 Seung-Yeol Nah
Seung-Yeol Nah- Ginsentology Research Laboratory, Department of Physiology, College of Veterinary Medicine and Bio/Molecular Informatics Center, Konkuk University, Seoul, South Korea
Ginseng, the root of Panax ginseng C.A. Meyer, is one of the oldest traditional medicines and is thought to be a tonic. It has been claimed that ginseng may improve vitality and health. Recent studies have advanced ginseng pharmacology and shown that ginseng has various pharmacological effects in the nervous system. Ginsenosides, steroid glycosides extracted from ginseng, were one of the first class of biologically active plant glycosides identified. The diverse pharmacological effects of ginsenosides have been investigated through the regulation of various types of ion channels and receptors in neuronal cells and heterologous expression systems. Ginsenoside Rg3 regulates voltage-gated ion channels such as Ca2+, K+, and Na+ channels, and ligand-gated ion channels such as GABAA, 5-HT3, nicotinic acetylcholine, and N-methyl-D-aspartate (NMDA) receptors through interactions with various sites including channel blocker binding sites, toxin-binding sites, channel gating regions, and allosteric channel regulator binding sites when the respective ion channels or receptors are stimulated with depolarization or ligand treatment. Treatment with ginsenoside Rg3 has been found to stabilize excitable cells by blocking influxes of cations such as Ca2+ and Na+, or by enhancing Cl− influx. The aim of this review is to present recent findings on the pharmacological functions of the ginsenosides through the interactions with ion channels and receptors. This review will detail the pharmacological applications of ginsenosides as neuroprotective drugs that target ion channels and ligand-gated ion channels.
Introduction
Ginseng, the root of Panax ginseng C.A. Meyer, contains a variety of ingredients useful in herbal medicines (Tyler, 1995). Ginseng glycosides, also called ginsenosides or ginseng saponin, are derivatives of triterpenoid dammarane, which consists of 30 carbon atoms. Each ginsenoside has a common hydrophobic four ring steroid-like structure with carbohydrate moieties attached (Nah et al., 2007). Several types of ginsenosides have been isolated and identified from the roots of various ginseng species from America, China, and Korea. They are mainly classified as protopanaxadiol (PD), protopanaxatriol (PT), oleanolic ginsenosides, and ginsenoside metabolites according to the position of different carbohydrate moieties at the carbon-3 and carbon-6 positions, as well as the aliphatic side chain (Figure 1). Recent studies have demonstrated that ginsenosides exhibit a variety of pharmacological effects in nervous and non-nervous systems. A line of accumulating evidence shows that ginsenoside Rg3, the most active ginsenoside, interacts and regulates voltage-gated ion channels and ligand-gated ion channel activity through interaction with specific amino acid(s) at channel entryways or channel pore regions that are associated with ion influx or efflux (Lee et al., 2004b). This review will describe the physiology and pharmacology of ginseng ginsenoside in the regulation of voltage-gated ion and ligand-gated ion channel activities through interactions with specific amino acids of channel proteins and receptors.
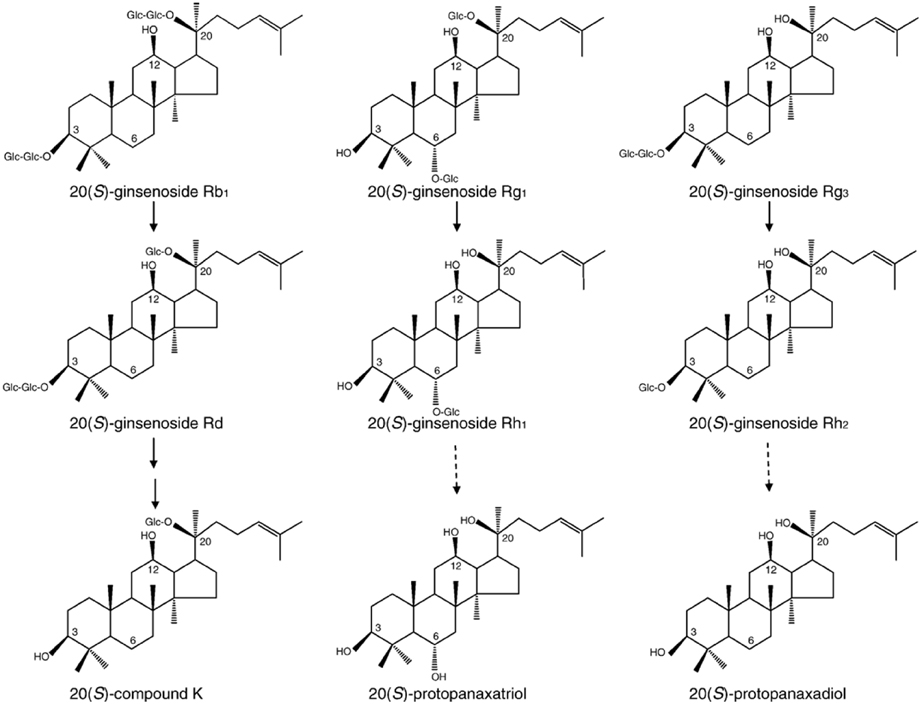
Figure 1. Structures and main metabolic pathways of 20(S)-ginsenoside Rb1, 20(S)- ginsenoside Rg1, and 20(S)- ginsenoside Rg3. Ginsenosides are known to be metabolized by human intestinal microflora. This scheme represents the structures and proposed metabolic pathways of 20(S)-ginsenoside Rb1, 20(S)-ginsenoside Rg1, and 20(S)-ginsenoside Rg3. Bold arrow: main pathway. Dotted arrow: minor or weak pathway. Adapted from Nah et al. (2007).
Ginsenoside Pharmacology through the Interaction with Voltage-Gated Ion Channels
Regulation of Voltage-Gated Ca2+ Channels by Ginsenosides
Ginseng extract and ginsenosides inhibit Ca2+ channel currents in sensory neurons. Among the various ginsenosides such as Rb1, Rc, Re, Rf, and Rg1, ginsenoside Rf was found to be the most effective in inhibiting Ca2+ channel activities (Nah et al., 1995). Ginsenosides also inhibit Ca2+ channels in rat chromaffin cells, which are one of the representative neurosecretory cells involved in catecholamine release under various stress conditions (Kim et al., 1998a). The ginsenosides listed in order of inhibitory potency on Ca2+ channels in rat chromaffin cells are: ginsenoside Rc > Re > Rf > Rg1 > Rb1. Ginsenosides showed a selectivity in Ca2+ channel regulation, inhibiting N-, P-, and Q/R-, but not L-type Ca2+ channels in bovine chromaffin cells (Choi et al., 2001). Ginsenoside Rg3 more potently inhibits L-, N-, and P-type Ca2+ channels than other ginsenosides tested in rat sensory neurons (Rhim et al., 2002). Ginsenoside Rb1 selectively inhibits L-type Ca2+ channel activity in cultured rat hippocampal neurons (Lin et al., 2012). Besides neuronal Ca2+ channels, ginsenoside Re also selectively inhibits L-type Ca2+ channel activity in guinea pig cardiomyocytes (Bai et al., 2003, 2004). In addition, ginsenosides attenuated the stimulation of membrane capacitance increase (ΔCm) in rat chromaffin cells. The ginsenosides listed in order of inhibitory potency on ΔCm are: ginsenoside Rf > Rc > Re > Rg1 > Rb1 (Kim et al., 1998a). The regulation of Ca2+ channel activity and membrane capacitance by ginsenosides indicates that ginsenosides are closely involved in the regulation of neurotransmitter release from presynaptic nerve terminal(s) (Duan and Nicholson, 2008).
Identification of Ginsenoside Interaction sites in Voltage-Gated Ca2+ Channel Regulation
Among the various domains of the L-type Ca2+ channel protein, mutations in L427R, N428R, and L431K in transmembrane domain-I-segment 6 (IS6) of the channel significantly attenuated the action of ginsenoside Rg3, resulting in a shift to the right in dose-response curves, although the inhibitory effects of ginsenoside Rg3 on Ca2+ channel currents was not completely abolished (Choi et al., 2009). In addition, while ginsenoside Rg3 treatment produced a negative shift in the inactivation voltage, it did not alter the steady-state activation voltage, and none of the mutant channels affected the ginsenoside Rg3-induced negative shift in inactivation voltage. Ginsenoside Rg3 had no effect on the inactivation time constant in wild-type and mutant channels. Thus, mutations in L427R, N428R, and L431K in transmembrane domain-I-segment 6 (IS6) of the channel partially attenuated ginsenoside Rg3 action. The Leu427, Asn428, and Leu431 residues of the transmembrane domain-I-segment 6 of L-type Ca2+ channels play important roles in the regulation of L-type Ca2+ channels by ginsenoside Rg3 (Choi et al., 2009) (Figure 2).
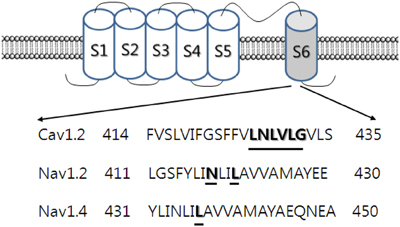
Figure 2. Topology and sequence alignment of Cav1.2, Nav1.2, and Nav1.4 channels. Ca2+ and Na+ channels consist of the main α subunit and other auxiliary subunits. α subunit has 4 domains and each domains contain 6 segments. Each segment 4 plays a membrane voltage sensor and segments 5 and 6 form channel pore. The mutated amino acid residues are underlined in the pore-forming segment 6. The underlined specific amino acid residues in these channels are thought to interact with 20(S)-ginsenoside Rg3 (Rg3) (Lee et al., 2008b; Choi et al., 2009).
Regulation of Voltage-Gated K+ Channels by Ginsenosides
Ginseng total saponins and ginsenoside Rg3 have been shown to activate Ca2+-activated K+ and ATP-sensitive K+ channels in rabbit coronary artery smooth muscle cells (Chung and Kim, 1999; Chung and Lee, 1999). Ginsenosides activate Ca2+-activated K+ channels in vascular smooth muscle cells (Li et al., 2001). In addition, the external application of ginseng total saponin fraction and ginsenoside Rg3 to rabbit coronary artery smooth muscle cells has been shown to increase the amplitude of whole-cell BKCa currents (Chung and Kim, 1999). Treatment with ginseng total extract relaxed rabbit vaginal tissue via hyperpolarization of BKCa channel activation (Kim et al., 2008a). In guinea pig cardiomyocytes, ginsenoside Re enhanced the delayed rectifier K+ channel (IKs) (Bai et al., 2003, 2004). Ginsenoside-induced relaxation of blood vessels and other smooth muscles may be achieved via activation of K+ channels (Kim et al., 1999).
