 Vernon A. Ruffin
Vernon A. Ruffin- Department of Physiology and Biophysics, Case Western Reserve University, OH, USA
Intracellular pH (pHi) regulation in the brain is important in both physiological and physiopathological conditions because changes in pHi generally result in altered neuronal excitability. In this review, we will cover 4 major areas: (1) The effect of pHi on cellular processes in the brain, including channel activity and neuronal excitability. (2) pHi homeostasis and how it is determined by the balance between rates of acid loading (JL) and extrusion (JE). The balance between JE and JL determine steady-state pHi, as well as the ability of the cell to defend pHi in the face of extracellular acid-base disturbances (e.g., metabolic acidosis). (3) The properties and importance of members of the SLC4 and SLC9 families of acid-base transporters expressed in the brain that contribute to JL (namely the Cl-HCO3 exchanger AE3) and JE (the Na-H exchangers NHE1, NHE3, and NHE5 as well as the Na+- coupled HCO3− transporters NBCe1, NBCn1, NDCBE, and NBCn2). (4) The effect of acid-base disturbances on neuronal function and the roles of acid-base transporters in defending neuronal pHi under physiopathologic conditions.
pH and Neuronal Excitability
The excitability of neurons is especially sensitive to changes in intracellular pH (pHi) and extracellular pH (pHo) due to the pH-sensitivity of intracellular and extracellular moieties on membrane proteins such as channels (Tombaugh and Somjen, 1996; Duprat et al., 1997; Waldmann et al., 1997; Ruffin et al., 2008), transporters (Irwin et al., 1994; Park et al., 2010; Adijanto and Philp, 2012), receptors (Giffard et al., 1990; Tang et al., 1990; Traynelis and Cull-Candy, 1990; McDonald et al., 1998), and ATPase pumps (Pick and Karlish, 1982; Wolosker et al., 1997). Together these proteins (1) govern the resting membrane potential of neurons, (2) affect neuronal responsiveness to agonists and antagonists, (3) set the threshold for firing an action potential, (4) influence the duration/amplitude of the action potential, (5) determine the length of the refractory period, and (6) synchronize neuronal network activity. These properties endow neurons with the ability to communicate with other neurons and glial cells within the nervous system (for functions such as learning, behavior, conscious thought, and unconscious homeostatic regulation), and with cells outside the nervous system (for functions such as motor control and endocrine regulation). Table 1 list examples of pH sensitive proteins and activities involved in setting neuronal excitability. The relationship between pH and neuronal excitability has been extensively reviewed by others (Balestrino and Somjen, 1988; Church, 1992; Tombaugh and Somjen, 1996; Dean et al., 2001; Makani and Chesler, 2007; Pavlov et al., 2013). Note that the link between pH and neuronal excitability is not a simple one: some neurons (e.g., chemosensitive neurons) exhibit enhanced excitability in response to an acid-load, whereas others (e.g., hippocampal neurons) may exhibit reduced excitability. The direction of the response presumably depends on the pH-responsiveness of the individual channels, transporters, and receptors that are responsible for dictating overall excitability in each neuron.
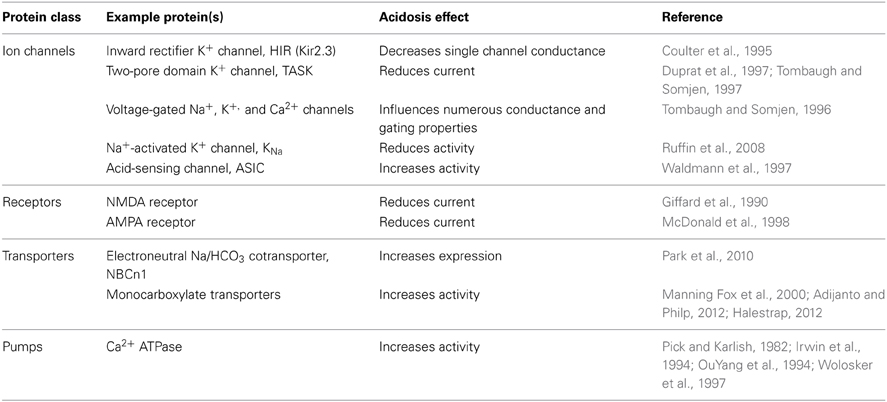
Table 1. Examples of pH-sensitive membrane proteins expressed in neurons.
Glial cells are not excitable cells and experience only small changes in membrane potential compared to neurons. Although glial cells are not the focus of this review, it is important to address their critical role in optimal brain function. Traditionally, glial cells have been called neural supportive cells because they produce growth factors and recycle neurotransmitters (astrocytes), assist in rapid electrical transmission (oligodendrocytes), and scavenge compromised cells in addition to cellular debris (microglia). Glia, together with choroid plexus epithelia (Schmitt et al., 2000; Damkier et al., 2010a; Christensen et al., 2013), control the composition—including the pH—of the extracellular cerebrospinal fluid (CSF) that bathes neurons (Chesler and Kraig, 1989; Deitmer and Rose, 1996; Bevensee et al., 1997; Brookes, 1997; Chesler, 2003; Ro and Carson, 2004; Ekdahl et al., 2009).
Models of seizure indicate that the acidification that follows intense firing (Chesler and Kaila, 1992; Jacobs et al., 2008), a phenomenon that likely forms part of a mechanism that prevents excessive firing by dampening neuronal excitability (Hormuzdi et al., 2004), is a major challenge to neuronal pH. Intriguingly, the main clinical presentations of several neurodegenerative disease states include signs of decreased brain pH. Examples include Alzheimer's disease (Demetrius and Simon, 2012), Parkinson's disease (Mattson et al., 1999), and multiple sclerosis (Vergo et al., 2011). Even disease states originating outside of the brain (e.g., metabolic acidosis) can affect the pH of the brain. Given the link between pH and neuronal function, it is probable that such alterations in pH compromise brain function, contributing to the neurological symptoms of these diseases.
The regulation of cytosolic pH in most cells, including neurons, is an active process, since H+ ions are not passively distributed across the cell membrane (Roos and Boron, 1981). In this brief review, we will provide an overview of the nature, function, and importance of the major acid-loading and acid-extruding proteins that contribute to neuronal pH homeostasis. We will also consider the pathologies that are associated with defective neuronal acid-base homeostasis and how the homeostatic systems respond in the face of pathological acid-base disturbances.
Neuronal pH Homeostasis
Pioneering work in invertebrate models first identified the importance of neuronal pH regulation (Boron and De Weer, 1976a,b; Thomas, 1976). In vertebrates, extensive research has been performed on pH regulation in hippocampal neurons. The typical resting or “steady-state” pHi of a hippocampal neuron in CO2/HCO3−-containing media is ~7.03–7.46. depending on the preparation, whereas the pHo is ~7.35 (Raley-Susman et al., 1991, 1993; Schwiening and Boron, 1994; Baxter and Church, 1996; Bevensee et al., 1996; Church et al., 1998; Smith et al., 1998; Vincent et al., 1999). Steady-state pHi is dependent on the balance between the rate of acid loading (JL, i.e., rate of acid influx/generation or alkali efflux/consumption) and the rate of acid extrusion (JE, i.e., rate of acid efflux/consumption or alkali influx/generation). Steady state pHi is achieved when JE = JL (intersection of red and blue lines in Figure 1A). It is important to note that, at steady-state, the opposing acid-loading and acid-extruding processes are not stopped but are proceeding at equal rates, thus their combined action results in no pH change.
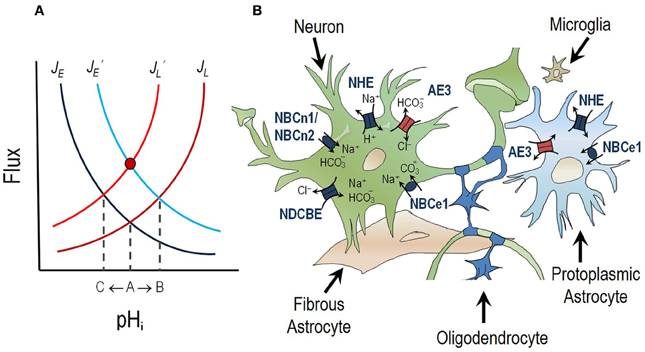
Figure 1. pH regulation in the central nervous system. (A) Steady-state pHi is dependent on the balance between the rate of acid extrusion (JE) and the rate of acid loading (JL). Steady-state pHi is achieved when JE = JL (intersection of dark-blue and dark-red lines: point A). If JE increases (J′E) the steady-state pHi will shift to a more alkaline value (intersection of light-blue and dark-red lines: point B). If JL increases (J′L) the steady-state pH will shift to a more acidic value (intersection of dark-blue and light-red lines: point C). If the rise in JE is matched by an equal increase in JL (J′E = J′L) there will be no net change in pHi (red circle). This is known as a compensatory response. (B) Neurons, astrocytes and oligodendrocytes express two classes of acid-base transporting proteins; acid loaders (red) and acid extruders (dark blue).
Acid loading in neurons—a process that tends to lower pHi—predominantly results from the accumulation of metabolically generated H+ (such as that produced by aerobic or anaerobic metabolism during neuronal firing: Chesler, 2003) and the extrusion of HCO3− from cells via a Cl-HCO3 (anion) exchanger of the SLC4 solute carrier family (see section titled The Chloride-Bicarbonate Exchanger AE3 and Figure 1B). Acid-loading processes tend to restore steady-state pHi after an alkali load.
Acid extrusion—a process that tends to raise pHi—in neurons is typically achieved by the action of the SLC4 and SLC9 families of Na+-coupled transport proteins. Both transporter families that take advantage of the inwardly directed Na+ gradient established by the Na+, K+ ATPase to either extrude H+ from the cell (in the case of Na-H exchangers (NHEs), see section titled Sodium-Hydrogen Exchangers NHE1, NHE3, and NHE5 and Figure 1B) or to accumulate a weak base, such as HCO3− (in the case of Na/HCO3 cotransporters, see section titled Sodium-coupled Bicarbonate Transporters and Figure 1B). Acid-extruding processes tend to restore steady-state pHi after an acid load, such as that which might result from intense neuronal activity (Chesler and Kraig, 1989; Kaila et al., 1991).
