 Ira Kurtz
Ira Kurtz Quansheng Zhu
Quansheng Zhu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 19 December 2013
Sec. Membrane Physiology and Membrane Biophysics
Volume 4 - 2013 | https://doi.org/10.3389/fphys.2013.00350
This article is part of the Research TopicAcid-base sensing and regulation: molecular mechanisms and functional implications in health and diseaseView all 27 articles
NBCe1 belongs to the SLC4 family of base transporting membrane proteins that plays a significant role in renal, extrarenal, and systemic acid-base homeostasis. Recent progress has been made in characterizing the structure-function properties of NBCe1 (encoded by the SLC4A4 gene), and those factors that regulate its function. In the kidney, the NBCe1-A variant that is expressed on the basolateral membrane of proximal tubule is the key transporter responsible for overall transepithelial bicarbonate absorption in this nephron segment. NBCe1 mutations impair transepithelial bicarbonate absorption causing the syndrome of proximal renal tubular acidosis (pRTA). Studies of naturally occurring NBCe1 mutant proteins in heterologous expression systems have been very helpful in elucidation the structure-functional properties of the transporter. NBCe1 mutations are now known to cause pRTA by various mechanisms including the alteration of the transporter function (substrate ion interaction, electrogenicity), abnormal processing to the plasma membrane, and a perturbation in its structural properties. The elucidation of how NBCe1 mutations cause pRTA in addition to the recent studies which have provided further insight into the topology of the transporter have played an important role in uncovering its critically important structural-function properties.
Renal tubular acidosis (RTA) can be divided clinically into proximal RTA (pRTA) caused by defective proximal tubule bicarbonate absorption (Haque et al., 2012), and distal RTA (dRTA) resulting from impaired collecting duct net acid excretion (Battle and Haque, 2012). This review highlights the structure-function abnormalities in NBCe1-A caused by mutations in the transporter that result in autosomal recessive pRTA.
Transepithelial bicarbonate absorption in the proximal tubule is an indirect two-step process driven by the coupled transport of the apical Na+/H+ exchanger NHE3 (and a quantitatively less important apical H+-ATPase), in parallel with basolateral electrogenic Na+-base transport mediated by NBCe1-A (Figure 1; Boron, 2006; Hamm et al., 2013). According to current concepts, luminal bicarbonate is initially protonated via NHE3 and is ultimately via a dehydration reaction (accelerated by GPI anchored CAIV) converted to CO2 which is transferred across the apical membrane down its concentration gradient into the cytoplasm. In the cytoplasm, the reverse hydration of CO2 to HCO−3 and CO2−3 is catalyzed by cytoplasmic CAII. The basolateral membrane potential generated by the basolateral Na+-K+-ATPase and TASK2 K+ channels (Warth et al., 2004), drive electrogenic Na+-base transport via NBCe1-A. Interestingly, mutations in NHE3 have not been documented clinically; mutant CAIV causes retinitis pigmentosa (RP17) without pRTA having been reported in patients (Rebello et al., 2004) nor in mice with targeted CAIV disruption (Shah et al., 2005); and in patients with CAII mutations (discussed in greater detail below) causing a mixed pRTA and dRTA, the pRTA component can be mild suggesting that compensatory processes may be involved (Sly et al., 1985). Of the known proteins that play an important role in transepithelial bicarbonate absorption, only mutations in NBCe1-A that is the focus of this review have been documented to a cause a clinically important defect in proximal tubule transepithelial bicarbonate absorption.
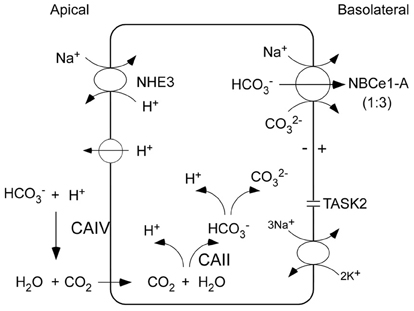
Figure 1. Coupled apical and basolateral H+/base transport in proximal tubule cells mediates trancellular transport of bicarbonate. The charge transport stoichiometry of NBCe1-A in the human proximal tubule is unknown and is depicted as 1:3. Luminal bicarbonate is initially protonated via NHE3 (and to a lesser extent an apical V-ATPase) and is converted to CO2 via a dehydration reaction that is accelerated by GPI anchored CAIV. Dissolved CO2 permeates the apical membrane passively through the lipid bilayer (or possibly through AQP1) down its concentration gradient into the cell. In the cytoplasm, the reverse hydration reaction catalyzed by cytoplasmic CAII converts CO2 to HCO−3 which is in equilibrium with CO2−3. The basolateral Na+-K+-ATPase coupled with TASK2 K+ channels generate the basolateral membrane potential, creating an electrical driving force for NBCe1-A mediated electrogenic Na+-base efflux.
NBCe1 is expressed in various extrarenal organs and accordingly patients with autosomal recessive pRTA due to NBCe1 mutations can be diagnosed clinically (without the need for genetic testing) because of the specific systemic phenotypic abnormalities that include, growth and mental retardation, glaucoma, cataracts, corneal opacities (band keratopathy), basal ganglia calcification, elevated serum lipase and amylase, and enamel defects (Tables 1 and 2; Igarashi et al., 1999; Kurtz, 2013). The loss of NBCe1 in mice results in an even more severe phenotype that includes colonic abnormalities, volume deletion, and decreased survival (Gawenis et al., 2007; Lacruz et al., 2010). Whether heterozygous family members also have subtle defects in proximal tubule bicarbonate transport and/or mild extrarenal (ocular, brain, growth, and enzymatic abnormalities) has not been determined. Interestingly, migraine headaches have been reported in patients with the R510H, L522P, and R881C missense mutations, 2311 delA, and a homozygous C-terminal 65 base-pair deletion (Suzuki et al., 2010). Headaches are hypothesized to be due to abnormal NMDA-mediated neuronal hyperactivity due to ER retained misfolded NBCe1-B in brain astrocytes. Headaches have also been reported in heterozygous family members of a patient with a 65 base-pair C-terminal deletion and the L522P mutation thought to be due to hetero-oligomer formation resulting in ER retention of wild-type transporters (Suzuki et al., 2010; Yamazaki et al., 2013). The possibility that single nucleotide polymorphisms (SNPs) alter the function of NBCe1-A in otherwise normal individuals was examined by Yamazaki et al. who studied the function of 4 SNPs in E122G, S356Y, K558R, and N640I, and reported that the base transport function of K558R was decreased 41-47% (Yamazaki et al., 2011).
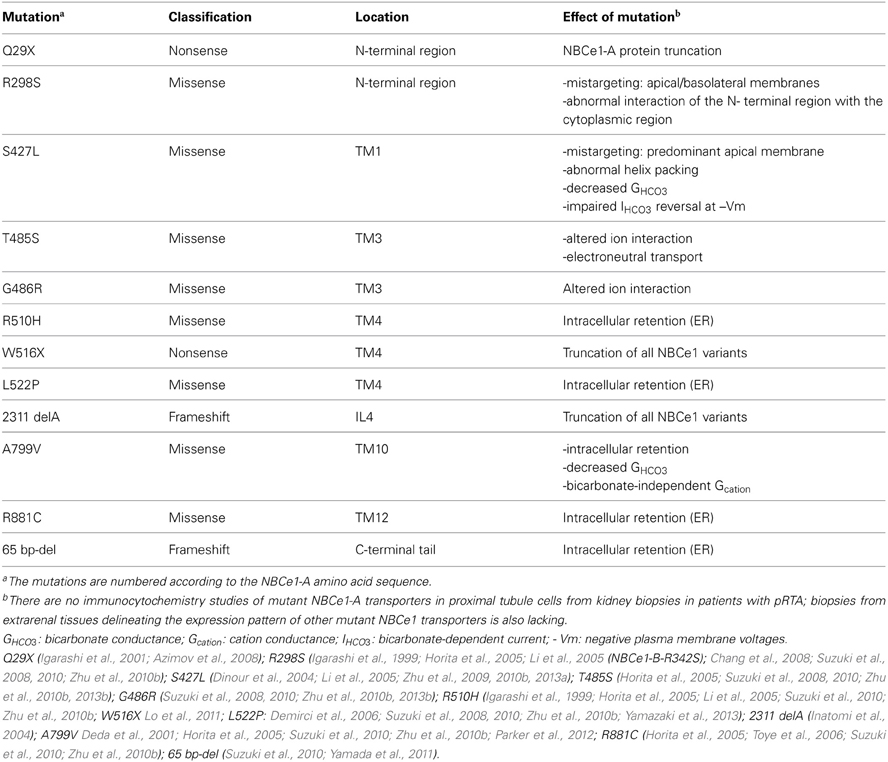
Table 1. NBCe1-A pRTA causing mutations.