By contrast, ginsenoside Rg3 inhibits voltage-dependent Kv1.4 and Kv4.2 in human cells, but not Kv1.3, Kv1.5, and Kv2.1 expressed in Xenopus laevis oocytes (Jeong et al., 2004; Lee et al., 2013c), indicating that ginsenoside Rg3 differentially regulates Kv channel subtypes. The regulatory effect of ginsenoside Rg3 on Kv1.4 channel activity has been found to be strongly dependent on the extracellular K+ concentration, by shifting the ginsenoside Rg3 concentration-response curve to the right, indicating that ginsenoside Rg3 competes with extracellular [K+] for the same interaction site(s) (Lee et al., 2008a). Further study showed that the inhibitory effects of ginsenoside Rg3 on Kv1.4 channel currents were abolished by K+ activation, which is induced by increasing extracellular K+ concentrations. In addition, some subsets of Kv channel currents are also affected by extracellular and intracellular tetraethylammonium (TEA), which is a well-known K+ channel blocker. The wild-type Kv1.4 channel, however, is nearly insensitive to TEA (Pardo et al., 1992; Lee et al., 2008a). Thus, although extracellular TEA treatment did not inhibit the wild-type Kv1.4 channel, it appeared that extracellular TEA competed with ginsenoside Rg3 to inhibit Kv1.4 channel currents by shifting the ginsenoside Rg3 concentration-response curve to the right (Lee et al., 2008a). Based on these results, ginsenoside Rg3 may have specific interaction site(s) for Kv1.4 channel activity regulation.
Ginsenoside Rg3 Interacts with the Extracellular Tea Binding Site to Regulate Kv1.4 Channel Activity
The K+ activation site of the Kv1.4 channel, which is located at the outer pore entry, consists of several amino acids including lysine 531 (K531) (Claydon et al., 2004). In addition, one of the extracellular TEA binding sites also contains the K531 residue. Mutations in this K531 residue to tyrosine (i.e., K531Y) increased the sensitivity of the Kv1.4 channel to extracellular TEA, abolished K+ activation, and also abolished the effect of ginsenoside Rg3 on the Kv1.4 channel. Thus, ginsenoside Rg3-mediated action of Kv1.4 channel activity may occur through common interaction site(s) for K+ activation and TEA binding sites. Alternatively, the ginsenoside Rg3 interaction site(s) may overlap or share the K+ activation site or the TEA binding site, as shown by Lee et al. (2008a) using various Kv1.4 channel mutations, including the K531 residue. Mutations have been generated in channel pore sites (S510K, D513Q, V525L, and V535Q) and outer pore sites (K531A, P532A, I533A, T534A, and V535A) (Watanabe et al., 2004). Kv1.4 channel mutations have also been generated in the N-glycosylation site (N353Q) (Judge et al., 1999), the voltage sensor site (R447C and R450C) (Claydon et al., 2004), the voltage shift sites (L478F and G548P) (Bett and Rasmusson, 2004), the pH sensitive site (H507Q), and the C-type inactivation site (V560A) (Claydon et al., 2004). The K531A mutant, located in one of the outer pores, significantly attenuated ginsenoside Rg3 inhibition of Kv1.4 channel currents, while the other mutations had no significant effect. Thus, ginsenoside Rg3 regulates Kv1.4 channel activity by interacting with Lys531, which is also known to be one of the K+ activation sites and one of the extracellular TEA binding sites. Other mutant channels at the K531 residue, such as K531Y, I533M, and K531Y-I533M, showed that the K531Y substitution, but not I533M, and the K531Y-I533M double substitution mostly abolished ginsenoside Rg3 inhibition of Kv1.4 channel currents (Lee et al., 2008a). Ginsenoside Rg3-mediated action of Kv1.4 channel activity is closely associated with the Lys531 residue (Figure 3).
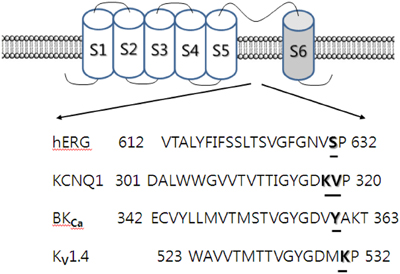
Figure 3. Topology and sequence alignment of BKCa, hERG, KCNQ1, and Kv1.4 K+ channels. BKCa, hERG, KCNQ1, and Kv1.4 K+ consist of the main α subunit and auxiliary β subunits. Tetramer of α subunit forms a functional K+ channel. α subunit contains 6 segments. Each segment 4 plays a membrane voltage sensor and segments 5 and 6 form channel pore. The mutated amino acid residues are underlined in the pore-forming segment 5 and 6. The underlined specific amino acid residues in those K+ channels are thought to interact with 20(S)-ginsenoside Rg3 near the K+ channel “signature sequence” of –TXGYGD– (Lee et al., 2008a; Choi et al., 2010, 2011a,b).
Using homology and virtual docking model methods, which give three-dimensional configurations, Lee et al. (2008a) demonstrated that ginsenoside Rg3 could bind to the Kv1.4 channel protein through various interactions such as hydrogen bonds or hydrophobic interactions. Therefore, the carbohydrate portion of ginsenoside Rg3 plays an important role in its interaction with the Kv1.4 channel. The second, but not the first, carbohydrate attached at carbon-3 of the ginsenoside Rg3 backbone forms six hydrogen bonds with amino acids in the pore entryway of the Kv1.4 channel. Among the amino acids forming hydrogen bonds with ginsenoside Rg3, K531 forms three bonds, and threonine and histidine form the other hydrogen bonds. In addition, the ginsenoside Rg3 backbone is located in the pore portion of the Kv1.4 channel, enabling it to block the pore and interrupt K+ efflux when the channel is stimulated by depolarization. The backbone of ginsenoside Rg3 may act as a physical plug or wedge in ginsenoside Rg3-mediated Kv1.4 channel regulation. However, the K531Y mutant in the Kv1.4 channel forms only two hydrogen bonds with ginsenoside Rg3. Thus, the use of site-directed mutagenesis, as well as homology and virtual docking model methods, are useful tools to confirm and identify ginsenoside Rg3 interaction sites on the Kv1.4 channel (Figure 3).
Ginsenoside Rg3 Regulates BKCa Channel Activity through Interaction with Amino Acid Residues Homologous to the Kv1.4 Channel
Ginsenoside Rg3 enhanced BKCa channel currents, in contrast to its inhibitory effect on Kv1.4 channel currents (Choi et al., 2011a). Interestingly, BAPTA, a Ca2+ chelator, did not block ginsenoside Rg3-induced enhancement of BKCa channel currents, indicating that ginsenoside Rg3-enhancement of BKCa channel currents was independent of intracellular Ca2+. The BKCa channel was sensitive to TEA, with a significant shift in the concentration response curve of ginsenoside Rg3 to the right in the presence of TEA, and vice versa in the wild-type channel, indicating that ginsenoside Rg3 competes for extracellular TEA binding site(s), similarly to mutant Kv1.4 channel interaction (Lee et al., 2008a). In addition, Choi et al. (2011a) reported that ginsenoside Rg3-induced enhancement of the BKCa channel current was independent of the β subunit, suggesting that ginsenoside directly interacts with the BKCa channel α subunit. At the channel pore entry of the α subunit in mutant BKCa channels, the effect of ginsenoside Rg3 on BKCa channel current enhancements was also almost abolished, for instance, in Y360I mutant channels, in which the residue is located at near K+ channel signature sequence, which is also known as the extracellular TEA binding site of the BKCa channel (Choi et al., 2011a) (Figure 3).
Ginsenoside Rg3 also Regulates hERG K+ (IKr) and KCNQ K+ Channel (IKs) activity through interaction with Amino Acid Residues Homologous to the Kv1.4 Channel
Ginsenosides have been shown to exhibit anti-hypertension and cardio-protective effects (Attele et al., 1999). Ginseng extract shortens action potential duration, whereas ginsenoside Re has been shown to regulate the IKr and IKs channel currents of guinea pig cardiomyocytes (Bai et al., 2003; Furukawa et al., 2006). However, relatively little is known at the molecular level about how ginseng extract and ginsenosides shorten action potential duration through the activation of hERG (IKr) and KCNQ (IKs) K+ channels. Ginsenoside Rg3 has been shown to regulate hERG (IKr) K+ channel by enhancing IhERG and Itail. Ginsenoside Rg3 not only caused a persistent Ideactivating−tail without decay but also decelerated deactivating time constants. The mutation of S631 to S631C in the hERG α subunit has been reported to abolish ginsenoside Rg3-mediated action of hERG K+ channels. Thus, ginsenoside Rg3 enhanced IhERG and Itail, and induced a persistent Ideactivating−tail with delayed deactivation of the hERG K+ channel through interaction with the S631 residue (Choi et al., 2011b) (Figure 3).
Ginsenoside Rg3 enhances IKs currents, indicating that it regulates the KCNQ (IKs) K+ channel, which consists of KCNQ1 and KCNE1 subunits (Choi et al., 2010). Site-directed mutagenesis has shown that the K318 and V319 residues of the KCNQ1 or KCNQ1 plus KCNE1 channels are involved in ginsenoside Rg3-mediated action of KCNQ1 plus KCNE1 channel activity (Figure 3). Homology docking models show that the K318 residue plays an important role in the interaction of ginsenoside Rg3 in the closed or open state of the channels (Choi et al., 2010). Ginsenoside Rg3 interacts with the S631 residue of the hERG K+ channel and the K318 and V319 residues of the KCNQ1 plus KCNE1 channel, respectively.
Regulation of Voltage-Gated Na+ Channel by Ginsenosides
Ginsenosides also regulate Na+ channel activities, for instance, inhibiting neuronal Na+ channels expressed in tsA201 cells and Xenopus laevis oocytes. Higher concentrations of ginseng extract and ginsenoside Rb1 than those used in other channel interaction experiments were required to inhibit Na+ channel currents (Liu et al., 2001). Ginsenoside Rg3 was highly potent in inhibiting Na+ channel currents compared to ginseng extract and the other ginsenosides tested (Jeong et al., 2004). In structure-activity relationships of the ginsenoside Rg3 stereoisomers, 20(S)-ginsenoside Rg3 but not 20(R)-ginsenoside Rg3 inhibited the neuronal Na+ channel currents in a dose- and voltage-dependent manner. The hydroxyl group at carbon-20 of 20(S)-ginsenoside Rg3 may be geometrically better aligned with the hydroxyl acceptor group in the ion channels than that of the 20(R)-ginsenoside Rg3 (Jeong et al., 2004).
A structure-activity relationship study of ginsenoside Rg3 investigated the role of the aliphatic side chain, [-CH2CH2CH = C(CH3)2], which is coupled to carbon-20 of the 20(S)-ginsenoside Rg3 backbone in Na+ channel interaction (Lee et al., 2008c). The reduction of the double bond in the aliphatic side chain of 20(S)-ginsenoside Rg3 strengthens the inhibitory effect on Na+ channel activity, shifting the concentration-response curve significantly to the left. However, deletion, hydroxylation, or oxygenation of the aliphatic side chain caused an attenuation or loss of Na+ channel current inhibition. The aliphatic side chain of 20(S)-ginsenoside Rg3, as well as the hydroxyl group of carbon-20 of ginsenoside Rg3 stereoisomers, plays an important role in Na+ channel regulation. Thus, the aliphatic side chain of ginsenoside Rg3 may be a future target for chemical modifications to tune regulation of Na+ channels by Rg3.
