 Marcel M. G. J. van Borren1
Marcel M. G. J. van Borren1 Marc A. Vos
Marc A. Vos Marien J. C. Houtman
Marien J. C. Houtman Gudrun Antoons
Gudrun Antoons Jan H. Ravesloot
Jan H. Ravesloot- 1Heart Failure Research Center, Academic Medical Center, University of Amsterdam, Netherlands
- 2Department of Medical Physiology, Division Heart and Lungs, University Medical Center Utrecht, Netherlands
Dogs with compensated biventricular hypertrophy due to chronic atrioventricular block (cAVB), are more susceptible to develop drug-induced Torsade-de-Pointes arrhythmias and sudden cardiac death. It has been suggested that the increased Na+ influx in hypertrophied cAVB ventricular myocytes contribute to these lethal arrhythmias. The increased Na+ influx was not mediated by Na+ channels, in fact the Na+ current proved reduced in cAVB myocytes. Here we tested the hypothesis that increased activity of the Na+/H+ exchanger type 1 (NHE-1), commonly observed in hypertrophic hearts, causes the elevated Na+ influx. Cardiac acid-base transport was studied with a pH-sensitive fluorescent dye in ventricular myocytes isolated from control and hypertrophied cAVB hearts; the H+ equivalent flux through NHE-1, Na+-HCO−3 cotransport (NBC), Cl−/OH− exchange (CHE), and Cl−/HCO−3 exchange (AE) were determined and normalized per liter cell water and corrected for surface-to-volume ratio. In cAVB, sarcolemmal NHE-1 flux was increased by 65 ± 6.3% in the pHi interval 6.3–7.2 and NBC, AE, and CHE fluxes remained unchanged. Accordingly, at steady-state intracellular pH the total sarcolemmal Na+ influx by NHE-1 + NBC increased from 8.5 ± 1.5 amol/μm2/min in normal myocytes to 15 ± 2.4 amol/μm2/min in hypertrophied cAVB myocytes. We conclude that compensated cardiac hypertrophy in cAVB dogs is accompanied with an increased sarcolemmal NHE-1 activity. This in conjunction with unchanged activity of the other acid-base transporters will raise the intracellular Na+ in hypertrophied cAVB myocytes.
Introduction
Patients with left ventricular hypertrophy are at a higher risk to develop ventricular arrhythmias and sudden cardiac death (Bikkina et al., 1993; Oikarinen et al., 2001). This observation has been confirmed in various animal models with cardiac hypertrophy (Tomaselli and Marban, 1999), including dogs with chronic complete atrioventricular block (cAVB). The cAVB dog has compensated biventricular hypertrophy and a high susceptibility for drug-induced Torsade-de-Pointes arrhythmias (Vos et al., 1998) and sudden cardiac death (van Opstal et al., 2001). These potential lethal arrhythmias have been related to increased spatial (Verduyn et al., 1997) and temporal dispersion (Thomsen et al., 2004) of repolarization (Volders et al., 1999; Antoons et al., 2007).
An important factor that potentially contributes to action potential (AP) changes is the elevated intracellular Na+ (Na+i) in hypertrophied cAVB ventricular myocytes (Verdonck et al., 2003). High Na+i is also reported in human hypertrophied myocardium (Gray et al., 2001). This condition reduces INCX inward current during the AP plateau phase thereby limiting AP duration lengthening. When Na+i is high enough it may even promote NCX-mediated Ca2+ loading. Indeed, an increase Ca2+ influx through NCX was observed in cAVB (Sipido et al., 2000). The high Na+i in hypertrophied cAVB myocytes has been attributed to an increased Na+ influx rather than impaired Na+ extrusion through the Na+/K+ pump(Verdonck et al., 2003). Recent studies indicate that the INa current (peak and late) is decreased and cannot underlie the increased Na+ influx in hypertrophied cAVB myocytes (Antoons et al., 2008).
