 Lilian I. Plotkin
Lilian I. Plotkin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 04 April 2014
Sec. Membrane Physiology and Membrane Biophysics
Volume 5 - 2014 | https://doi.org/10.3389/fphys.2014.00131
This article is part of the Research Topic Hemichannels; from the molecule to the function. View all 12 articles
Cell function and survival are controlled by intracellular signals, and modulated by surrounding cells and the extracellular environment. Connexin channels participate in these processes by mediating cell-to-cell communication. In bone cells, gap junction channels were detected in the early 1970s, and are present among bone resorbing osteoclasts, bone forming osteoblasts, and osteocytes - mature osteoblasts embedded in the mineralized matrix. These channels are composed mainly by Cx43, although the expression of other connexins (45, 46, and 37) has also been reported. It is now believed that undocked Cx43 hemichannels (connexons) formed in unopposed cell membranes facing the extracellular environment participate in the interaction of bone cells with the extracellular environment, and in their communication with neighboring cells. Thus, we and others demonstrated the presence of active hemichannels in osteoblastic and osteocytic cells. These hemichannels open in response to pharmacological and mechanical stimulation. In particular, preservation of the viability of osteoblasts and osteocytes by the anti-osteoporotic drugs bisphosphonates depends on Cx43 expression in vitro and in vivo, and is mediated by undocked hemichannels. Cx43 hemichannels are also required for the release of prostaglandins and ATP by osteocytes, and for cell survival induced by mechanical stimulation in vitro. Moreover, they are required for the anti-apoptotic effect of parathyroid hormone in osteoblastic cells. This review summarizes the current knowledge on the presence and function of undocked connexons, and the role of hemichannel regulation for the maintenance of bone cell viability and, potentially, bone health.
The amount of bone and its strength is maintained throughout life by the concerted actions of osteoblasts, the bone forming cells, and osteoclasts, the bone removing cells (Figure 1A). The action of these two cell types is coordinated by signals derived from osteocytes, the bone resident cells that derived from osteoblasts and are embedded in the bone matrix (Figures 1A–D). Osteocytes are highly communicated among themselves and with cells of the bone surface through cytoplasmic projections (Figure 1C).
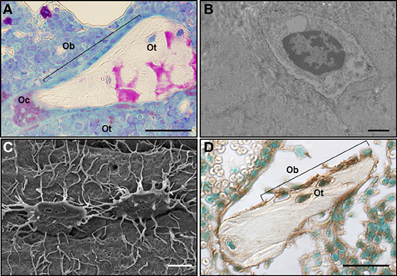
Figure 1. Cell-to-cell interactions among osteoclasts, osteoblasts, and osteocytes in close proximity maintain bone homeostasis. (A) An osteoclast (Oc), a team of osteoblasts (Ob) and bone-embedded osteocytes (Ot) are shown in a rat bone section, in which osteoclasts were stained for the osteoclast-specific enzyme tartrate resistant acid phosphatase (TRAPase) (red) and counterstained using Toluidine blue. Scale bar indicates 50 μm. Picture contributed by Keith W. Condon (Indiana University School of Medicine). (B) Transmission electron microscope image of an osteocyte embedded in the bone matrix. Image was obtained at the Electron Microscopy Center of the Department of Anatomy and Cell Biology (Indiana University School of Medicine). Scale bar indicates 5 μm (C) Scanning electron microscope image of an acid-etched bone showing two osteocytes highly communicated through their cytoplasmic projections (Bellido et al., 2014). Scale bar indicates 1 μm. (D) Cx43 immunostaining of a section of cancellous bone showing osteoblasts (Ob) on the bone surface and osteocytes (Ot) embedded in the bone matrix stained for Cx43 (brown) and counterstained with Methyl green (Plotkin et al., 2008). Scale bar indicates 50 μm.
The existence of gap junctions between osteocytes and osteoblasts on the bone surface was first suggested by electron microscopy observations of calvaria bones from newborn mice (Weinger and Holtrop, 1974). Although the level of resolution of the images did not allow for these authors to conclusive demonstrate the presence of gaps separating the cell surfaces of osteoblasts and osteocytes, they concluded that the structures seen were consistent with gap junctions. This was later confirmed at the ultramicroscopy level by the groups of Doty (1981) and Marotti (Palumbo et al., 1990). Further studies showed that Cx43 is the most abundant connexin expressed in vitro and in vivo in all type of bone cells: osteoblasts, osteocytes and osteoclasts (Schirrmacher et al., 1992; Civitelli et al., 1993; Jones et al., 1993; Donahue et al., 1995). This is exemplified on a murine bone section stained for Cx43 (Figure 1D).
The small molecules that are transferred through connexin channels, and might act as second messengers in bone cells have not been completely identified (see Stains et al., 2014 for a recent revision). Second messengers such as ATP and Ca2+ can move from one cell to another through gap junctions, or can be released to the extracellular media through hemichannels in osteoblastic cells (Jorgensen et al., 1997; Genetos et al., 2007). In addition, cAMP production induced by parathyroid hormone requires Cx43 expression in osteoblastic cells (Vander Molen et al., 1996; Bivi et al., 2011), and Cx43-mediated amplification of FGF2 effect on the osteoblast gene RUNX2 depends on the production of water-soluble inositol polyphosphates (Niger et al., 2013). Taken together, these pieces of evidence suggest that Cx43 not only can control the movement of second messengers, but also their synthesis.
The expression of Cx45, Cx46 and, more recently, Cx37 has also been demonstrated in bone cells (Kruger et al., 2000; Stains and Civitelli, 2005; Paic et al., 2009; Chaible et al., 2011; Pacheco-Costa et al., 2014). In particular, Cx37 is required for osteoclast differentiation and mice lacking Cx37 exhibit high bone mass due to defective bone resorption (Pacheco-Costa et al., 2014).
In addition to be part of gap junctions, connexin channels can be found in unopposed cell membranes forming undocked connexons or hemichannels. Although it was long known that bone cells express connexins, the presence of hemichannels in osteoblastic cells was not reported until 2001 (Romanello and D'Andrea, 2001). In the current review, the demonstration of the presence and function of connexin hemichannels in osteoblasts and osteocytes is discussed.
The importance of Cx43 expression in osteoblasts and osteocytes for bone development, as well as for osteoblast and osteocyte differentiation, survival and function has been clearly established (for recent reviews see Civitelli, 2008; Loiselle et al., 2013; Plotkin and Bellido, 2013). Thus, global deletion of Cx43 results in delayed ossification and impaired osteoblast differentiation in the embryos (Lecanda et al., 2000). In addition, studies with tissue specific deletion of Cx43 have demonstrated that the adult skeleton is also affected by the absence of the connexin (Chung et al., 2006; Watkins et al., 2011; Zhang et al., 2011; Bivi et al., 2012a,b). Cx43 is also important for osteoclast differentiation, as demonstrated in mice in which the connexin was deleted from osteoclast precursors (Sternlieb et al., 2012). Because these studies were performed by deleting the whole Cx43 molecule, it is not possible to determine whether absence of cell-to-cell gap junction communication or the function of Cx43 in undocked hemichannels present in cell membranes (or even channel-independent functions of the connexin), or a combination of these functions, are responsible for the observed phenotypes. Nevertheless, recent developments discussed below support the presence and functionality of Cx43 hemichannels in bone cells in vivo.