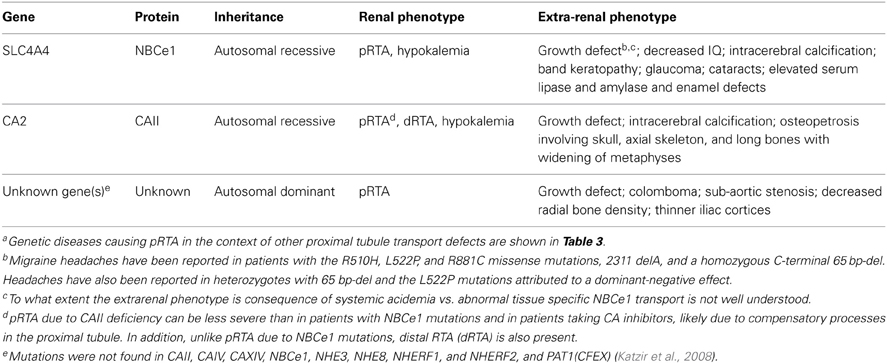
Table 2. Hereditarya proximal renal tubular acidosis.
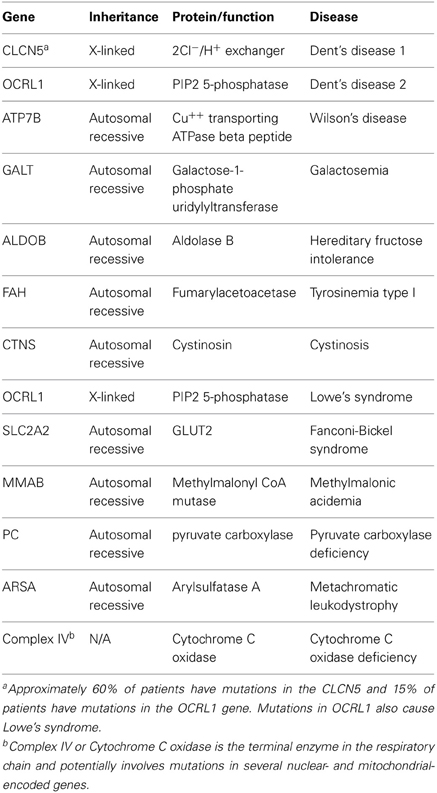
Table 3. Genetic causes of pRTA with additional proximal tubule transport abnormalities.
Mutations throughout the transporter including 2 nonsense mutations (Q29X, W516X), a frameshift deletion at nucleotide 2311 (2311 delA), a C-terminal 65 base-pair deletion from exon 23 to intron 23 (predicted to truncate the C-terminus), and 8 missense mutations are reported (Table 1; Figure 2; Zhu et al., 2010b). Based on the most recent topology of NBCe1 (Zhu et al., 2010b) other than the cytosolic Q29X, R298S, and C-terminal 65 base-pair deletion mutations, and the 2311 delA frameshift stop codon mutation in intracellular loop 4 (IL4), the remaining pRTA mutations reside in the transmembrane region. This finding is clinically relevant since the human SLC4A4 gene (Abuladze et al., 2000) encodes 3 variants (-A, -B, and -C) with additional variants (-D and -E) having been reported in mouse (Liu et al., 2011). All 5 mammalian NBCe1 variants have the identical transmembrane region differing in their N- and C-terminal sequences. NBCe1-A/-B/-C have been shown to mediate electrogenic Na+-base transport (McAlear et al., 2006) but differ in their tissue expression, intrinsic activity, and regulation.
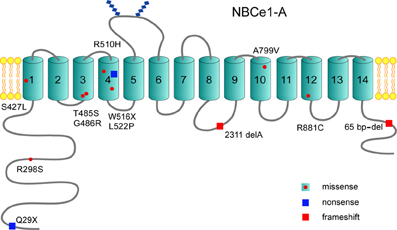
Figure 2. Structural topology of NBCe1-A: The NBCe1-A variant has been the most thoroughly studied and is depicted. Thus far, 12 NBCe1 mutations have been reported that include 8 missense mutations, 2 nonsense mutations, and 2 frameshift mutations. Of the reported missense mutations, the majority are localized to the transmembrane region. All NBCe1 variants share the identical transmembrane region where ion transport is mediated, and have a large cytoplasmic N-terminal region with a shorter cytoplasmic C-terminal tail.
NBCe1-A is predominantly expressed in S1 and S2 proximal tubules where it mediates basolateral Na+-base efflux contributing to the reabsorption of ~80% of the filtered bicarbonate (Skelton et al., 2010). NBCe1-B was originally cloned from pancreas and unlike NBCe1-A and NBCe1-C is widely expressed in multiple organs with a unique N-terminus wherein 85 aa replaces the 41 aa in NBCe1-A) (Abuladze et al., 1998). NBCe1-C originally cloned from rat brain has a unique C terminus (61 aa replaces 46 aa in NBCe1-A or B) ending in a type I PDZ-binding motif (McAlear et al., 2006). NBCe1-D and NBCe1-E transcripts more recently identified in mouse reproductive tissues are otherwise identical to NBCe1-A and NBCe1-B respectively, and within the cytosolic N-terminus lack a predicted nine amino-acid sequence (Liu et al., 2011). A long-standing unanswered question is whether the extrarenal symptoms in patients with pRTA result from defective locally expressed NBCe1 protein variants, or as a consequence of systemic acidemia due to pRTA per se.
NBCe1-A exists as a homodimer in the plasma membrane, of which each monomer functions independently (Kao et al., 2008; Sergeev et al., 2012). Extensive topological analysis has shown that each NBCe1-A monomer contains a large N-terminal cytoplasmic region, a plasma membrane embedded transmembrane region, and a short C-terminal cytoplasmic tail (Figure 2) (Zhu et al., 2010a,b, 2013a). Unlike the highly aqueous exposed C-terminal cytoplasmic tail, the N-terminal cytoplasmic region is modeled to form a domain structure and is tightly folded (Zhu et al., 2013a). The oligomerization of the N-terminal region appears to be pH and/or bicarbonate dependent (Gill, 2012). The transmembrane region has been determined to contain 14 lipid embedded transmembrane helices (Zhu et al., 2010a,b). TM5 and 6 bracket a large extracellular loop 3 that contains two glycosylated sites (Choi et al., 2003). Although NBCe1 variants differ in the sequence of their N-terminal regions and/or C-terminal tails, they all are predicted to share the same topographic structure as NBCe1-A.
The unique N-terminus of NBCe1-A functions as an autostimulatory domain (ASD) because of its ability to stimulate transporter function through an unknown mechanism (McAlear et al., 2006). The autostimulation has been hypothesized to be mediated by specific residues in the N-terminus of NBCe1-A which can potentially interact with a region(s) in the ion permeation pathway. Recent evidence suggests that that the N-terminal cytoplasmic region of NBCe1-A can interact with the transmembrane region (Zhu et al., 2013a). Studies of the cytoplasmic N-terminally localized R298S pRTA mutation indicate that it resides in an aqueous inaccessible tightly folded region that forms a “HCO−3 tunnel” whose structure is disrupted in the mutant transporter (Igarashi et al., 1999; Horita et al., 2005; Chang et al., 2008; Suzuki et al., 2010; Zhu et al., 2010b). By preventing the putative interaction of the N-terminus with the transmembrane region, the efficient delivery of base to the ion permeation pathway in the transmembrane region may be perturbed. An additional mechanism is suggested from studies of the NBCe1-A-R298S and NBCe1-B-R342S mutants expressed in MDCK cells. Both mutants were mistargeted to both the apical and basolateral membranes (Li et al., 2005; Suzuki et al., 2008) suggesting that targeting signal(s) may also be affected. The nonsense Q29X mutation in NBCe1-A is unique in that it affects the expression of the NBCe1-A variant specifically (Igarashi et al., 2001; Azimov et al., 2008). The mouse NBCe1-D variant which shares the same N-terminus if expressed in humans would be predicted to also be involved (Liu et al., 2011). There are currently no reported NBCe1 mutations that are specific for other NBCe1 variants. Previous studies in HEK 293-H cells expressing the NBCe1-A-Q29X mutant have shown that in the presence of the aminoglycoside G418 which induces ribosomal read-though, full-length functional NBCe1-A protein can be produced (Azimov et al., 2008).