Of course, transmembrane acid-loading processes tend to alkalinize the cell surface (raise pHs) and transmembrane acid-extruding processes tend to acidify the cell surface (lower pHs), thereby also potentially impacting the function of membrane proteins in the brain with extracellular pH-sensitive moieties. In practice, extracellular membrane-associated carbonic anhydrases catalyze the interconversion of H+ + HCO3− ↔ CO2 + H2O at the cell surface, which would tend to promote dissipation of pH gradients. A consideration of the CO2/HCO3− buffering system and whole-body pH homeostasis is provided elsewhere (Giebisch and Windhager, 2009; Boron, 2012; Bevensee and Boron, 2013).
Neuronal Acid-Base Transporters
Acid Loaders
The chloride-bicarbonate exchanger AE3
Molecular identity. The main acid loader that is predicted to contribute to JL in neurons is the Cl-HCO3 exchanger AE3 (Anion Exchanger 3, encoded by the SLC4A3 gene). AE3 was the third member of the 10 members of the SLC4 solute carrier family to be cloned and characterized and, like its close relatives AE1 and AE2 (Alper, 2009), mediates the stilbene-sensitive, electroneutral exchange of one Cl− for one HCO3− (Sterling and Casey, 1999). As we will see later, not all SLC4 members are acid loaders: indeed the majority are acid extruders (see Na+-coupled HCO3− transporters, SLC4A4-8 in Section titled Sodium-coupled Bicarbonate Transporters). In mammals SLC4A3 encodes two alternative gene products: bAE3 (abundant in the brain, often referred to as AE3fl, full-length) and cAE3 (abundant in cardiac tissue). bAE3 is expressed from a different promoter than cAE3 and includes a longer and different Nt appendage. Artificial truncation of the bAE3-specific sequence appears to confer a lesser functional expression on the transporter, consistent with the hypothesis that this sequence is autostimulatory. The appendage has also been reported to include two SH3 domains and a PKC site indicating a possible role for modulation by extrinsic signals (Sterling and Casey, 1999; Alvarez et al., 2001).
Distribution. bAE3 transcripts and protein are expressed throughout the central nervous system. In mice, bAE3 transcripts are especially abundant in pyramidal neurons of the hippocampal (HC) formation (Kopito et al., 1989; Hentschke et al., 2006) although western blotting of brain regions shows AE3 protein to be similarly abundant in the cerebral cortex (CX), cerebellum (CB), and brainstem-diencephalon (BD): (Xue et al., 2003). Western blotting of fractionated cells suggests that AE3 protein expression in the brain is mainly in neurons rather than in astrocytes (Hentschke et al., 2006). However, in the retina of rats, bAE3 is located in the basal end feet of Müller cells (glia) and it is cAE3 that is expressed in horizontal cells (neurons): (Kobayashi et al., 1994; Alvarez et al., 2007).
Influence on neuronal pHi. Cl-HCO3 exchange is a feature of adult neurons and AE3 is the sole Cl-HCO3 exchanger in neurons as evidenced by the absence of Cl-HCO3 exchange activity in AE3-null mice (Hentschke et al., 2006). Interestingly, despite evidence for the presence of AE3 transcripts in embryonic mouse and rat brain, (Hentschke et al., 2006) neurons from fetal mice exhibit no substantial Cl-HCO3 exchange activity as if AE3 protein is absent or otherwise indisposed (Raley-Susman et al., 1993; Vilas et al., 2009). However, as mentioned below, AE3-like activity is evident as a damping mechanism when acid-extruders are in robust operation. Due to the prevailing ion gradients and probably also due to the relative substrate affinities of the intracellular and extracellular ion translocation sites, AE3 typically extrudes HCO3− in exchange for extracellular Cl−, thereby tending to lower pHi (raise pHs) and raise [Cl−]i. Evidence for the role of AE3 as an acid loader is provided by the observations that (1) COS cells expressing AE3 exhibit a markedly lower pHi than control cells that do not express AE3 (Kopito et al., 1989) and (2) hippocampal neurons from embryonic mice exhibit an enhanced rate of acid-extrusion in the presence of DIDS or in the absence of extracellular Cl−: both maneuvers that would block AE3 action (Svichar et al., 2009). Furthermore, HC neurons from adult Ae3-null mice exhibit a slightly higher pHi than wild-type neurons, although the authors of the study note that the pH difference did not achieve statistical significance in their study (Hentschke et al., 2006). A Cl-HCO3 exchanger, likely AE3, also contributes to JL in non-chemosensitive and some chemosensitive neurons of the medulla oblongata (Ritucci et al., 1998; Meier et al., 2007).
Importance for neuronal function. Mice lacking acid-extruders of the SLC4 family tend to exhibit evidence of reduced neuronal excitability (see section titled Sodium-coupled Bicarbonate Transporters), thus it seems fitting that a missense mutation in AE3, an acid-loader, is associated with idiopathic generalized epilepsy (Sander et al., 2002). Subsequent work has shown that the mutant AE3 is functionally defective in a heterologous system (Vilas et al., 2009). Moreover, a strain of Ae3-null mouse exhibits lower seizure threshold in response to proconvulsants and a greater seizure-mortality consistent with enhanced neuronal excitability (Hentschke et al., 2006). Ae3-null mice exhibit a reduced respiratory rate consistent with a contribution to the resting pH in chemosensitive neurons that, unlike other neurons, are stimulated by lowered pHi (Meier et al., 2007). Finally, in the mouse retina, the importance of AE3 for maintaining appropriate electrical excitability is indicated by the association of AE3-deficiency with blindness (Alvarez et al., 2007). However, it is not clear if any of these indicators of altered neuronal excitability are entirely due to defective pH regulation, or if they are related to altered Cl− accumulation, another factor known to influence neuronal excitability (Irie et al., 1998; Kahle et al., 2005).
Acid Extruders
In neurons and astrocytes, the main acid extruders contributing to JE are the NHEs in the SLC9 family of solute carriers (recently reviewed by Donowitz et al., 2013) and the Na+-coupled HCO3− transporters (NCBTs) in the SLC4 family (recently reviewed by Parker and Boron, 2013).
Sodium-hydrogen exchangers NHE1, NHE3, and NHE5
Molecular identity. The main HCO3-independent acid loaders that are predicted to contribute to JE in neurons and astrocytes are the NHEs (Orlowski and Grinstein, 2004; Donowitz et al., 2013). NHE1 (neurons and astrocytes), NHE3 (chemosensitive neurons), and NHE5 (neurons) are encoded by the SLC9A1, SLC9A3, and SLC9A5 genes, respectively. Three of five members of the SLC9 family mediate the electroneutral exchange of one Na+ for one H+ across the plasma membrane. The other four other members, NHE6-NHE9, are intracellular K-H exchangers. The general topology and relatedness of family members are show in Figures 2A,B. SLC9 family members have a short Nt and an extensive Ct that plays a regulatory role (Orlowski and Grinstein, 2004; Donowitz et al., 2013). NHE1 is far more sensitive to amiloride derivatives than either NHE3 or NHE5 (Counillon et al., 1993; Orlowski, 1993).
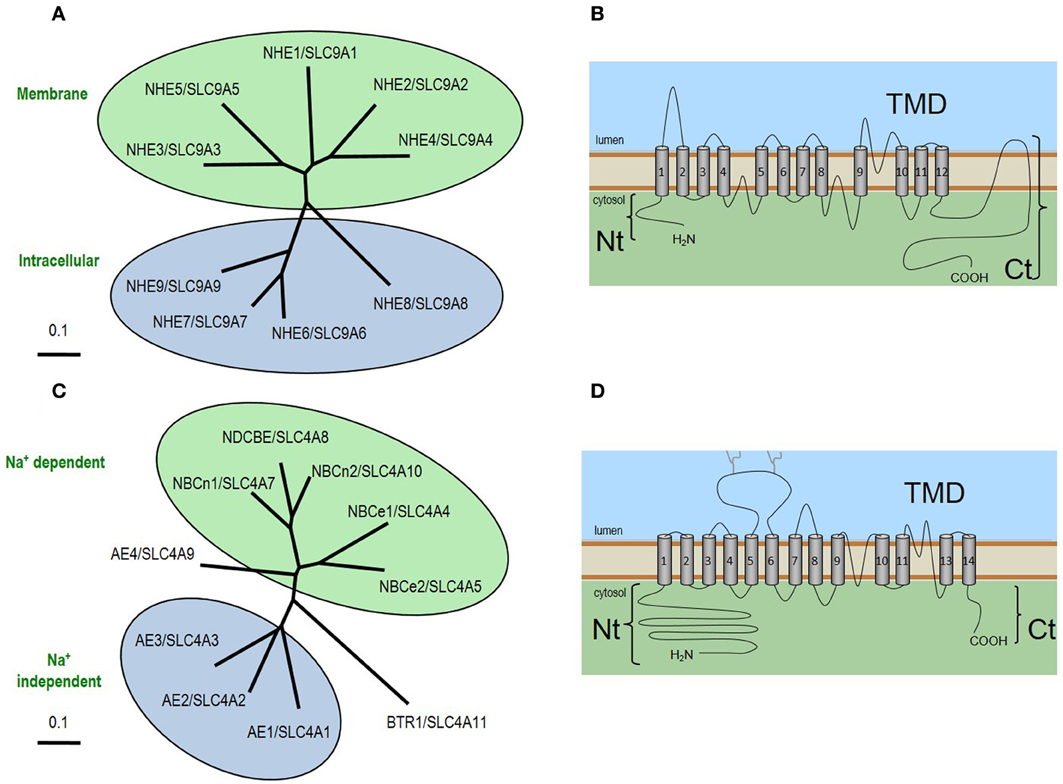
Figure 2. Neuronal acid-base transporters of the SLC9 and SLC4 families. (A) The relatedness of SLC9 family proteins. (B) The general topology of SLC9 proteins. The human SLC9 gene family of solute carriers consists of 9 members which encode proteins containing a ~450 aa N-terminus composed of 12 membrane spans that form the transmembrane domain (TMD) where the exchange of extracellular Na+ and intracellular H+ occurs, and a C-terminus of varying length (~125–440 aa) depending on isoform, that is involved in the regulation of exchange activity. (C) The relatedness of SLC4 family proteins. (D) The general topology of SLC4 proteins. The extended N-terminus and C-terminus are linked via a TMD that includes 14 transmembrane spans, one of which (transmembrane span 12) is believed to be an extended non-helical region. Each NCBT gene encodes multiple products that differ from each other in their extreme Nt or Ct sequence (Boron et al., 2009).