Mutations of the Cx43 gene are associated with occulodentodigital dysplasia (ODDD), a condition with skeletal malformation that include craniofacial abnormalities and broad long bones (Paznekas et al., 2002). Interestingly, some of the Cx43 mutations leading to ODDD result not only in decreased gap junction communication, but also in enhanced hemichannel activity (Dobrowolski et al., 2007), suggesting that part of the phenotype of the patients might be due to exacerbated hemichannel function. Consistent with this, a study reported in the form of an abstract showed that osteocytic expression of the Cx43 mutant R76W, which does not form gap junctions but has the ability to form hemichannels, leads to decreased bone mineral density (Jiang et al., 2010). The phenotype of these mouse models might be due to the lack of intercellular gap junction communication or, alternatively, to excessive hemichannel activity in bone cells, resulting in skeletal defects.
Interestingly, stimuli that increase bone mass have been shown to increase Cx43 expression, its localization in the cell membrane, and the activation of gap junction and hemichannel activity. Thus, estrogen, increases the expression of Cx43 and gap junction communication in the osteocytic cell line MLO-Y4 (Ren et al., 2013); and the effect of sex steroid removal on the cortical bone is partially prevented in mice lacking Cx43 in osteoblastic cells (Watkins et al., 2012). However, we have shown that the anti-apoptotic effect of sex steroids on osteocytic cells does not require Cx43 function (Plotkin et al., 2002). On the other hand, as it will be discussed below, Cx43, and in particular, hemichannels, mediate at least in part the effect of the anti-osteoporotic drugs bisphosphonates (Plotkin et al., 2002, 2008), mechanical stimulation (Cherian et al., 2005; Zhang et al., 2011; Grimston et al., 2012; Bivi et al., 2013), and parathyroid hormone (Bivi et al., 2011).
Bisphosphonates, a family of drugs that include alendronate, have been used over the past 40 years to treat conditions with low bone mass such as osteoporosis, and to prevent bone fractures (Russell, 2011). Bisphosphonates block osteoclastic bone resorption, therefore preserving the amount of bone. However, stopping bone resorption cannot completely explain the ability of these drugs to prevent bone fractures. We therefore proposed that, in addition to inhibiting bone resorption, bisphosphonates have a positive effect on osteoblasts and osteocytes that can contribute to the anti-fracture properties of the drugs. Preservation of osteoblast life span should lead to prolonged matrix synthesizing activity, whereas maintenance of osteocyte viability should preserve their mechanosensory function, therefore improving bone strength. We found that bisphosphonates protect osteoblasts and osteocytes from apoptosis induced by several agents (Figure 2A) in vitro using osteoblastic cell isolated from neonatal calvaria bone and osteocytic MLO-Y4 cells, and by glucocorticoid excess in vivo using a mouse model of glucocorticoid-induced bone disease (Plotkin et al., 1999). Although osteoblasts and osteocytes have distinct functions, they respond in similar fashion to bisphosphonates. Therefore, the studies described in this section were performed with both cell types.
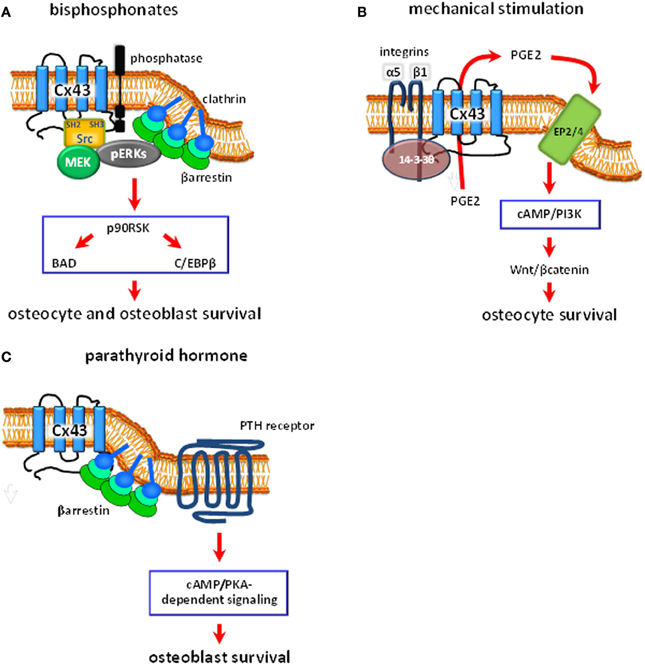
Figure 2. Schematic representation of the proposed intracellular signaling pathways regulated by Cx43 hemichannels in bone cells. (A) Bisphosphonates bind to phosphatases present in the cell membrane. This induces Cx43 hemichannel opening, followed by activation of the kinases Src and MEK, and ERKs. ERKs activated downstream of Cx43 hemichannel opening are retained in the cytoplasm by a complex formed by βarrestin and clathrins. This leads to the phosphorylation of the cytoplasmic targets p90RSK, BAD and C/EBPβ, which results in osteoblast and osteocyte survival. (B) Mechanical stimulation induces α5β1 integrin engagement and the association of the integrins with Cx43, by a mechanism that requires the protein 14-3-3θ. This results in hemichannel opening and the release of PGE2. PGE2, in turn, activates EP2/4 prostaglandin receptor by an autocrine/paracrine mechanism, leading to activation of the cAMP/PI3K signaling pathways, accumulation of βcatenin with the consequent activation of Wnt signaling, and inhibition of osteocyte apoptosis. (C) Parathyroid hormone (PTH), through binding to the PTH receptor, induces activation of the cAMP/PKA signaling pathway. Cx43, by sequestering β-arrestin away from the PTH receptor, facilitate cAMP/PKA-mediated downstream signaling and osteoblast survival.
The survival effect of bisphosphonates is mediated by the activation of the extracellular signal regulated kinases ERKs in cell cultures and in vivo (Figure 2A) (Plotkin et al., 1999, 2011). Thus, phosphorylation of ERKs is increased upon treatment of cells or mice with the bisphosphonate alendronate. Importantly, ERK activation is required for the protective effect of bisphosphonates since their effect was abolished by ERK pharmacological inhibitors and by transfection of a dominant negative form of MEK, the kinase that activates ERKs (Plotkin et al., 1999, 2002).