A possible pathway for increased Na+ influx in hypertrophied myocardium is the Na+/H+ exchanger (NHE-1). The activity of NHE-1 is increased in a number of cardiovascular diseases and has been shown to be the major cause for the increased Na+i concentration commonly observed in the hypertrophied failing hearts (Baartscheer, 2006; Bers et al., 2006). Inhibition of NHE-1 not only lowers Na+i but also prevents cellular hypertrophy, ionic remodeling, delayed after depolarizations and ultimately the development of heart failure (Baartscheer et al., 2005). Although less-well studied and species dependent, the Na+-HCO−3 cotransporter (NBC) is another potential pathway for increased Na+ influx (van Borren et al., 2006; Yamamoto et al., 2007). In addition, the extent of Na+i loading by NHE-1 and NBC largely depends on the supply of acid by the Cl−-dependent acid loaders such as, the Cl−/OH− exchanger (CHE) and Cl−/HCO−3 exchanger (AE) (Chiappe de Cingolani et al., 2001; van Borren et al., 2006). However, as yet there is no literature on the identity and characteristics of the cardiac acid-base transporters in dog.
In this study we first confirm the presence of NHE-1, NBC, AE, and CHE in dog ventricular myocytes before we tested the hypothesis that increased sarcolemmal NHE-1 activity underlies the elevated Na+ influx in cAVB dogs with compensated biventricular hypertrophy. Here we report that dog ventricular myocytes exhibit NHE-1, NBC, AE, and CHE and that compensated cardiac hypertrophy in cAVB dogs is accompanied with an increased sarcolemmal NHE-1 activity. This together with unchanged sarcolemmal NBC, AE, and CHE activities will raise intracellular Na+ concentrations without significant consequences for the resting pHi.
Materials and Methods
Animal handling was performed in accordance with the “European Directive for the Protection of Vertebrate Animals used for Experimental and Scientific Purpose, European Community Directive 86/609/CEE” and under the regulations of “The Committee for Experiments on Animals” of the University of Utrecht, The Netherlands. All research was performed in accordance with the American PhysiologicalSociety's “Guiding Principles in the Care and Use of Animals.”
Animal Model and Cell Isolation
Total atrioventricular (AV) block (AVB) was created in adult dogs (Marshall, USA) of either sex (N = 6) by radiofrequency ablation of the AV node as previously described (Schoenmakers et al., 2003). At the time of sacrifice, AVB duration was 48 ± 7 days and body weight 24 ± 1 kg; normal control animals were weight matched (N = 5, 27 ± 2 kg). Normal and chronic AVB (cAVB) dogs received full anesthesia. After premedication (1 ml/5 kg: 10 mg oxycodone HCl, 1 mg acepromazine and 0.5 mg atropine, i.m.), sodiumpentobarbital (20 mg/kg i.v.) was given. Dogs were artificially ventilated with a mixture of oxygen, nitrous oxide (40:60%), and halothane (0.5–1% vapor concentration). Upon thoracotomy, heparin was administered i.v. The hearts were quickly excised and washed in cold cardioplegic solution. Heart weight/body weight was significantly larger in cAVB dogs (12.1 ± 0.4 g/kg, vs. 8.6 ± 0.3 g/kg in controls, P < 0.05). Single myocytes were enzymatically isolated from the midmyocardial layer of the left and right ventricular free wall, as previously described (Volders et al., 1999).
Solutions
Tyrode's solution consisted of (mM) 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1.0 MgCl2, and 5.5 glucose at pH 7.4 (37°C). The normal variant was buffered with 5.0 mM HEPES. In the CO2/HCO−3-buffered variant, 22.4 mM NaCl was replaced by NaHCO3 and the saline was gassed with 5% CO2 balanced with 95% air. For ammonium prepulses, 20 mM NaCl was replaced by NH4Cl. For acetate prepulses, 40 or 80 mM NaCl was replaced by NaAcetate (NaAc). In Na+-free Tyrode's solutions, Na+ was replaced by N-methyl-D-glucammonium (NMDG+) and Ca2+ was omitted to prevent Ca2+ loading via reverse mode NCX. In Cl−-free Tyrode's solutions Cl− was replaced by gluconate. All salts were purchased from Merck (Darmstadt, Germany). The Na+/H+ exchanger inhibitor cariporide was kindly provided by Dr. Pünter (Aventis, Germany). Cariporide was prepared as 1000× stock in water.