Unlike NBCe1-A, the unique N- terminus of the -B and -C variants (and possible the -E variant) have an autoinhibitory domain (AID) that inhibits NBCe1 transport (McAlear et al., 2006; Lee et al., 2012). Studies of heterologously expressed NBCe1 variants in cultured cell and Xenopus oocyte expression systems have revealed the regulatory mechanisms involved. Amino acids 37–65 (a positively charged motif) in the N-terminus of NBCe1-B (absent in NBCe1-A and NBCe1-D) mediate the interaction of NBCe1-B with IRBIT and PIP2 (Hong et al., 2013). IRBIT activates NBCe1-B by preventing the inhibition by AID through recruitment of protein phosphatase 1 (PP1), which dephosphorylates the transporter thereby blocking the inhibition by the WNK/SPAK pathway (Yang et al., 2011; Lee et al., 2012). The autoinhibition by AID is stabilized by recruitment of SPAK by the WNK kinases (Hong et al., 2013). A staurosporine-sensitive kinase appears to be involved in the PIP2 mediated activation of NBCe1-B and -C (Thornell et al., 2012). The mechanism by which PIP2 activates NBCe1-A remains to be determined (Wu et al., 2009).
Intracellular Mg2+ inhibits bovine NBCe1-B currents heterologously expressed in HEK 293 cells, and the inhibition was decreased by truncation of NBCe1-B specific N-terminal residues (Yamaguchi and Ishikawa, 2008). Expression of IRBIT lowered the sensitivity of NBCe1-B currents to Mg2+ inhibition (Yamaguchi and Ishikawa, 2012) via an unknown mechanism. Mg2+ also inhibits the NBCe1-A variant that has been hypothesized to involve a Mg2+-dependent phosphatase (5'-lipid phosphatase) dephosphorylation of PIP2to PIP (Wu et al., 2009). Whether this effect is mediated by the NBCe1-A cytoplasmic N-terminus or C-terminal tail is unknown. The inhibition of NBCe1 variants by cytosolic Mg2+ has been postulated to provide a mechanism for reducing cellular dysfunction in ischemia (Wu et al., 2009).
Distal to the IRBIT interaction site in NBCe1-B, the Hsp70-like stress 70 protein chaperone STCH interacts with the residues amino acids 96–440 in the amino terminus and significantly increases the plasma membrane expression of the transporter (Bae et al., 2013). Whether the plasma membrane expression of NBCe1-A, -C, -D, and -E also increases after STCH binding (since this region is common to all known variants) has not been studied. Although the biologic role of this interaction has not been determined, it has been proposed that during acidemic conditions, enhanced NBCe1 transport would improve the efficiency of intracellular pH recovery (Bae et al., 2013).
NBCe1-A like all SLC4 proteins has a large transmembrane region that mediates ion transport. The current topology model of NBCe1-A indicates that it contains 14 transmembrane segments (TMs) with various lengths, which may resemble certain topologic characteristics with the vGLUT and LeuT prokaryotic Na+-coupled transporters (Yamashita et al., 2005; Watanabe et al., 2010; Zhu et al., 2010a,b). Topological analysis shows that NBCe1-A has a large glycosylated extracellular loop 3 (EL3) and a smaller loop 4 (EL4), and the extracellular surface is compactly folded. Various pRTA missense mutations reside in the transmembrane region thereby altering its structure-functional properties. The role of each TM in the structure-functional properties of NBCe1-A and the perturbations induced by specific pRTA mutations will now summarized in detail.
Evidence for the importance of TM1 is derived from structure-function studies of the wild-type transporter and from experiments that have addressed the abnormalities caused by the TM1 S427L pRTA mutation (Dinour et al., 2004; Li et al., 2005; Zhu et al., 2009, 2010b, 2013a). NBCe1-A-TM1 has unique properties that have been detailed in recent structural studies (Zhu et al., 2009, 2010b, 2013a). Specifically, TM1 is longer than a standard TM and contains 31 amino acids with an N-terminal cytosolic portion that has a helical conformation connecting the plasma membrane portion and the remaining cytoplasmic portion. In addition, TM1 contains several key residues including Asp416, Gln424, Tyr433, and Asn439 whose substitution with cysteine causes intracellular retention likely due to protein misfolding. Ala428, Ala435, and Thr442 are functionally important residues that line the ion permeation pathway. Thr442 also forms part of an extracellular gate involved in ion entry to the permeation pathway.
The importance of TM1 is further highlighted by the S427L pRTA mutation which decreases transporter function by 90% coupled with inability to reverse the direction of transport at very negative membrane potentials (Dinour et al., 2004). Recent studies have shown that Ser427 resides in a space-confined region and that the hydrophobicity of the serine side chain plays an important role in helix packing (Zhu et al., 2013a). Accordingly, the S427L mutation alters the conformation of TM1 by abolishing the potential ionic interactions between helices resulting in collapsed or an altered configuration of NBCe1-A ion permeation pathway. When expressed in MDCK cells, the S427L mutant is preferentially mistargeted to the apical membrane providing an additional mechanism for inducing pRTA (Li et al., 2005).
NBCe1-A-TM3 contains two adjacent mutations, T485S and R486R that cause pRTA (Horita et al., 2005; Suzuki et al., 2008, 2010; Zhu et al., 2010b, 2013b). The T485S mutation is unusual in that serine and threonine are similar structurally and chemically, and their substitution would not be expected to alter the function of the transporter. A recent study addressing this question has shown that Thr485 is located in an aqueous confined region accessible to NEM labeling that undergoes substrate-driven intra- and extracellular facing conformational changes (Zhu et al., 2013b). These results suggested that Thr485 resides in an NBCe1-A ion interaction site. This interpretation was further supported by the finding that the adjacent G486R mutation perturbs the function of the transporter and causes pRTA by altering the orientation of Thr485 (Zhu et al., 2013b).
The effect of the T485S mutation on transporter function is complex. Base transport is decreased by ~50% (Horita et al., 2005; Suzuki et al., 2008, 2010; Zhu et al., 2010b, 2013b) and in addition, the mutant transporter is electroneutral because of a change in its charge transport stoichiometry (positive charge to negative charge transport ratio) (Zhu et al., 2013b). The mechanism of the change in charge transport stoichiometry has recently been elucidated. Experiments using NO−3 as a surrogate for CO2−3 have shown that human wt-NBCe1-A functioning with a charge transport stoichiometry of 1:2 in HEK 293 cells mediates electrogenic Na+-CO2−3 cotransport (Zhu et al., 2013b). Furthermore, the electroneutral T485S mutant fails to transport Na+-NO−3. To account for these finding two pRTA proximal tubule transport models have been proposed (Figure 3). In model 1, assuming wt-NBCe1-A normally has a 1:2 charge transport stoichiometry in vivo, the T485S mutant loses its electrogenecity because of the preferential transport of Na+-HCO−3 rather Na+-CO2−3. In model 2, assuming a wt-NBCe1-A charge transport stoichiometry of 1:3 in vivo (1 Na+ + 1 HCO−3 + 1 CO2−3 cotransport), the T485S mutant becomes electroneutral because of loss of CO2−3 interaction resulting in Na+-HCO−3 cotransport. Independent of the initial stoichoimetry, the T485S mutant differs from other pRTA mutations in that transepithelial bicarbonate transport would be predicted to be impaired by a fundamentally different mechanism: the basolateral cellular influx of Na+ and HCO−3 down their respective concentration gradients.
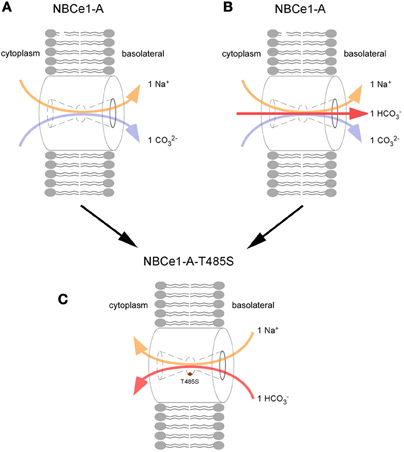
Figure 3. Loss of electrogenicity caused by the T485S pRTA mutation. The charge transport stoichiometry of NBCe1-A in the human proximal tubule is unknown and 2 potential models are depicted: In the 1:2 charge transport stoichiometry mode (A), wt-NBCe1-A is modeled to lose its electrogenecity because the T485S mutant (C) preferentially transports Na+-HCO−3 rather Na+-CO2−3. i.e., the anion interaction site favors HCO−3 rather than CO2−3. (B) wt-NBCe1-A transporting 1 Na+ + 1 HCO−3 + 1 CO2−3 (1:3 charge transport stoichiometry), following the loss of CO2−3 interaction, the T485S mutant is converted into an electroneutral Na+-HCO−3 transporter (C).