ERKs can be activated by an array of extracellular stimuli, including binding of growth factors and cytokines to their receptors. We then explored the possibility that bisphosphonates activate ERKs by acting on membrane channels. Because connexin channels are abundant in osteoblasts and osteocytes, and bisphosphonates are molecules of low molecular size (approximately 400 daltons), we examined whether inhibition of connexin channels affected the protective response to the bisphosphonate alendronate. We found that inhibiting connexin channels with 18 α-glycyrrhetinic acid (AGA) abolished the anti-apoptotic effect of alendronate, whereas the inactive analog of AGA, glycyrrizic acid (GA), did not modify this response (Plotkin et al., 2002). Similar to anti-apoptosis, AGA, but not GA, prevented ERK activation induced by alendronate. These results suggested that the connexin channels are required for the ERK-mediated anti-apoptotic effect of bisphosphonates. However, prevention of apoptosis by alendronate does not require cell-to-cell contact, since the bisphosphonate inhibited apoptosis in cells plated at low density or maintained in suspension, suggesting that gap junctions do not mediate the effect of bisphosphonates and that hemichannels are involved. Moreover, treatment with alendronate did not affect gap junction communication. Instead, alendronate induced opening of hemichannels in MLO-Y4 cells measured as uptake of the fluorescent small dye Lucifer Yellow. Because Cx43 is the main connexin protein expressed in osteocytic MLO-Y4 cells, we next determined whether responsiveness to alendronate depends on its expression using several cell models (Plotkin et al., 2002). We found that the drug was able to protect from apoptosis embryonic fibroblasts derived from wild type mice, but it was unable to do so in cells derived from Cx43 deficient mice, and that transfecting Cx43 to deficient cells rescued responsiveness to alendronate. Similarly, ROS17/2.8 osteoblastic cells that express Cx43 were protected from apoptosis by alendronate, but UMR 106 cells which do not express Cx43 were not. And Cx43 transfection to UMR106 cells rescued responsiveness to alendronate. Moreover, authentic osteoblasts derived from Cx43 deficient mice did not respond to alendronate, whereas apoptosis was prevented by this agent in osteoblasts derived from wild type mice. Cx43, but not other connexins tested as representative of the three connexin subfamilies (26, 31, 32, 37, 40, and 45), was able to confer responsiveness to alendronate in HeLa cells, which do not express connexins (Plotkin et al., 2002). These results established the requirement of Cx43 for the anti-apoptotic effect of bisphosphonates in vitro.
The cytoplasmic C-terminal domains of connexins differ considerably, suggesting that the specific requirement of Cx43 for the effect of alendronate could be due to this region. Indeed, a Cx43 truncated mutant that lacks the cytoplasmic tail was unable to confer responsiveness to alendronate, response that was recovered by co-transfecting the truncated mutant and the cytoplasmic tail (Plotkin et al., 2002). However, the C-terminus tail alone was not able to mediate anti-apoptosis. These results suggest that the C-terminus of Cx43 contains domains required for anti-apoptosis by bisphosphonates and that the pore-forming region also contributes to the effect.
Further studies showed that bisphosphonates induce the transient opening of undocked Cx43 hemichannels resulting in activation of Src kinase, which is associated with Cx43, leading to ERK activation and cell survival (Figure 2A) (Plotkin et al., 2002). Since receptors for bisphosphonates have not been described, the requirement of Cx43 for bisphosphonate actions raised the possibility that Cx43 would be such receptor. However, we found that bisphosphonates not only bind to cells that express Cx43 but also to Cx43-deficient cells (Lezcano et al., 2012). Moreover, binding of labeled bisphosphonate to osteoblastic cells is displaced by unlabeled bisphosphonates, as expected, but also by protein phosphatase substrates. Furthermore, these substrates inhibit the anti-apoptotic effect of bisphosphonates (Morelli et al., 2011) Changes in the phosphorylation status of Cx43 C-terminus tail are associated with channel activity (reviewed in Herve and Sarrouilhe, 2006), suggesting that bisphosphonates might induce opening of Cx43 hemichannels thought their interaction with membrane bound phosphatases. However, the identity of the protein phosphatase that binds to Cx43, and whether it can dephosphorylate Cx43 and induce hemichannel opening remain to be determined. Taken together, these pieces of evidence indicate that although Cx43 is required for survival signaling, it is dispensable for bisphosphonate binding; and raises the possibility that bisphosphonates bind to a protein phosphatase that interact with Cx43 in the cell membrane.
An interesting feature of ERKs activated by bisphosphonates and the Cx43/Src pathway is that instead of undergoing nuclear translocation like ERKs activated by most stimuli, they are retained in the cytoplasm where ERKs modify cytoplasmic substrates (Plotkin et al., 2005a). We found that this phenomenon is due to the ability of Cx43 of interacting with β-arrestins (Plotkin et al., 2006b). Consistent with a role of β-arrestin in the retention of bisphosphonate-activated ERKs in the cytoplasm, ERKs stay in the cytoplasm in cells expressing endogenous β-arrestin or transfected with wild type β-arrestin. However, in the presence of a dominant negative from of β-arrestin, ERKs activated by alendronate translocate to the nucleus. Because cytoplasmic localization of ERKs is required for survival induced by bisphosphonates (Plotkin et al., 2005a), the dominant negative β-arrestin reversed anti-apoptosis induced by alendronate. Thus, Cx43 regulates the ERK signaling pathway due to its ability of function as a scaffold that foster interaction between β-arrestin and Src/ERKs.
Further support for the role of Cx43 on the survival effect of bisphosphonates was obtained in vivo, by the demonstration that administration of alendronate does not prevents glucocorticoid-induced osteoblast and osteocyte apoptosis in mice lacking Cx43 in these cells (Plotkin et al., 2008). However, the bisphosphonate was still able to prevent bone loss induced by glucocorticoids due to its potent inhibition of bone resorption. Similarly, bisphosphonates prevented bone loss induced by sex steroid removal in mice lacking Cx43 in osteoblasts and osteocytes, by reducing bone resorption (Watkins et al., 2012). On the other hand, IG9402, a bisphosphonate that does not affect osteoclast function by prevents osteoblast and osteocyte apoptosis, prevents bone loss induced by glucocorticoids in mice (Plotkin et al., 2006a, 2011). This suggest that in the absence of anti-resorptive effects, the protective effect of preventing osteocyte and osteocyte apoptosis through opening Cx43 hemichannels can be revealed. Thus, preservation of osteoblast and osteocyte apoptosis mediated by hemichannel opening might contribute to the anti-fracture efficacy of bisphosphonates.
Adequate mechanical stimulation is required for maintaining bone mass and strength throughout life. Indeed, reduced or absent loading leads to decrease bone mass and elevated risk of fractures in the elderly, in immobilized patients, and in astronauts (Bikle et al., 1997; Marcus, 2002; Dolbow et al., 2011). Osteocytes are ideally positioned for detecting and responding to changes in mechanical strains imposed on bone. Since their description at the microscopy level, connexin channels were thought to be the means by which the cells buried in the bone communicate with the cells on the bone surface and transmit the need for bone formation or removal.