Distribution. NHE1 exhibits the broadest distribution of all the NHE isoforms throughout the body and has been identified in multiple brain regions (Ma and Haddad, 1997; Kanaan et al., 2007), both in neurons (e.g., cultured mouse HC and neocortical neurons: Sin et al., 2009; Diering et al., 2011) and astrocytes (e.g., cultured rat HC astrocytes: Pizzonia et al., 1996). NHE3 exhibits robust expression in cerebellar Purkinje cells (Ma and Haddad, 1997) and also in chemosensitive, ventrolateral neurons in the brainstem/medulla oblongata (Wiemann et al., 1999, 2005; Kiwull-Schöne et al., 2001, 2007). NHE5 expression is predominantly detected in the brain (Klanke et al., 1995; Attaphitaya et al., 1999) with robust expression in multiple brain regions (Attaphitaya et al., 1999; Baird et al., 1999). At the subcellular level, NHE5 protein has been detected in the synapses of HC pyramidal neurons of mice in both a subset of inhibitory and excitable synapses (Diering et al., 2011).
Influence on neuronal pHi. Due to the prevailing ion gradients, NHEs typically extrude H+ in exchange for extracellular Na+ (taking advantage of the inwardly directed gradient for Na+), thereby tending to raise pHi (e.g., while restoring steady-state pHi after an acid load) and lower pHs. Evidence for the role of NHE1 as an acid extruder is provided by the observation that CA1 neurons from Nhe1-null mice exhibit a significantly lower steady-state pHi than wild-type neurons (7.17 vs 7.25) and a slower recovery from NH4-induced acid-loads: with some individual neurons being completely unable to recover from the acid-load in the absence of HCO3− (Yao et al., 1999). NHE1 is also a major contributor to pHi recovery in chemosensitive neurons of the retrotrapezoid nucleus (RTN) and nucleus tractus solitarius (NTS) (Kersh et al., 2009). In addition to the direct effects of NHE1 absence, loss of NHE1 also has indirect effects on the mechanism of neuronal pHi regulation due to the compensatory downregulation of AE3 in the HC and upregulation of NHE3 (in the CB) and NBCe1 (in the BD), all of which would tend to compensate for the loss of NHE1-mediated JE (Xue et al., 2003).
The importance of NHE3 to pH regulation in chemosensitive neurons is indicated by the following studies: (1) inhibition of NHE3 lowers the steady-state pHi of medullary raphé chemosensitive neurons by 0.1 unit (Wiemann et al., 1999) and (2) pharmacological data suggests that NHE3 contributes, along with NHE1, to JE in RTN neurons (Kersh et al., 2009).
One study of cultured HC neurons revealed that NHE5 contributes to acid-extrusion in the dendritic spine during enhanced neuronal activity (effected by NMDA receptor activation), a process that would tend to acidify the synaptic cleft (pHs): (Diering et al., 2011).
Importance for neuronal function. A spontaneous slow wave epilepsy was mapped to a null-mutation in the Nhe1 gene locus in one strain of mouse, (Cox et al., 1997) a finding that was bolstered by the exhibition by targeted-null mice of ataxia, apparent absence-seizures, and a post-mortem appearance consistent with seizure-related death (Bell et al., 1999). Indeed, CA1 neurons from spontaneously Nhe1-null mice are demonstrated to have enhanced excitability compared to wild-type neurons (Gu et al., 2001; Xia et al., 2003). However, the underlying cause is complex because other changes are detected in these neurons that would tend to enhance excitability, such as increased Na+ channel density (Gu et al., 2001; Xia et al., 2003), and a reduction of delta-opioid receptor expression (Zhao et al., 2005).
Chemosensitive neurons are unusually stimulated by acidification, a phenomenon that serves to stimulate exhalation of CO2 (a potential acid) during acidosis. Three lines of evidence suggest that NHE3 plays a role in maintaining steady-state pHi in these cells, thereby setting resting ventilation rate: (1) in rats, NHE3 blockade results in a lowering of pHi, causing a great increase in bioelectric activity (Wiemann et al., 1999; Kiwull-Schöne et al., 2001), (2) systemic application of an NHE3 blocker to rats causes an increased respiratory frequency (Ribas-Salgueiro et al., 2009), and (3) rabbits with lower NHE3 mRNA abundance tend to exhibit greater ventilation rates than these with a higher NHE3 mRNA abundance (Wiemann et al., 2005).
It has been suggested that NHE5 could play a critical role in the synaptic pH regulation during the firing of action potentials. In cultured hippocampal neurons, the activation of NMDA receptors recruits NHE5 protein to the membrane surface where it not only fosters focal-synaptic-cleft acidification, but also suppresses activity-induced dendritic spine growth by an autocrine feedback mechanism (Diering et al., 2011). Accordingly, knockdown of NHE5 or overexpression of a dominant-negative mutant of NHE5 in cultured hippocampal neurons causes dendritic spine overgrowth (Diering et al., 2011).
Sodium-coupled bicarbonate transporters
Molecular identity. The main HCO3-dependent acid loaders that are predicted to contribute to JE in neurons and astrocytes are the Na+-coupled HCO3 transporters (NCBTs): NBCe1, NBCn1, NDCBE, and NBCn2. NCBTs—like the acid loader AE3— are members of the SLC4 solute carrier family (Figure 2C) and share the same general topology (Figure 2D). However, unlike AE3, and more like NHEs, the common molecular action of NCBTs takes advantage of the inwardly directed Na+ gradient to promote the influx of HCO3−, thereby tending to raise pHi. NBCe1 (encoded by SLC4A4) is an electrogenic Na/HCO3 cotransporter that mediates the coupled influx of 1 Na+ plus 2 HCO3− equivalents (Romero et al., 1997). NBCn1 (encoded by SLC4A7) is an electroneutral NCBT that mediates the coupled influx of 1 Na+ and 1 HCO3− equivalent; NBCn1 is unique among the NCBTs inasmuch as it exhibits a pronounced HCO3−-independent Na+ flux (Choi et al., 2000). NDCBE (SLC4A8) is a Na+-driven Cl-HCO3 exchanger that mediates the electroneutral exchange of 1 Na+ plus 2 HCO3− equivalents for 1 Cl−(Grichtchenko et al., 2001). NBCn2/NCBE (SLC4A10) has a controversial molecular action in as much it is appears capable of NDCBE-like activity in under certain assay conditions (Wang et al., 2000; Parker et al., 2008; Damkier et al., 2010b) yet the protein expressed in Xenopus oocytes mediates NBCn1-like electroneutral Na+ and HCO3− cotransport alongside futile Cl-Cl exchange cycles that result in no net movement of Cl−(Parker et al., 2008). The majority of SLC4 members are inhibited by disulfonic stilbene derivatives such as DIDS, although NBCn1 is relatively poorly inhibited by DIDS (Choi et al., 2000).
Distribution. NBCe1 transcripts and protein are expressed throughout the central nervous system in both neurons and astrocytes (Majumdar et al., 2008). The three electroneutral NCBTs (NBCn1, NDCBE, and NBCn2) are also expressed throughout the brain—in fact the brain is the major site of expression for NDCBE and NBCn2 (Grichtchenko et al., 2001; Parker et al., 2008)—and are especially abundant in HC neurons (Damkier et al., 2007; Boedtkjer et al., 2008; Chen et al., 2008a,b; Jacobs et al., 2008; Cooper et al., 2009). All chemosensitive neurons from the medullary raphe appear to express all NCBTs in culture (Coley et al., 2013). In situ, NBCn1 expression has been detected in GABAergic and non-GABAergic HC neurons (Cooper et al., 2005) as well as in the post-synaptic dendritic spines of embryonic rat neurons (Cooper et al., 2005; Park et al., 2010). In addition, an NBCn1-like activity is present in locus coeruleus neurons (Kersh et al., 2009). NDCBE expression has been detected in presynaptic nerve endings of glutamatergic neurons, with a lesser presence in GABAergic neurons (Sinning et al., 2011; Burette et al., 2012). NBCn2 expression has been detected in pre- and post-synaptic compartments of GABAergic HC neurons (Sinning and Hübner, 2013).
Influence on neuronal pHi. It has long been recognized that the pHi and the excitability of HC neurons is enhanced in the presence vs. the absence of CO2/HCO3 (Yao et al., 1999) and also that neuronal JE is enhanced in the presence of CO2/HCO3 (Bevensee et al., 1996). Being expressed in neurons, all four of the NCBTs mentioned the in the previous section likely contribute to JE. Neurons from mice lacking NBCe1 (Svichar et al., 2011) and brain slices from mice lacking NBCn2 (Jacobs et al., 2008) exhibit substantial deficits in JE. However, we are not aware of any studies that directly address the role of NBCn1 or NDCBE in setting steady-state pHi, or contributing to JE.
