RES.0000260809.15393.fa
Davis, J., Wen, H., Edwards, T., and Metzger, J. M. (2007). Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ. Res. 100, 1494–1502. doi: 10.1161/01.RES.0000268412.34364.50
Deb, C., Chatterjee, S., and Boral, M. C. (1974). Body fluid and hematological changes in toads following heat exposure. Am. J. Physiol. 226, 408–410.
Di Lisa, F., De Tullio, R., Salamino, F., Barbato, R., Melloni, E., Siliprandi, N., et al. (1995). Specific degradation of troponin T and I by mu-calpain and its modulation by substrate phosphorylation. Biochem. J. 308, 57.
Du, J., Liu, J., Feng, H. Z., Hossain, M. M., Gobara, N., Zhang, C., et al. (2008). Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am. J. Physiol. Heart Circ. Physiol. 294, H2604–H2613. doi: 10.1152/ajpheart.91506.2007
Farah, C. S., Miyamoto, C. A., Ramos, C. H., Da Silva, A. C., Quaggio, R. B., Fujimori, K., et al. (1994). Structural and regulatory functions of the NH2- and COOH-terminal regions of skeletal muscle troponin I. J. Biol. Chem. 269, 5230–5240.
Feng, H. Z., Biesiadecki, B. J., Yu, Z. B., Hossain, M. M., and Jin, J. P. (2008a). Restricted N-terminal truncation of cardiac troponin T: a novel mechanism for functional adaptation to energetic crisis. J. Physiol. 586, 3537–3550. doi: 10.1113/jphysiol.2008.153577
Feng, H. Z., Chen, M., Weinstein, L. S., and Jin, J. P. (2008b). Removal of the N-terminal extension of cardiac troponin I as a functional compensation for impaired myocardial beta-adrenergic signaling. J. Biol. Chem. 283, 33384–33393. doi: 10.1074/jbc.M803302200
Feng, H. Z., Chen, X., Hossain, M. M., and Jin, J. P. (2012). Toad heart utilizes exclusively slow skeletal muscle troponin T: an evolutionary adaptation with potential functional benefits. J. Biol. Chem. 287, 29753–29764. doi: 10.1074/jbc.M112.373191
Feng, H. Z., Hossain, M. M., Huang, X. P., and Jin, J. P. (2009a). Myofilament incorporation determines the stoichiometry of troponin I in transgenic expression and the rescue of a null mutation. Arch. Biochem. Biophys. 487, 36–41. doi: 10.1016/j.abb.2009.05.001
Feng, H. Z., and Jin, J. P. (2010). Coexistence of cardiac troponin T variants reduces heart efficiency. Am. J. Physiol. Heart Circ. Physiol. 299, H97–H105. doi: 10.1152/ajpheart.01105.2009
Feng, H. Z., Wei, B., and Jin, J. P. (2009b). Deletion of a genomic segment containing the cardiac troponin I gene knocks down expression of the slow troponin T gene and impairs fatigue tolerance of diaphragm muscle. J. Biol. Chem. 284, 31798–31806. doi: 10.1074/jbc.M109.020826
Feng, J., Schaus, B. J., Fallavollita, J. A., Lee, T. C., and Canty, J. M. Jr., (2001). Preload induces troponin I degradation independently of myocardial ischemia. Circulation 103, 2035–2037. doi: 10.1161/01.CIR.103.16.2035
Fentzke, R. C., Buck, S. H., Patel, J. R., Lin, H., Wolska, B. M., Stojanovic, M. O., et al. (1999). Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J. Physiol. 517, 143–157. doi: 10.1111/j.1469-7793.1999.0143z.x
Finley, N., Abbott, M. B., Abusamhadneh, E., Gaponenko, V., Dong, W., Gasmi-Seabrook, G., et al. (1999). NMR analysis of cardiac troponin C-troponin I complexes: effects of phosphorylation. FEBS Lett. 453, 107–112. doi: 10.1016/S0014-5793(99)00693-6
Flicker, P. F., Phillips, G. N. Jr., and Cohen, C. (1982). Troponin and its interactions with tropomyosin: an electron microscope study. J. Mol. Biol. 162, 495–501. doi: 10.1016/0022-2836(82)90540-X
Foster, D. B., Noguchi, T., Vanburen, P., Murphy, A. M., and Van Eyk, J. E. (2003). C-terminal truncation of cardiac troponin I causes divergent effects on ATPase and force implications for the pathophysiology of myocardial stunning. Circ. Res. 93, 917–924. doi: 10.1161/01.RES.0000099889.35340.6F
Galiñska, A., Hatch, V., Craig, R., Murphy, A. M., Van Eyk, J. E., Wang, C. L. A., et al. (2010). The C terminus of cardiac troponin I stabilizes the Ca2+-activated state of tropomyosin on actin filaments. Circ. Res. 106, 705–711. doi: 10.1161/CIRCRESAHA.109.210047
Gomes, A. V., Guzman, G., Zhao, J., and Potter, J. D. (2002). Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development. Insights into the role of troponin T isoforms in the heart. J. Biol. Chem. 277, 35341–35349. doi: 10.1074/jbc.M204118200
Gomes, A. V., Liang, J., and Potter, J. D. (2005). Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J. Biol. Chem. 280, 30909–30915. doi: 10.1074/jbc.M500287200
Gomes, A. V., and Potter, J. D. (2004). Cellular and molecular aspects of familial hypertrophic cardiomyopathy caused by mutations in the cardiac troponin I gene. Mol. Cell. Biochem. 263, 99–114. doi: 10.1023/B:MCBI.0000041852.42291.aa
Gordon, A., Homsher, E., and Regnier, M. (2000). Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924.
Grabarek, Z., Tao, T., and Gergely, J. (1992). Molecular mechanism of troponin-C function. J. Muscle Res. Cell Motil. 13, 383–393. doi: 10.1007/BF01738034
Greaser, M., and Gergely, J. (1971). Reconstitution of troponin activity from three protein components. J. Biol. Chem. 246, 4226–4233.