Consistent with a role of connexin channels on mechanotransduction in bone, Cx43 expression and intercellular communication is increased by loading in vitro and in vivo (Ziambaras et al., 1998; Cheng et al., 2001b; Robinson et al., 2006; Tu et al., 2012). Early work by Donahue's group show that mechanical stimulation of osteoblastic cells induced by oscillatory fluid flow leads to release of prostaglandin (PG) E2 by a mechanism that requires intact Cx43 channels (Figure 2B) (Saunders et al., 2001). Thus, the release of PGE2 was inhibited in cells expressing a dominant negative form of Cx43 with reduced channel permeability. Moreover, PGE2 release induced by mechanical stimulation further increased gap junction communication (Cheng et al., 2001a) by a mechanism mediated by the prostaglandin EP2 receptor (Cherian et al., 2003) suggesting the existence of a positive feedback loop. This effect of mechanical loading was originally ascribed to a role of Cx43 in intercellular gap junction communication. However, based on previous studies showing that mechanical stimulation increases hemichannel opening in osteoblastic cells in vitro (Romanello et al., 2003), work from Jiang's group demonstrated that PGE2 released induced by mechanical stimulation requires opening of Cx43 hemichannels in osteocytic cells (Figure 2B) (Cherian et al., 2005). Interestingly, hemichannels present in the osteocytic cell body, and not in the dendritic cytoplasmic projections, are opened by mechanical stimulation (Burra et al., 2010). The release of PGE2 induced by fluid flow in osteocytic MLO-Y4 cells was prevented by using pharmacologic inhibitors of connexin channels and by an anti-sense oligonucleotide for Cx43 (Cherian et al., 2005). Moreover, prostaglandin release was enhanced when the cells were seeded at low density, which reduces intercellular gap junction communication. Intracellular levels of PGE2 production were not affected by inhibition of connexin channels, suggesting that PGE2 release but not its synthesis requires Cx43 hemichannel activity. Other authors have postulated, however, that P2X7 receptors (Li et al., 2005) and/or pannexin1 (Thi et al., 2012) are involved in the release of PGE2 induced by mechanical stimulation in bone cells. This suggests that the combined actions of these channels are required for prostaglandin release induced by mechanical stimulation in osteoblastic and osteocytic cells.
Further studies by Jiang's group demonstrated that engagement of α5β1 integrin in required for opening of Cx43 hemichannels in osteocytic cells (Figure 2B) (Batra et al., 2012). Thus, Cx43 interacts with α5β1 integrin, an interaction enhanced by mechanical stimulation (Batra et al., 2012, 2014). Cx43/α5β1 association is required for Cx43 hemichannel opening induced by loading and is mediated by activation of phosphatidylinositol-3 kinase (PI3K) and AKT. Consistent with a role of the integrins on mechanotransduction, we have shown that mechanical stimulation leads to the engagement of integrins α5 and β1, which in turn activate FAK/Src and the ERK pathway promoting osteocyte survival (Plotkin et al., 2005b).
More recently, Jiang's group showed that the scaffolding molecule 14-3-3θ is required for the interaction between integrin α5 and Cx43 and for plasma membrane delivery and function of Cx43 hemichannels (Figure 2B) (Batra et al., 2013). In particular, silencing of 14-3-3θ prevents the accumulation of Cx43 on the cell membrane and opening of hemichannels induced by fluid flow. Taken together, these studies support a role of Cx43 hemichannels on PGE2 release.
PGE2 release by osteocytes through Cx43 hemichannels subjected to mechanical forces is required for maintaining cell viability (Figure 2B). Indeed, prostaglandins prevent osteoblastic apoptosis via activation of EP4 receptors (Machwate et al., 2001); and mechanical stimulation prevents osteocyte apoptosis in vitro (Plotkin et al., 2005b; Aguirre et al., 2007; Kitase et al., 2010). Moreover, inhibition of glucocorticoid-induced apoptosis by mechanical stimulation is abolished by inhibiting prostaglandin synthesis using indomethacin (Kitase et al., 2010). The pro-survival effect of PGE2 release by osteocytes subjected to mechanical stimulation in mediated by activation of the EP2 and EP4 receptors, via cAMP/PKA signaling pathway. In addition, the study by Kitase and colleagues showed that activation of Wnt/βcatenin signaling downstream of PI3K/Akt contributes to PGE2/EP2/4-induced osteocyte survival (Figure 2B).
The participation of the Cx43/PGE2 survival pathway in skeletal homeostasis in vivo is not known. However, this potential role of Cx43 is supported by our work (Plotkin et al., 2008; Bivi et al., 2012a), later confirmed by others (Lloyd et al., 2013) showing that deletion of Cx43 from osteoblastic cells results in increased osteocyte apoptosis. Interestingly, recent work from Jiang's group (Xu et al., 2013) showed that transgenic mice expressing in osteocytes a dominant negative Cx43 mutant with impaired permeability (Cx43Δ130-136), which lacks both hemichannel and gap junction functions (Krutovskikh et al., 1998), also exhibit increased osteocyte apoptosis (Jean X. Jiang, personal communication). On the other hand, mice with osteocytic expression of the R76W Cx43 mutant, in which the ability to form gap junction channels is impaired, but that maintains hemichannel activity (Xu et al., 2013) did not exhibit increased osteocyte apoptosis compared to wild type controls (Jean X. Jiang, personal communication). This evidence supports the role of Cx43 hemichannels for maintain osteocyte survival in vivo; and suggests that in the absence of Cx43 hemichannels in osteocytes, the mechanical loading that occurs during normal ambulatory conditions cannot protect the cells from undergoing apoptosis. Indeed, one of the earliest effects of skeletal unloading is the accumulation of apoptotic osteocytes (Aguirre et al., 2006).
However, Cx43 is not required for the increased bone mass induced by mechanical stimulation in murine models. On the contrary, removal of Cx43 from osteoblasts and/or osteocytes results in increased anabolic response to mechanical stimulation of osteoblast on the periosteal bone surface (Zhang et al., 2011; Grimston et al., 2012; Bivi et al., 2013). The cause of this exacerbated response is not known. We have found that Cx43-deficient osteocytes exhibit elevated Wnt/βcatenin signaling (Bivi et al., 2013), a known mediator of mechanical signals in osteocytes (Robinson et al., 2006). This higher basal activation of Wnt signaling could explain the exacerbated response to loading in Cx43-deficient mice. Nevertheless, mounting evidence indicates that Cx43 hemichannel opening and the release of PGE2 mediate the survival effect of mechanical stimulation on osteocytes, suggesting that the increase in bone formation induce by mechanical stimulation depends on signaling pathway different from those involved in loading-induced osteocyte survival.