In further studies on ginsenoside Rg3-mediated voltage-gated neuronal Na+ channel interaction, two main characteristics have been identified. One is that ginsenoside Rg3 treatment causes a depolarizing shift in the activation voltage step in wild-type Na+ channels, indicating that ginsenoside Rg3 binding to the Na+ channel does not allow the Na+ channel to easily open at a given voltage step, requiring greater depolarizing stimulation compared to untreated channels (Lee et al., 2005). The other characteristic is that ginsenoside Rg3 induces use-dependent inhibition, meaning that the channel pore-blocking actions of ginsenoside Rg3 are enhanced by rapid, repeated stimulation over a very short time period, indicating that ginsenoside Rg3 may be a kind of open channel blocker.
The idea that ginsenoside Rg3 is a kind of open channel blocker of Na+ channel is supported by experiments using inactivation-deficient Na+ channel mutants, in which the inactivation gate has been mutated from IFM to Q3, and transient inward currents are converted into long-lasting inward currents (Lee et al., 2005). Ginsenoside Rg3 more potently inhibited the plateau than peak INa, and facilitated channel closing in inactivation-deficient channel mutants. Interestingly, mutations in one amino acid (Lys859 to Glu859 in brain NaV1.2 channel) in the voltage-sensor domain in the S4 helix abolished the ginsenoside Rg3-mediated depolarizing shift without affecting the ginsenoside Rg3-mediated inhibition of peak current (INa). These results indicate that ginsenoside Rg3 may have interaction sites for the brain Nav1.2 channel regulation.
Identification of Ginsenoside Interaction Sites in Voltage-Gated Na+ Channel Regulation
Although ginsenoside Rg3 regulates neuronal Na+ channels as a kind of open channel blocker, showing use-dependent inhibition and a depolarizing shift in the activation curve of wild-type Na+ channels, the exact interaction sites in the Na+ channel proteins have not been identified. Batrachotoxin (BTX) is a neurotoxin that acts on Na+ channels. BTX toxin was first found in the skin of the South American frog Phyllobates terribilis, and persistently activates brain NaV1.2 and skeletal muscle NaV1.4 channels, rather than inhibiting Na+ currents as lidocaine and tetrodotoxin do (Wang and Wang, 1998). In addition, BTX is a steroidal alkaloid toxin, with a backbone structure similar to that of ginsenoside Rg3. Interestingly, ginsenoside Rh2 inhibited [3H]BTX-B binding in rat brain membrane fractions and attenuated glutamate release (Duan et al., 2006; Duan and Nicholson, 2008), showing that the ginsenoside Rg3-induced interaction with rat brain NaV1.2 channel activity may involve the BTX binding sites, and that the interference by ginsenoside of [3H]BTX-B binding in rat brain membrane fractions is relevant to ginsenoside Rg3-induced Na+ channel regulation. BTX interaction sites are located at the I433, N434, and L437 residues of the NaV1.4 channel and equivalent residues such as I417, N418, and L421 of brain NaV1.2 channels in domain-I segment 6 (IS6).
Channel mutations in BTX binding sites, such as N418K and L421K in rat brain NaV1.2, and L437K in the mouse skeletal muscle NaV1.4 channel, have been shown to attenuate or abolish ginsenoside Rg3 inhibition of Na+ currents (Figure 2). In addition, channel mutations in BTX binding sites also greatly attenuate the ginsenoside Rg3-mediated depolarizing shift in the activation voltage observed in wild-type channels. Moreover, ginsenoside Rg3-mediated use-dependent inhibition was almost abolished in these mutant channels. BTX binding sites in brain- and muscle-type Na+ channels play important roles in ginsenoside Rg3-mediated Na+ channel interaction at cellular and molecular levels (Lee et al., 2008b).
Ginsenoside Pharmacology in the Interaction with Membrane Receptor Ligand-Gated Ion Channels
Regulation of GABAA and Glycine Receptor Channel Activity by Ginsenosides
Ginseng has been shown to have an anxiolytic-like effect in animal model studies (Kim et al., 2009). Recent studies have shown that ginsenosides interact with the GABAA receptor, and may regulate the binding of the ligand with the GABAA receptor. Ginsenosides differentially regulate the binding of [3H]-flunitrazepam or [3H]-muscimol to the GABAA receptor in a rat brain membrane fraction (Kimura et al., 1994). On the other hand, prolonged infusion with ginsenoside Rc in the rat brain elevates [3H]-muscimol binding to the GABAA receptor in a brain region-specific manner, while ginsenoside Rg1 has no effect (Kim et al., 2001). Thus, ginsenosides may regulate the GABAA receptor by affecting the binding affinities of its ligands.
In a GABAA receptor channel activity study, ginsenosides were also shown to regulate GABAA receptor channel activity by enhancing GABA-mediated channel activity (Choi et al., 2003a). Thus, in studies using Xenopus oocytes expressing human recombinant GABAA receptor, ginsenosides Rb1, Rb2, Rc, Rd, Re, Rf, Rg1, and Rg2 affected GABAA receptor channel activity, and ginsenoside Rc most potently enhanced the GABA-induced inward peak current (IGABA). Bicuculline, a GABAA receptor antagonist, and picrotoxin, a GABAA channel blocker, blocked the stimulatory effect of ginsenoside Rc on IGABA. Niflumic acid (NFA) and 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid, both Cl− channel blockers, attenuated the effect of ginsenoside Rc on GABA-induced inward peak current.
Compared to the GABAA receptor, few investigations have been carried out into the ginsenoside-mediated action on glycine receptor channel activity. In a study using human glycine α1 receptor channel expressed in Xenopus oocytes, Noh et al. (2003) demonstrated that although treatment with ginsenoside Rf enhanced the glycine-induced inward peak current in a dose-dependent and reversible manner, ginsenoside Rf itself did not elicit membrane currents. The effect of ginsenoside Rf action on glycine receptor channel activity was blocked by strychnine, a glycine receptor antagonist, and 4,4′-disothiocyanostilbene-2,2′-disulfonic acid (DIDS), a Cl−channel blocker. The various ginsenosides, listed in order of potency for the enhancement of the glycine-induced inward Cl− current, were ginsenoside Rb1 > >Rb2 > Rg2 = Rc > Rf > Rg1 > Re. Further study will be helpful for elucidation of the interaction of ginsenosides with glycine receptor proteins to enhance the glycine-induced inward Cl− current.
Ginsenoside Rg3 Interacts with the γ2 Subunit to Regulate GABAA Receptor Channel Activity
Two characteristics of ginsenoside Rg3-induced GABAA receptor interaction have been described. First, ginsenoside Rg3 itself evoked an inward current in a concentration-dependent manner in Xenopus oocytes expressing human recombinant GABAA receptor (α1β1γ2S) in the absence of GABA. Ginsenoside Rg3-elicited inward currents were blocked by a GABAA receptor antagonist, indicating that ginsenoside Rg3 itself activates GABAA receptors (Lee et al., 2013b). Ginsenoside Rg3-elicited inward currents were not observed in the absence of the γ2S or γ2L subunits. The magnitude of the ginsenoside Rg3-elicited inward current was dependent on the expression ratio of the γ2S subunit. However, ginsenoside Rg3 exerted no effects in oocytes expressing other subunits, such as γ1, γ3, δ, and ε, with α1β1.
The γ2 subunit of the GABAA receptor plays an important role in the action of human epilepsy (Bowser et al., 2002). The γ2 subunit of the GABAA receptor is also essential for the formation of high-affinity benzodiazepine-binding sites, and mutations in benzodiazepine-binding sites greatly attenuated the benzodiazepine-induced potentiation of IGABA (Buhr et al., 1997). In addition, increased expression of the γ2 subunit compared to other subunits further potentiated benzodiazepine-induced IGABA (Boileau et al., 2003). However, GABAA receptors with mutant γ2 subunit benzodiazepine-binding sites did not affect the action of ginsenoside Rg3 (Lee et al., 2013b). Thus, it is unlikely that the ginsenoside Rg3-induced activation of α1β1γ2 GABA receptors is achieved through interaction with benzodiazepine-binding sites.
Second, co-treatment of ginsenoside Rg3 with GABA further potentiated IGABA in oocytes expressing GABAA receptor (α1β1γ2S) (Lee et al., 2013b). However, the potentiating effect of ginsenoside Rg3 on IGABA was not specific to the γ2 subunit, and co-expression of other subunits such as γ1, γ3, δ, and ε also enhanced IGABA. The degree of potentiation of IGABA by ginsenoside Rg3 was not significantly different in the presence of different subunits. Even ginsenoside Rg3 enhanced IGABA in oocytes expressing α1β1 subunits and ginsenoside Rg3 itself did not elicit inward currents in oocytes expressing α1β1 GABAA receptor in the absence of GABA. Thus, ginsenoside Rg3 may have dual binding sites for GABAA receptor modifications, one for GABA binding sites in the presence of GABA and the other one for γ2 subunit in the absence of GABA. However, ginsenoside Rg3 had no effects on GABAc receptor channel activity (Lee et al., 2013b).
Regulation of 5-HT3 Receptor Channel Activity by Ginsenosides
Ginsenoside Rg2 and ginsenoside metabolites inhibit 5-HT3 receptor-mediated ion currents (I5−HT) in Xenopus oocytes expressing 5-HT3 receptors (Choi et al., 2003b; Lee et al., 2004a). The inhibitory effect of ginsenoside Rg2 on 5-HT-induced inward currents was non-competitive and voltage-independent, similar to the ginsenoside-induced modulation of heteromeric nicotinic acetylcholine receptor. In addition, the inhibitory effect of ginsenoside Rg3 on serotonin-induced currents (I5−HT) is observed when applied extracellularly but not intracellularly (Lee et al., 2004b), indicating that ginsenoside Rg3 regulates 5-HT3 receptors outside the cell.