Guo, X., Wattanapermpool, J., Palmiter, K. A., Murphy, A. M., and Solaro, R. J. (1994). Mutagenesis of cardiac troponin I. Role of the unique NH2-terminal peptide in myofilament activation. J. Biol. Chem. 269, 15210–15216.
Harada, K., and Potter, J. D. (2004). Familial hypertrophic cardiomyopathy mutations from different functional regions of Troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J. Biol. Chem. 279, 14488–14495. doi: 10.1074/jbc.M309355200
Hastings, K. (1997). Molecular evolution of the vertebrate troponin I gene family. Cell Struct. Funct. 22:205. doi: 10.1247/csf.22.205
Haworth, R. S., Cuello, F., Herron, T. J., Franzen, G., Kentish, J. C., Gautel, M., et al. (2004). Protein kinase D is a novel mediator of cardiac troponin I phosphorylation and regulates myofilament function. Circ. Res. 95, 1091–1099. doi: 10.1161/01.RES.0000149299.34793.3c
He, X., Liu, Y., Sharma, V., Dirksen, R. T., Waugh, R., Sheu, S. S., et al. (2003). ASK1 associates with troponin T and induces troponin T phosphorylation and contractile dysfunction in cardiomyocytes. Am. J. Pathol. 163, 243–251. doi: 10.1016/S0002-9440(10)63647-4
Heeley, D., Golosinska, K., and Smillie, L. B. (1987). The effects of troponin T fragments T1 and T2 on the binding of nonpolymerizable tropomyosin to F-actin in the presence and absence of troponin I and troponin C. J. Biol. Chem. 262, 9971–9978.
Herzberg, O., and James, M. N. G. (1985). Structure of the calcium regulatory muscle protein troponin-C at 2.8 Å resolution. Nature 313, 653–659 doi: 10.1038/313653a0
Hinkle, A., Goranson, A., Butters, C. A., and Tobacman, L. S. (1999). Roles for the troponin tail domain in thin filament assembly and regulation: a deletion study of cardiac troponin T. J. Biol. Chem. 274, 7157–7164. doi: 10.1074/jbc.274.11.7157
Hoffmann, B., Schmidt-Traub, H., Perrot, A., Osterziel, K. J., and Gessner, R. (2001). First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Hum. Mutat. 17, 524–524. doi: 10.1002/humu.1143
Howarth, J. W., Meller, J., Solaro, R. J., Trewhella, J., and Rosevear, P. R. (2007). Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J. Mol. Biol. 373, 706–722. doi: 10.1016/j.jmb.2007.08.035
Huang, Q. Q., Feng, H. Z., Liu, J., Du, J., Stull, L. B., Moravec, C. S., et al. (2008). Co-expression of skeletal and cardiac troponin T decreases mouse cardiac function. Am. J. Physiol. Cell Physiol. 294, C213–C222. doi: 10.1152/ajpcell.00146.2007
Iorga, B., Blaudeck, N., Solzin, J., Neulen, A., Stehle, I., Davila, A. J. L., et al. (2008). Lys184 deletion in troponin I impairs relaxation kinetics and induces hypercontractility in murine cardiac myofibrils. Cardiovasc. Res. 77, 676–686. doi: 10.1093/cvr/cvm113
Jeong, E. M., Wang, X., Xu, K., Hossain, M. M., and Jin, J. P. (2009). Nonmyofilament-associated troponin T fragments induce apoptosis. Am. J. Physiol. Heart Circ. Physiol. 297, H283–H292. doi: 10.1152/ajpheart.01200.2008
Jideama, N. M., Crawford, B. H., Hussain, A. K. M. A., and Raynor, R. L. (2006). Dephosphorylation specificities of protein phosphatase for cardiac troponin I, troponin T, and sites within troponin T. Int. J. Biol. Sci. 2:1. doi: 10.7150/ijbs.2.1
Jideama, N. M., Noland, T. A. Jr. Raynor, R. L., Blobe, G. C., Fabbro, D., Kazanietz, M. G., et al. (1996). Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J. Biol. Chem. 271, 23277–23283. doi: 10.1074/jbc.271.38.23277
Jin, J. P. (1996). Alternative RNA splicing-generated cardiac troponin T isoform switching: a non-heart-restricted genetic programming synchronized in developing cardiac and skeletal muscles. Biochem. Biophys. Res. Commun. 225, 883–889. doi: 10.1006/bbrc.1996.1267
Jin, J. P., Chen, A., Ogut, O., and Huang, Q. Q. (2000). Conformational modulation of slow skeletal muscle troponin T by an NH(2)-terminal metal-binding extension. Am. J. Physiol. Cell Physiol. 279, C1067–C1077.
Jin, J. P., and Chong, S. M. (2010). Localization of the two tropomyosin-binding sites of troponin T. Arch. Biochem. Biophys. 500, 144–150. doi: 10.1016/j.abb.2010.06.001
Jin, J. P., Huang, Q. Q., Yeh, H. I., and Lin, J. J. (1992). Complete nucleotide sequence and structural organization of rat cardiac troponin T gene. A single gene generates embryonic and adult isoforms via developmentally regulated alternative splicing. J. Mol. Biol. 227, 1269–1276. doi: 10.1016/0022-2836(92)90540-Z
Jin, J. P., and Lin, J. J. (1988). Rapid purification of mammalian cardiac troponin T and its isoform switching in rat hearts during development. J. Biol. Chem. 263, 7309–7315.
Jin, J. P., and Lin, J. J. (1989). Isolation and characterization of cDNA clones encoding embryonic and adult isoforms of rat cardiac troponin T. J. Biol. Chem. 264, 14471–14477.