The R510H and L522P pRTA mutations result in ER retention due to protein misfolding (Igarashi et al., 1999; Horita et al., 2005; Li et al., 2005; Demirci et al., 2006; Suzuki et al., 2008, 2010; Zhu et al., 2010b; Yamazaki et al., 2013). On this basis and because TM4 carries potential signal anchor and stop transfer sequences, it has been suggested that TM4 functions as a scaffolding helix that is essential for normal protein folding. Although not well studied, the finding that the histidine substitution at Arg510 leads to protein misfolding suggests the magnitude of the positive charge and/or the size of the side-chain of Arg510 is required for ionic interaction between TM4 and neighboring TMs. The L522P mutation has been more extensively studied where it has been shown that L522C and L522I are not ER retained and do not alter NBCe1-A plasma membrane processing (Zhu et al., 2010b; Yamazaki et al., 2013). These findings suggest that the proline likely as a result of increased flexibility causes helix disruption, significant protein misfolding, and subsequent intracellular retention. Finally W516X mutation located in TM4 results in a truncated and likely misfolded protein (Lo et al., 2011).
The stilbene inhibitor 4,4′-diisothiocyanatostilbene-2,2′-disulfonate (DIDS) binds to NBCe1-A from both the extracellular and intracellular surfaces. Extracellular DIDS binds to a KKMIK motif in TM5 resulting in inhibition of transporter function through a mechanism that is currently unknown (Lu and Boron, 2007). At more positive membrane voltages the apparent affinity of the interaction between DIDS and transporter is increased that has been attributed to membrane voltage dependent conformational changes (Yamaguchi and Ishikawa, 2005; Lu and Boron, 2007). The intracellular binding site(s) is currently undetermined (Heyer et al., 1999). Moreover, site(es) of the interaction of other inhibitors including tenidap (Ducoudret et al., 2001), benzamil (Ducoudret et al., 2001), and S0859 (Ch'en et al., 2008) and whether they interact with residues in TM5 remains to be addressed. In separate studies, the TM5- Asp555 residue that is near the proposed DIDS binding site has been identified as being functionally important and involved in anion selectivity (Yang et al., 2009). Substitution of Asp555 with glutamate induces outward rectifying Cl−, NO−3, and SCN− currents.
EL3 (56 amino acids), the largest loop in NBCe1 is glycosylated and contains 4 cysteine residues (Zhu et al., 2012). Structural studies have shown that in each monomer, the 4 cysteines are intramolecular disufided forming a highly ordered topologically domain (Zhu et al., 2012). Based on homology with Cys-loop ligand-gated ion channel superfamily, this region in EL3 may bind a ligand that regulates the function of the transporter. The next extracellular loop, EL4, binds plasma membrane CAIV (Alvarez et al., 2003) and CAIX (Orlowski et al., 2012). EL4 which contains a large number of proline residues is also involved in the electrogenic properties of the transporter (Chen et al., 2011). It is possible that the flexible EL4 loop (formed between TMs7-8) interacts with residues embedded in the lipid bilayer that alter ion interaction with NBCe1-A. Residues in TM8 which potentially form part of the ion permeation pathway (see below) may interact with EL4 to modulate the electrogenic properties of NBCe1.
Based on the finding that residues in TM8 were reported to form part of the ion permeation pathway in AE1 (Tang et al., 1999), McAlear et al. proposed that NBCe1-A-TM8 might have similar properties (McAlear and Bevensee, 2006). Using a cysteine scanning mutagenesis approach, residues in TM8 were identified to play a role in ion permeation with Leu750 being particularly involved. In further support of residues in TM8 being involved in ion permeation, pCMBS accessibility was decreased in the presence of substrate ions and the stilbene inhibitor 4.4-dinitro-stilbene-2,2'-disulfonate (DNDS).
Although more typically seen in patients with Fanconi's syndrome (Soriano, 2002), patients with isolated pRTA can be hypokalemic as a result of initial enhanced collecting duct HCO−3 delivery (prior to achieving a steady state), an elevation of serum aldosterone due to volume depletion, and enhanced collecting duct HCO−3 delivery during bicarbonate therapy. In addition to the known renal and extrarenal phenotype of patients with NBCe1 mutations, Deda et al reported a patient with extra-renal K+ loss (diarrhea, and vomiting) that resulting in severe acute hypokalemia (Deda et al., 2001). The patient was subsequently shown to have a new A799V pRTA mutation that significantly decreased the function of mutant NBCe1-A (Horita et al., 2005; Suzuki et al., 2010; Zhu et al., 2010b). Further studies on the NBCe1-A799V demonstrated that the mutant transporter had an associated HCO−3-independent cation leak conductance (Parker et al., 2012). It was hypothesized that since there is evidence that NBCe1 is expressed in skeletal muscles (sarcollema and possibly t-tubules), during severe hypokalemia, the HCO−3-independent cation leak conductance would result in exacerbated muscle weakness when compared to patients with other NBCe1 pRTA mutations.
The TM12 R881C pRTA mutation (Horita et al., 2005; Toye et al., 2006; Suzuki et al., 2010; Zhu et al., 2010a,b) induces ER retention when expressed in mammalian cells likely due to protein misfolding, suggesting that TM12 is involved in helix packing (Zhu et al., 2010a). In Xenopus oocytes where plasma membrane expression is decreased but present (Horita et al., 2005; Toye et al., 2006), when corrected for the plasma membrane expression level, the transporter appears to function normally (Toye et al., 2006).
Recent evidence suggests that the entire C-terminal transmembrane region from TM10-14 (Ala800 and Lys967), plays an important role helix packing and protein folding (Zhu et al., 2010a). Specifically, 18 residues clustered on the surface of each TM form intramolecular hydrogen bonds contributing to helix packing thereby stabilizing the structure of NBCe1. The loops connecting TMs11-14 are tightly folded rather than being aqueous exposed. At the beginning of TM11, 5 residues likely function as a topogenic signal guiding TM11 into the lipid bilayer. TM11 and 12 are abruptly bent into the lipid bilayer at Met858 which is bracketed by Pro857 and Pro858. Cryptic intracellular loop 6 (IL6) that connects TM12 and 13 is also tightly folded and appears to interact with the cytoplasmic region. TM12-Lys924 appears to act as a counter-ion countributing to helix packing. The final extracellular 7 (EL7; Thr926-Ala929) is only slightly exposed to the aqueous media, suggesting that it is folded in the transmembrane region.
It has also been hypothesized based on domain swapping experiments that TM12 and TM6 form a “functional unit” required for plasma membrane processing (Chen et al., 2012). Specifically, in an NBCe1-A chimera formed by replacing TM6 and what was referred to as “TM12” (according to the most recent topologic model of NBCe1 containing residues from TM12, intracellular loop 6 (IL6) and TM13) with corresponding regions from electroneutral NBCn1, membrane processing was significantly impaired. This interpretation of the data is likely premature given that mixed chimera proteins can be improperly folded resulting in ER retention (Fujinaga et al., 2003). In addition, specific residues in TM12 have been shown to play a role in helix packing (Zhu et al., 2010a).
The intracellular lipid/aqueous interface of TM14 is marked at Asp960 and Pro963 likely induces a kink exposing the C-terminal cytoplasmic tail to the aqueous cytoplasm (Zhu et al., 2010a). In the NBCe1-A dimer, there is a close association between the C-terminal cytoplasmic tail of each monomer raising the question as to whether this grouping of two stretches of strongly charged amino acids plays an important functional regulatory role (Zhu et al., 2010c). Two pieces of evidence indicate that the C-terminal tail plays a role in membrane processing and targeting. First, in the pRTA 65 bp-del frame shift mutation which truncates the C-terminal tail, the mutant transporter is retained intracellularly in the ER in mammalian cells (though not in Xenopus oocytes) (Suzuki et al., 2010). Secondly, when expressed in MDCK cells, a C-terminal tail 1010QQPFLS1015 motif functions as a basolateral targeting signal (Li et al., 2004).