Ginsenoside Rg3 Regulates 5-HT3 Receptor Channel Activity through Interaction with Amino Acids at the Channel Gating Region
Mutations in V291A, F292A, and I295A in the transmembrane domain 2 (TM2) greatly attenuated or abolished ginsenoside Rg3-induced inhibition of peak I5−HT. Thus, ginsenoside Rg3 acts through the 5-HT3 receptor protein, and alterations in TM2 of the 5-HT3 receptor could affect the action of ginsenoside Rg3 (Lee et al., 2007). Interestingly, the V291A mutation, although not the F292A or I295A mutations, induced constitutively active ion currents, with a decreased current decay rate. Ginsenoside Rg3 treatment of this mutant receptor accelerated the rate of current decay in the presence of 5-HT, suggesting that the presence of ginsenoside Rg3 caused channel closure rather than opening. Thus, ginsenoside Rg3 and TMB-8, an open channel blocker, inhibited the constitutively active ion currents. Diltiazem, another open channel blocker, did not prevent ginsenoside Rg3-induced inhibition of the constitutively active ion currents in occlusion experiments (Lee et al., 2007). This report provides the following insight: first, ginsenoside Rg3 inhibits 5-HT3A receptor channel activity through interaction with residues V291, F292, and I295 in the channel gating region of TM2, and second, ginsenoside Rg3 regulates 5-HT3A receptor channel activity in the open state at different site(s) from those used by TMB-8 and diltiazem. Thus, ginsenoside Rg3 inhibits the 5-HT3 receptor in the open state through interactions with V291, F292, and I295. The identification of ginsenoside Rg3 interacting sites in the 5-HT3 receptor indicates that ginsenoside Rg3 achieves its pharmacological actions via a specific interaction with the 5-HT3 receptor (Figure 4).
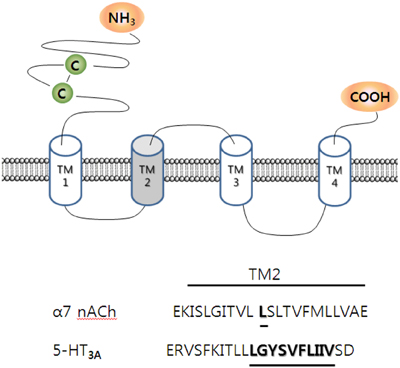
Figure 4. Topology and sequence alignment of α7 nicotinic acetylcholine (nACh) and 5-HT3A receptors at transmembrane domain 2 (TM2). α7 nACh and 5-HT3A receptors consist homomeric pentamer. TM2 of each segment forms a channel pore. The underlined specific amino acid residues in those ligand-gated channels are thought to interact with 20(S)-ginsenoside Rg3 (Lee et al., 2007, 2009).
Regulation of Nicotinic Acetylcholine Receptor Channel Activity by Ginsenosides
Ginsenosides inhibit acetylcholine-stimulated catecholamine release from chromaffin cells, which mainly contain α3β4 nicotinic acetylcholine receptors involved in catecholamine release (Tachikawa et al., 2001). Furthermore, ginsenosides also inhibited acetylcholine-induced inward currents in oocytes expressing nicotinic receptor α1β1δε or α3β4 subunits, suggesting that ginsenosides directly regulate nicotinic acetylcholine receptor channel activities (Choi et al., 2002). However, ginsenosides themselves had no effect on basal currents in oocytes expressing nicotinic acetylcholine receptor αβδε or α3β4 subunits. The inhibition of acetylcholine-induced inward currents by ginsenosides in oocytes expressing nicotinic acetylcholine receptor αβδε or α3β4 subunits was reversible, voltage-independent, and non-competitive, indicating that ginsenosides may bind to the nicotinic acetylcholine receptor when channels are open, but do not compete with acetylcholine to regulate these receptors (Choi et al., 2002). Interestingly, it appears that protopanaxatriol ginsenosides such as Re, Rf, Rg1, or Rg2 were more potent than protopanaxadiol ginsenosides such as Rb1, Rb2, Rc, and Rd in inhibiting acetylcholine-induced inward currents (Choi et al., 2002). On the other hand, ginsenoside Rg2 reduced the peak current and increased the desensitization of acetylcholine-induced inward currents in oocytes expressing human neuronal nicotinic acetylcholine receptors such as α3β4, α3β2, α4β4, and α4β2 (Sala et al., 2002). Thus, the inhibitory effects of ginsenosides on acetylcholine-stimulated attenuation of catecholamine release are achieved through nicotinic acetylcholine receptor channel activity.
In Xenopus oocytes heterologously expressing the α9α10 nicotinic acetylcholine receptor, ginsenosides blocked acetylcholine-induced inward currents (IACh), listed in order of potency, with ginsenoside Rg3 > Rb2 > CK > Re = Rg2 > Rf > Rc > Rb1 > Rg1 in a reversible manner, and the blocking effect of ginsenoside Rg3 on IACh was the same after pre-application compared to co-application of ginsenoside Rg3. Ginsenoside Rg3-induced IACh inhibition was not affected by acetylcholine concentration and was independent of membrane holding potential. The inhibitory effect of ginsenoside Rg3 on IACh was not observed in oocytes expressing the α9 subunit alone, indicating that the presence of the α10 subunit is required for ginsenoside Rg3-induced regulation of α9α10 nicotinic acetylcholine receptor channel activity (Lee et al., 2013a). The α10 subunit of the α9α10 nicotinic acetylcholine receptor may play an important role in ginsenoside Rg3-induced interaction with the α9α10 nicotinic acetylcholine receptor.
Identification of Ginsenoside Interaction Sites in the Regulation of the Nicotinic Acetylcholine Receptor
In contrast to heteromeric acetylcholine receptors, ginsenosides had no effect on wild-type homomeric α7 nicotinic acetylcholine receptor-mediated ion currents (Lee et al., 2009). The homomeric α7 nicotinic acetylcholine receptors, which are predominantly expressed in the cortical and limbic areas, are the major binding sites for α-bungarotoxin in the mammalian central nervous system and are Ca2+ permeable (Gotti et al., 2009). The α7 nicotinic acetylcholine receptor and the 5-HT3A receptors are both homomeric ligand-gated ion channels. Interestingly, single point mutations of Leu247 to Thr247 in the highly conserved TM2, which forms the channel pore region, creates gain-of-function alterations, including slower desensitization, increased acetylcholine affinity, and a linear current-voltage relationship, as well as altering pharmacological properties such as the conversion of various α7 nicotinic acetylcholine receptor antagonists into agonists (Revah et al., 1991; Bertrand et al., 1992). Thus, the L247 residue of the α7 nicotinic acetylcholine receptor could be a useful target for drug development and a focal point in the investigation of channel gating of the acetylcholine receptor (Palma et al., 2006). In addition, the Leu residue corresponding to position 247 of the chick α7 nicotinic acetylcholine receptor channel is highly conserved in all nicotinic, GABAA, 5-HT3, and glycine receptors, and is believed to be positioned at the gate (Lester et al., 2004). Moreover, recently acquired high resolution structures of the nicotinic acetylcholine receptor channel show that the conserved Leu is located at the narrowest part of the channel, and that the side chain of the amino acid head points toward the lumen of the pore (Miyazawa et al., 2003). The importance of position 247 for gating and conductance has also been demonstrated in a functional study (Bertrand et al., 1992).
The mutation of Leu247 to various other amino acid residues induces changes in the receptor sensitivity to ginsenoside Rg3. Interestingly, mutations in L247 to L247A, L247D, L247E, L247I, L247S, and L247T, although not L247K, rendered mutant receptors sensitive to ginsenoside Rg3 (Lee et al., 2009). Ginsenoside Rg3 inhibition of the mutant α7 nicotinic acetylcholine receptor channel currents was reversible and concentration-dependent. In addition, ginsenoside Rg3 inhibition of the mutant α7 nicotinic acetylcholine receptor was strongly voltage-dependent and non-competitive. The homology docking model between ginsenoside Rg3 and the mutant receptor revealed that ginsenoside Rg3 forms hydrogen bonds with amino acids, such as Ser240 of subunit I and Thr244 of subunits II and V at the channel pore, whereas the wild-type receptor ginsenoside Rg3 localizes at the interface of the two wild-type receptor subunits. Thus, the mutation of Leu247 to Thr247 may induce conformational changes in the wild-type receptor, creating a binding pocket for ginsenoside Rg3 at the channel pore (Figure 4).
Regulation of N-Methyl-D-Aspartate (NMDA) Receptor Channel Activity by Ginsenosides
Ginsenosides Rb1 and Rg3 have been shown to attenuate glutamate- and NMDA-induced neurotoxicity by inhibiting the overproduction of nitric oxide, the formation of malondialdehyde, and the influx of Ca2+ in rat cortical cultures (Kim et al., 1998b). In addition, in rat hippocampal cultures, ginsenosides and ginsenoside Rg3 attenuated high K+-, glutamate-, and NMDA-induced Ca2+ influx (Kim et al., 2002). Seong et al. (1995) showed that ginsenosides attenuated glutamate-induced swelling of cultured rat astrocytes. Notoginsenoside R1 prevents glutamate-mediated neurotoxicity in cultured mouse cortical neurons (Gu et al., 2009). On the other hand, in an in vivo study using anesthetized rats, intracerebroventricular administration of ginsenoside Rb1, and not Rg1,significantly inhibited the magnitude of long term potentiation induced by strong tetanus in the dentate gyrus, although ginsenoside Rb1 did not affect the basal synaptic responses evoked by low-frequency tests (Abe et al., 1994). Pretreatment with ginsenosides via the intrathecal route attenuated NMDA- or substance P-induced nociceptive behavior, but had no effect on glutamate-induced behavior (Yoon et al., 1998; Shin et al., 1999). Pretreatment of ginsenosides via the intraperitoneal route also attenuated cell death of hippocampal neurons induced by kainate (Lee et al., 2002). Ginsenosides Rh2 and Rg3 inhibit NMDA receptor channel currents in cultured rat hippocampal neurons (Lee et al., 2006a). Regarding the effects of ginsenoside metabolites such as compound K (CK), protopanaxadiol (PPD), and protopanaxatriol (PPT) on NMDA receptor channel activity, PPT, unlike CK and PPD, reversibly inhibited NMDA-mediated inward currents (INMDA) in a concentration-dependent manner. PPT inhibition of INMDA was non-competitive with NMDA, and was independent of the membrane holding potential (Shin et al., 2012), even though ginsenoside Rh2, Rg3, and PPT interact with the NMDA receptor.
Identification of Ginsenoside Interaction sites in the Regulation of the NMDA Receptor
Ginsenoside Rg3 does not compete with NMDA in NMDA receptor-mediated ion current inhibition in rat hippocampal neuron cultures (Kim et al., 2004b; Lee et al., 2006a). In addition, ginsenoside Rg3 does not interact with Mg2+ or phencyclidine (PCP, MK-801, ketamine) binding sites. However, the inhibitory effect of ginsenoside Rg3 on NMDA receptor-mediated ion currents was attenuated by increasing glycine concentrations, whereas ginsenoside Rh2-mediated inhibition of NMDA receptor-mediated ion currents were attenuated by increasing spermine concentrations. Thus, ginsenoside Rg3 and ginsenoside Rh2 selectively targeted NMDA receptors with different NMDA receptor regulatory sites (Lee et al., 2006a). These reports show the possibility that ginsenoside Rg3 is a competitive antagonist at the glycine- and polyamine-binding site, although the specific amino acid(s) involved in ginsenoside Rg3 binding have not been identified. Site-directed mutagenesis may provide further information about the interaction of ginsenoside Rg3 with glycine binding site(s).