Jin, J. P., and Root, D. D. (2000). Modulation of troponin T molecular conformation and flexibility by metal ion binding to the NH2-terminal variable region. Biochemistry 39, 11702–11713. doi: 10.1021/bi9927437
Jin, J. P., and Smillie, L. B. (1994). An unusual metal-binding cluster found exclusively in the avian breast muscle troponin T of Galliformes and Craciformes. FEBS Lett. 341, 135–140. doi: 10.1016/0014-5793(94)80256-4
Jin, J. P., Wang, J., and Zhang, J. (1996). Expression of cDNAs encoding mouse cardiac troponin T isoforms: characterization of a large sample of independent clones. Gene 168, 217–221. doi: 10.1016/0378-1119(95)00803-9
Jin, J. P., Yang, F. W., Yu, Z. B., Ruse, C. I., Bond, M., and Chen, A. (2001). The highly conserved COOH terminus of troponin I forms a Ca2+-modulated allosteric domain in the troponin complex. Biochemistry 40, 2623–2631. doi: 10.1021/bi002423j
Jin, J. P., Zhang, Z., and Bautista, J. A. (2008). Isoform diversity, regulation, and functional adaptation of troponin and calponin. Crit. Rev. Eukaryot Gene Expr. 18, 93–124. doi: 10.1615/CritRevEukarGeneExpr.v18.i2.10
Kamisago, M., Sharma, S. D., Depalma, S. R., Solomon, S., Sharma, P., McDonough, B., et al. (2000). Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 343, 1688–1696. doi: 10.1056/NEJM200012073432304
Kawasaki, H., Nakayama, S., and Kretsinger, R. (1998). Classification and evolution of EF-hand proteins. Biometals 11, 277–295. doi: 10.1023/A:1009282307967
Ke, Y., Lei, M., Wang, X., and Solaro, R. J. (2012). Novel roles of PAK1 in the heart. Cell. Logist. 2, 89–94. doi: 10.4161/cl.21497
Ke, Y., Wang, L., Pyle, W. G., De Tombe, P. P., and Solaro, R. J. (2004). Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circ. Res. 94, 194–200. doi: 10.1161/01.RES.0000111522.02730.56
Kim, S. J., Kudej, R. K., Yatani, A., Kim, Y. K., Takagi, G., Honda, R., et al. (2001). A novel mechanism for myocardial stunning involving impaired Ca2+ handling. Circ. Res. 89, 831–837. doi: 10.1161/hh2101.098547
Kimura, A., Harada, H., Park, J. E., Nishi, H., Satoh, M., Takahashi, M., et al. (1997). Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat. Genet. 16, 379–382. doi: 10.1038/ng0897-379
Kirkpatrick, K. P., Robertson, A. S., Klaiman, J. M., and Gillis, T. E. (2011). The influence of trout cardiac troponin I and PKA phosphorylation on the Ca2+ affinity of the cardiac troponin complex. J. Exp. Biol. 214, 1981–1988. doi: 10.1242/jeb.052860
Kobayashi, T., Yang, X., Walker, L. A., Van Breemen, R. B., and Solaro, R. J. (2005). A non-equilibrium isoelectric focusing method to determine states of phosphorylation of cardiac troponin I: identification of Ser-23 and Ser-24 as significant sites of phosphorylation by protein kinase C. J. Mol. Cell. Cardiol. 38, 213–218. doi: 10.1016/j.yjmcc.2004.10.014
Kooij, V., Stienen, G. J. M., and Van Der Velden, J. (2011). The role of protein kinase C-mediated phosphorylation of sarcomeric proteins in the heart—detrimental or beneficial? Biophys. Rev. 3, 107–117. doi: 10.1007/s12551-011-0050-y
Kracklauer, M. P., Feng, H. Z., Jiang, W., Lin, J. L., Lin, J. J., and Jin, J. P. (2013). Discontinuous thoracic venous cardiomyocytes and heart exhibit synchronized developmental switch of troponin isoforms. FEBS J. 280, 880–891. doi: 10.1111/febs.12076
Lakdawala, N. K., Thune, J. J., Colan, S. D., Cirino, A. L., Farrohi, F., Rivero, J., et al. (2012). Subtle abnormalities in contractile function are an early manifestation of sarcomere mutations in dilated cardiomyopathy. Circ. Cardiovasc. Genet. 5, 503–510. doi: 10.1161/CIRCGENETICS.112.962761
Landstrom, A. P., Parvatiyar, M. S., Pinto, J. R., Marquardt, M. L., Bos, J. M., Tester, D. J., et al. (2008). Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol. 45, 281–288. doi: 10.1016/j.yjmcc.2008.05.003
Layland, J., Li, J. M., and Shah, A. M. (2002). Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J. Physiol. 540, 457–467. doi: 10.1113/jphysiol.2001.014126
Layland, J., Solaro, R. J., and Shah, A. M. (2005). Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc. Res. 66, 12–21. doi: 10.1016/j.cardiores.2004.12.022
Li, D., Czernuszewicz, G. Z., Gonzalez, O., Tapscott, T., Karibe, A., Durand, J.-B., et al. (2001). Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy. Circulation 104, 2188–2193. doi: 10.1161/hc4301.098285
Li, M. X., Wang, X., Lindhout, D. A., Buscemi, N., Van Eyk, J. E., and Sykes, B. D. (2003). Phosphorylation and mutation of human cardiac troponin I deferentially destabilize the interaction of the functional regions of troponin I with troponin C. Biochemistry 42, 14460–14468. doi: 10.1021/bi035408y
Li, M. X., Wang, X., and Sykes, B. D. (2004). Structural based insights into the role of troponin in cardiac muscle pathophysiology. J. Muscle Res. Cell Motil. 25, 559–579. doi: 10.1007/s10974-004-5879-2