Intermittent administration of parathyroid hormone (PTH) is the only FDA-approved treatment to increase bone mass. Cx43 expression appears to be required to obtain a full anabolic response to intermittent PTH administration in mice (Castro et al., 2003; Chung et al., 2006). Thus, PTH does not increase bone mass, bone formation and osteoblast number when administered to heterozygous Cx43 deficient mice (Cx43+/−) (Castro et al., 2003). Moreover, PTH-induced mineral appositional rate, a measure of the work of osteoblast teams, is reduced in mice lacking Cx43 in osteoblastic cells (in Cx43fl/−; Col1a1-2.3kb-Cre mice) (Chung et al., 2006). Studies in mice have shown that the anabolic effect of intermittent PTH administration in cancellous bone is due, at least in part, to inhibition of osteoblast apoptosis (Figure 2C) (Jilka et al., 1999). Similarly, PTH related protein (PTHrP), the other ligand of the PTH receptor, as well as constitutive activation of this receptor in transgenic mice, also increases osteoblast number and decreases the prevalence of osteoblast apoptosis (Calvi et al., 2001; Martin, 2005; Miao et al., 2005; O'Brien et al., 2008). Taken together, these pieces of evidence suggest that intermittent PTH administration does not result in a full anabolic response in mice lacking Cx43 due to the inability of the hormone to prevent apoptosis of osteoblastic cells in the absence of Cx43.
In vitro mechanistic studies showed that, indeed, PTH does not prevent apoptosis in osteoblastic cells lacking Cx43, whereas overexpression of wild type Cx43 rescues the survival effect of the hormone (Figure 2C) (Bivi et al., 2011). A similar result is obtained by transfecting a Cx43 mutant in which cysteine residues of the extracellular domain have been mutated, rendering it unable to dock with other connexin molecules to form gap junction channels (Bao et al., 2004). On the other hand, a Cx43 mutant with impaired permeability was not able to confer responsiveness to PTH, suggesting that active Cx43 hemichannels are required for the survival effect of the hormone.
In addition, the interaction of Cx43 with β-arrestin modulates the response of osteoblasts to PTH (Figure 2C) (Bivi et al., 2011). Thus, PTH-induced anti-apoptosis is rescued by transfecting a dominant negative form of β-arrestin to cells lacking Cx43; and overexpression of β-arrestin induces the same inhibition on PTH-induced osteoblast survival as removing Cx43. Moreover, transfection of Cx43 decreases the association between the PTH receptor and β-arrestin, suggesting that Cx43 binds to β-arrestin, thus competing with the PTH receptor. Interestingly, Cx43 mutants lacking the C-terminus domain or lacking the phosphorylation site in serine 368 decrease the interaction between PTHR and β-arrestin, suggesting that Cx43 binds β-arrestin at the phosphorylated serine 368.
We found that PTH does not increase the expression of cAMP-target genes in osteoblastic cells lacking Cx43 (Bivi et al., 2011). Consistent with this, it was previously shown that the response to PTH on cAMP production is blunted in osteoblastic cells in which Cx43 expression has been reduced using anti-sense cDNA (Vander Molen et al., 1996). These findings, together with evidence that β-arrestin reduces cAMP responses of the PTH receptor (Premont and Gainetdinov, 2007), support the hypothesis that Cx43 interacts with β-arrestin decreasing β-arrestin binding to the PTH receptor, then facilitating cAMP dependent signaling induced by PTH. Thus, Cx43 hemichannel activity and the ability of the connexin to interact with intracellular signaling molecules, through specific phosphorylation sites in its cytoplasmic tail, regulates survival signaling induced by PTH in osteoblastic cells.
Since the first description of gap junction channels at the structural level in bone cells, substantial advances on our understanding of the role of connexins on bone cell physiology have been made (Civitelli, 2008; Loiselle et al., 2013; Plotkin and Bellido, 2013). The requirement of connexins, and in particular Cx43, for bone cell differentiation and function, as well as for the response of the cells to bone-acting stimuli has been clearly established in vitro and in vivo. Moreover, the role of the Cx43 hemichannels in vivo has begun to be unveiled. Indeed, while studies with genetically modified mice appear to agree that sustained opening of Cx43 hemichannels is deleterious for bone (Dobrowolski et al., 2007; Jiang et al., 2010), transient opening of hemichannels is beneficial for osteoblast and osteocyte survival (Plotkin et al., 2002, 2008, 2011; Kitase et al., 2010; Bivi et al., 2011). Studies using genetically modified mice currently underway will allow to conclusively demonstrate the role of Cx43 hemichannels on bone development and cell function.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by National Institutes of Health (R01-AR053643) and by a Biomedical Research Grant and a Developing Diverse Researchers with InVestigative Expertise (DRIVE) Grant from Indiana University School of Medicine.
Aguirre, J. I., Plotkin, L. I., Gortazar, A. R., O'Brien, C. A., Manolagas, S. C., and Bellido, T. (2007). A novel ligand-independent function of the estrogen receptor is essential for osteocyte and osteoblast mechanotransduction. J. Biol. Chem. 282, 25501–25508. doi: 10.1074/jbc.M702231200
Aguirre, J. I., Plotkin, L. I., Stewart, S. A., Weinstein, R. S., Parfitt, A. M., Manolagas, S. C., et al. (2006). Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Miner. Res. 21, 605–615. doi: 10.1359/jbmr.060107
Bao, X., Chen, Y., Reuss, L., and Altenberg, G. A. (2004). Functional expression in xenopus oocytes of gap-junctional hemichannels formed by a cysteine-less connexin 43. J. Biol. Chem. 279, 9689–9692. doi: 10.1074/jbc.M311438200
Batra, N., Burra, S., Siller-Jackson, A. J., Gu, S., Xia, X., Weber, G. F., et al. (2012). Mechanical stress-activated integrin alpha5beta1 induces opening of connexin 43 hemichannels. Proc. Natl. Acad. Sci. U.S.A. 109, 3359–3364. doi: 10.1073/pnas.1115967109
Batra, N., Riquelme, M. A., Burra, S., and Jiang, J. X. (2013). 14-3-3theta facilitates plasma membrane delivery and function of mechanosensitive connexin 43 hemichannels. J. Cell Sci. 127, 137–146. doi: 10.1242/jcs.133553
Batra, N., Riquelme, M. A., Burra, S., Rekha, K., Gu, S., and Jiang, J. X. (2014). Direct regulation of osteocytic connexin 43 hemichannels through AKT kinase activated by mechanical stimulation. J. Biol. Chem. doi: 10.1074/jbc.M114.550608. [Epub ahead of print].
Bellido, T., Plotkin, L. I., and Bruzzaniti, A. (2014). “Bone cells” in Basic and Applied Bone Biology, eds D. B. Burr, and M. R. Allen (Academic Press/Elsevier), 27–45.
Bikle, D. D., Halloran, B. P., and Morey-Holton, E. (1997). Spaceflight and the skeleton: lessons for the earthbound. Gravit. Space Biol. Bull. 10, 119–135.