Importance for neuronal function. Individuals with mutations in NBCe1 often exhibit intellectual impairments that could be a result of dysfunctional neuronal pHi regulation (Igarashi et al., 2001, 2002; Horita et al., 2005; Demirci et al., 2006). However, these individuals also exhibit a severe metabolic acidosis (NBCe1 is required in the kidney maintain plasma HCO3−) that could itself disturb brain pH (see section titled Metabolic Acidosis).
Mice lacking NDCBE (Sinning et al., 2011) and NBCn2 (Jacobs et al., 2008) exhibit greater resistance to seizure-induction consistent with the hypothesis that these transporters are normally required to contribute to JE and maintain neuronal excitability. Furthermore, in humans, the SLC4A10 (NBCn2) gene-locus is linked with epilepsy and autism (Sebat et al., 2007; Gurnett et al., 2008; Krepischi et al., 2010). However, as mentioned above in relation to AE3-null mice, both transporters are capable of influencing Cl− accumulation which may itself impact excitability. Another factor that cannot be ignored is that NBCn2 is expressed in the choroid plexus where it is a key player in a pathway that controls [HCO3−] in the CSF (Jacobs et al., 2008), thus the effect of NBCn2 loss on neuronal excitability could have an indirect component. One other NCBT that we have not considered above is NBCe2, the second electrogenic NCBT, which is encoded by SLC4A5 (Sassani et al., 2002; Virkki et al., 2002). NBCe2 is another key player in CSF secretion by the choroid plexus (Bouzinova et al., 2005; Millar and Brown, 2008), a factor that likely underlies the reduced neuronal excitability evident in brain slices from Nbce2-null mice (Kao et al., 2011); expression of NBCe2 has not been demonstrated in neurons.
Other Plasma Membrane Proteins that Influence Neuronal pH
Although this review focuses on the major pH regulating protein in the brain, other factors contribute either directly or indirectly to pHi, pHs, and pHo through the influx or efflux of acid-base equivalents. For instance, the H+-coupled monocarboxylate transporters (MCT1-4) play major role in transporting carboxylic acids, e.g., lactate and pyruvate, between neurons and astrocytes which are then used as a source of energy (Adijanto and Philp, 2012; Choi et al., 2012). The activation of NMDA receptors induces a Ca2+ dependent pHi acidification in rat HC neurons (Irwin et al., 1994) that is likely due to Ca-H exchange mediated by the Ca2+-ATPase (Makani and Chesler, 2010). The extent of the drop in neuronal pHi due to Ca2+-ATPase action during electrical activity is limited by a depolarization-induced alkalization that is likely mediated by the proton-efflux channel HV1(Meech and Thomas, 1987; Cheng et al., 2008; Meech, 2012). Finally, we must not discount the contribution of acid-base transporters in the astrocytes (Chesler, 2003) and choroid plexus epithelia (Damkier et al., 2010a, 2013), cells that control the composition of the brain extracellular fluid and thus indirectly influence the pH of neurons.
Physiopathological Acid-Base Disturbances
The four major acid base disturbances in the body are respiratory acidosis, respiratory alkalosis, metabolic acidosis, and metabolic alkalosis. In the following section we will discuss (1) the cause of each of these disturbances, (2) the clinical presentation of these disturbances, (3) the effect of these disturbances on pHi, (4) the effect of these pH changes on the acid base transporter activity, and (5) the way the body compensates for these disturbances using the respiratory and renal systems.
Respiratory Acidosis (Hypercapnia)
Respiratory acidosis results from inability to eliminate, from the body, the CO2 that is produced from cellular respiration. As a consequence the partial pressure of CO2 (pCO2) in the blood rises and the pH of the blood decreases as described by the Henderson-Hasselbalch equation (Hills, 1973; Hurn et al., 1991). Some of the causes of respiratory acidosis are CNS depression, neuromuscular disease, chronic obstructive pulmonary disease (COPD), sleep apnea, alveolar hypoventilation, ischemia, lung disease, and obesity. Clinical symptoms of hypercapnia include anxiety, pulmonary hypertension, tachypnea, extrasystoles, muscle twitches, and reduced neural activity. Prolonged hypercapnia results in disorientation, convulsions, unconsciousness, and eventually death (Ayers and Warrington, 2008).
Specialized neuronal groups (respiratory chemoreceptors) within the brain protect against the compromised neuronal function associated with ECF (extracellular fluid) acidosis. Respiratory chemoreceptors were first identified on the surface of the medulla (Mitchell et al., 1963; Loeschcke et al., 1970; Schlaefke et al., 1970), and later throughout the brainstem and hypothalamus (Berquin et al., 2000). Respiratory chemoreceptors chemically sense increased CO2 or H+ production. In response to the stimulus of lowered pHi that accompanies this increased CO2 or H+ production, the excitability of these cells is enhanced, which increases respiratory drive, thereby appropriately adjusting the pH of the ECF (blood, CSF, and interstitial fluid): (Douglas et al., 2001; Putnam, 2001; Bouyer et al., 2004; Richerson, 2004; Hodges and Richerson, 2010).
Longer term exposure to elevated pCO2 increases the expression of acid extruders (NHE1 and NBCn1) and decreases the expression of the acid loader AE3 throughout the mouse brain, mostly prominently in the cortex, and especially in neonates (Kanaan et al., 2007). This may reflect a compensatory mechanism that would counter acidosis and tend to maintain neuronal excitability. Recent data points to a potential genetic link between NHE3, breathing control, and sudden infant death syndrome (Wiemann et al., 2005, 2008; Poetsch et al., 2010).
Respiratory Alkalosis (Hypocapnia)
Respiratory alkalosis results from the excess elimination of CO2 from the body. As a result, pCO2 decreases and the pH of the blood increases. Some of the causes of respiratory alkalosis are hypoxia, hyperventilation, CNS disorders (meningitis), and drugs. The decrease in CO2 is usually well tolerated, although there are typical clinical symptoms that include confusion, dizziness, muscle cramps, chest wall tightness, and tetany in the extremities (Ayers and Warrington, 2008).
The main stimulus for ventilation is increased CO2, and as a result the decreased pCO2 present in respiratory alkalosis can suppress breathing. As breathing decreases, the pCO2 rises and returns the blood pH to lower values. The decreased breathing also results in decreased O2 intake resulting in hypoxia secondary to hypocapnia. In addition, respiratory alkalosis also causes cerebral vasoconstriction with concomitant cerebral hypoxia. Clinical treatment for respiratory alkalosis include supplemental oxygen or drug removal if the alkalosis is drug induced (Ayers and Warrington, 2008).
Metabolic Acidosis
Metabolic acidosis results from an increase in metabolic acid production or an inability to remove acid/reabsorb base in the kidneys. As a result there is a decrease in blood pH. Some of the causes of metabolic acidosis include diarrhea, severe renal failure, lactic acidosis, ketoacidosis, and drug intoxication. Clinical symptoms include a low blood pH (<7.35), chest pain, decreased cardiac output, hypotension, increased calcium release, and muscle weakness. Patients often display deep, labored breathing patterns (Kussmaul respiration) described as “air hunger.” Metabolic acidosis can lead to coma and death (Ayers and Warrington, 2008).
Similar to respiratory acidosis, the overall result of this insult is decreased blood pH and consequently increased ventilation rate. Metabolic acidosis causes upregulation of NHE3 (Kiwull-Schöne et al., 2007) and increase expression of NBCn1 in several brain regions (Cooper et al., 2009; Park et al., 2010). This predicted increase in JE would facilitate extra protection against intracellular and extracellular acid overload.
As an acute compensation, the body regulates the bicarbonate buffering system to drive the production of CO2 which can be eliminated through increased ventilation. The chemoreceptors in the brainstem and hypothalamus are activated and stimulate respiratory structures to increase breathing rate and elimination of CO2. The increased acid is also intrinsically buffered by proteins, phosphates, and carbonate in the bone. As a chronic compensation, the renal system increases the secretion of H+ (Giebisch and Windhager, 2009). Clinical treatment for metabolic acidosis includes normalizing blood volume and cardiac output. For more severe cases bicarbonate and acetate are administered and pCO2 is decreased (Ayers and Warrington, 2008).
Metabolic Alkalosis
Metabolic alkalosis results from an increase bicarbonate in the blood. This increase may be due to either a primary increase in bicarbonate reabsorption or be secondary to decreased production or increased loss of H+. As a result there is an increase in blood pH. Some of the causes of metabolic alkalosis include vomiting, diuretics, or increased urinary excretion of Cl−. Clinical symptoms include arteriolar constriction, reduced coronary blood flow, hypokalemia, tetany, and seizures (Ayers and Warrington, 2008).
The increased blood pH reduces the respiratory stimulus for breathing and hypoventilation occurs. As a result CO2 is retained and shuttled through the carbonic anhydrase buffering system and H+ is produced which lowers blood pH. A more chronic compensation occurs when the renal system decreases H+ secretion (Giebisch and Windhager, 2009). Clinical treatment for metabolic alkalosis includes volume replacement, use of carbonic anhydrase inhibitors (acetazolamide), and correction of potassium depletion (Ayers and Warrington, 2008). We are unaware of any reports of compensatory alterations in acid base transporter activity or expression in the brain under metabolic alkalosis.
Summary
Neuronal excitability is highly susceptible to fluctuations in intra- and extracellular pH. It is the delicate balance between the actions of the acid-base transporters that contributes to JL and JE, maintaining a permissive neuronal pH in the face of physiological and pathophysiological acid-base disturbances. Loss of any of these transporters is associated with profound neuronal abnormalities and conversely, disturbance in pH are associated with many different physiopathological conditions such as Alzheimer's and Parkinson's diseases. The contribution of acid-base transporters to the severity of the signs of neurological disorders is a promising area of investigation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Mark D. Parker is supported by start-up funds from the Dean of the School of Medicine and Biomedical Sciences and from the Department of Physiology and Biophysics at SUNY Buffalo. This work was also funded by grants from the National Institutes of Health to Walter F. Boron (NS18400) and to Michael L. Jennings and Mark D. Parker (EY021646).