Li, Y., Charles, P. Y., Nan, C., Pinto, J. R., Wang, Y., Liang, J., et al. (2010). Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J. Mol. Cell. Cardiol. 49, 402–411. doi: 10.1016/j.yjmcc.2010.04.017
Li, Y., Zhang, L., Jean-Charles, P. Y., Nan, C., Chen, G., Tian, J., et al. (2013). Dose-dependent diastolic dysfunction and early death in a mouse model with cardiac troponin mutations. J. Mol. Cell. Cardiol. 62, 227–236. doi: 10.1016/j.yjmcc.2013.06.007
Liang, B., Chung, F., Qu, Y., Pavlov, D., Gillis, T. E., Tikunova, S. B., et al. (2008). Familial hypertrophic cardiomyopathy-related cardiac troponin C mutation L29Q affects Ca2+ binding and myofilament contractility. Physiol. Genomics 33, 257–266. doi: 10.1152/physiolgenomics.00154.2007
Lim, C. C., Yang, H., Yang, M., Wang, C. K., Shi, J., Berg, E. A., et al. (2008). A novel mutant cardiac troponin C disrupts molecular motions critical for calcium binding affinity and cardiomyocyte contractility. Biophys. J. 94, 3577–3589. doi: 10.1529/biophysj.107.112896
Lindhout, D. A., Li, M. X., Schieve, D., and Sykes, B. D. (2002). Effects of T142 phosphorylation and mutation R145G on the interaction of the inhibitory region of human cardiac troponin I with the C-domain of human cardiac troponin C. Biochemistry 41, 7267–7274. doi: 10.1021/bi020100c
Liu, B., Lee, R. S., Biesiadecki, B. J., Tikunova, S. B., and Davis, J. P. (2012). Engineered Troponin C constructs correct disease-related cardiac myofilament calcium sensitivity. J. Biol. Chem. 287, 20027–20036. doi: 10.1074/jbc.M111.334953
Liu, R., Feng, H. Z., and Jin, J. P. (2014). Physiological contractility of cardiomyocytes in the wall of mouse and rat azygos vein. Am. J. Physiol. Cell Physiol. 306, C697–C704. doi: 10.1152/ajpcell.00004.2014
Lu, Q. W., Hinken, A. C., Patrick, S. E., Solaro, R. J., and Kobayashi, T. (2010). Phosphorylation of cardiac troponin I at protein kinase C site threonine 144 depresses cooperative activation of thin filaments. J. Biol. Chem. 285, 11810–11817. doi: 10.1074/jbc.M109.055657
Lüss, H., Meissner, A., Rolf, N., Van Aken, H., Bokník, P., Kirchhefer, U., et al. (2000). Biochemical mechanism (s) of stunning in conscious dogs. Am. J. Physiol. Heart Circ. Physiol. 279, H176–H184.
Macgowan, G. A., Du, C., Cowan, D. B., Stamm, C., McGowan, F. X., Solaro, R. J., et al. (2001). Ischemic dysfunction in transgenic mice expressing troponin I lacking protein kinase C phosphorylation sites. Am. J. Physiol. Heart Circ. Physiol. 280, H835–H843.
Maron, B. J., Shirani, J., Poliac, L. C., Mathenge, R., Roberts, W. C., and Mueller, F. O. (1996). Sudden death in young competitive athletes. JAMA 276, 199–204. doi: 10.1001/jama.1996.03540030033028
Martin, A. F. (1981). Turnover of cardiac troponin subunits. Kinetic evidence for a precursor pool of troponin-I. J. Biol. Chem. 256, 964–968.
McConnell, B. K., Moravec, C. S., and Bond, M. (1998). Troponin I phosphorylation and myofilament calcium sensitivity during decompensated cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 274, H385–H396.
McDonough, J. L., Arrell, D. K., and Van Eyk, J. E. (1999). Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ. Res. 84, 9–20. doi: 10.1161/01.RES.84.1.9
McDonough, J., Labugger, R., Pickett, W., Tse, M., Mackenzie, S., Pang, S., et al. (2001). Cardiac troponin I is modified in the myocardium of bypass patients. Circulation 103, 58–64. doi: 10.1161/01.CIR.103.1.58
Mesnard, L., Logeart, D., Taviaux, S., Diriong, S., Mercadier, J. J., and Samson, F. (1995). Human cardiac troponin T: cloning and expression of new isoforms in the normal and failing heart. Circ. Res. 76, 687–692. doi: 10.1161/01.RES.76.4.687
Messer, A. E., Jacques, A. M., and Marston, S. B. (2007). Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J. Mol. Cell. Cardiol. 42, 247–259. doi: 10.1016/j.yjmcc.2006.08.017
Mirza, M., Marston, S., Willott, R., Ashley, C., Mogensen, J., McKenna, W., et al. (2005). Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J. Biol. Chem. 280, 28498–28506. doi: 10.1074/jbc.M412281200
Mogensen, J., Murphy, R. T., Shaw, T., Bahl, A., Redwood, C., Watkins, H., et al. (2004). Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 44, 2033–2040. doi: 10.1016/j.jacc.2004.08.027
Monasky, M. M., Taglieri, D. M., Patel, B. G., Chernoff, J., Wolska, B. M., Ke, Y., et al. (2012). p21-activated kinase improves cardiac contractility during ischemia-reperfusion concomitant with changes in troponin-T and myosin light chain 2 phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 302, H224–H230. doi: 10.1152/ajpheart.00612.2011
Montgomery, D. E., Chandra, M., Huang, Q., Jin, J., and Solaro, R. J. (2001). Transgenic incorporation of skeletal TnT into cardiac myofilaments blunts PKC-mediated depression of force. Am. J. Physiol. Heart Circ. Physiol. 280, H1011–H1018.