The C-terminal tail may also have a functional role. Using isothermal titration calorimetry CAII was shown to bind to the cytosolic C-terminus of NBCe1 at a D986NDD989 motif (Gross et al., 2002). Based on studies of CAII interaction with AE1, it was proposed that NBCe1 and CAII form a transport metabolon wherein NBCe1 transfers ions intra-molecularly to CAII. Subsequent studies provided additional support for a functional interaction between NBCe1 and CAII (Pushkin et al., 2004; Becker and Deitmer, 2007). In addition, other CA isoforms including plasma membrane CAIV and CAIX were also shown to bind to NBCe1-EL4 (Alvarez et al., 2003; Orlowski et al., 2012). However, not all groups were able to document a functional interaction with CAII (Lu et al., 2006; Piermarini et al., 2007; Yamada et al., 2011). In comparing the conflicting data between these studies, there is a need to consider the various techniques/preparations used including differences in aqueous binding vs. solid phase interaction, sensitivity, specificity, signal/noise, and artifacts introduced by the various assays employed. In support of a lack of clinically important interaction between CAII and NBCe1 is the finding that some patients with loss of function CAII mutations, and mice with targeted disruption of CAII do not have as severe defect in proximal tubule bicarbonate absorption as predicted (Sly et al., 1985; Lewis et al., 1988). In addition, the systemic acidemia due to loss of CAII is not as severe as the metabolic acidosis in patients with NBCe1 mutations and mice with loss of NBCe1 function. Whether compensatory mechanisms account for these findings and the potential utility of performing conditional targeted disruption of CAII in mice remains to be determined. A renal phenotype has also not been reported in patients with CAIV mutations with retinitis pigmentosa (RP17) (Rebello et al., 2004). The important question of whether carbonic anhydrase can bind and modulate the function of NBCe1 has recently been re-examined by Schueler who showed that CAI, CAII, and CAIII stimulate NBCe1-A transport in Xenopus oocytes (Schueler et al., 2011). The increase in NBCe1-A transport was attributed to CA enzymatic activity rather than an intramolecular proton shuttle reaction.
In 1977, a family from Costa Rica was reported of whom 9 members had isolated pRTA and short stature with dominant inheritance (Table 2; Luis et al., 1977). A follow-up study on 2 of the family members documented decreased bone density (Lemann et al., 2000). A second family with autosomal dominant isolated pRTA and short stature has been subsequently reported (Katzir et al., 2008). The authors ruled out mutations in the coding regions and splice sites of 9 proteins involved in proximal tubule H+/base transport including CAII, CAIV, CAXIV, NBCe1, NHE3, NHE8, NHERF1, and NHERF2, and PAT1(CFEX). Importantly, intron sequence and promoter abnormalities in the genes coding for these proteins were not completely excluded. Determining the molecular basis for the cause of pRTA in these patients will potentially uncover new transport and/or regulatory mechanisms involved in mammalian proximal tubule bicarbonate absorption.
Thermodynamics dictates that when the electrochemical driving force (μ) across NBCe1-A in the basolateral membrane of the proximal tubule has a positive value, the transporter will mediate Na+-coupled base efflux in the direction of cytoplasm to peritubular blood (Kurtz et al., 2004; Zhu et al., 2013b). The charge transport stoichiometry of NBCe1 is an important determinant of the value of the electrochemical driving force. Although the charge transport stoichiometry of NBCe1-A in the human proximal tubule has not been measured, it has been implicitly assumed to be 1:3, largely based on the belief that a 1:2 charge transport stoichiometry would not result in a positive value for μ. The literature on the charge transport stoichiometry of NBCe1-A is somewhat confusing given that species-specific, preparation specific, cell-specific, and perhaps technique-specific factors may play a role. In the in vivo rat proximal tubule Yoshitomi et al obtained a value for the charge transport stoichiometry of 1:3 (Yoshitomi et al., 1985). In the isolated perfused rabbit proximal tubule, the charge transport stoichiometry appears to vary with the properties of the solutions used: i.e., 1:2 in Ringers solution (Seki et al., 1993) and 1:2.7 (Müller-Berger et al., 1997) in tubules bathed in norepinephrine and DMEM. In Necturus proximal tubules in vivo, the charge transport stoichiometry was found to decrease from 1:3 to 1:2 during respiratory acidosis (Planelles et al., 1993). In heterologously expressed NBCe1-A, Ca2+ (Müller-Berger et al., 2001), phosphorylation state, (Gross et al., 2001b) and cell-type (Gross et al., 2001a) appear to modulate the charge transport stoichiometry. Specifically, PKA dependent phosphorylation of NBCe1-A-Ser982 was reported to shift the charge transport stoichiometry from 1:3 to 1:2 (Gross et al., 2001b). It has been postulated that Ca2+ also induces a shift in charge transport stoichiometry from 1:2 to 1:3 via a change in the phosphorylation state the transporter (Müller-Berger et al., 2001). Whether Ca2+ modulation of the transporter stoichiometry can be detected in mammalian expression systems is unknown. Finally, whether NBCe1-A can change the stoichiometry in the in vivo human proximal tubule remains an unresolved question.
It is generally accepted that NBCe1-A functions as a Na+-CO2−3-HCO−3 cotransporter (charge transport stoichiometry of 1:3) thereby mediating Na+-coupled base efflux, despite the uncertainty as to the exact electrochemical driving force across the basolateral membrane in the human proximal tubule. An important unanswered question is whether NBCe1-A in the human proximal tubule has a charge transport stoichiometry of 1:2, as has been clearly demonstrated when the human transporter is heterologously expressed in mammalian HEK 293 cells (Zhu et al., 2013b). Although a charge transport stoichiomtery of 1:2 is compatible with a 1 Na+: 2 HCO−3 transport mode, recent data using NO−3 as a surrogate for CO−3 transport is more compatible with a Na+-CO2−3 cotransport mode (Zhu et al., 2013b). In addition, preliminary data using surface pH electrodes in the Xenopus oocyte expression system also suggests that rat NBCe1-A mediates Na+-CO2−3 cotransport (Lee et al., 2011). Unfortunately, given that the basolateral membrane potential and the in vivo gradients of the transported ions in the human proximal tubule are unknown, whether the electrochemical driving force is sufficient to drive NBCe1-A mediated basolateral Na+-coupled CO2−3 efflux is uncertain. Using data from the rat proximal tubule, a species where the necessary data is available, it has recently been calculated that NBCe1-A would indeed be capable of mediating basolateral Na+-CO2−3 efflux while at the same time being sensitive to small changes in the electrochemical chemical potential of the transported ions (Zhu et al., 2013b). If indeed future studies confirm that NBCe1-A normally functions in the proximal as a Na+-CO2−3 cotransporter, one could argue that its name should be changed for example to NCCe1-A (sodium carbonate cotransporter electrogenic 1-A).
In this regard, it is instructive to again consider how the loss of electrogenicity in the context of the T485S mutation causes pRTA. Unlike all other known pRTA mutations, NBCe1-A-T485S would be insensitive to the basolateral membrane potential. As shown in Figure 3, given the inwardly directed initial chemical gradients (peritubular to cell), the mutant transporter would be predicted to transport Na+-HCO−3 intracellularly across the basolateral membrane impairing proximal tubule HCO−3 absorption. If this is in fact is what is occurring in vivo, it would represent an entirely novel mechanism for generating pRTA. In this regard, transgenic mice expressing NBCe1-A-T485S would be an important tool for studying various aspects of this sequence of events in greater detail.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by the NIH grants DK077162 and DK058563 (to Ira Kurtz) and Coplon grant from Satellite Healthcare (to Quansheng Zhu).