References
Adijanto, J., and Philp, N. J. (2012). The SLC16A family of monocarboxylate transporters (MCTs)—physiology and function in cellular metabolism, pH homeostasis, and fluid transport. Curr. Top. Membr. 70, 275–311. doi: 10.1016/B978-0-12-394316-3.00009-0
Alper, S. L. (2009). Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J. Exp. Biol. 212, 1672–1683. doi: 10.1242/jeb.029454
Alvarez, B. V., Fujinaga, J., and Casey, J. R. (2001). Molecular basis for angiotensin II-induced increase of chloride/bicarbonate exchange in the myocardium. Circ. Res. 89, 1246–1253. doi: 10.1161/hh2401.101907
Alvarez, B. V., Gilmour, G. S., Mema, S. C., Martin, B. T., Shull, G. E., Casey, J. R., et al. (2007). Blindness caused by deficiency in AE3 chloride/bicarbonate exchanger. PLoS ONE 2:e839. doi: 10.1371/journal.pone.0000839
Attaphitaya, S., Park, K., and Melvin, J. E. (1999). Molecular cloning and functional expression of a rat Na+/H+ exchanger (NHE5) highly expressed in brain. J. Biol. Chem. 274, 4383–4388. doi: 10.1074/jbc.274.7.4383
Ayers, P., and Warrington, L. (2008). Diagnosis and treatment of simple acid-base disorders. Nutr. Clin. Pract. 23, 122–127. doi: 10.1177/0884533608314534
Baird, N. R., Orlowski, J., Szabó, E. Z., Zaun, H. C., Schultheis, P. J., Menon, A. G., et al. (1999). Molecular cloning, genomic organization, and functional expression of Na+/H+ exchanger isoform 5 (NHE5) from human brain. J. Biol. Chem. 274, 4377–4382. doi: 10.1074/jbc.274.7.4377
Balestrino, M., and Somjen, G. G. (1988). Concentration of carbon dioxide, interstitial pH and synaptic transmission in hippocampal formation of the rat. J. Physiol. 396, 247–266.
Baxter, K. A., and Church, J. (1996). Characterization of acid extrusion mechanisms in cultured fetal rat hippocampal neurones. J. Physiol. 493(Pt 2), 457–470.
Bell, S. M., Schreiner, C. M., Schultheis, P. J., Miller, M. L., Evans, R. L., Vorhees, C. V., et al. (1999). Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures. Am. J. Physiol. 276, C788–795.
Berquin, P., Bodineau, L., Gros, F., and Larnicol, N. (2000). Brainstem and hypothalamic areas involved in respiratory chemoreflexes: a Fos study in adult rats. Brain Res. 857, 30–40. doi: 10.1016/S0006-8993(99)02304-5
Bevensee, M. O., and Boron, W. F. (2013). “Control of intracellular pH,” in Seldin and Giebisch's The Kidney: Physiology and Pathophysiology, eds R. J. Alpern, M. J. Caplan, and O. W. Moe (San Diego, CA: Academic Press), 1773–1835. doi: 10.1016/B978-0-12-381462-3.00052-5
Bevensee, M. O., Cummins, T. R., Haddad, G. G., Boron, W. F., and Boyarsky, G. (1996). pH regulation in single CA1 neurons acutely isolated from the hippocampi of immature and mature rats. J. Physiol. 494(Pt 2), 315–328.
Bevensee, M. O., Weed, R. A., and Boron, W. F. (1997). Intracellular pH regulation in cultured astrocytes from rat hippocampus. I. Role of HCO3−. J. Gen. Physiol. 110, 453–465. doi: 10.1085/jgp.110.4.453
Boedtkjer, E., Praetorius, J., Füchtbauer, E.-M., and Aalkjaer, C. (2008). Antibody-independent localization of the electroneutral Na+−HCO3− cotransporter NBCn1 (slc4a7) in mice. Am. J. Physiol. Cell Physiol. 294, C591–C603. doi: 10.1152/ajpcell.00281.2007
Boron, W. (2012). “Acid-base physiology,” in Medical Physiology, eds W. Boron and E. Boulpaep (Philadelphia, PA: Saunders Elsevier), 652–671.
Boron, W. F., Chen, L., and Parker, M. D. (2009). Modular structure of sodium-coupled bicarbonate transporters. J. Exp. Biol. 212, 1697–1706. doi: 10.1242/jeb.028563
Boron, W. F., and De Weer, P. (1976a). Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. J. Gen. Physiol. 67, 91–112. doi: 10.1085/jgp.67.1.91
Boron, W. F., and De Weer, P. (1976b). Active proton transport stimulated by CO2/HCO3−, blocked by cyanide. Nature 259, 240–241. doi: 10.1038/259240a0
Bouyer, P., Bradley, S. R., Zhao, J., Wang, W., Richerson, G. B., and Boron, W. F. (2004). Effect of extracellular acid-base disturbances on the intracellular pH of neurones cultured from rat medullary raphe or hippocampus. J. Physiol. 559, 85–101. doi: 10.1113/jphysiol.2004.067793
Bouzinova, E. V., Praetorius, J., Virkki, L. V., Nielsen, S., Boron, W. F., and Aalkjaer, C. (2005). Na+-dependent HCO3− uptake into the rat choroid plexus epithelium is partially DIDS sensitive. Am. J. Physiol. Cell Physiol. 289, C1448–C1456. doi: 10.1152/ajpcell.00313.2005
Brookes, N. (1997). Intracellular pH as a regulatory signal in astrocyte metabolism. Glia 21, 64–73. doi: 10.1002/(SICI)1098-1136(199709)21:1%3C64::AID-GLIA7%3E3.3.CO;2-Z
Burette, A. C., Weinberg, R. J., Sassani, P., Abuladze, N., Kao, L., and Kurtz, I. (2012). The sodium-driven chloride/bicarbonate exchanger in presynaptic terminals. J. Comp. Neurol. 520, 1481–1492. doi: 10.1002/cne.22806
Chen, L.-M., Kelly, M. L., Parker, M. D., Bouyer, P., Gill, H. S., Felie, J. M., et al. (2008a). Expression and localization of Na-driven Cl-HCO3− exchanger (SLC4A8) in rodent CNS. Neuroscience 153, 162–174. doi: 10.1016/j.neuroscience.2008.02.018
Chen, L.-M., Kelly, M. L., Rojas, J. D., Parker, M. D., Gill, H. S., Davis, B. A., et al. (2008b). Use of a new polyclonal antibody to study the distribution and glycosylation of the sodium-coupled bicarbonate transporter NCBE in rodent brain. Neuroscience 151, 374–385. doi: 10.1016/j.neuroscience.2007.10.015
Cheng, Y. M., Kelly, T., and Church, J. (2008). Potential contribution of a voltage-activated proton conductance to acid extrusion from rat hippocampal neurons. Neuroscience 151, 1084–1098. doi: 10.1016/j.neuroscience.2007.12.007
Chesler, M. (2003). Regulation and modulation of pH in the brain. Physiol. Rev. 83, 1183–1221. doi: 10.1152/physrev.00010.2003
Chesler, M., and Kaila, K. (1992). Modulation of pH by neuronal activity. Trends Neurosci. 15, 396–402. doi: 10.1016/0166-2236(92)90191-A
Chesler, M., and Kraig, R. P. (1989). Intracellular pH transients of mammalian astrocytes. J. Neurosci. 9, 2011–2019.
Choi, H. B., Gordon, G. R. J., Zhou, N., Tai, C., Rungta, R. L., Martinez, J., et al. (2012). Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron 75, 1094–1104. doi: 10.1016/j.neuron.2012.08.032
Choi, I., Aalkjaer, C., Boulpaep, E. L., and Boron, W. F. (2000). An electroneutral sodium/bicarbonate cotransporter NBCn1 and associated sodium channel. Nature 405, 571–575. doi: 10.1038/35014615
Christensen, H. L., Nguyen, A. T., Pedersen, F. D., and Damkier, H. H. (2013). Na+ dependent acid-base transporters in the choroid plexus; insights from slc4 and slc9 gene deletion studies. Front. Physiol. 4:304. doi: 10.3389/fphys.2013.00304
Church, J. (1992). A change from HCO3− CO2 to hepes-buffered medium modifies membrane properties of rat CA1 pyramidal neurones in vitro. J. Physiol. 455, 51–71.