Morris, E. P., and Lehrer, S. S. (1984). Troponin-tropomyosin interactions. Fluorescence studies of the binding of troponin, troponin T and chymotryptic troponin T fragments to specifically labeled tropomyosin. Biochemistry 23, 2214–2220. doi: 10.1021/bi00305a018
Murphy, A. M. (2006). Heart failure, myocardial stunning, and troponin: a key regulator of the cardiac myofilament. Congest Heart Fail 12, 32–40. doi: 10.1111/j.1527-5299.2006.04320.x
Murphy, A. M., Kögler, H., Georgakopoulos, D., McDonough, J. L., Kass, D. A., Van Eyk, J. E., et al. (2000). Transgenic mouse model of stunned myocardium. Science 287, 488–491. doi: 10.1126/science.287.5452.488
Narolska, N. A., Piroddi, N., Belus, A., Boontje, N. M., Scellini, B., Deppermann, S., et al. (2006). Impaired diastolic function after exchange of endogenous troponin I with C-terminal truncated troponin I in human cardiac muscle. Circ. Res. 99, 1012–1020. doi: 10.1161/01.RES.0000248753.30340.af
Nixon, B. R., Thawornkaiwong, A., Jin, J., Brundage, E. A., Little, S. C., Davis, J. P., et al. (2012). AMP-activated protein kinase phosphorylates cardiac Troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. J. Biol. Chem. 287, 19136–19147. doi: 10.1074/jbc.M111.323048
Noland, T. A. Jr., and Kuo, J. F. (1991). Protein kinase C phosphorylation of cardiac troponin I or troponin T inhibits Ca2+-stimulated actomyosin MgATPase activity. J. Biol. Chem. 266, 4974–4978.
Noland, T. A. Jr., and Kuo, J. F. (1993). Protein kinase C phosphorylation of cardiac troponin I and troponin T inhibits Ca2+-stimulated MgATPase activity in reconstituted actomyosin and isolated myofibrils, and decreases actin-myosin interactions. J. Mol. Cell. Cardiol. 25, 53–65. doi: 10.1006/jmcc.1993.1007
Ogut, O., and Jin, J. P. (1996). Expression, zinc-affinity purification, and characterization of a novel metal-binding cluster in troponin T: metal-stabilized alpha-helical structure and effects of the NH2-terminal variable region on the conformation of intact troponin T and its association with tropomyosin. Biochemistry 35, 16581–16590. doi: 10.1021/bi961712y
Oliveira, S. M., Zhang, Y. H., Solis, R. S., Isackson, H., Bellahcene, M., Yavari, A., et al. (2012). AMP-activated protein kinase phosphorylates cardiac Troponin I and alters contractility of murine ventricular MyocytesNovelty and significance. Circ. Res. 110, 1192–1201. doi: 10.1161/CIRCRESAHA.111.259952
Palpant, N. J., Houang, E. M., Delport, W., Hastings, K. E. M., Onufriev, A. V., Sham, Y. Y., et al. (2010). Pathogenic peptide deviations support a model of adaptive evolution of chordate cardiac performance by troponin mutations. Physiol. Genomics 42, 287–299. doi: 10.1152/physiolgenomics.00033.2010
Parvatiyar, M. S., Landstrom, A. P., Figueiredo-Freitas, C., Potter, J. D., Ackerman, M. J., and Pinto, J. R. (2012). A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation. J. Biol. Chem. 287, 31845–31855. doi: 10.1074/jbc.M112.377713
Perry, S. (1998). Troponin T: genetics, properties and function. J. Muscle Res. Cell Motil. 19, 575–602. doi: 10.1023/A:1005397501968
Perry, S. (1999). Troponin I: inhibitor or facilitator. Mol. Cell. Biochem. 190, 9–32. doi: 10.1023/A:1006939307715
Pfleiderer, P., Sumandea, M. P., Rybin, V. O., Wang, C., and Steinberg, S. F. (2009). Raf-1: a novel cardiac troponin T kinase. J. Muscle Res. Cell Motil. 30, 67–72. doi: 10.1007/s10974-009-9176-y
Pi, Y. Q., Kemnitz, K. R., Zhang, D., Kranias, E. G., and Walker, J. W. (2002). Phosphorylation of troponin I controls cardiac twitch dynamics evidence from phosphorylation site mutants expressed on a troponin I-null background in mice. Circ. Res. 90, 649–656. doi: 10.1161/01.RES.0000014080.82861.5F
Pi, Y. Q., Zhang, D., Kemnitz, K. R., Wang, H., and Walker, J. W. (2003). Protein kinase C and A sites on troponin I regulate myofilament Ca2+ sensitivity and ATPase activity in the mouse myocardium. J. Physiol. 552, 845–857. doi: 10.1113/jphysiol.2003.045260
Pinto, J. R., Parvatiyar, M. S., Jones, M. A., Liang, J., Ackerman, M. J., and Potter, J. D. (2009). A functional and structural study of Troponin C mutations related to hypertrophic cardiomyopathy. J. Biol. Chem. 284, 19090–19100. doi: 10.1074/jbc.M109.007021
Prigozy, T. I., Dalrymple, K., Shuler, C., and Kedes, L. (1997). Differential expression of troponin C genes during tongue myogenesis. Dev. Dyn. 209, 36–44.
Quirk, P. G., Patchell, V. B., Gao, Y., Levine, B. A., and Victor Perry, S. (1995). Sequential phosphorylation of adjacent serine residues on the N-terminal region of cardiac troponin-I: Structure-activity implications of ordered phosphorylation. FEBS Lett. 370, 175–178. doi: 10.1016/0014-5793(95)00812-N
Rao, V. S., Korte, F. S., Razumova, M. V., Feest, E. R., Hsu, H., Irving, T. C., et al. (2012). N-terminal phosphorylation of cardiac troponin-I reduces length dependent calcium sensitivity of contraction in cardiac muscle. J. Physiol. 591, 475–490. doi: 10.1113/jphysiol.2012.241604
Ricchiuti, V., and Apple, F. S. (1999). RNA expression of cardiac troponin T isoforms in diseased human skeletal muscle. Clin. Chem. 45, 2129–2135.