Abuladze, N., Lee, I., Newman, D., Hwang, J., Boorer, K., Pushkin, A., et al. (1998). Molecular cloning, chromosomal localization, tissue distribution, and functional expression of the human pancreatic sodium bicarbonate cotransporter. J. Biol. Chem. 273, 17689–17695. doi: 10.1074/jbc.273.28.17689
Abuladze, N., Song, M., Pushkin, A., Newman, D., Lee, I., Nicholas, S., et al. (2000). Structural organization of the human NBC1 gene: kNBC1 is transcribed from an alternative promoter in intron 3. Gene 251, 109–122. doi: 10.1016/S0378-1119(00)00204-3
Alvarez, B. V., Loiselle, F. B., Supuran, C. T., Schwartz, G. J., and Casey, J. R. (2003). Direct extracellular interaction between carbonic anhydrase IV and the human NBC1 sodium/bicarbonate co-transporter. Biochemistry 42, 12321–12329. doi: 10.1021/bi0353124
Azimov, R., Abuladze, N., Sassani, P., Newman, D., Kao, L., Liu, W., et al. (2008). G418-mediated ribosomal read-through of a nonsense mutation causing autosomal recessive proximal renal tubular acidosis. Am. J. Physiol. Renal Physiol. 295, F633–F641. doi: 10.1152/ajprenal.00015.2008
Bae, J. S., Koo, N. Y., Namkoong, E., Davies, A. J., Choi, S. K., Shin, Y., et al. (2013). Chaperone stress 70 protein (STCH) binds and regulates two acid/base transporters NBCe1-B and NHE1. J. Biol. Chem. 288, 6295–6305. doi: 10.1074/jbc.M112.392001
Battle, D., and Haque, S. K. (2012). Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol. Dial. Transplant. 27, 3691–3704. doi: 10.1093/ndt/gfs442
Becker, H. M., and Deitmer, J. W. (2007). Carbonic anhydrase II increases the activity of the human electrogenic Na+/HCO−3 cotransporter. J. Biol. Chem. 282, 13508–13521. doi: 10.1074/jbc.M700066200
Boron, W. F. (2006). Acid-base transport by the renal proximal tubule. J. Am. Soc. Nephrol. 17, 2368–2382. doi: 10.1681/ASN.2006060620
Ch'en, F. F., Villafuerte, F. C., Swietach, P., Cobden, P. M., and Vaughan-Jones, R. D. (2008). S0859, an N-cyanosulphonamide inhibitor of sodium-bicarbonate cotransport in the heart. Br. J. Pharm. 153, 972–982. doi: 10.1038/sj.bjp.0707667
Chang, M. H., DiPiero, J., Sonnichsen, F. D., and Romero, M. F. (2008). Entry to “formula tunnel” revealed by SLC4A4 human mutation and structural model. J. Biol. Chem. 283, 18402–18410. doi: 10.1074/jbc.M709819200
Chen, L. M., Liu, Y., and Boron, W. F. (2011). Role of an extracellular loop in determining the stoichiometry of Na+-HCO−3 cotransporters. J. Physiol. 589, 877–890. doi: 10.1113/jphysiol.2010.198713
Chen, L. M., Qin, X., Moss, F. J., Liu, Y., and Boron, W. F. (2012). Effect of simultaneously replacing putative TM6 and TM12 of human NBCe1-A with those from NBCn1 on surface abundance in Xenopus oocytes. J. Membr. Biol. 245, 131–140. doi: 10.1007/s00232-012-9421-y
Choi, I., Hu, L., Rojas, J. D., Schmitt, B. M., and Boron, W. F. (2003). Role of glycosylation in the renal electrogenic Na+-HCO−3 cotransporter (NBCe1). Am. J. Physiol. Renal Physiol. 284, F1199–1206.
Deda, G., Ekim, M., Güven, A., Karagöl, U., and Tümer, N. (2001). Hypopotassemic paralysis: a rare presentation of proximal renal tubular acidosis. J. Child Neurol. 16, 770–771. doi: 10.1177/088307380101601013
Demirci, F. Y., Chang, M. H., Mah, T. S., Romero, M. F., and Gorin, M. B. (2006). Proximal renal tubular acidosis and ocular pathology: a novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1). Mol. Vis. 12, 324–330.
Dinour, D., Chang, M. H., Satoh, J., Smith, B. L., Angle, N., Knecht, A., et al. (2004). A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J. Biol. Chem. 279, 52238–52246. doi: 10.1074/jbc.M406591200
Ducoudret, O., Diakov, A., Müller-Berger, S., Romero, M. F., and Frömter, E. (2001). The renal Na-HCO3-cotransporter expressed in Xenopus laevis oocytes: inhibition by tenidap and benzamil and effect of temperature on transport rate and stoichiometry. Pflugers Arch. 442, 709–717. doi: 10.1007/s004240100594
Fujinaga, J., Loiselle, F. B., and Casey, J. R. (2003). Transport activity of chimaeric AE2-AE3 chloride/bicarbonate anion exchange proteins. Biochem J. 371, 687–696. doi: 10.1042/BJ20030007
Gawenis, L. R., Bradford, E. M., Prasad, V., Lorenz, J. N., Simpson, J. E., Clarke, L. L., et al. (2007). Colonic anion secretory defects and metabolic acidosis in mice lacking the NBC1 Na+/HCO−3 cotransporter. J. Biol. Chem. 282, 9042–9052. doi: 10.1074/jbc.M607041200
Gill, H. S. (2012). pH-sensitive self-associations of the N-terminal domain of NBCe1-A suggest a compact conformation under acidic intracellular conditions. Prot. Pep. Let. 19, 1054–1063. doi: 10.2174/092986612802762642
Gross, E., Hawkins, K., Abuladze, N., Pushkin, A., Cotton, C. U., Hopfer, U., et al. (2001a). The stoichiometry of the electrogenic sodium bicarbonate cotransporter NBC1 is cell-type dependent. J. Physiol. 531, 597–603. doi: 10.1111/j.1469-7793.2001.0597h.x
Gross, E., Hawkins, K., Pushkin, A., Sassani, P., Dukkipati, R., Abuladze, N., et al. (2001b). Phosphorylation of Ser982 in the sodium bicarbonate cotransporter kNBC1 shifts the HCO−3: Na+ stoichiometry from 3:1 to 2:1 in murine proximal tubule cells. J. Physiol. 537, 659–665. doi: 10.1113/jphysiol.2001.012956
Gross, E., Pushkin, A., Abuladze, N., Fedotoff, O., and Kurtz, I. (2002). Regulation of the sodium bicarbonate cotransporter kNBC1 function: role of Asp986, Asp988 and kNBC1-carbonic anhydrase II binding. J. Physiol. 544, 679–685. doi: 10.1113/jphysiol.2002.029777
Hamm, L. L., Alperin, R. J., and Preisig, P. A. (2013). “Cellular mechanisms of unrinary acidification,” in Seldin and Giebisch's The Kidney: Physiology and Pathophysiology, eds R. J. Alperin, M. Caplan, and O.W. Moe (Amsterdam; Boston: Elsevier/Academic Press), 1917–1978. doi: 10.1016/B978-0-12-381462-3.00055-0
Haque, S. K., Ariceta, G., and Batlle, D. (2012). Proximal renal tubular acidosis: a not so rare disorder of multiple etiologies. Nephrol. Dial. Transplant. 27, 4273–4287. doi: 10.1093/ndt/gfs493
Heyer, M., Müller-Berger, S., Romero, M. F., Boron, W. F., and Frömter, E. (1999). Stoichiometry of the rat kidney Na+-HCO−3 cotransporter expressed in Xenopus laevis oocytes. Pflugers Arch. 438, 322–329. doi: 10.1007/s004240050916
Hong, J. H., Yang, D., Shcheynikov, N., Ohana, E., Shin, D. M., and Muallem, S. (2013). Convergence of IRBIT, phosphatidylinositol (4,5) bisphosphate, and WNK/SPAK kinases in regulation of the Na+-HCO−3 cotransporters family. Proc. Natl. Acad. Sci. U.S.A. 110, 4105–4110. doi: 10.1073/pnas.1221410110
Horita, S., Yamada, H., Inatomi, J., Moriyama, N., Sekine, T., Igarashi, T., et al. (2005). Functional analysis of NBC1 mutants associated with proximal renal tubular acidosis and ocular abnormalities. J. Am. Soc. Nephrol. 16, 2270–2278. doi: 10.1681/ASN.2004080667
Igarashi, T., Inatomi, J., Sekine, T., Cha, S. H., Kanai, Y., Kunimi, M., et al. (1999). Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat. Genet. 23, 264–266. doi: 10.1038/15440
Igarashi, T., Inatomi, J., Sekine, T., Seki, G., Shimadzu, M., Tozawa, F., et al. (2001). Novel nonsense mutation in the Na+/HCO−3 cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. J. Am. Soc. Nephrol. 12, 713–718.
Inatomi, J., Horita, S., Braverman, N., Sekine, T., Yamada, H., Suzuki, Y., et al. (2004). Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflug. Arch. 448, 438–444. doi: 10.1007/s00424-004-1278-1
Kao, L., Sassani, P., Azimov, R., Pushkin, A., Abuladze, N., Peti-Peterdi, J., et al. (2008). Oligomeric structure and minimal functional unit of the electrogenic sodium bicarbonate cotransporter NBCe1-A. J. Biol. Chem. 283, 26782–26794. doi: 10.1074/jbc.M804006200
Katzir, Z., Dinour, D., Reznik-Wolf, H., Nissenkorn, A., and Holtzman, E. (2008). Familial pure proximal renal tubular acidosis-a clinical and genetic study. Nephrol. Dial. Transplant. 23, 1211–1215. doi: 10.1093/ndt/gfm583
Kurtz, I. (2013). “SLC4 Sodium-driven bicarbonate transporters,” in Seldin and Giebisch's The Kidney: Physiology and Pathophysiology, ed Alperin, R.J., Caplan, M., Moe, O.W. (Amsterdam; Boston: Elsevier/Academic Press), 1837–1860. doi: 10.1016/B978-0-12-381462-3.00053-7
Kurtz, I., Petrasek, D., and Tatishchev, S. (2004). Molecular mechanisms of electrogenic sodium bicarbonate cotransport: structural and equilibrium thermodynamic considerations. J. Membr. Biol. 197, 77–90. doi: 10.1007/s00232-003-0643-x
Lacruz, R. S., Nanci, A., White, S. N., Wen, X., Wang, H., Zalzal, S. F., et al. (2010). The sodium bicarbonate cotransporter (NBCe1) is essential for normal development of mouse dentition. J. Biol. Chem. 285, 24432–24438. doi: 10.1074/jbc.M110.115188
Lee, S. K., Boron, W. F., and Parker, M. D. (2012). Relief of autoinhibition of the electrogenic Na-HCO3 cotransporter NBCe1-B: role of IRBIT vs. amino-terminal truncation. Am. J. Physiol. Cell. Physiol. 302, C518–526. doi: 10.1152/ajpcell.00352.2011
Lee, S. K., Grichtchenko, I. I., and Boron, W. F. (2011). Distinguishing HCO−3 from CO2−3 transport by NBCe1-A. FASEB J. 25, 656.9.