Church, J., Baxter, K. A., and McLarnon, J. G. (1998). pH modulation of Ca2+ responses and a Ca2+-dependent K+ channel in cultured rat hippocampal neurones. J. Physiol. 511(Pt 1), 119–132. doi: 10.1111/j.1469-7793.1998.119bi.x
Coley, A. A., Ruffin, V. A., Moss, F. J., Hopfer, U., and Boron, W. F. (2013). Immunocytochemical identification of electroneutral Na+-coupled transporters in freshly dissociated mouse medullary raphé neurons. Neuroscience 246, 451–467. doi: 10.1016/j.neuroscience.2013.02.064
Cooper, D. S., Saxena, N. C., Yang, H. S., Lee, H. J., Moring, A. G., Lee, A., et al. (2005). Molecular and functional characterization of the electroneutral Na/HCO3 cotransporter NBCn1 in rat hippocampal neurons. J. Biol. Chem. 280, 17823–17830. doi: 10.1074/jbc.M408646200
Cooper, D. S., Yang, H. S., He, P., Kim, E., Rajbhandari, I., Yun, C. C., et al. (2009). Sodium/bicarbonate cotransporter NBCn1/slc4a7 increases cytotoxicity in magnesium depletion in primary cultures of hippocampal neurons. Eur. J. Neurosci. 29, 437–446. doi: 10.1111/j.1460-9568.2008.06611.x
Coulter, K. L., Périer, F., Redeke, C. M., and Vanderberg, C. A. (1995). Identification and molecular localization of a pH-sensing domain for the inward rectifier potassium channel HIR. Neuron 15, 1157–1168. doi: 10.1016/0896-6273(95)90103-5
Counillon, L., Franchi, A., and Pouysségur, J. (1993). A point mutation of the Na+/H+ exchanger gene (NHE1) and amplification of the mutated allele confer amiloride resistance upon chronic acidosis. PNAS 90, 4508–4512. doi: 10.1073/pnas.90.10.4508
Cox, G. A., Lutz, C. M., Yang, C. L., Biemesderfer, D., Bronson, R. T., Fu, A., et al. (1997). Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice. Cell 91, 139–148. doi: 10.1016/S0092-8674(01)80016-7
Damkier, H. H., Brown, P. D., and Praetorius, J. (2010a). Epithelial pathways in choroid plexus electrolyte transport. Physiology (Bethesda) 25, 239–249. doi: 10.1152/physiol.00011.2010
Damkier, H. H., Aalkjaer, C., and Praetorius, J. (2010b). Na+-dependent HCO3− import by the slc4a10 gene product involves Cl− export. J. Biol. Chem. 285, 26998–27007. doi: 10.1074/jbc.M110.108712
Damkier, H. H., Brown, P. D., and Praetorius, J. (2013). Cerebrospinal fluid secretion by the choroid plexus. Physiol. Rev. 93, 1847–1892. doi: 10.1152/physrev.00004.2013
Damkier, H. H., Nielsen, S., and Praetorius, J. (2007). Molecular expression of SLC4-derived Na+-dependent anion transporters in selected human tissues. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R2136–R2146. doi: 10.1152/ajpregu.00356.2007
Dean, J. B., Kinkade, E. A., and Putnam, R. W. (2001). Cell-cell coupling in CO2/H+-excited neurons in brainstem slices. Respir. Physiol. 129, 83–100. doi: 10.1016/S0034-5687(01)00284-5
Deitmer, J. W., and Rose, C. R. (1996). pH regulation and proton signalling by glial cells. Prog. Neurobiol. 48, 73–103. doi: 10.1016/0301-0082(95)00039-9
Demetrius, L. A., and Simon, D. K. (2012). An inverse-Warburg effect and the origin of Alzheimer's disease. Biogerontology 13, 583–594. doi: 10.1007/s10522-012-9403-6
Demirci, F. Y. K., Chang, M.-H., Mah, T. S., Romero, M. F., and Gorin, M. B. (2006). Proximal renal tubular acidosis and ocular pathology: a novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1). Mol. Vis. 12, 324–330.
Diering, G. H., Mills, F., Bamji, S. X., and Numata, M. (2011). Regulation of dendritic spine growth through activity-dependent recruitment of the brain-enriched Na+/H+ exchanger NHE5. Mol. Biol. Cell 22, 2246–2257. doi: 10.1091/mbc.E11-01-0066
Donowitz, M., Ming Tse, C., and Fuster, D. (2013). SLC9/NHE gene family, a plasma membrane and organellar family of Na+/H+ exchangers. Mol. Aspects Med. 34, 236–251. doi: 10.1016/j.mam.2012.05.001
Douglas, R. M., Trouth, C. O., James, S. D., Sexcius, L. M., Kc, P., Dehkordi, O., et al. (2001). Decreased CSF pH at ventral brain stem induces widespread c-Fos immunoreactivity in rat brain neurons. J. Appl. Physiol. 90, 475–485.
Duprat, F., Lesage, F., Fink, M., Reyes, R., Heurteaux, C., and Lazdunski, M. (1997). TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 16, 5464–5471. doi: 10.1093/emboj/16.17.5464
Ekdahl, C. T., Kokaia, Z., and Lindvall, O. (2009). Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158, 1021–1029. doi: 10.1016/j.neuroscience.2008.06.052
Giebisch, G., and Windhager, E. E. (2009). “Transport of acids and bases,” in Medical Physiology. A Cellular and Molecular Approach, eds W. F. Boron and E. L. Boulpaep (Philadelphia, PA: Elsevier Saunders), 851–865.
Giffard, R. G., Monyer, H., Christine, C. W., and Choi, D. W. (1990). Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res. 506, 339–342. doi: 10.1016/0006-8993(90)91276-M
Grichtchenko, I. I., Choi, I., Zhong, X., Bray-Ward, P., Russell, J. M., and Boron, W. F. (2001). Cloning, characterization, and chromosomal mapping of a human electroneutral Na+-driven Cl-HCO3 exchanger. J. Biol. Chem. 276, 8358–8363. doi: 10.1074/jbc.C000716200
Gu, X. Q., Yao, H., and Haddad, G. G. (2001). Increased neuronal excitability and seizures in the Na+/H+ exchanger null mutant mouse. Am. J. Physiol. Cell Physiol. 281, C496–503.
Gurnett, C. A., Veile, R., Zempel, J., Blackburn, L., Lovett, M., and Bowcock, A. (2008). Disruption of sodium bicarbonate transporter SLC4A10 in a patient with complex partial epilepsy and mental retardation. Arch. Neurol. 65, 550–553. doi: 10.1001/archneur.65.4.550
Halestrap, A. P. (2012). The monocarboxylate transporter family—structure and functional characterization. IUBMB Life 64, 1–9. doi: 10.1002/iub.573
Hentschke, M., Wiemann, M., Hentschke, S., Kurth, I., Hermans-Borgmeyer, I., Seidenbecher, T., et al. (2006). Mice with a targeted disruption of the Cl−/HCO3− exchanger AE3 display a reduced seizure threshold. Mol. Cell. Biol. 26, 182–191. doi: 10.1128/MCB.26.1.182-191.2006
Hills, A. G. (1973). pH and the Henderson-Hasselbalch equation. Am. J. Med. 55, 131–133. doi: 10.1016/0002-9343(73)90160-5
Hodges, M. R., and Richerson, G. B. (2010). Medullary serotonin neurons and their roles in central respiratory chemoreception. Respir. Physiol. Neurobiol. 173, 256–263. doi: 10.1016/j.resp.2010.03.006
Horita, S., Yamada, H., Inatomi, J., Moriyama, N., Sekine, T., Igarashi, T., et al. (2005). Functional analysis of NBC1 mutants associated with proximal renal tubular acidosis and ocular abnormalities. J. Am. Soc. Nephrol. 16, 2270–2278. doi: 10.1681/ASN.2004080667
Hormuzdi, S. G., Filippov, M. A., Mitropoulou, G., Monyer, H., and Bruzzone, R. (2004). Electrical synapses: a dynamic signaling system that shapes the activity of neuronal networks. Biochim. Biophys. Acta 1662, 113–137. doi: 10.1016/j.bbamem.2003.10.023
Hurn, P. D., Koehler, R. C., Norris, S. E., Schwentker, A. E., and Traystman, R. J. (1991). Bicarbonate conservation during incomplete cerebral ischemia with superimposed hypercapnia. Am. J. Physiol. 261, H853–859.
Igarashi, T., Inatomi, J., Sekine, T., Seki, G., Shimadzu, M., Tozawa, F., et al. (2001). Novel nonsense mutation in the Na+/HCO3− cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. J. Am. Soc. Nephrol. 12, 713–718.
Igarashi, T., Sekine, T., Inatomi, J., and Seki, G. (2002). Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. JASN 13, 2171–2177. doi: 10.1097/01.ASN.0000025281.70901.30
Irie, T., Hara, M., Yasukura, T., Minamino, M., Omori, K., Matsuda, H., et al. (1998). Chloride concentration in cultured hippocampal neurons increases during long-term exposure to ammonia through enhanced expression of an anion exchanger. Brain Res. 806, 246–256. doi: 10.1016/S0006-8993(98)00700-8
Irwin, R. P., Lin, S. Z., Long, R. T., and Paul, S. M. (1994). N-methyl-D-aspartate induces a rapid, reversible, and calcium-dependent intracellular acidosis in cultured fetal rat hippocampal neurons. J. Neurosci. 14, 1352–1357.
Jacobs, S., Ruusuvuori, E., Sipilä, S. T., Haapanen, A., Damkier, H. H., Kurth, I., et al. (2008). Mice with targeted Slc4a10 gene disruption have small brain ventricles and show reduced neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 105, 311–316. doi: 10.1073/pnas.0705487105
Kahle, K. T., Rinehart, J., de los Heros, P., Louvi, A., Meade, P., Vazquez, N., et al. (2005). WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. PNAS 102, 16783–16788. doi: 10.1073/pnas.0508307102
Kaila, K., Panula, P., Karhunen, T., and Heinonen, E. (1991). Fall in intracellular pH mediated by GABAA receptors in cultured rat astrocytes. Neurosci. Lett. 126, 9–12. doi: 10.1016/0304-3940(91)90358-Z
Kanaan, A., Douglas, R. M., Alper, S. L., Boron, W. F., and Haddad, G. G. (2007). Effect of chronic elevated carbon dioxide on the expression of acid-base transporters in the neonatal and adult mouse. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1294–R1302. doi: 10.1152/ajpregu.00261.2007
Kao, L., Kurtz, L. M., Shao, X., Papadopoulos, M. C., Liu, L., Bok, D., et al. (2011). Severe neurologic impairment in mice with targeted disruption of the electrogenic sodium bicarbonate cotransporter NBCe2 (Slc4a5 Gene). J. Biol. Chem. 286, 32563–32574. doi: 10.1074/jbc.M111.249961
Kersh, A. E., Hartzler, L. K., Havlin, K., Hubbell, B. B., Nanagas, V., Kalra, A., et al. (2009). pH regulating transporters in neurons from various chemosensitive brainstem regions in neonatal rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1409–R1420. doi: 10.1152/ajpregu.91038.2008
Kiwull-Schöne, H., Kiwull, P., Frede, S., and Wiemann, M. (2007). Role of brainstem sodium/proton exchanger 3 for breathing control during chronic acid base imbalance. Am. J. Respir. Crit. Care Med. 176, 513–519. doi: 10.1164/rccm.200703-347OC
Kiwull-Schöne, H., Wiemann, M., Frede, S., Bingmann, D., Wirth, K. J., Heinelt, U., et al. (2001). A novel inhibitor of the Na+/H+ exchanger type 3 activates the central respiratory CO2 response and lowers the apneic threshold. Am. J. Respir. Crit. Care Med. 164, 1303–1311. doi: 10.1164/ajrccm.164.7.2010147
Klanke, C. A., Su, Y. R., Callen, D. F., Wang, Z., Meneton, P., Baird, N., et al. (1995). Molecular cloning and physical and genetic mapping of a novel human Na+/H+ exchanger (NHE5/SLC9A5) to chromosome 16q22.1. Genomics 25, 615–622. doi: 10.1016/0888-7543(95)80002-4
Kobayashi, S., Morgans, C. W., Casey, J. R., and Kopito, R. R. (1994). AE3 anion exchanger isoforms in the vertebrate retina: developmental regulation and differential expression in neurons and glia. J. Neurosci. 14, 6266–6279.