Robertson, S., Johnson, J. D., and Potter, J. (1981). The time-course of Ca2+ exchange with calmodulin, troponin, parvalbumin, and myosin in response to transient increases in Ca2+. Biophys. J. 34, 559–569. doi: 10.1016/S0006-3495(81)84868-0
Saggin, L., Gorza, L., Ausoni, S., and Schiaffino, S. (1989). Troponin I switching in the developing heart. J. Biol. Chem. 264, 16299–16302.
Sakthivel, S., Finley, N. L., Rosevear, P. R., Lorenz, J. N., Gulick, J., Kim, S., et al. (2005). In vivo and in vitro analysis of cardiac troponin I phosphorylation. J. Biol. Chem. 280, 703–714. doi: 10.1074/jbc.M409513200
Sancho Solis, R., Ge, Y., and Walker, J. W. (2008). Single amino acid sequence polymorphisms in rat cardiac troponin revealed by top–down tandem mass spectrometry. J. Muscle Res. Cell Motil. 29, 203–212. doi: 10.1007/s10974-009-9168-y
Sancho Solis, R., Ge, Y., and Walker, J. W. (2011). A preferred AMPK phosphorylation site adjacent to the inhibitory loop of cardiac and skeletal troponin I. Protein Sci. 20, 894–907. doi: 10.1002/pro.623
Sasse, S., Brand, N., Kyprianou, P., Dhoot, G., Wade, R., Arai, M., et al. (1993). Troponin I gene expression during human cardiac development and in end-stage heart failure. Circ. Res. 72, 932–938. doi: 10.1161/01.RES.72.5.932
Schaertl, S., Lehrer, S., and Geeves, M. (1995). Separation and characterization of the two functional regions of troponin involved in muscle thin filament regulation. Biochemistry 34, 15890–15894. doi: 10.1021/bi00049a003
Schmidtmann, A., Lindow, C., Villard, S., Heuser, A., Mügge, A., Geßner, R., et al. (2005). Cardiac troponin C-L29Q, related to hypertrophic cardiomyopathy, hinders the transduction of the protein kinase A dependent phosphorylation signal from cardiac troponin I to C. FEBS J. 272, 6087–6097. doi: 10.1111/j.1742-4658.2005.05001.x
Schreier, T., Kedes, L., and Gahlmann, R. (1990). Cloning, structural analysis, and expression of the human slow twitch skeletal muscle/cardiac troponin C gene. J. Biol. Chem. 265, 21247–21253.
Sehnert, A. J., Huq, A., Weinstein, B. M., Walker, C., Fishman, M., and Stainier, D. Y. R. (2002). Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nat. Genet. 31, 106–110. doi: 10.1038/ng875
Seidman, J. G., and Seidman, C. (2001). The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104, 557–567. doi: 10.1016/S0092-8674(01)00242-2
Sfichi-Duke, L., Garcia-Cazarin, M. L., Sumandea, C. A., Sievert, G. A., Balke, C. W., Zhan, D. Y., et al. (2010). Cardiomyopathy-causing deletion K210 in cardiac troponin T alters phosphorylation propensity of sarcomeric proteins. J. Mol. Cell. Cardiol. 48, 934–942. doi: 10.1016/j.yjmcc.2010.01.005
Sheng, Z., Strauss, W. L., Francois, J. M., and Potter, J. D. (1990). Evidence that both Ca2+-specific sites of skeletal muscle TnC are required for full activity. J. Biol. Chem. 265, 21554–21560.
Sherman, A. J., Klocke, F. J., Decker, R. S., Decker, M. L., Kozlowski, K. A., Harris, K. R., et al. (2000). Myofibrillar disruption in hypocontractile myocardium showing perfusion-contraction matches and mismatches. Am. J. Physiol. Heart Circ. Physiol. 278, H1320–H1334.
Solaro, R. J. (2010). Sarcomere control mechanisms and the dynamics of the cardiac cycle. J. Biomed. Biotechnol. 2010:105648. doi: 10.1155/2010/105648
Solaro, R. J., and Kobayashi, T. (2011). Protein phosphorylation and signal transduction in cardiac thin filaments. J. Biol. Chem. 286, 9935–9940. doi: 10.1074/jbc.R110.197731
Solaro, R. J., Lee, J. A., Kentish, J. C., and Allen, D. G. (1988). Effects of acidosis on ventricular muscle from adult and neonatal rats. Circ. Res. 63, 779–787. doi: 10.1161/01.RES.63.4.779
Solaro, R. J., Rosevear, P., and Kobayashi, T. (2008). The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochem. Biophys. Res. Commun. 369, 82–87. doi: 10.1016/j.bbrc.2007.12.114
Solaro, R. J., and Van Der Velden, J. (2010). Why does troponin I have so many phosphorylation sites? Fact and fancy. J. Mol. Cell. Cardiol. 48, 810–816. doi: 10.1016/j.yjmcc.2010.02.014
Stelzer, J. E., Patel, J. R., Walker, J. W., and Moss, R. L. (2007). Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ. Res. 101, 503–511. doi: 10.1161/CIRCRESAHA.107.153650
Stevens, L., Bastide, B., Kischel, P., Pette, D., and Mounier, Y. (2002). Time-dependent changes in expression of troponin subunit isoforms in unloaded rat soleus muscle. Am. J. Physiol. Cell Physiol. 282, C1025–C1030. doi: 10.1152/ajpcell.00252.2001
Sumandea, M. P., Pyle, W. G., Kobayashi, T., De Tombe, P. P., and Solaro, R. J. (2003). Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J. Biol. Chem. 278, 35135–35144. doi: 10.1074/jbc.M306325200
Sumandea, M. P., Vahebi, S., Sumandea, C. A., Garcia-Cazarin, M. L., Staidle, J., and Homsher, E. (2009). Impact of cardiac troponin T N-terminal deletion and phosphorylation on myofilament function. Biochemistry 48, 7722–7731. doi: 10.1021/bi900516n
Sweeney, H. L., Brito, R., Rosevear, P. R., and Putkey, J. A. (1990). The low-affinity Ca2+-binding sites in cardiac/slow skeletal muscle troponin C perform distinct functions: site I alone cannot trigger contraction. Proc. Natl. Acad. Sci. U.S.A. 87, 9538–9542. doi: 10.1073/pnas.87.24.9538
Taglieri, D. M., Monasky, M. M., Knezevic, I., Sheehan, K. A., Lei, M., Wang, X., et al. (2011). Ablation of p21-activated kinase-1 in mice promotes isoproterenol-induced cardiac hypertrophy in association with activation of Erk1/2 and inhibition of protein phosphatase 2A. J. Mol. Cell. Cardiol. 51, 988–996. doi: 10.1016/j.yjmcc.2011.09.016
Takeda, S., Yamashita, A., Maeda, K., and Maeda, Y. (2003). Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424, 35–41. doi: 10.1038/nature01780
Tanokura, M., Tawada, Y., Ono, A., and Ohtsuki, I. (1983). Chymotryptic subfragments of troponin T from rabbit skeletal muscle. Interaction with tropomyosin, troponin I and troponin C. J. Biochem. (Tokyo) 93, 331–337.