Chacterizations of Ginsenoside Rg3-Mediated Actons on Ion Channels and Receptors
Ginsenosides exhibit several principles in their actions of a variety of ion channels and receptors. Firstly, ginsenosides do not affect most of ion channel and receptor activities, when they are at rest or without ligand stimulations (Nah et al., 2007). But, when voltage-gated ion channels are depolarized or receptors are activated by the relevant receptor agonists, ginsenosides affect ion channel or ligand-gated ion channel activities. Thus, conformational changes to ion channels or receptor proteins induced by depolarization or receptor ligand binding may provide an opportunity for ginsenoside Rg3 to bind ion channels or receptors to exert its action (i.e., Na+ channels and 5-HT3 receptor) (Lee et al., 2007, 2008c).
Secondly, in ginsenoside Rg3-induced voltage-gated ion channel regulations, the ginsenoside Rg3 effects on voltage-gated Ca2+, K+, and Na+ channel currents were achieved via interaction with transmembrane domain-I-segment 6, which is known to form a channel pore or pore entryway. Ginsenoside Rg3-induced inhibition of L-type Ca2+ channel currents was attenuated by mutations of Leu427, Asn428, and Leu431 in transmembrane domain-I-segment 6 residues (Choi et al., 2009). Similarly, N418K and L421K mutant rat brain NaV1.2, and L437K mutant mouse skeletal muscle NaV1.4 channel attenuated or abolished ginsenoside Rg3 inhibition of Na+ currents. Another interesting observation was the consistent pattern exhibited in ginsenoside Rg3-mediated action on various K+ channel subtypes. As shown in Figure 3, K+ channels have a common feature, in that they all have a pore-lining P-loop with a consensus amino acid sequence –TXGYGD–, which is called the K+ channel “signature sequence” (Heginbotham et al., 1994). These residues, repeated in each of the 4 α subunits, form the K+ selectivity filter. Ginsenoside Rg3-mediated human Kv1.4, KCNQ (IKs), hERG, and BKCa K+ channels are regulated through interaction with K531 and K318, V319, S631, and Y360 residues respectively, all of which are the first or second amino acid after –TXGYGD–. They are all located at the channel pore entry (Lee et al., 2008a; Choi et al., 2010, 2011a,b) (Figure 2). Thus, ginsenoside Rg3 has a common interaction site near the “signature sequence” in the subsets of the K+ channels examined. However, the interaction of ginsenoside Rg3 with these K+ channel subtypes exhibits differential effects (i.e., activation of BKCa, IKr, IKs or inhibition of Kv1.2 and Kv4.2). Another important characteristic in Kv1.4 and BKCa channel interaction with ginsenoside Rg3 is the sharing of other K+ channel regulator binding sites such as the extracellular TEA binding site, although TEA structure is different from that of ginsenoside (Lee et al., 2008a).
Thirdly, ginsenoside Rg3 exhibits its effects via differential interactions with ligand-gated ion channels. Ginsenoside Rg3-induced actions of 5-HT3 and mutant α7 nicotinic acetylcholine receptor are achieved via interaction with amino acids in TM2, which forms channel pore (Choi et al., 2009). Ginsenoside Rg3 regulates GABAA receptor channel activity via γ2 subunit, whereas ginsenoside Rg3 regulate NMDA receptor channel activity via allosteric interaction sites such as co-agonist, glycine or polyamines, binding site.
Finally, there might be a compensatory action by ginsenoside action in ion channel regulations. For example, ginsenoside Rg3 stabilizes membrane potentials through the inhibition of both Ca2+ and Na+ channels, but ginsenoside Rg3 might induce a depolarization of neuronal cells by inhibiting Kv1.4 and Kv4.2 channel activity. Thus, ginsenoside Rg3 action is interesting because the same inhibitory effects on Ca2+, Kv1.4 and Kv4.2, and Na+ channels could result in opposite effects in neurons. One possible speculation on the actions of ginsenoside Rg3 on nervous system is that suppression of neuronal excitability by ginsenoside Rg3-mediated Ca2+ and Na+ channel inhibition could be to some extent compensated by inhibition of Kv1.4 and Kv4.2 channel activity. On the other hand, there is no evidence that ginsenoside directly binds to ion channels or receptors, since most of studies are indirectly obtained from mutation experiments. In future studies, it will be needed to obtain direct evidence(s) to confirm ginsenoside binding to ion channel or ligand-gated ion channel proteins.
A Linkage of the Ginsenoside-Mediated Ion Channels and Receptor Interactions between Ginseng Pharmacology
In traditional medicine, ginseng was utilized as a tonic to invigorate body (Nah et al., 2007). Ginsenoside-mediated actions of ion channels and receptors could underlie molecular bases for the explanations of traditional ginseng pharmacology. Here, this review illustrates representative examples how ginsenoside-mediated actions of ion channels and receptors link to ginseng pharmacology. In normal Ca2+ homeostatic state, cytosolic calcium plays a key role for learning, memory and other many cellular events (Berridge et al., 1998). In contrast, Ca2+ dyshomeostasis is one of important events in brain ischemic and traumatic brain injury (Wojda et al., 2008). Ginsenoside might be helpful for neuroprotection by negative coupling of [Ca2+]i in Ca2+ dyshomeostasis of nervous systems. Brain injury will cause ATP exhaustion by mitochondrial dysfunction and also subsequently couples to the slow secondary excitotoxicity by glutamate (Pang and Geddes, 1997). The secondary glutamate-induced excitotoxicity in ATP deficient neurons is initiated by voltage-dependent Na+ channel activation, which is coupled to membrane depolarization, Ca2+ channel activation, and subsequent NMDA receptor activation (Alzheimer, 2002). Moreover, pathologically elevated Na+ and Ca2+ levels in the cytosol are likely to trigger a cascade of molecular events that eventually lead to neuronal death (e.g., formation of reactive oxygen species, lipid peroxidation, mitochondrial dysfunction, activation of calpain and caspases, etc.). As described above, ginsenosides inhibit cytosolic Ca2+ and Na+ elevation through these channel activations as well as NMDA receptor inhibition (Lee et al., 2006a). Thus, negative couplings of ginsenosides to Ca2+ and Na+ channels and NMDA receptors might be helpful for the attenuation of excitotoxity under the brain ischemic and traumatic brain injury. Figure 5 illustrate a schematic example based on reports how ginsenoside-mediated interactions with Ca2+ and Na+ channels and NMDA receptors may be coupled to neuroprotective effects against Ca2+ dyshomeostasis induced by uncontrolled Ca2+ and Na+ channels or NMDA receptors (Lee et al., 2002, 2006a; Kim and Rhim, 2004; Park et al., 2004; Kim et al., 2005c; Lian et al., 2005; Tian et al., 2005; Kim et al., 2007, 2008b; Zhang et al., 2008; Cheon et al., 2013).
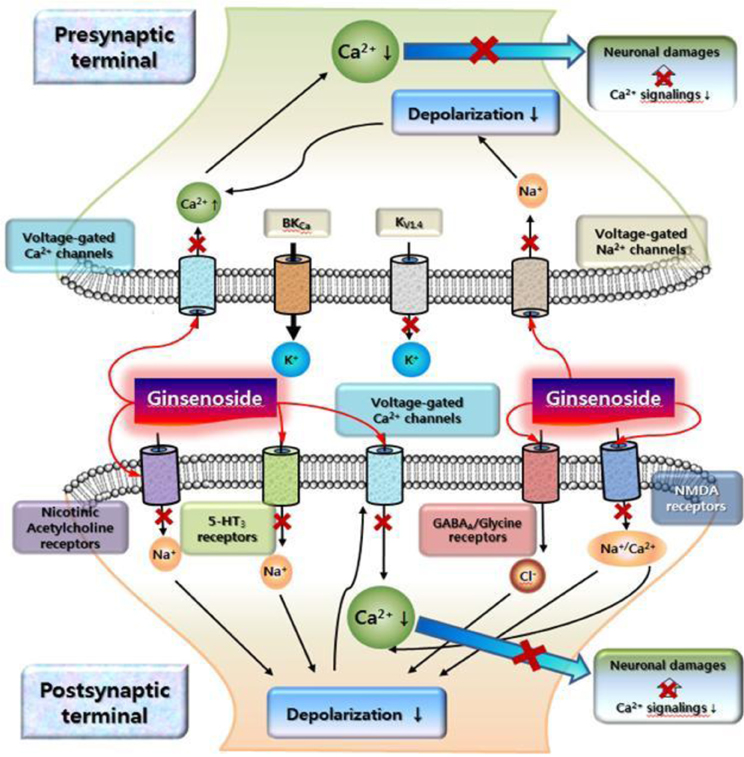
Figure 5. A schematic illustration on cytosolic Ca2+ overload signalings and ginsenoside-mediated attenuation against cytosolic Ca2+ overload. This schematic drawing shows that cytosolic Ca2+ levels could be elevated in ischemic or traumatic brain injury. The elevation of cytosolic Ca2+ levels occurs either via direct activation of voltage-gated Ca2+ channels or via depolarization caused by voltage-dependent Na+ channel activation and other excitatory ligand-gated ion channels at pre- and post-synaptic sites. The overload of cytosolic Ca2+ levels caused by excitatory neurotransmitters or neurotoxins may initiate the persistent activation of Ca2+-dependent signaling, resulting in damage, for instance through apoptosis or necrosis. Although ginsenoside has no effects on ion channels and ligand-gated ion channels at rest state of neurons, ginsenoside might exhibit its effects by attenuation of cytosolic Ca2+ elevation by abnormal conditions by inhibiting ion channels and receptors (as indicated with “x” in arrow). The detailed explanations were described in text. Ginsenoside exhibits differential regulations on subsets of K+ channels. So, one possible hypothesis is that K+ channels regulated by ginsenoside might play a balancing role on Ca2+ and Na+ channel inhibitions by ginsenoside.
Ginseng and ginsenosides have also been reported to alleviate stress-induced symptoms and lesions (Attele et al., 1999). Stress is one of main causes for development of almost all types of diseases. The adrenal gland is one of the peripheral organs responding to stress. In stress situation, the adrenal gland secrets catecholamines from the medulla and renders the organs to cope with stress. α3β4 nicotinic acetylcholine receptors in adrenal medulla play a key role in catecholamine secretion. Ginsenosides and their metabolites inhibit α3β4 nicotinic acetylcholine receptor channel activity and catecholamine release (Tachikawa et al., 2001; Choi et al., 2002). Thus, ginsenosides might be helpful for alleviation of stress by controlling catecholamine secretion during over-stress situation.