Lemann, Jr. J., Adams, N. D., Wilz, D. R., and Brenes, L. G. (2000). Acid and mineral balances and bone in familial proximal renal tubular acidosis. Kid. Int. 58, 1267–1277. doi: 10.1046/j.1523-1755.2000.00282.x
Lewis, S. E., Erickson, R. P., Barnett, L. B., Venta, P. J., and Tashian, R. E. (1988). N-ethyl-N-nitrosourea-induced null mutation at the mouse Car-2 locus: an animal model for human carbonic anhydrase II deficiency syndrome. Proc. Natl. Acad. Sci. U.S.A. 85, 1962–1966. doi: 10.1073/pnas.85.6.1962
Li, H. C., Worrell, R. T., Matthews, J. B., Husseinzadeh, H., Neumeier, L., Petrovic, S., et al. (2004). Identification of a carboxyl-terminal motif essential for the targeting of Na+-HCO−3 cotransporter NBC1 to the basolateral membrane. J. Biol. Chem. 279, 43190–43197. doi: 10.1074/jbc.M405780200
Li, H. C., Szigligeti, P., Worrell, R. T., Matthews, J. B., Conforti, L., and Soleimani, M. (2005). Missense mutations in Na+-HCO−3 cotransporter NBC1 show abnormal trafficking in polarized kidney cells: a basis of proximal renal tubular acidosis. Am. J. Physiol. Renal Physiol. 289, F61–F71. doi: 10.1152/ajprenal.00032.2005
Liu, Y., Xu, J. Y., Wang, D. K., Wang, L., and Chen, L. M. (2011). Cloning and identification of two novel NBCe1 splice variants from mouse reproductive tract tissues: a comparative study of NCBT genes. Genomics 98, 112–119. doi: 10.1016/j.ygeno.2011.04.010
Lo, Y. F., Yang, S. S., Seki, G., Yamada, H., Horita, S., Yamazaki, O., et al. (2011). Severe metabolic acidosis causes early lethality in NBC1 W516X knock-in mice as a model of human isolated proximal renal tubular acidosis. Kid. Int. 79, 730–741. doi: 10.1038/ki.2010.523
Lu, J., and Boron, W. F. (2007). Reversible and irreversible interactions of DIDS with the human electrogenic Na/HCO3 cotransporter NBCe1-A: role of lysines in the KKMIK motif of TM5. Am. J. Physiol. Cell. Physiol. 292, C1787–1798. doi: 10.1152/ajpcell.00267.2006
Lu, J., Daly, C. M., Parker, M. D., Gill, H. S., Piermarini, P. M., Pelletier, M. F., et al. (2006). Effect of human carbonic anhydrase II on the activity of the human electrogenic Na/HCO3 cotransporter NBCe1-A in Xenopus oocytes. J. Biol. Chem. 281, 19241–19250. doi: 10.1074/jbc.M602181200
Luis, G., Brenes, J. N., Brenes, M., and Hernandez, M. (1977). Familial proximal renal tubular acidosis a distinct clinical entity. Am. J. Med. 63, 244–252. doi: 10.1016/0002-9343(77)90238-8
McAlear, S. D., and Bevensee, M. O. (2006). A cysteine-scanning mutagenesis study of transmembrane domain 8 of the electrogenic sodium/bicarbonate cotransporter NBCe1. J. Biol. Chem. 281, 32417–32427. doi: 10.1074/jbc.M607253200
McAlear, S. D., Liu, X., Williams, J. B., McNicholas-Bevensee, C. M., and Bevensee, M. O. (2006). Electrogenic Na/HCO3 cotransporter (NBCe1) variants expressed in Xenopus oocytes: functional comparison and roles of the amino and carboxy termini. J. Gen. Physiol. 127, 639–658. doi: 10.1085/jgp.200609520
Müller-Berger, S., Ducoudret, O., Diakov, A., and Frömter, E. (2001). The renal Na-HCO−3 cotransporter expressed in Xenopus laevis oocytes: change in stoichiometry in response to elevation of cytosolic Ca2+ concentration. Pflugers Arch. 442, 718–728. doi: 10.1007/s004240100592
Müller-Berger, S., Nesterov, V. V., and Frömter, E. (1997). Partial recovery of in vivo function by improved incubation conditions of isolated renal proximal tubule. II. Change of Na-HCO3 cotransport stoichiometry and of response to acetazolamide. Pflugers Arch. 434, 383–391. doi: 10.1007/s004240050411
Orlowski, A., De Giusti, V. C., Morgan, P. E., Aiello, E. A., and Alvarez, B. V. (2012). Binding of carbonic anhydrase IX to extracellular loop 4 of the NBCe1 Na+/HCO−3 cotransporter enhances NBCe1-mediated HCO−3 influx in the rat heart. Am. J. Physiol. Cell Physiol. 303, C69–C80. doi: 10.1152/ajpcell.00431.2011
Parker, M. D., Qin, X., Qin, X., Williamson, R. C., Toye, A. M., and Boron, W. F. (2012). HCO−3-independent conductance with a mutant Na+/HCO−3 cotransporter (SLC4A4) in a case of proximal renal tubular acidosis with hypokalaemic paralysis. J. Physiol. 590, 2009–2034. doi: 10.1113/jphysiol.2011.224733
Piermarini, P. M., Kim, E., and Boron, W. F. (2007). Evidence against a direct interaction between intracellular carbonic anhydrase II and pure C-terminal domains of SLC4 bicarbonate transporters. J. Biol. Chem. 282, 1409–1421. doi: 10.1074/jbc.M608261200
Planelles, G., Thomas, S. R., and Anagnostopoulos, T. (1993). Change of apparent stoichiometry of proximal-tubule Na+-HCO−3 cotransport upon experimental reversal of its orientation. Proc. Natl. Acad. Sci. U.S.A. 90, 7406–7410. doi: 10.1073/pnas.90.15.7406
Pushkin, A., Abuladze, N., Gross, E., Newman, D., Tatishchev, S., Lee, I., et al. (2004). Molecular mechanism of kNBC1–carbonic anhydrase II interaction in proximal tubule cells. J. Physiol. 559(Pt.1), 55–65. doi: 10.1113/jphysiol.2004.065110
Rebello, G., Ramesar, R., Vorster, A., Roberts, L., Ehrenreich, L., Oppon, E., et al. (2004). Apoptosis-inducing signal sequence mutation in carbonic anhydrase IV identified in patients with the RP17 form of retinitis pigmentosa. Proc. Natl. Acad. Sci. U.S.A. 101, 6617–6622. doi: 10.1073/pnas.0401529101
Schueler, C., Becker, H. M., McKenna, R., and Deitmer, J. W. (2011). Transport activity of the sodium bicarbonate cotransporter NBCe1 is enhanced by different isoforms of carbonic anhydrase. PLoS ONE 6:e27167. doi: 10.1371/journal.pone.0027167
Seki, G., Coppola, S., and Frömter, E. (1993). The Na+-HCO−3 cotransporter operates with a coupling ratio of 2 HCO−3 to 1 Na+ in isolated rabbit renal proximal tubule. Pflug. Arch. 425, 409–416. doi: 10.1007/BF00374866
Sergeev, M., Godin, A. G., Kao, L., Abuladze, N., Wiseman, P. W., and Kurtz, I. (2012). Determination of membrane protein transporter oligomerization in native tissue using spatial fluorescence intensity fluctuation analysis. PLoS ONE 7:e36215. doi: 10.1371/journal.pone.0036215
Shah, G. N., Ulmasov, B., Waheed, A., Becker, T., Makani, S., Svichar, N., et al. (2005). Carbonic anhydrase IV and XIV knockout mice: roles of the respective carbonic anhydrases in buffering the extracellular space in brain. Proc. Natl. Acad. Sci. U.S.A. 102, 16771–16776. doi: 10.1073/pnas.0508449102
Skelton, L. A., Boron, W. F., and Zhou, Y. (2010). Acid-base transport by the renal proximal tubule. J. Nephrol. 23(Suppl. 16), S4–18.