Tardiff, J. C., Factor, S. M., Tompkins, B. D., Hewett, T. E., Palmer, B. M., Moore, R. L., et al. (1998). A truncated cardiac troponin T molecule in transgenic mice suggests multiple cellular mechanisms for familial hypertrophic cardiomyopathy. J. Clin. Invest. 101, 2800. doi: 10.1172/JCI2389
Thierfelder, L., Watkins, H., Macrae, C., Lamas, R., McKenna, W., Vosberg, H.-P., et al. (1994). α-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 77, 701–712. doi: 10.1016/0092-8674(94)90054-X
Thomas, S. A., Fallavollita, J. A., Lee, T. C., Feng, J., and Canty, J. M. (1999). Absence of troponin I degradation or altered sarcoplasmic reticulum uptake protein expression after reversible ischemia in swine. Circ. Res. 85, 446–456. doi: 10.1161/01.RES.85.5.446
Townsend, P. J., Barton, P. J., Yacoub, M. H., and Farza, H. (1995). Molecular cloning of human cardiac troponin T isoforms: expression in developing and failing heart. J. Mol. Cell. Cardiol. 27, 2223–2236. doi: 10.1016/S0022-2828(95)91587-7
Vahebi, S., Kobayashi, T., Warren, C. M., De Tombe, P. P., and Solaro, R. J. (2005). Functional effects of rho-kinase–dependent phosphorylation of specific sites on cardiac troponin. Circ. Res. 96, 740–747. doi: 10.1161/01.RES.0000162457.56568.7d
Van Der Velden, J., Merkus, D., Klarenbeek, B., James, A., Boontje, N., Dekkers, D., et al. (2004). Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ. Res. 95, e85–e95. doi: 10.1161/01.RES.0000149531.02904.09
Van Eerd, J. P., and Takahashi, K. (1976). Determination of the complete amino acid sequence of bovine cardiac troponin C. Biochemistry 15, 1171–1180. doi: 10.1021/bi00650a033
Varnava, A. M., Elliott, P. M., Baboonian, C., Davison, F., Davies, M. J., and McKenna, W. J. (2001). Hypertrophic cardiomyopathy histopathological features of sudden death in cardiac Troponin T disease. Circulation 104, 1380–1384. doi: 10.1161/hc3701.095952
Vinogradova, M. V., Stone, D. B., Malanina, G. G., Karatzaferi, C., Cooke, R., Mendelson, R. A., et al. (2005). Ca2+-regulated structural changes in troponin. Proc. Natl. Acad. Sci. U.S.A. 102, 5038–5043. doi: 10.1073/pnas.0408882102
Wang, D., Robertson, I. M., Li, M. X., McCully, M. E., Crane, M. L., Luo, Z., et al. (2012a). Structural and functional consequences of the cardiac Troponin C L48Q Ca2+-sensitizing mutation. Biochemistry 51, 4473–4487. doi: 10.1021/bi3003007
Wang, H., Grant, J. E., Doede, C. M., Sadayappan, S., Robbins, J., and Walker, J. W. (2006). PKC-betaII sensitizes cardiac myofilaments to Ca2+ by phosphorylating troponin I on threonine-144. J. Mol. Cell. Cardiol. 41, 823. doi: 10.1016/j.yjmcc.2006.08.016
Wang, J., and Jin, J. P. (1998). Conformational modulation of troponin T by configuration of the NH2-terminal variable region and functional effects. Biochemistry 37, 14519–14528. doi: 10.1021/bi9812322
Wang, X., Huang, Q. Q., Breckenridge, M. T., Chen, A., Crawford, T. O., Morton, D. H., et al. (2005). Cellular fate of truncated slow skeletal muscle troponin T produced by Glu180 nonsense mutation in amish nemaline myopathy. J. Biol. Chem. 280, 13241–13249. doi: 10.1074/jbc.M413696200
Wang, Y., Pinto, J. R., Solis, R. S., Dweck, D., Liang, J., Diaz-Perez, Z., et al. (2012b). Generation and functional characterization of knock-in mice harboring the cardiac Troponin I-R21C mutation associated with hypertrophic cardiomyopathy. J. Biol. Chem. 287, 2156–2167. doi: 10.1074/jbc.M111.294306
Ward, A. J., and Cooper, T. A. (2010). The pathobiology of splicing. J. Pathol. 220, 152–163. doi: 10.1002/path.2649
Watkins, H., McKenna, W. J., Thierfelder, L., Suk, H. J., Anan, R., O'donoghue, A., et al. (1995). Mutations in the genes for cardiac troponin T and α-tropomyosin in hypertrophic cardiomyopathy. N. Engl. J. Med. 332, 1058–1065.