On the other hand, traditional medicine also showed that ginseng is utilized for the alleviation of emesis, which includes nausea and vomiting. Nausea and vomiting are significant adverse effects of anti-cancer agents such as cisplatin, and cause significant patient morbidity. 5-HT3 receptors are involved in vomiting and irritable bowel syndrome (Kim et al., 2005a,b). Ginsenoside-mediated action of 5-HT3A receptor channel activity may be the molecular basis of the anti-vomiting action of ginseng. Ginsenoside might be used for attenuation of anticancer agent-induced side effects of vomiting and nausea.
There are multiple cardiovascular effects attributed to ginseng, including cardioprotection, antihypertensive effects, and attenuation of myocardial hypertrophy, heart failure, and the ischemic and reperfused heart in a variety of experimental models (Attele et al., 1999). Ginseng total saponins and ginsenoside Rg3 regulate L-type Ca2+ and various K+ channel activities. Especially, ginsenoside Re enhances IKr and IKs channel currents in guinea myocytes (Bai et al., 2003, 2004). Ginsenoside Rg3 also delays deactivation of IKs and enhances IKr. In heart, IKr and IKs channels are clinically important and are target for drug development, since dysfunction of these channels is associated with sudden death in human (Bai et al., 2004). Thus, ginsenosides-mediated actions of IKr and IKs channels as well as K+ channels might contribute to cardioprotective effects of ginseng.
Summary
The pharmacological behaviors of ginseng are diverse rather than unique, since ginsenosides, as one of active ingredients of ginseng, exhibit differential pharmacological actions in ion channels or ligand-gated ion channel regulations. Therefore, it is unlikely that ginseng ginsenoside achieves its diverse effects on ion channels and receptors via mediation of its own receptor activations on plasma membrane for the following reasons. Firstly, ginsenoside affinity for ion channels and receptors is very low compared to other receptor-specific ligands or toxins (Table 1). Rather, ginsenosides directly interact with various ion channel proteins and receptors without selectivity (Table 1). Secondly, since ginsenoside does not have its receptor on plasma membrane, ginsenoside does not induce spontaneous cellular responses in ion channel or receptor activity without stimuli such as depolarization or receptor ligands. Thus, diverse actions of ginsenoside on ion channels and receptors without its specific receptor mediations could be a limitation on its therapeutic use. Nevertheless, since ginseng influences various body functions as a traditional medicine, a pharmacological role of ginsenosides through the interactions with diverse ion channels and receptors might have potential as a prevention or treatment for a variety of nervous system disorders.
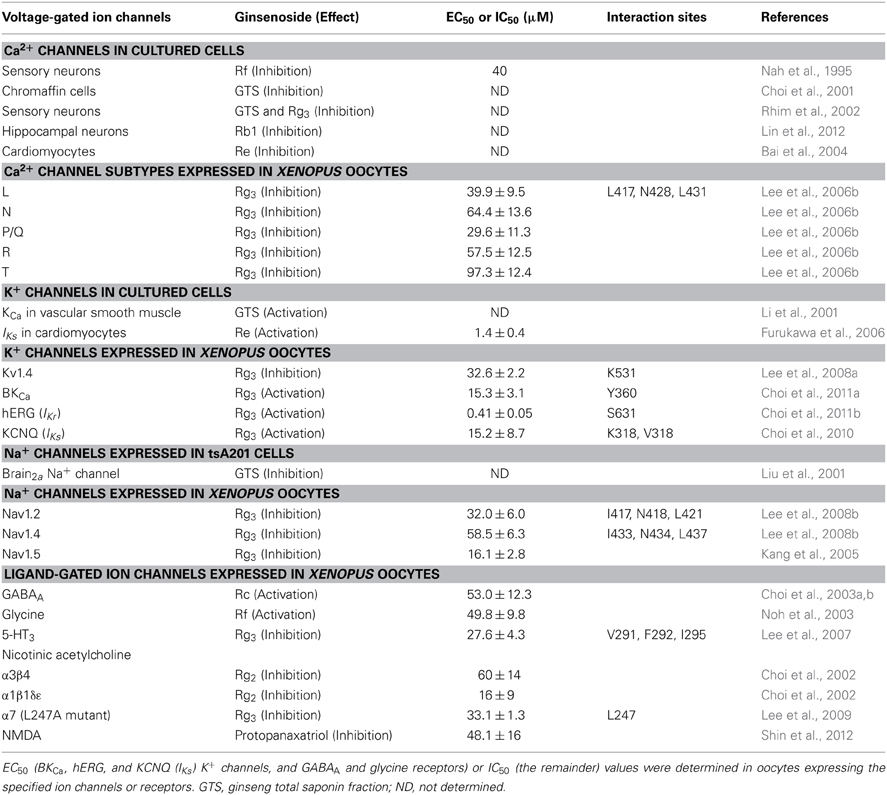
Table 1. Summary of EC50 or IC50 on ginsenoside-induced inhibition or stimulation of various voltage-gated ion or ligand-gated ion channel activities.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Basic Science Research Program (2011-0021144) and the Priority Research Centers Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education, Science, and Technology (2012-0006686) and by the BK21 plus project fund for Seung-Yeol Nah.
References
Abe, K., Cho, S. I., Kitagawa, I., Nishiyama, N., and Saito, H. (1994). Differential effects of ginsenoside Rb1 and malonylginsenoside Rb1 on long-term potentiation in the dentate gyrus of rats. Brain Res. 649, 7–11. doi: 10.1016/0006-8993(94)91042-1
Alzheimer, C. (2002). Na+ channels and Ca2+ channels of the cell membrane as targets of neuroprotective substances. Adv. Exp. Med. Biol. 513, 161–181. doi: 10.1007/978-1-4615-0123-7_5
Attele, A. S., Wu, J. A., and Yuan, C. S. (1999). Ginseng pharmacology: multiple constituents and multiple actions. Biochem. Pharmacol. 58, 1685–1693. doi: 10.1016/S0006-2952(99)00212-9
Bai, C. X., Sunami, A., Namiki, T., Sawanobori, T., and Furukawa, T. (2003). Electrophysiological effects of ginseng and ginsenoside Re in guinea pig ventricular myocytes. Eur. J. Pharmacol. 476, 35–44. doi: 10.1016/S0014-2999(03)02174-5
Bai, C. X., Takahashi, K., Masumiya, H., Sawanobori, T., and Furukawa, T. (2004). Nitric oxide-dependent modulation of the delayed rectifier K+ current and the L-type Ca2+ current by ginsenoside Re, an ingredient of Panax ginseng, in guinea-pig cardiomyocytes. Br. J. Pharmacol. 142, 567–575. doi: 10.1038/sj.bjp.0705814
Berridge, M. J., Bootman, M. D., and Lipp, P. (1998). Calcium—a life and death signal. Nature 395, 645–668. doi: 10.1038/27094
Bertrand, D., Devillers-Thiéry, A., Revah, F., Galzi, J. L., Hussy, N., Mulle, C., et al. (1992). Unconventional pharmacology of a neuronal nicotinic receptor mutated in the channel domain. Proc. Natl. Acad. Sci. U.S.A. 89, 1261–1265. doi: 10.1073/pnas.89.4.1261
Bett, G. C., and Rasmusson, R. L. (2004). Inactivation and recovery in Kv1.4 K+ channels: lipophilic interactions at the intracellular mouth of the pore. J. Physiol. 556, 109–120. doi: 10.1113/jphysiol.2003.055012
Boileau, A. J., Li, T., Benkwitz, C., Czajkowski, C., and Pearce, R. A. (2003). Effects of gamma2S subunit incorporation on GABAA receptor macroscopic kinetics. Neuropharmacology 44, 1003–1012. doi: 10.1016/S0028-3908(03)00114-X
Bowser, D. N., Wagner, D. A., Czajkowski, C., Cromer, B. A., Parker, M. W., Wallace, R. H., et al. (2002). Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation [gamma 2(R43Q)] found in human epilepsy. Proc. Natl. Acad. Sci. U.S.A. 99, 15170–15175. doi: 10.1073/pnas.212320199
Buhr, A., Baur, R., and Sigel, E. (1997). Subtle changes in residue 77 of the gamma subunit of alpha1beta2gamma2 GABAA receptors drastically alter the affinity for ligands of the benzodiazepine binding site. J. Biol. Chem. 272, 11799–117804. doi: 10.1074/jbc.272.18.11799
Cheon, S. Y., Cho, K. J., Lee, J. E., Kim, H. W., Lee, S. K., Kim, H. J., et al. (2013). Cerebroprotective effects of red ginseng extract pretreatment against ischemia-induced oxidative stress and apoptosis. Int. J. Neurosci. 123, 269–277. doi: 10.3109/00207454.2012.758120
Choi, S. E., Choi, S., Lee, J. H., Whiting, P. J., Lee, S. M., and Nah, S. Y. (2003a). Effects of ginsenosides on GABAA receptor channels expressed in Xenopus oocytes. Arch. Pharm. Res. 26, 28–33. doi: 10.1007/BF03179927
Choi, S. H., Lee, J. H., Pyo, M. K., Lee, B. H., Shin, T. J., Hwang, S. H., et al. (2009). Mutations of Leu427, Asn428, and Leu431 residues within transmembrane domain-1-segment 6 attenuate ginsenoside-mediated L-type Ca2+ channel current inhibitions. Biol. Pharm. Bull. 32, 1224–1230. doi: 10.1248/bpb.32.1224
Choi, S. H., Shin, T. J., Hwang, S. H., Lee, B. H., Kang, J., Kim, H. J., et al. (2011b). Ginsenoside Rg3 decelerates hERG K+ channel deactivation through Ser631 residue interaction. Eur. J. Pharmacol. 663, 59–67. doi: 10.1016/j.ejphar.2011.05.006
Choi, S. H., Shin, T. J., Lee, B. H., Chu, D. H., Choe, H., Pyo, M. K., et al. (2010). Ginsenoside Rg3 activates human KCNQ1 K+ channel currents through interacting with the K318 and V319 residues: a role of KCNE1 subunit. Eur. J. Pharmacol. 63, 138–147. doi: 10.1016/j.ejphar.2010.04.001
Choi, S. H., Shin, T. J., Lee, B. H., Hwang, S. H., Lee, S. M., Lee, B. C., et al. (2011a). Ginsenoside Rg3 enhances large conductance Ca2+-activated potassium channel currents: a role of Tyr360 residue. Mol. Cells 31, 133–140. doi: 10.1007/s10059-011-0017-7
Choi, S., Jung, S. Y., Kim, C. H., Kim, H. S., Rhim, H., Kim, S. C., et al. (2001). Effect of ginsenosides on voltage-dependent Ca2+ channel subtypes in bovine chromaffin cells. J. Ethnopharmacol. 74, 75–81. doi: 10.1016/S0378-8741(00)00353-6
Choi, S., Jung, S. Y., Lee, J. H., Sala, F., Criado, M., Mulet, J., et al. (2002). Effects of ginsenosides, active components of ginseng, on nicotinic acetylcholine receptors expressed in Xenopus oocytes. Eur. J. Pharmacol. 442, 37–45. doi: 10.1016/S0014-2999(02)01508-X
Choi, S., Lee, J. H., Oh, S., Rhim, H., Lee, S. M., and Nah, S. Y. (2003b). Effects of ginsenoside Rg2 on the 5-HT3A receptor-mediated ion current in Xenopus oocytes. Mol. Cells 15, 108–113.