Sly, W. S., Whyte, M. P., Sundaram, V., Tashian, R. E., Hewett-Emmett, D., Guibaud, P., et al. (1985). Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. N. Engl. J. Med. 313, 139–145. doi: 10.1056/NEJM198507183130302
Soriano, J. R. (2002). Renal tubular acidosis: the clinical entity. J. Am. Soc. Nephrol. 13, 2160–2170. doi: 10.1097/01.ASN.0000023430.92674.E5
Suzuki, M., Vaisbich, M. H., Yamada, H., Horita, S., Li, Y., Sekine, T., et al. (2008). Functional analysis of a novel missense NBC1 mutation and of other mutations causing proximal renal tubular acidosis. Pflugers Arch. 455, 583–593. doi: 10.1007/s00424-007-0319-y
Suzuki, M., Van Paesschen, W., Stalmans, I., Horita, S., Yamada, H., Bergmans, B. A., et al. (2010). Defective membrane expression of the Na+-HCO−3 cotransporter NBCe1 is associated with familial migraine. Proc. Natl. Acad. Sci. U.S.A. 107, 15963–15968. doi: 10.1073/pnas.1008705107
Tang, X. B., Kovacs, M., Sterling, D., and Casey, J. R. (1999). Identification of residues lining the translocation pore of human AE1, plasma membrane anion exchange protein. J. Biol. Chem. 274, 3557–3564. doi: 10.1074/jbc.274.6.3557
Thornell, I. M., Wu, J., Liu, X., and Bevensee, M. O. (2012). PIP2 hydrolysis stimulates the electrogenic Na+-bicarbonate cotransporter NBCe1-B and -C variants expressed in Xenopus laevis oocytes. J. Physiol. 590, 5993–6011. doi: 10.1113/jphysiol.2012.242479
Toye, A. M., Parker, M. D., Daly, C. M., Lu, J., Virkki, L. V., Pelletier, M. F., et al. (2006). The human NBCe1-A mutant R881C, associated with proximal renal tubular acidosis, retains function but is mistargeted in polarized renal epithelia. Am. J. Physiol. Cell. Physiol. 291, C788-801. doi: 10.1152/ajpcell.00094.2006
Warth, R., Barriere, H., Meneton, P., Bloch, M., Thomas, J., Tauc, M., et al. (2004). Proximal renal tubular acidosis in TASK2 K+ channel-deficient mice reveals a mechanism for stabilizing bicarbonate transport. Proc. Natl. Acad. Sci. U.S.A. 101: 8215–8220. doi: 10.1073/pnas.0400081101
Watanabe, A., Choe, S., Chaptal, V., Rosenberg, J. M., Wright, E. M., Grabe, M., et al. (2010). The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature. 468, 988–991. doi: 10.1038/nature09580
Wu, J., McNicholas, C. M., and Bevensee, M. O. (2009). Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates the electrogenic Na/HCO3 cotransporter NBCe1-A expressed in Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 106, 14150–14155. doi: 10.1073/pnas.0906303106
Yamada, H., Horita, S., Suzuki, M., Fujita, T., and Seki, G. (2011). Functional role of a putative carbonic anhydrase II-binding domain in the electrogenic Na+-HCO−3 cotransporter NBCe1 expressed in Xenopus oocytes. Channels 5, 106–109. doi: 10.4161/chan.5.2.14341
Yamaguchi, S., and Ishikawa, T. (2005). Electrophysiological characterization of native Na+-HCO−3 cotransporter current in bovine parotid acinar cells. J. Physiol. 568, 181–197. doi: 10.1113/jphysiol.2005.088633
Yamaguchi, S., and Ishikawa, T. (2008). The electrogenic Na+-HCO−3 cotransporter NBCe1-B is regulated by intracellular Mg2+. Biochem. Biophys. Res. Commun. 376, 100–104. doi: 10.1016/j.bbrc.2008.08.104
Yamaguchi, S., and Ishikawa, T. (2012). IRBIT reduces the apparent affinity for intracellular Mg2+ in inhibition of the electrogenic Na+-HCO−3 cotransporter NBCe1-B. Biochem. Biophys. Res. Commun. 424, 433–438. doi: 10.1016/j.bbrc.2012.06.127
Yamashita, A., Singh, S. K., Kawate, T., Jin, Y., and Gouaux, E. (2005). Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223. doi: 10.1038/nature03978
Yamazaki, O., Yamada, H., Suzuki, M., Horita, S., Shirai, A., Nakamura, M., et al. (2011). Functional characterization of nonsynonymous single nucleotide polymorphisms in the electrogenic Na+-HCO−3 cotransporter NBCe1A. Pflug. Arch. 461, 249–259. doi: 10.1007/s00424-010-0918-x
Yamazaki, O., Yamada, H., Suzuki, M., Horita, S., Shirai, A., Nakamura, M., et al. (2013). Identification of dominant negative effect of L522P mutation in the electrogenic Na+-HCO−3 cotransporter NBCe1. Pflugers Arch. 465, 1281–1291. doi: 10.1007/s00424-013-1277-1
Yang, D., Li, Q., So, I., Huang, C. L., Ando, H., Mizutani, A., et al. (2011). IRBIT governs epithelial secretion in mice by antagonizing the WNK/SPAK kinase pathway. J. Clin. Invest. 121, 956–965. doi: 10.1172/JCI43475
Yang, H. S., Kim, E., Lee, S., Park, H. J., Cooper, D. S., Rajbhandari, I., et al. (2009). Mutation of Aspartate 555 of the sodium/bicarbonate transporter SLC4A4/NBCe1 induces chloride transport. J. Biol. Chem. 284, 15970–15979. doi: 10.1074/jbc.M109.001438
Yoshitomi, K., Burckhardt, B. C., and Frömter, E. (1985). Rheogenic sodium-bicarbonate cotransport in the peritubular cell membrane of rat renal proximal tubule. Pflugers Arch. 405, 360–366. doi: 10.1007/BF00595689
Zhu, Q., Azimov, R., Kao, L., Newman, D., Liu, W., Abuladze, N., et al. (2009). NBCe1-A transmembrane segment 1 lines the ion translocation pathway. J. Biol. Chem. 284, 8918–8929. doi: 10.1074/jbc.M806674200
Zhu, Q., Kao, L., Azimov, R.,. Abuladze, N., Newman, D., Pushkin, A., et al. (2010a). Structural and functional characterization of the C-terminal transmembrane region of NBCe1-A. J. Biol. Chem. 285, 37178–37187.
Zhu, Q., Kao, L., Azimov, R., Newman, D., Liu, W., Pushkin, A., et al. (2010b). Topological location and structural importance of the NBCe1-A residues mutated in proximal renal tubular acidosis. J. Biol. Chem. 285, 13416–13426. doi: 10.1074/jbc.M109.093286
Zhu, Q., Kao, L., Newman, D., and Kurtz, I. (2010c) The C-terminal tail of the NBCe1-A dimer is structured: relevance to CAII binding. J. Am. Soc. Nephrol. 21, 251A.
Zhu, Q., Kao, L., Liu, W., Newman, D., Azimov, R., and Kurtz, I. (2012). Extracellular loop 3 forms domain-like structure on the surface of NBCe1-A. J. Am. Soc. Nephrol. 23, 31A.
Zhu, Q., Liu, W., Kao, L., Azimov, R., Newman, D., Abuladze, N., et al. (2013a). Topology of NBCe1 protein transmembrane segment 1 and structural effect of proximal renal tubular acidosis (pRTA) S427L mutation. J. Biol. Chem. 288, 7894–7906. doi: 10.1074/jbc.M112.404533
Keywords: NBCe1, bicarbonate, carbonate, transport, proximal tubule, kidney, proximal renal tubular acidosis
Citation: Kurtz I and Zhu Q (2013) Proximal renal tubular acidosis mediated by mutations in NBCe1-A: unraveling the transporter's structure-functional properties. Front. Physiol. 4:350. doi: 10.3389/fphys.2013.00350
Received: 24 September 2013; Accepted: 13 November 2013;
Published online: 19 December 2013.
Edited by:
Ebbe Boedtkjer, Aarhus University, DenmarkCopyright © 2013 Kurtz and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ira Kurtz, Division of Nephrology, David Geffen School of Medicine and Brain Research Institute, UCLA, 7-155 Factor Bldg, 10833 Le Conte Ave., Los Angeles, CA 90035, USA e-mail:aWt1cnR6QG1lZG5ldC51Y2xhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.