Bivi, N., Condon, K. W., Allen, M. R., Farlow, N., Passeri, G., Brun, L., et al. (2012a). Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J. Bone Miner. Res. 27, 374–389. doi: 10.1002/jbmr.548
Bivi, N., Lezcano, V., Romanello, M., Bellido, T., and Plotkin, L. I. (2011). Connexin43 interacts with âarrestin: a pre-requisite for osteoblast survival induced by parathyroid hormone. J. Cell. Biochem. 112, 2920–2930. doi: 10.1002/jcb.23208
Bivi, N., Nelson, M. T., Faillace, M. E., Li, J., Miller, L. M., and Plotkin, L. I. (2012b). Deletion of Cx43 from osteocytes results in defective bone material properties but does not decrease extrinsic strength in cortical bone. Calcif. Tissue Int. 91, 215–224. doi: 10.1007/s00223-012-9628-z
Bivi, N., Pacheco-Costa, R., Brun, L. R., Murphy, T. R., Farlow, N. R., Robling, A. G., et al. (2013). Absence of Cx43 selectively from osteocytes enhances responsiveness to mechanical force in mice. J. Orthop. Res. 31, 1075–1081. doi: 10.1002/jor.22341
Burra, S., Nicolella, D. P., Francis, W. L., Freitas, C. J., Mueschke, N. J., Poole, K., et al. (2010). Dendritic processes of osteocytes are mechanotransducers that induce the opening of hemichannels. Proc. Natl. Acad. Sci. U.S.A. 107, 13648–13653. doi: 10.1073/pnas.1009382107
Calvi, L. M., Sims, N. A., Hunzelman, J. L., Knight, M. C., Giovannetti, A., Saxton, J. M., et al. (2001). Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J. Clin. Invest. 107, 277–286. doi: 10.1172/JCI11296
Castro, C. H. M., Stains, J. P., and Civitelli, R. (2003). The anabolic response to intermittent PTH (1-34) requires connexin43 (Cx43) mediated gap junctional communication. J. Bone Miner. Res. 18:S162.
Chaible, L. M., Sanches, D. S., Cogliati, B., Mennecier, G., and Dagli, M. L. (2011). Delayed osteoblastic differentiation and bone development in Cx43 knockout mice. Toxicol. Pathol. 39, 1046–1055. doi: 10.1177/0192623311422075
Cheng, B., Kato, Y., Zhao, S., Luo, J., Sprague, E., Bonewald, L. F., et al. (2001a). PGE(2) is essential for gap junction-mediated intercellular communication between osteocyte-like MLO-Y4 cells in response to mechanical strain. Endocrinology 142, 3464–3473. doi: 10.1210/endo.142.8.8338
Cheng, B., Zhao, S., Luo, J., Sprague, E., Bonewald, L. F., and Jiang, J. X. (2001b). Expression of functional gap junctions and regulation by fluid flow in osteocyte-like MLO-Y4 cells. J. Bone. Miner. Res. 16, 249–259. doi: 10.1359/jbmr.2001.16.2.249
Cherian, P. P., Cheng, B., Gu, S., Sprague, E., Bonewald, L. F., and Jiang, J. X. (2003). Effects of mechanical strain on the function of gap junctions in osteocytes are mediated through the prostaglandin EP2 receptor. J. Biol. Chem. 278, 43146–43156. doi: 10.1074/jbc.M302993200
Cherian, P. P., Siller-Jackson, A. J., Gu, S., Wang, X., Bonewald, L. F., Sprague, E., et al. (2005). Mechanical strain opens connexin 43 hemichannels in osteocytes: a novel mechanism for the release of prostaglandin. Mol. Biol. Cell. 16, 3100–3106. doi: 10.1091/mbc.E04-10-0912
Chung, D., Castro, C. H., Watkins, M., Stains, J. P., Chung, M. Y., Szejnfeld, V. L., et al. (2006). Low peak bone mass and attenuated anabolic response to parathyroid hormone in mice with an osteoblast-specific deletion of connexin43. J. Cell Sci. 119, 4187–4198. doi: 10.1242/jcs.03162
Civitelli, R. (2008). Cell-cell communication in the osteoblast/osteocyte lineage. Arch. Biochem. Biophys. 473, 188–192. doi: 10.1016/j.abb.2008.04.005
Civitelli, R., Beyer, E. C., Warlow, P. M., Robertson, A. J., Geist, S. T., and Steinberg, T. H. (1993). Connexin43 mediates direct intercellular communication in human osteoblastic cell networks. J. Clin. Invest. 91, 1888–1896. doi: 10.1172/JCI116406
Dobrowolski, R., Sommershof, A., and Willecke, K. (2007). Some oculodentodigital dysplasia-associated cx43 mutations cause increased hemichannel activity in addition to deficient gap junction channels. J. Membr. Biol. 219, 9–17. doi: 10.1007/s00232-007-9055-7
Dolbow, D. R., Gorgey, A. S., Daniels, J. A., Adler, R. A., Moore, J. R., and Gater, D. R. Jr. (2011). The effects of spinal cord injury and exercise on bone mass: a literature review. Neurorehabilitation 29, 261–269. doi: 10.3233/NRE-2011-0702
Donahue, H. J., McLeod, K. J., Rubin, C. T., Andersen, J., Grine, E. A., Hertzberg, E. L., et al. (1995). Cell-to-cell communication in osteoblastic networks: cell line- dependent hormonal regulation of gap junction function. J. Bone Miner. Res. 10, 881–889. doi: 10.1002/jbmr.5650100609
Doty, S. B. (1981). Morphological evidence of gap junctions between bone cells. Calcif. Tissue Int. 33, 509–512. doi: 10.1007/BF02409482
Genetos, D. C., Kephart, C. J., Zhang, Y., Yellowley, C. E., and Donahue, H. J. (2007). Oscillating fluid flow activation of gap junction hemichannels induces ATP release from MLO-Y4 osteocytes. J. Cell Physiol. 212, 207–214. doi: 10.1002/jcp.21021
Grimston, S. K., Watkins, M. P., Brodt, M. D., Silva, M. J., and Civitelli, R. (2012). Enhanced periosteal and endocortical responses to axial tibial compression loading in conditional connexin43 deficient mice. PLoS ONE 7:e44222. doi: 10.1371/journal.pone.0044222
Herve, J. C., and Sarrouilhe, D. (2006). Protein phosphatase modulation of the intercellular junctional communication: importance in cardiac myocytes. Prog. Biophys. Mol. Biol. 90, 225–248. doi: 10.1016/j.pbiomolbio.2005.06.005
Jiang, J., Burra, S., Harris, S., Zhao, H., Johnson, M., and Bonewald, L. (2010). Inhibiting connexin 43 gap junction function in osteocytes, but not connexin 43 hemmichannel function results in defects in skeletal structure and bone mass. J. Bone Miner. Res. 25:S12.