Chung, I., and Kim, N. D. (1999). Ginseng saponins enhance maxi Ca2+-activated K+ currents of the rabbit coronary artery smooth muscle cells. J. Ginseng Res. 23, 230–234.
Chung, I., and Lee, J. S. (1999). Ginsenoside Rg3 increases the ATP-sensitive K+ channel activity in the smooth muscle of the rabbit coronary artery. J. Ginseng Res. 23, 235–238.
Claydon, T. W., Makary, S. Y., Dibb, K. M., and Boyett, M. R. (2004). K+ activation of kir3.1/kir3.4 and Kv1.4 K+ channels is regulated by extracellular charges. Biophys. J. 87, 2407–2418. doi: 10.1529/biophysj.103.039073
Duan, Y., and Nicholson, R. A. (2008). 20(S)-protopanaxadiol and the ginsenoside Rh2 inhibit Na+ channel-activated depolarization and Na+ channel-dependent amino acid neurotransmitter release in synaptic fractions isolated from mammalian brain. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 147, 351–356. doi: 10.1016/j.cbpc.2008.01.001
Duan, Y., Zheng, J., Law, V., and Nicholson, R. (2006). Natural products from ginseng inhibit [3H]batrachotoxinin A 20-alpha-benzoate binding to Na+ channels in mammalian brain. Eur. J. Pharmacol. 530, 9–14. doi: 10.1016/j.ejphar.2005.11.022
Furukawa, T., Bai, C. X., Kaihara, A., Ozaki, E., Kawano, T., Nakaya, Y., et al. (2006). Ginsenoside Re, a main phytosterol of Panax ginseng, activates cardiac potassium channels via a nongenomic pathway of sex hormones. Mol. Pharmacol. 70, 1916–1924. doi: 10.1124/mol.106.028134
Gotti, C., Clementi, F., Fornari, A., Gaimarri, A., Guiducci, S., Manfredi, I., et al. (2009). Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 78, 703–711. doi: 10.1016/j.bcp.2009.05.024
Gu, B., Nakamichi, N., Zhang, W. S., Nakamura, Y., Kambe, Y., Fukumori, R., et al. (2009). Possible protection by notoginsenoside R1 against glutamate neurotoxicity mediated by N-methyl-D-aspartate receptors composed of an NR1/NR2B subunit assembly. J. Neurosci. Res. 87, 2145–2156. doi: 10.1002/jnr.22021
Heginbotham, L., Lu, Z., Abramson, T., and Mackinnon, R. (1994). Mutations in the K+ channel signature sequence. Biophys. J. 66, 1061–1067. doi: 10.1016/S0006-3495(94)80887-2
Jeong, S. M., Lee, J. H., Kim, J. H., Lee, B. H., Yoon, I. S., Lee, J. H., et al. (2004). Stereospecificity of ginsenoside Rg3 action on ion channels. Mol. Cells 18, 383–389.
Judge, S. I., Monteiro, M. J., Yeh, J. Z., and Bever, C. T. (1999). Inactivation gating and 4-AP sensitivity in human brain Kv1.4 potassium channel. Brain Res. 831, 43–54. doi: 10.1016/S0006-8993(99)01391-8
Kang, D. I., Lee, J. Y., Yang, J. Y., Jeong, S. M., Lee, J. H., Nah, S. Y., et al. (2005). Evidence that the tertiary structure of 20(S)-ginsenoside Rg3 with tight hydrophobic packing near the chiral center is important for Na+ channel regulation. Biochem. Biophys. Res. Commun. 333, 1194–1201. doi: 10.1016/j.bbrc.2005.06.026
Kim, H. S., Hwang, S. L., Nah, S. Y., and Oh, S. (2001). Changes of [3H]MK-801, [3H]muscimol and [3H]flunitrazepam binding in rat brain by the prolonged ventricular infusion of ginsenoside Rc and Rg1. Pharmacol. Res. 43, 473–479. doi: 10.1006/phrs.2001.0809
Kim, H. S., Lee, J. H., Goo, Y. S., and Nah, S. Y. (1998a). Effects of ginsenosides on Ca2+ channels and membrane capacitance in rat adrenal chromaffin cells. Brain Res. Bull. 46, 245–251. doi: 10.1016/S0361-9230(98)00014-8
Kim, J. H., Cho, S. Y., Lee, J. H., Jeong, S. M., Yoon, I. S., Lee, B. H., et al. (2007). Neuroprotective effects of ginsenoside Rg3 against homocysteine-induced excitotoxicity in rat hippocampus. Brain Res. 1136, 190–199. doi: 10.1016/j.brainres.2006.12.047
Kim, J. H., Kim, S., Yoon, I. S., Lee, J. H., Jang, B. J., Jeong, S. M., et al. (2005c). Protective effects of ginseng saponins on 3-nitropropionic acid-induced striatal degeneration in rats. Neuropharmacology 48, 743–756. doi: 10.1016/j.neuropharm.2004.12.013
Kim, J. H., Lee, J. H., Jeong, S. M., Lee, B. H., Yoon, I. S., Lee, J. H., et al. (2005a). Effect of ginseng saponins on a rat visceral hypersensitivity model. Biol. Pharm. Bull. 11, 2120–2124. doi: 10.1248/bpb.28.2120
Kim, J. H., Yoon, I. S., Lee, B. H., Choi, S. H., Lee, J. H., Lee, J. H., et al. (2005b). Effects of Korean red ginseng extract on cisplatin-induced nausea and vomiting. Arch. Pharm. Res. 6, 680–684. doi: 10.1007/BF02969358
Kim, N. D., Kang, S. Y., Park, J. H., and Schini-Kerth, V. B. (1999). Ginsenoside Rg3 mediates endothelium-dependent relaxation in response to ginsenosides in rat aorta: role of K+ channels. Eur. J. Pharmacol. 367, 41–49. doi: 10.1016/S0014-2999(98)00898-X
Kim, S., Ahn, K., Oh, T. H., Nah, S. Y., and Rhim, H. (2002). Inhibitory effect of ginsenosides on NMDA receptor-mediated signals in rat hippocampal neurons. Biochem. Biophys. Res. Commun. 296, 247–254. doi: 10.1016/S0006-291X(02)00870-7
Kim, S., Kim, T., Ahn, K., Park, W. K., Nah, S. Y., and Rhim, H. (2004b). Ginsenoside Rg3 antagonizes NMDA receptors through a glycine modulatory site in rat cultured hippocampal neurons. Biochem. Biophys. Res. Commun. 323, 416–424. doi: 10.1016/j.bbrc.2004.08.106
Kim, S., Nah, S. Y., and Rhim, H. (2008b). Neuroprotective effects of ginseng saponins against L-type Ca2+ channel-mediated cell death in rat cortical neurons. Biochem. Biophys. Res. Commun. 365, 399–405. doi: 10.1016/j.bbrc.2007.10.048
Kim, S. O., Kim, M. K., Lee, H. S., Park, J. K., and Park, K. (2008a). The effect of Korean red ginseng extract on the relaxation response in isolated rabbit vaginal tissue and its mechanism. J. Sex. Med. 5, 2079–2084. doi: 10.1111/j.1743-6109.2008.00946.x
Kim, S., and Rhim, H. (2004). Ginsenosides inhibit NMDA receptor-mediated epileptic discharges in cultured hippocampal neurons. Arch. Pharm. Res. 27, 524–530. doi: 10.1007/BF02980126
Kim, T. W., Choi, H. J., Kim, N. J., and Kim, D. H. (2009). Anxiolytic-like effects of ginsenoside Rg3 and Rh2 from red ginseng in the elevated plus-maze model. Planta Med. 75, 836–839. doi: 10.1055/s-0029-1185402
Kim, Y. C., Kim, S. R., Markelonis, G. J., and Oh, T. H. (1998b). Ginsenosides Rb1 and Rg3 protect cultured rat cortical cells from glutamate-induced neurodegeranation. J. Neurosci. Res. 53, 426–432.
Kimura, T., Saunders, P. A., Kim, H. S., Rheu, H. M., Oh, K. W., and Ho, I. K. (1994). Interactions of ginsenosides with ligand bindings of GABAA and GABAB receptors. Gen. Pharmacol. 25, 193–199. doi: 10.1016/0306-3623(94)90032-9
Lee, B. H., Choi, S. H., Hwang, S. H., Kim, H. J., Lee, S. M., Kim, H. C., et al. (2013a). Effects of ginsenoside Rg3 on α9α10 nicotinic acetylcholine receptor-mediated ion currents. Biol. Pharm. Bull. 36, 812–818. doi: 10.1248/bpb.b12-01009
Lee, B. H., Choi, S. H., Pyo, M. K., Shin, T. J., Hwang, S. H., Kim, B. R., et al. (2009). A role for Leu247 residue within transmembrane domain 2 in ginsenoside-mediated alpha7 nicotinic acetylcholine receptor regulation. Mol. Cells 27, 591–599. doi: 10.1007/s10059-009-0073-4
Lee, B. H., Jeong, S. M., Ha, T. S., Park, C. S., Lee, J. H., Kim, J. H., et al. (2004b). Ginsenosides regulate ligand-gated ion channels from the outside. Mol. Cells 18, 115–121.
Lee, B. H., Jeong, S. M., Lee, J. H., Kim, D. H., Kim, J. H., Kim, J. I., et al. (2004a). Differential effect of ginsenoside metabolites on the 5-HT3A receptor-mediated ion current in Xenopus oocytes. Mol. Cells 17, 51–56.