Wei, B., Gao, J., Huang, X. P., and Jin, J. P. (2010). Mutual rescues between two dominant negative mutations in cardiac troponin I and cardiac troponin T. J. Biol. Chem. 285, 27806–27816. doi: 10.1074/jbc.M110.137844
Wei, B., and Jin, J. P. (2011). Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch. Biochem. Biophys. 505, 144–154. doi: 10.1016/j.abb.2010.10.013
Wei, H., and Jin, J. P. (2013). N-terminal truncated cardiac troponin I enhanced the contractility of isolated cardiomyocytes. Biophys. J. 104, 154a–155a. doi: 10.1016/j.bpj.2012.11.875
Westfall, M. V., Albayya, F. P., and Metzger, J. M. (1999). Functional analysis of troponin I regulatory domains in the intact myofilament of adult single cardiac myocytes. J. Biol. Chem. 274, 22508–22516. doi: 10.1074/jbc.274.32.22508
Westfall, M. V., Albayya, F. P., Turner, I. I., and Metzger, J. M. (2000). Chimera analysis of troponin I domains that influence Ca2+-activated myofilament tension in adult cardiac myocytes. Circ. Res. 86, 470–477. doi: 10.1161/01.RES.86.4.470
Westfall, M. V., Lee, A. M., and Robinson, D. A. (2005). Differential contribution of Troponin I phosphorylation sites to the endothelin-modulated contractile response. J. Biol. Chem. 280, 41324–41331. doi: 10.1074/jbc.M506043200
White, S. P., Cohen, C., and Phillips, G. N. Jr. (1987). Structure of co-crystals of tropomyosin and troponin. Nature 325, 826–828 doi: 10.1038/325826a0
Willott, R. H., Gomes, A. V., Chang, A. N., Parvatiyar, M. S., Pinto, J. R., and Potter, J. D. (2010). Mutations in Troponin that cause HCM, DCM AND RCM: What can we learn about thin filament function? J. Mol. Cell. Cardiol. 48, 882–892. doi: 10.1016/j.yjmcc.2009.10.031
You, B., Yan, G., Zhang, Z., Yan, L., Li, J., Ge, Q., et al. (2009). Phosphorylation of cardiac troponin I by mammalian sterile 20-like kinase 1. Biochem. J. 418, 93–101. doi: 10.1042/BJ20081340
Yu, Z. B., Gao, F., Feng, H. Z., and Jin, J. P. (2007). Differential regulation of myofilament protein isoforms underlying the contractility changes in skeletal muscle unloading. Am. J. Physiol. Cell Physiol. 292, C1192–C1203. doi: 10.1152/ajpcell.00462.2006
Yu, Z. B., Wei, H., and Jin, J. P. (2012). Chronic coexistence of two troponin T isoforms in adult transgenic mouse cardiomyocytes decreased contractile kinetics and caused dilatative remodeling. Am. J. Physiol. Cell Physiol. 303, C24–C32. doi: 10.1152/ajpcell.00026.2012
Yu, Z. B., Zhang, L. F., and Jin, J. P. (2001). A proteolytic NH2-terminal truncation of cardiac troponin I that is up-regulated in simulated microgravity. J. Biol. Chem. 276, 15753–15760. doi: 10.1074/jbc.M011048200
Yumoto, F., Lu, Q. W., Morimoto, S., Tanaka, H., Kono, N., Nagata, K., et al. (2005). Drastic Ca2+ sensitization of myofilament associated with a small structural change in troponin I in inherited restrictive cardiomyopathy. Biochem. Biophys. Res. Commun. 338, 1519–1526. doi: 10.1016/j.bbrc.2005.10.116
Zakhary, D. R., Moravec, C. S., Stewart, R. W., and Bond, M. (1999). Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation 99, 505–510. doi: 10.1161/01.CIR.99.4.505
Zhang, P., Kirk, J. A., Ji, W., Dos Remedios, C. G., Kass, D. A., Van Eyk, J. E., et al. (2012). Multiple reaction monitoring to identify site-specific Troponin I phosphorylated residues in the failing human heart/clinical perspective. Circulation 126, 1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388
Zhang, R., Zhao, J., Mandveno, A., and Potter, J. D. (1995a). Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ. Res. 76, 1028–1035. doi: 10.1161/01.RES.76.6.1028
Zhang, R., Zhao, J. J., and Potter, J. D. (1995b). Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca affinity of cardiac troponin C. J. Biol. Chem. 270, 30773–30780. doi: 10.1074/jbc.270.51.30773
Zhang, Z., Akhter, S., Mottl, S., and Jin, J. P. (2011a). Calcium-regulated conformational change in the C-terminal end segment of troponin I and its binding to tropomyosin. FEBS J. 278, 3348–3359. doi: 10.1111/j.1742-4658.2011.08250.x
Zhang, Z., Biesiadecki, B. J., and Jin, J. P. (2006). Selective deletion of the NH2-terminal variable region of cardiac troponin T in ischemia reperfusion by myofibril-associated mu-calpain cleavage. Biochemistry 45, 11681–11694. doi: 10.1021/bi060273s
Zhang, Z., Feng, H. Z., and Jin, J. P. (2011b). Structure of the NH2-terminal variable region of cardiac troponin T determines its sensitivity to restrictive cleavage in pathophysiological adaptation. Arch. Biochem. Biophys. 515, 37–45. doi: 10.1016/j.abb.2011.08.013
Keywords: isoforms, cardiac muscle, myofilament proteins, posttranslational modification, cardiomyopathy
Citation: Sheng J-J and Jin J-P (2014) Gene regulation, alternative splicing, and posttranslational modification of troponin subunits in cardiac development and adaptation: a focused review. Front. Physiol. 5:165. doi: 10.3389/fphys.2014.00165
Received: 08 January 2014; Accepted: 10 April 2014;
Published online: 30 April 2014.
Edited by:
Li Zuo, Ohio State University, USACopyright © 2014 Sheng and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-Ping Jin, Department of Physiology, Wayne State University School of Medicine, 540 E. Canfield, Detroit, MI 48201, USA e-mail:amppbkBtZWQud2F5bmUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.