Jilka, R. L., Weinstein, R. S., Bellido, T., Roberson, P., Parfitt, A. M., and Manolagas, S. C. (1999). Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Invest. 104, 439–446. doi: 10.1172/JCI6610
Jones, S. J., Gray, C., Sakamaki, H., Arora, M., Boyde, A., Gourdie, R., et al. (1993). The incidence and size of gap junctions between the bone cells in rat calvaria. Anat. Embryol. (Berl.) 187, 343–352. doi: 10.1007/BF00185892
Jorgensen, N. R., Geist, S. T., Civitelli, R., and Steinberg, T. H. (1997). ATP- and gap junction-dependent intercellular calcium signaling in osteoblastic cells. J. Cell Biol. 139, 497–506. doi: 10.1083/jcb.139.2.497
Kitase, Y., Barragan, L., Jiang, J. X., Johnson, M. L., and Bonewald, L. F. (2010). Mechanical induction of PGE(2) in osteocytes blocks glucocorticoid induced apoptosis through both the beta-catenin and PKA pathways. J. Bone Miner. Res. 25, 2657–2668. doi: 10.1002/jbmr.168
Kruger, O., Plum, A., Kim, J. S., Winterhager, E., Maxeiner, S., Hallas, G., et al. (2000). Defective vascular development in connexin 45-deficient mice. Development 127, 4179–4193.
Krutovskikh, V. A., Yamasaki, H., Tsuda, H., and Asamoto, M. (1998). Inhibition of intrinsic gap-junction intercellular communication and enhancement of tumorigenicity of the rat bladder carcinoma cell line BC31 by a dominant-negative connexin 43 mutant. Mol. Carcinog. 23, 254–261. doi: 10.1002/(SICI)1098-2744(199812)23:4<254::AID-MC9>3.0.CO;2-4
Lecanda, F., Warlow, P. M., Sheikh, S., Furlan, F., Steinberg, T. H., and Civitelli, R. (2000). Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J. Cell Biol. 151, 931–944. doi: 10.1083/jcb.151.4.931
Lezcano, V., Bellido, T., Plotkin, L. I., Boland, R., and Morelli, S. (2012). Role of connexin 43 in the mechanism of action of alendronate: dissociation of anti-apoptotic and proliferative signaling pathways. Arch. Biochem. Biophys. 518, 95–102. doi: 10.1016/j.abb.2011.12.022
Li, J., Liu, D., Ke, H. Z., Duncan, R. L., and Turner, C. H. (2005). The P2X7 nucleotide receptor mediates skeletal mechanotransduction. J. Biol. Chem. 280, 42952–42959. doi: 10.1074/jbc.M506415200
Lloyd, S. A., Loiselle, A. E., Zhang, Y., and Donahue, H. J. (2013). Connexin 43 deficiency desensitizes bone to the effects of mechanical unloading through modulation of both arms of bone remodeling. Bone 57, 76–83. doi: 10.1016/j.bone.2013.07.022
Loiselle, A. E., Jiang, J. X., and Donahue, H. J. (2013). Gap junction and hemichannel functions in osteocytes. Bone 54, 205–212. doi: 10.1016/j.bone.2012.08.132
Machwate, M., Harada, S., Leu, C. T., Seedor, G., Labelle, M., Gallant, M., et al. (2001). Prostaglandin receptor EP(4) mediates the bone anabolic effects of PGE(2). Mol. Pharmacol. 60, 36–41. doi: 10.1124/mol.60.1.36
Martin, T. J. (2005). Osteoblast-derived PTHrP is a physiological regulator of bone formation. J. Clin. Invest. 115, 2322–2324. doi: 10.1172/JCI26239
Miao, D., He, B., Jiang, Y., Kobayashi, T., Soroceanu, M. A., Zhao, J., et al. (2005). Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1-34. J. Clin. Invest. 115, 2402–2411. doi: 10.1172/JCI24918
Morelli, S., Bilbao, P. S., Katz, S., Lezcano, V., Roldan, E., Boland, R., et al. (2011). Protein phosphatases: possible bisphosphonate binding sites mediating stimulation of osteoblast proliferation. Arch. Biochem. Biophys. 507, 248–253. doi: 10.1016/j.abb.2010.12.013
Niger, C., Luciotti, M. A., Buo, A. M., Hebert, C., Ma, V., and Stains, J. P. (2013). The regulation of runt-related transcription factor 2 by fibroblast growth factor-2 and connexin43 requires the inositol polyphosphate/protein kinase Cdelta cascade. J. Bone Miner. Res. 28, 1468–1477. doi: 10.1002/jbmr.1867
O'Brien, C. A., Plotkin, L. I., Galli, C., Goellner, J., Gortazar, A. R., Allen, M. R., et al. (2008). Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 3:e2942. doi: 10.1371/journal.pone.0002942
Pacheco-Costa, R., Hassan, I., Reginato, R. D., Davis, H. M., Bruzzaniti, A., Allen, M. R., et al. (2014). High bone mass in mice lacking Cx37 due to defective osteoclast differentiation. J. Biol. Chem. 289, 8508–8520. doi: 10.1074/jbc.M113.529735
Paic, F., Igwe, J. C., Nori, R., Kronenberg, M. S., Franceschetti, T., Harrington, P., et al. (2009). Identification of differentially expressed genes between osteoblasts and osteocytes. Bone 45, 682–692. doi: 10.1016/j.bone.2009.06.010
Palumbo, C., Palazzini, S., and Marotti, G. (1990). Morphological study of intercellular junctions during osteocyte differentiation. Bone 11, 401–406. doi: 10.1016/8756-3282(90)90134-K
Paznekas, W. A., Boyadjiev, S. A., Shapiro, R. E., Daniels, O., Wollnik, B., Keegan, C. E., et al. (2002). Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am. J. Hum. Genet. 72, 408–418. doi: 10.1086/346090
Plotkin, L. I., Aguirre, J. I., Kousteni, S., Manolagas, S. C., and Bellido, T. (2005a). Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of ERK activation. J. Biol. Chem. 280, 7317–7325. doi: 10.1074/jbc.M412817200
Plotkin, L. I., and Bellido, T. (2013). Beyond gap junctions: connexin43 and bone cell signaling. Bone 52, 157–166. doi: 10.1016/j.bone.2012.09.030
Plotkin, L. I., Bivi, N., and Bellido, T. (2011). A bisphosphonate that does not affect osteoclasts prevents osteoblast and osteocyte apoptosis and the loss of bone strength induced by glucocorticoids in mice. Bone 49, 122–127. doi: 10.1016/j.bone.2010.08.011
Plotkin, L. I., Lezcano, V., Thostenson, J., Weinstein, R. S., Manolagas, S. C., and Bellido, T. (2008). Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. J. Bone Miner. Res. 23, 1712–1721. doi: 10.1359/jbmr.080617
Plotkin, L. I., Manolagas, S. C., and Bellido, T. (2002). Transduction of cell survival signals by connexin-43 hemichannels. J. Biol. Chem. 277, 8648–8657. doi: 10.1074/jbc.M108625200
Plotkin, L. I., Manolagas, S. C., and Bellido, T. (2006a). Dissociation of the pro-apoptotic effects of bisphosphonates on osteoclasts from their anti-apoptotic effects on osteoblasts/osteocytes with novel analogs. Bone 39, 443–452. doi: 10.1016/j.bone.2006.02.060
Plotkin, L. I., Mathov, I., Aguirre, J. I., Parfitt, A. M., Manolagas, S. C., and Bellido, T. (2005b). Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases and ERKs. Am. J. Physiol. Cell Physiol. 289, C633–C643. doi: 10.1152/ajpcell.00278.2004
Plotkin, L. I., Vyas, K., Gortazar, A. R., Manolagas, S. C., and Bellido, T. (2006b). βarrestin complexes with connexin (Cx) 43 and anchors ERKs outside the nucleus: a requirement for the Cx43/ERK-mediated anti-apoptotic effect of bisphosphonates in osteocytes. J. Bone Miner. Res. 21:S65.