Lee, B. H., Kim, H. J., Chung, L., and Nah, S. Y. (2013b). Ginsenoside Rg3regulates GABAA receptor channel activity: involvement of interaction with the γ2subunit. Eur. J. Pharmacol. 705, 119–125. doi: 10.1016/j.ejphar.2013.02.040
Lee, B. H., Lee, J. H., Lee, S. M., Jeong, S. M., Yoon, I. S., Lee, J. H., et al. (2007). Identification of ginsenoside interaction sites in 5-HT3A receptors. Neuropharmacology 2, 1139–1150. doi: 10.1016/j.neuropharm.2006.12.001
Lee, E., Kim, S., Chung, K. C., Choo, M. K., Kim, D. H., Nam, G., et al. (2006a). 20(S)-ginsenoside Rh2, a newly identified active ingredient of ginseng, inhibits NMDA receptors in cultured rat hippocampal neurons. Eur. J. Pharmacol. 536, 69–77. doi: 10.1016/j.ejphar.2006.02.038
Lee, J. H., Choi, S. H., Lee, B. H., Hwang, S. H., Kim, H. J., Rhee, J., et al. (2013c). Activation of lysophosphatidic acid receptor by gintonin inhibits Kv1.2 channel activity: involvement of tyrosine kinase and receptor protein tyrosine phosphatase α. Neurosci. Lett. 548, 143–148. doi: 10.1016/j.neulet.2013.05.048
Lee, J. H., Choi, S. H., Lee, B. H., Yoon, I. S., Shin, T. J., Pyo, M. K., et al. (2008c). Modifications of aliphatic side chain of 20(S)-ginsenoside Rg3 cause an enhancement or loss of brain Na+ channel current inhibitions. Biol. Pharm. Bull. 31, 480–486. doi: 10.1248/bpb.31.480
Lee, J. H., Jeong, S. M., Kim, J. H., Lee, B. H., Yoon, I. S., Lee, J. H., et al. (2005). Characteristics of ginsenoside Rg3-mediated brain Na+ current inhibition. Mol. Pharmacol. 68, 1114–1126. doi: 10.1124/mol.105.015115
Lee, J. H., Jeong, S. M., Kim, J. H., Lee, B. H., Yoon, I. S., Lee, J. H., et al. (2006b). Effects of ginsenosides and their metabolites on voltage-dependent Ca2+ channel subtypes. Mol. Cells 21, 52–62.
Lee, J. H., Kim, S. H., Kim, D., Hong, H. N., and Nah, S. Y. (2002). Protective effect of ginsenosides, active ingredients of Panax ginseng, on kainate-induced neurotoxicity in rat hippocampus. Neurosci. Lett. 325, 129–133. doi: 10.1016/S0304-3940(02)00256-2
Lee, J. H., Lee, B. H., Choi, S. H., Yoon, I. S., Pyo, M. K., Shin, T. J., et al. (2008a). Ginsenoside Rg3 inhibits human Kv1.4 channel currents by interacting with the Lys531 residue. Mol. Pharmacol. 73, 619–626. doi: 10.1124/mol.107.040360
Lee, J. H., Lee, B. H., Choi, S. H., Yoon, I. S., Shin, T. J., Pyo, M. K., et al. (2008b). Involvement of batrachotoxin binding sites in ginsenoside-mediated voltage-gated Na+ channel regulation. Brain Res. 1203, 61–67. doi: 10.1016/j.brainres.2008.01.078
Lester, H. A., Dibas, M. I., Dahan, D. S., Leite, J. F., and Dougherty, D. A. (2004). Cys-loop receptors: new twists and turns. Trends Neurosci. 27, 329–236. doi: 10.1016/j.tins.2004.04.002
Li, Z., Chen, X., Niwa, Y., Sakamoto, S., and Nakaya, Y. (2001). Involvement of Ca2+-activated K+ channels in ginsenosides-induced aortic relaxation in rats. J. Cardiovasc. Pharmacol. 37, 41–47. doi: 10.1097/00005344-200101000-00005
Lian, X. Y., Zhang, Z., and Stringer, J. L. (2005). Protective effects of ginseng components in a rodent model of neurodegeneration. Ann. Neurol. 57, 642–648. doi: 10.1002/ana.20450
Lin, Z. Y., Chen, L. M., Zhang, J., Pan, X. D., Zhu, Y. G., Ye, Q. Y., et al. (2012). Ginsenoside Rb1 selectively inhibits the activity of L-type voltage-gated calcium channels in cultured rat hippocampal neurons. Acta Pharmacol. Sin. 33, 438–444. doi: 10.1038/aps.2011.181
Liu, D., Li, B., Liu, Y., Attele, A. S., Kyle, J. W., and Yuan, C. S. (2001). Voltage-dependent inhibition of brain Na+ channels by American ginseng. Eur. J. Pharmacol. 413, 47–54. doi: 10.1016/S0014-2999(01)00735-X
Miyazawa, A., Fujiyoshi, Y., and Unwin, N. (2003). Structure and gating mechanism of the acetylcholine receptor pore. Nature 423, 949–955. doi: 10.1038/nature01748
Nah, S. Y., Kim, D. H., and Rhim, H. (2007). Ginsenosides: are any of them candidates for drugs acting on the central nervous system? CNS Drug Rev. 13, 381–404. doi: 10.1111/j.1527-3458.2007.00023.x
Nah, S. Y., Park, H. J., and McCleskey, E. W. (1995). A trace component of ginseng that inhibit Ca2+ channels through a pertussis toxin-sensitive G protein. Proc. Natl. Acad. Sci. U.S.A. 92, 8739–8743. doi: 10.1073/pnas.92.19.8739
Noh, J. H., Choi, S., Lee, J. H., Betz, H., Kim, J. I., Park, C. S., et al. (2003). Effects of ginsenosides on glycine receptor alpha1 channels expressed in Xenopus oocytes. Mol. Cells 15, 34–39.
Palma, E., Mileo, A. M., Eusebi, F., and Miledi, R. (2006). Threonine-for-leucine mutation within domain M2 of the neuronal alpha(7) nicotinic receptor converts 5-hydroxytryptamine from antagonist to agonist. Proc. Natl. Acad. Sci. U.S.A. 93, 11231–11235. doi: 10.1073/pnas.93.20.11231
Pang, Z., and Geddes, J. W. (1997). Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J. Neurosci. 17, 3064–3073.
Pardo, L. A., Heinemann, S. H., Terlau, H., Ludewig, U., Lorra, C., Pongs, O., et al. (1992). Extracellular K+ specifically modulates a rat brain K+ channel. Proc. Natl. Acad. Sci. U.S.A. 89, 2466–2470. doi: 10.1073/pnas.89.6.2466
Park, E. K., Choo, M. K., Oh, J. K., Ryu, J. H., and Kim, D. H. (2004). Ginsenoside Rh2 reduces ischemic brain injury in rats. Biol. Pharm. Bull. 27, 433–436. doi: 10.1248/bpb.27.433
Revah, F., Bertrand, D., Galzi, J. L., Devillers-Thiéry, A., Mulle, C., Hussy, N., et al. (1991). Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 353, 846–849. doi: 10.1038/353846a0
Rhim, H., Kim, H., Lee, D. Y., Oh, T. H., and Nah, S. Y. (2002). Ginseng and ginsenoside Rg3, a newly identified active ingredient of ginseng, modulate Ca2+ channel currents in rat sensory neurons. Eur. J. Pharmacol. 436, 151–158. doi: 10.1016/S0014-2999(01)01613-2
Sala, F., Mulet, J., Choi, S., Jung, S. Y., Nah, S. Y., Rhim, H., et al. (2002). Effects of ginsenoside Rg2 on human neuronal nicotinic acetylcholine receptors. J. Pharm. Exp. Ther. 301, 1052–1059. doi: 10.1124/jpet.301.3.1052
Seong, Y. H., Shin, C. S., Kim, H. S., and Baba, A. (1995). Inhibitory effect of ginseng total saponins on glutamate-induced swelling of cultured atrocytes. Biol. Pharm. Bull. 18, 1776–1778. doi: 10.1248/bpb.18.1776
Shin, T. J., Hwang, S. H., Choi, S. H., Lee, B. H., Kang, J., Kim, H. J., et al. (2012). Effects of protopanaxatriol-ginsenoside metabolites on rat N-methyl-d-aspartic Acid receptor-mediated ion currents. Korean J. Physiol. Pharmacol. 16, 113–118. doi: 10.4196/kjpp.2012.16.2.113
Shin, Y. H., Jung, O. M., Nah, J. J., Nam, K. Y., Kim, C. Y., and Nah, S. Y. (1999). Ginsenosides that produce differential antinociception in mice. Gen. Pharmacol. 32, 653–659. doi: 10.1016/S0306-3623(98)00239-0
Tachikawa, E., Kudo, K., Nunokawa, M., Kashimoto, T., Takahashi, E., and Kitagawa, S. (2001). Characterization of ginseng saponin ginsenoside-Rg3 inhibition of catecholamine secretion in bovine adrenal chromaffin cells. Biochem. Pharmacol. 62, 943–951. doi: 10.1016/S0006-2952(01)00743-2
Tian, J., Fu, F., Geng, M., Jiang, Y., Yang, J., Jiang, W., et al. (2005). Neuroprotective effect of 20(S)-ginsenoside Rg3 on cerebral ischemia in rats. Neurosci. Lett. 374, 92–97. doi: 10.1016/j.neulet.2004.10.030
Wang, S. Y., and Wang, G. K. (1998). Point mutations in segment I-S6 render voltage-gated Na+ channels resistant to batrachotoxin. Proc. Natl. Acad. Sci. U.S.A. 95, 2653–2658. doi: 10.1073/pnas.95.5.2653
Watanabe, I., Zhu, J., Recio-Pinto, E., and Thornhill, W. B. (2004). Glycosylation affects the protein stability and cell surface expression of Kv1.4 but Not Kv1.1 potassium channels. A pore region determinant dictates the effect of glycosylation on trafficking. J. Biol. Chem. 279, 8879–8885. doi: 10.1074/jbc.M309802200
Wojda, U., Salinska, E., and Kuznicki, J. (2008). Calcium ions in neuronal degeneration. IUBMB Life 60, 575–90. doi: 10.1002/iub.91
Yoon, S. R., Nah, J. J., Shin, Y. H., Kim, S. K., Nam, K. Y., Choi, H. S., et al. (1998). Ginsenosides induce differential antinocicepion and inhibit substance P induced-nociceptive response in mice. Life Sci. 62, PL319–PL325. doi: 10.1016/S0024-3205(98)00168-4
Zhang, Y. F., Fan, X. J., Li, X., Peng, L. L., Wang, G. H., Ke, K. F., et al. (2008). Ginsenoside Rg1 protects neurons from hypoxic-ischemic injury possibly by inhibiting Ca2+ influx through NMDA receptors and L-type voltage-dependent Ca2+ channels. Eur. J. Pharmacol. 586, 90–99. doi: 10.1016/j.ejphar.2007.12.037
Keywords: ginseng, ginsenosides, ion channels and receptors, interaction site(s), nervous system
Citation: Nah S-Y (2014) Ginseng ginsenoside pharmacology in the nervous system: involvement in the regulation of ion channels and receptors. Front. Physiol. 5:98. doi: 10.3389/fphys.2014.00098
Received: 16 November 2013; Accepted: 27 February 2014;
Published online: 19 March 2014.
Edited by:
Sebastian Brauchi, Universidad Austral de Chile, ChileReviewed by:
Richard Barrett-Jolley, University of Liverpool, UKLuis A. Pardo, Max-Planck-Institute of Experimental Medicine, Germany
Harley Takatsuna Kurata, University of British Columbia, Canada
Copyright © 2014 Nah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seung-Yeol Nah, Ginsentology Research Laboratory, Department of Physiology, College of Veterinary Medicine and Bio/Molecular Informatics Center, Konkuk University, 120 Neungdong-ro, Seoul 143-701, South Korea e-mail:synah@konkuk.ac.kr