Kopito, R. R., Lee, B. S., Simmons, D. M., Lindsey, A. E., Morgans, C. W., and Schneider, K. (1989). Regulation of intracellular pH by a neuronal homolog of the erythrocyte anion exchanger. Cell 59, 927–937. doi: 10.1016/0092-8674(89)90615-6
Krepischi, A. C. V., Knijnenburg, J., Bertola, D. R., Kim, C. A., Pearson, P. L., Bijlsma, E., et al. (2010). Two distinct regions in 2q24.2-q24.3 associated with idiopathic epilepsy. Epilepsia 51, 2457–2460. doi: 10.1111/j.1528-1167.2010.02742.x
Loeschcke, H. H., De Lattre, J., Schläfke, M. E., and Trouth, C. O. (1970). Effects on respiration and circulation of electrically stimulating the ventral surface of the medulla oblongata. Respir. Physiol. 10, 184–197. doi: 10.1016/0034-5687(70)90082-4
Ma, E., and Haddad, G. G. (1997). Expression and localization of Na+/H+ exchangers in rat central nervous system. Neuroscience 79, 591–603. doi: 10.1016/S0306-4522(96)00674-4
Majumdar, D., Maunsbach, A. B., Shacka, J. J., Williams, J. B., Berger, U. V., Schultz, K. P., et al. (2008). Localization of electrogenic Na/bicarbonate cotransporter NBCe1 variants in rat brain. Neuroscience 155, 818–832. doi: 10.1016/j.neuroscience.2008.05.037
Makani, S., and Chesler, M. (2007). Endogenous alkaline transients boost postsynaptic NMDA receptor responses in hippocampal CA1 pyramidal neurons. J. Neurosci. 27, 7438–7446. doi: 10.1523/JNEUROSCI.2304-07.2007
Makani, S., and Chesler, M. (2010). Rapid rise of extracellular pH evoked by neural activity is generated by the plasma membrane calcium ATPase. J. Neurophysiol. 103, 667–676. doi: 10.1152/jn.00948.2009
Manning Fox, J. E., Meredith, D., and Halestrap, A. P. (2000). Characterisation of human monocarboxylate transporter 4 substantiates its role in lactic acid efflux from skeletal muscle. J. Physiol. 529, 285–293. doi: 10.1111/j.1469-7793.2000.00285.x
Mattson, M. P., Pedersen, W. A., Duan, W., Culmsee, C., and Camandola, S. (1999). Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann. N.Y. Acad. Sci. 893, 154–175. doi: 10.1111/j.1749-6632.1999.tb07824.x
McDonald, J. W., Bhattacharyya, T., Sensi, S. L., Lobner, D., Ying, H. S., Canzoniero, L. M. T., et al. (1998). Extracellular acidity potentiates AMPA receptor-mediated cortical neuronal death. J. Neurosci. 18, 6290–6299.
Meech, R. (2012). A contribution to the history of the proton channel. Wiley Interdiscip. Rev. Membr. Transp. Signal. 1, 533–557. doi: 10.1002/wmts.59
Meech, R. W., and Thomas, R. C. (1987). Voltage-dependent intracellular pH in Helix aspersa neurones. J. Physiol. 390, 433–452.
Meier, S., Hübner, C. A., Groeben, H., Peters, J., Bingmann, D., and Wiemann, M. (2007). Expression of anion exchanger 3 influences respiratory rate in awake and isoflurane anesthetized mice. J. Physiol. Pharmacol. 58(Suppl. 5), 371–378.
Millar, I. D., and Brown, P. D. (2008). NBCe2 exhibits a 3 HCO3−:1 Na+ stoichiometry in mouse choroid plexus epithelial cells. Biochem. Biophys. Res. Commun. 373, 550–554. doi: 10.1016/j.bbrc.2008.06.053
Mitchell, R. A., Loeschcke, H. H., Severinghaus, J. W., Richardson, B. W., and Massion, W. H. (1963). Regions of respiratory chemosensitivity on the surface of the medulla. Ann. N.Y. Acad. Sci. 109, 661–681. doi: 10.1111/j.1749-6632.1963.tb13496.x
Orlowski, J. (1993). Heterologous expression and functional properties of amiloride high affinity (NHE-1) and low affinity (NHE-3) isoforms of the rat Na/H exchanger. J. Biol. Chem. 268, 16369–16377.
Orlowski, J., and Grinstein, S. (2004). Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch. 447, 549–565. doi: 10.1007/s00424-003-1110-3
OuYang, Y. B., Mellergård, P., Kristián, T., Kristiánova, V., and Siesjö, B. K. (1994). Influence of acid-base changes on the intracellular calcium concentration of neurons in primary culture. Exp. Brain Res. 101, 265–271. doi: 10.1007/BF00228746
Park, H. J., Rajbhandari, I., Yang, H. S., Lee, S., Cucoranu, D., Cooper, D. S., et al. (2010). Neuronal expression of sodium/bicarbonate cotransporter NBCn1 (SLC4A7) and its response to chronic metabolic acidosis. Am. J. Physiol. Cell Physiol. 298, C1018–C1028. doi: 10.1152/ajpcell.00492.2009
Parker, M. D., and Boron, W. F. (2013). The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol. Rev. 93, 803–959. doi: 10.1152/physrev.00023.2012
Parker, M. D., Musa-Aziz, R., Rojas, J. D., Choi, I., Daly, C. M., and Boron, W. F. (2008). Characterization of human SLC4A10 as an electroneutral Na/HCO3 cotransporter (NBCn2) with Cl− self-exchange activity. J. Biol. Chem. 283, 12777–12788. doi: 10.1074/jbc.M707829200
Pavlov, I., Kaila, K., Kullmann, D. M., and Miles, R. (2013). Cortical inhibition, pH and cell excitability in epilepsy: what are optimal targets for antiepileptic interventions? J. Physiol. 591, 765–774. doi: 10.1113/jphysiol.2012.237958
Pick, U., and Karlish, S. J. (1982). Regulation of the conformation transition in the Ca-ATPase from sarcoplasmic reticulum by pH, temperature, and calcium ions. J. Biol. Chem. 257, 6120–6126.
Pizzonia, J. H., Ransom, B. R., and Pappas, C. A. (1996). Characterization of Na+/H+ exchange activity in cultured rat hippocampal astrocytes. J. Neurosci. Res. 44, 191–198. doi: 10.1002/(SICI)1097-4547(19960415)44:2%3C191::AID-JNR12%3E3.3.CO;2-B
Poetsch, M., Nottebaum, B. J., Wingenfeld, L., Frede, S., Vennemann, M., and Bajanowski, T. (2010). Impact of sodium/proton exchanger 3 gene variants on sudden infant death syndrome. J. Pediatry 156, 44–48.e1. doi: 10.1016/j.jpeds.2009.07.018
Putnam, R. W. (2001). Intracellular pH regulation of neurons in chemosensitive and nonchemosensitive areas of brain slices. Respir. Physiol. 129, 37–56. doi: 10.1016/S0034-5687(01)00281-X
Raley-Susman, K. M., Cragoe, E. J. Jr., Sapolsky, R. M., and Kopito, R. R. (1991). Regulation of intracellular pH in cultured hippocampal neurons by an amiloride-insensitive Na+/H+ exchanger. J. Biol. Chem. 266, 2739–2745.
Raley-Susman, K. M., Sapolsky, R. M., and Kopito, R. R. (1993). Cl−/HCO3− exchange function differs in adult and fetal rat hippocampal neurons. Brain Res. 614, 308–314. doi: 10.1016/0006-8993(93)91049-X
Ribas-Salgueiro, J. L., Matarredona, E. R., Sarmiento, M., Ribas, J., and Pásaro, R. (2009). Respiratory response to systemic inhibition of the Na+/H+ exchanger type 3 in intact rats. Respir. Physiol. Neurobiol. 165, 254–260. doi: 10.1016/j.resp.2008.12.011
Richerson, G. B. (2004). Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat. Rev. Neurosci. 5, 449–461. doi: 10.1038/nrn1409
Ritucci, N. A., Chambers-Kersh, L., Dean, J. B., and Putnam, R. W. (1998). Intracellular pH regulation in neurons from chemosensitive and nonchemosensitive areas of the medulla. Am. J. Physiol. 275, R1152–R1163.