Plotkin, L. I., Weinstein, R. S., Parfitt, A. M., Roberson, P. K., Manolagas, S. C., and Bellido, T. (1999). Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J. Clin. Invest. 104, 1363–1374. doi: 10.1172/JCI6800
Premont, R. T., and Gainetdinov, R. R. (2007). Physiological roles of G protein-coupled receptor kinases and arrestins. Annu. Rev. Physiol. 69, 511–534. doi: 10.1146/annurev.physiol.69.022405.154731
Ren, J., Wang, X. H., Wang, G. C., and Wu, J. H. (2013). 17beta estradiol regulation of connexin 43-based gap junction and mechanosensitivity through classical estrogen receptor pathway in osteocyte-like MLO-Y4 cells. Bone 53, 587–596. doi: 10.1016/j.bone.2012.12.004
Robinson, J. A., Chatterjee-Kishore, M., Yaworsky, P. J., Cullen, D. M., Zhao, W., Li, C., et al. (2006). WNT/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J. Biol. Chem. 281, 31720–31728. doi: 10.1074/jbc.M602308200
Romanello, M., and D'Andrea, P. (2001). Dual mechanism of intercellular communication in HOBIT osteoblastic cells: a role for gap-junctional hemichannels. J. Bone Miner. Res. 16, 1465–1476. doi: 10.1359/jbmr.2001.16.8.1465
Romanello, M., Veronesi, V., and D'Andrea, P. (2003). Mechanosensitivity and intercellular communication in HOBIT osteoblastic cells: a possible role for gap junction hemichannels. Biorheology 40, 119–121.
Russell, R. G. (2011). Bisphosphonates: the first 40 years. Bone 49, 2–19. doi: 10.1016/j.bone.2011.04.022
Saunders, M. M., You, J., Trosko, J. E., Yamasaki, H., Li, Z., Donahue, H. J., et al. (2001). Gap junctions and fluid flow response in MC3T3-E1 cells. Am. J. Physiol. Cell Physiol. 281, C1917–C1925.
Schirrmacher, K., Schmitz, I., Winterhager, E., Traub, O., Brümmer, F., Jones, D., et al. (1992). Characterization of gap junctions between osteoblast-like cells in culture. Calcif. Tissue Int. 51, 285–290. doi: 10.1007/BF00334489
Stains, J. P., and Civitelli, R. (2005). Gap junctions in skeletal development and function. Biochim. Biophys. Acta. 1719, 69–81. doi: 10.1016/j.bbamem.2005.10.012
Stains, J. P., Watkins, M. P., Grimston, S. K., Hebert, C., and Civitelli, R. (2014). Molecular mechanisms of osteoblast/osteocyte regulation by connexin43. Calcif. Tissue Int. 94, 55–67. doi: 10.1007/s00223-013-9742-6
Sternlieb, M., Paul, E., Donahue, H. J., and ZhangY. (2012). Ablation of connexin 43 in osteoclasts leads to decreased in vivo osteoclastogenesis. J. Bone Miner. Res. 27:S53.
Thi, M. M., Islam, S., Suadicani, S. O., and Spray, D. C. (2012). Connexin43 and pannexin1 channels in osteoblasts: who is the “hemichannel”? J. Membr. Biol. 245, 401–409. doi: 10.1007/s00232-012-9462-2
Tu, X., Rhee, Y., Condon, K. W., Bivi, N., Allen, M. R., Dwyer, D., et al. (2012). Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 50, 209–217. doi: 10.1016/j.bone.2011.10.025
Vander Molen, M. A., Rubin, C. T., McLeod, K. J., McCauley, L. K., and Donahue, H. J. (1996). Gap junctional intercellular communication contributes to hormonal responsiveness in osteoblastic networks. J. Biol. Chem. 271, 12165–12171. doi: 10.1074/jbc.271.21.12165
Watkins, M. P., Norris, J. Y., Grimston, S. K., Zhang, X., Phipps, R. J., Ebetino, F. H., et al. (2012). Bisphosphonates improve trabecular bone mass and normalize cortical thickness in ovariectomized, osteoblast connexin43 deficient mice. Bone 51, 787–794. doi: 10.1016/j.bone.2012.06.018
Watkins, M., Grimston, S. K., Norris, J. Y., Guillotin, B., Shaw, A., Beniash, E., et al. (2011). Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol. Biol. Cell. 22, 1240–1251. doi: 10.1091/mbc.E10-07-0571
Weinger, J. M., and Holtrop, M. E. (1974). An ultrastructural study of bone cells: the occurrence of microtubules, microfilaments and tight junctions. Calcif. Tissue Res. 14, 15–29. doi: 10.1007/BF02060280
Xu, X., Gu, S., Riquelme, M. A., Burra, S., Fajardo, R., Shang, P., et al. (2013). Disruption of connexin 43 channel function in osteocytes differentially reduces bbone mass and imparis the tibial anabolic response to load. J. Bone Miner. Res. 25:S60.
Zhang, Y., Paul, E. M., Sathyendra, V., Davidson, A., Bronson, S., Srinivasan, S., et al. (2011). Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS ONE 6:e23516. doi: 10.1371/journal.pone.0023516
Keywords: connexin43 hemichannels, osteoblast, osteocyte, apoptosis, bone
Citation: Plotkin LI (2014) Connexin 43 hemichannels and intracellular signaling in bone cells. Front. Physiol. 5:131. doi: 10.3389/fphys.2014.00131
Received: 27 December 2013; Accepted: 15 March 2014;
Published online: 04 April 2014.
Edited by:
Juan C. Saez, Universidad Catolica de Chile, ChileReviewed by:
Martyn P. Mahaut-Smith, University of Leicester, UKCopyright © 2014 Plotkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lilian I. Plotkin, Department Anatomy and Cell Biology, Indiana University School of Medicine, 635 Barnhill Drive MS 5035, Indianapolis, IN 46202-5120, USA e-mail:bHBsb3RraW5AaXVwdWkuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.