Ro, H., and Carson, J. H. (2004). pH microdomains in oligodendrocytes. J. Biol. Chem. 279, 37115–37123. doi: 10.1074/jbc.M403099200
Romero, M. F., Hediger, M. A., Boulpaep, E. L., and Boron, W. F. (1997). Expression cloning and characterization of a renal electrogenic Na+/HCO3− cotransporter. Nature 387, 409–413. doi: 10.1038/387409a0
Ruffin, V. A., Gu, X. Q., Zhou, D., Douglas, R. M., Sun, X., Trouth, C. O., et al. (2008). The sodium-activated potassium channel Slack is modulated by hypercapnia and acidosis. Neuroscience 151, 410–418. doi: 10.1016/j.neuroscience.2007.10.031
Sander, T., Toliat, M. R., Heils, A., Leschik, G., Becker, C., Rüschendorf, F., et al. (2002). Association of the 867Asp variant of the human anion exchanger 3 gene with common subtypes of idiopathic generalized epilepsy. Epilepsy Res. 51, 249–255. doi: 10.1016/S0920-1211(02)00152-3
Sassani, P., Pushkin, A., Gross, E., Gomer, A., Abuladze, N., Dukkipati, R., et al. (2002). Functional characterization of NBC4: a new electrogenic sodium-bicarbonate cotransporter. Am. J. Physiol. Cell Physiol. 282, C408–C416. doi: 10.1152/ajpcell.00409.2001
Schlaefke, M. E., See, W. R., and Loeschcke, H. H. (1970). Ventilatory response to alterations of H+ ion concentration in small areas of the ventral medullary surface. Respir. Physiol. 10, 198–212. doi: 10.1016/0034-5687(70)90083-6
Schmitt, B. M., Berger, U. V., Douglas, R. M., Bevensee, M. O., Hediger, M. A., Haddad, G. G., et al. (2000). Na+CO3− cotransporters in rat brain: expression in glia, neurons, and choroid plexus. J. Neurosci. 20, 6839–6848.
Schwiening, C. J., and Boron, W. F. (1994). Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na+-dependent Cl−−HCO3− exchange. J. Physiol. 475, 59–67.
Sebat, J., Lakshmi, B., Malhotra, D., Troge, J., Lese-Martin, C., Walsh, T., et al. (2007). Strong association of de novo copy number mutations with autism. Science 316, 445–449. doi: 10.1126/science.1138659
Sin, W.-C., Moniz, D. M., Ozog, M. A., Tyler, J. E., Numata, M., and Church, J. (2009). Regulation of early neurite morphogenesis by the Na+/H+ exchanger NHE1. J. Neurosci. 29, 8946–8959. doi: 10.1523/JNEUROSCI.2030-09.2009
Sinning, A., and Hübner, C. A. (2013). Minireview: pH and synaptic transmission. FEBS Lett. 587, 1923–1928. doi: 10.1016/j.febslet.2013.04.045
Sinning, A., Liebmann, L., Kougioumtzes, A., Westermann, M., Bruehl, C., and Hübner, C. A. (2011). Synaptic glutamate release is modulated by the Na+-driven Cl−/HCO3− exchanger Slc4a8. J. Neurosci. 31, 7300–7311. doi: 10.1523/JNEUROSCI.0269-11.2011
Smith, G. A., Brett, C. L., and Church, J. (1998). Effects of noradrenaline on intracellular pH in acutely dissociated adult rat hippocampal CA1 neurones. J. Physiol. 512(Pt 2), 487–505. doi: 10.1111/j.1469-7793.1998.487be.x
Sterling, D., and Casey, J. R. (1999). Transport activity of AE3 chloride/bicarbonate anion-exchange proteins and their regulation by intracellular pH. Biochem. J. 344(Pt 1), 221–229. doi: 10.1042/0264-6021:3440221
Svichar, N., Esquenazi, S., Chen, H.-Y., and Chesler, M. (2011). Preemptive regulation of intracellular pH in hippocampal neurons by a dual mechanism of depolarization-induced alkalinization. J. Neurosci. 31, 6997–7004. doi: 10.1523/JNEUROSCI.6088-10.2011
Svichar, N., Waheed, A., Sly, W. S., Hennings, J. C., Hübner, C. A., and Chesler, M. (2009). Carbonic anhydrases CA4 and CA14 both enhance AE3-mediated Cl−−HCO3− exchange in hippocampal neurons. J. Neurosci. 29, 3252–3258. doi: 10.1523/JNEUROSCI.0036-09.2009
Tang, C. M., Dichter, M., and Morad, M. (1990). Modulation of the N-methyl-D-aspartate channel by extracellular H+. Proc. Natl. Acad. Sci. U.S.A. 87, 6445–6449. doi: 10.1073/pnas.87.16.6445
Thomas, R. C. (1976). The effect of carbon dioxide on the intracellular pH and buffering power of snail neurones. J. Physiol. 255, 715–735.
Tombaugh, G. C., and Somjen, G. G. (1996). Effects of extracellular pH on voltage-gated Na+, K+ and Ca2+ currents in isolated rat CA1 neurons. J. Physiol. 493, 719–732.
Tombaugh, G. C., and Somjen, G. G. (1997). Differential Sensitivity to intracellular pH among high- and low-threshold Ca2+ currents in isolated rat CA1 neurons. J. Neurophysiol. 77, 639–653.
Traynelis, S. F., and Cull-Candy, S. G. (1990). Proton inhibition of N-methyl-D-aspartate receptors in cerebellar neurons. Nature 345, 347–350. doi: 10.1038/345347a0
Vergo, S., Craner, M. J., Etzensperger, R., Attfield, K., Friese, M. A., Newcombe, J., et al. (2011). Acid-sensing ion channel 1 is involved in both axonal injury and demyelination in multiple sclerosis and its animal model. Brain 134, 571–584. doi: 10.1093/brain/awq337
Vilas, G. L., Johnson, D. E., Freund, P., and Casey, J. R. (2009). Characterization of an epilepsy-associated variant of the human Cl−/HCO3− exchanger AE3. Am. J. Physiol. Cell Physiol. 297, C526–C536. doi: 10.1152/ajpcell.00572.2008
Vincent, A. M., TenBroeke, M., and Maiese, K. (1999). Neuronal intracellular pH directly mediates nitric oxide-induced programmed cell death. J. Neurobiol. 40, 171–184. doi: 10.1002/(SICI)1097-4695(199908)40:2%3C171::AID-NEU4%3E3.0.CO;2-F
Virkki, L. V., Wilson, D. A., Vaughan-Jones, R. D., and Boron, W. F. (2002). Functional characterization of human NBC4 as an electrogenic Na+−HCO3− cotransporter (NBCe2). Am. J. Physiol. Cell Physiol. 282, C1278–C1289. doi: 10.1152/ajpcell.00589.2001
Waldmann, R., Champigny, G., Bassilana, F., Heurteaux, C., and Lazdunski, M. (1997). A proton-gated cation channel involved in acid-sensing. Nature 386, 173–177. doi: 10.1038/386173a0
Wang, C. Z., Yano, H., Nagashima, K., and Seino, S. (2000). The Na+-driven Cl−/HCO3− exchanger. Cloning, tissue distribution, and functional characterization. J. Biol. Chem. 275, 35486–35490. doi: 10.1074/jbc.C000456200
Wiemann, M., Frede, S., Bingmann, D., Kiwull, P., and Kiwull-Schöne, H. (2005). Sodium/Proton exchanger 3 in the medulla oblongata and set point of breathing control. Am. J. Respir. Crit. Care Med. 172, 244–249. doi: 10.1164/rccm.200411-1567OC
Wiemann, M., Frede, S., Tschentscher, F., Kiwull-Schöne, H., Kiwull, P., Bingmann, D., et al. (2008). NHE3 in the human brainstem: implication for the pathogenesis of the sudden infant death syndrome (SIDS)? Adv. Exp. Med. Biol. 605, 508–513. doi: 10.1007/978-0-387-73693-8_89
Wiemann, M., Schwark, J. R., Bonnet, U., Jansen, H. W., Grinstein, S., Baker, R. E., et al. (1999). Selective inhibition of the Na+/H+ exchanger type 3 activates CO2/H+-sensitive medullary neurones. Pflugers Arch. 438, 255–262. doi: 10.1007/s004240050907
Wolosker, H., Rocha, J. B., Engelender, S., Panizzutti, R., Miranda, J., and de Meis, L. (1997). Sarco/endoplasmic reticulum Ca2+-ATPase isoforms: diverse responses to acidosis. Biochem. J. 321, 545–550.
Xia, Y., Zhao, P., Xue, J., Gu, X. Q., Sun, X., Yao, H., et al. (2003). Na+ channel expression and neuronal function in the Na+/H+ exchanger 1 null mutant mouse. J. Neurophysiol. 89, 229–236. doi: 10.1152/jn.00488.2002
Xue, J., Douglas, R. M., Zhou, D., Lim, J. Y., Boron, W. F., and Haddad, G. G. (2003). Expression of Na+/H+ and HCO3− -dependent transporters in Na+/H+ exchanger isoform 1 null mutant mouse brain. Neuroscience 122, 37–46. doi: 10.1016/S0306-4522(03)00598-0
Yao, H., Ma, E., Gu, X. Q., and Haddad, G. G. (1999). Intracellular pH regulation of CA1 neurons in Na(+)/H(+) isoform 1 mutant mice. J. Clin. Invest. 104, 637–645. doi: 10.1172/JCI6785
Keywords: acidosis, alkalosis, brain, neurons, pH, NHE, NCBT
Citation: Ruffin VA, Salameh AI, Boron WF and Parker MD (2014) Intracellular pH regulation by acid-base transporters in mammalian neurons. Front. Physiol. 5:43. doi: 10.3389/fphys.2014.00043
Received: 01 November 2013; Accepted: 23 January 2014;
Published online: 13 February 2014.
Edited by:
Mario L. Diaz, Universidad de La Laguna, SpainReviewed by:
Alberto Passi, Università dell'Insubria, ItalyMitchell Chesler, New York University School of Medicine, USA
Copyright © 2014 Ruffin, Salameh, Boron and Parker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vernon A. Ruffin, Department of Physiology and Biophysics, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH 44106, USA e-mail:vernon.ruffin@case.edu
†Present address: Mark D. Parker, Department of Physiology and Biophysics, State University of New York at Buffalo, NY, USA