Intracellular H+ Measurements
Myocytes were loaded with the fluorescent pH indicator carboxy-seminaphthorhodafluor-1 (SNARF, Molecular Probes) by exposing them for 10 min to 10 μM of the acetoxy methyl ester at 35°C. The inverted microscope was equipped with an apparatus for epi-illumination. Dye-loaded myocytes were excited with light of wavelength of 515 nm for 50 ms once every 3 s (75 W Xenon arc lamp). Intensities of the emitted light at wavelengths of 580 (I580) and 640 nm (I640) were recorded by two photomultiplier tubes. A rectangular adjustable slit ensured negligible background fluorescence levels. The I580/I640 ratio was calibrated by a series of precisely set pH solutions that contained 140 mM K+ instead of Na+ and 10 μM nigericin (Sigma) (van Borren et al., 2004).
Computation of Cytoplasmic H+ Equivalent Flux (J+H) Per Liter Cell Water
Intrinsic buffering power (βint) was determined in left and right ventricular myocytes from normal and cAVB dogs. We used the stepwise reduction in NH3/NH+4 technique as previously described by van Borren et al. (2004). In short, myocytes were exposed to series of nominally Na+ and Ca2+ free (replaced with N-methyl-D-glucammonium ions), HEPES buffered (pH 7.4) Tyrode's solutions containing decreasing amounts of NH3/NH+4. To minimize NH+4 entry via K+ channels, 2.0 mM BaCl2 was added, whereas addition of 1.0 mM CdCl3 prevented Ba2+ influx through L-type Ca2+ channels. βint was calculated as –Δ[acid]i/ΔpHi and assigned to the mean of the two pHi values used for its calculation. The CO2/HCO−3 buffering power, βCO2, was computed according to: βCO2 = 2.3 × [HCO−3]o × 10(pHi − pHo), with [HCO−3]o representing the extracellular HCO−3 concentration. The total buffering power, βtot, is defined as the sum of βint and βCO2.
To study the H+ equivalent flux (J+H) of the acid-base transporters, we imposed an acute acid load followed by an acute alkaline load on the myocytes and allowed them to recover from both. We fitted exponential functions to the recovering pHi traces. From these functions we computed the time derivatives, dpHi/dt's, and multiplied these with the appropriate βint or βtot to obtain J+H. The first 4 min of the pHi recovery from an alkaline load under CO2/HCO−3-buffered conditions were ignored to exclude out-of-equilibrium effects of the cytoplasmic buffering systems.
Computation of Sarcolemmal H+ Equivalent Flux (J+H) Per Unit Membrane Area
The sarcolemmal J+H per unit area (amol/μm2/min) was computed by dividing the cytoplasmic J+H by the surface to volume ratios. Atto (symbol a) is an SI prefix representing 10−18. Cell surface was estimated from the membrane capacitance Cm using a specific membrane capacitance of 10 fF/μm2. Cm was recorded with the whole-cell ruptured patch-clamp technique, the Axopatch 200B patch-clamp amplifier (Molecular Devices Corporation, Sunnyvale, CA, USA) and borosilicate glass patch pipettes (2–5 MΩ) filled with pipette solution containing (mM): 130 KCl, 10 NaCl, 0.5 MgCl2, 5 MgATP, 10 HEPES; pH 7.2 (5.5 KOH). Cm of right ventricular cAVB myocytes were significantly increased from 182 ± 10 pF (n = 29) to 216 ± 13 pF (n = 33) (P < 0.05), whereas the increase in Cm of left ventricular cAVB myocytes did not reach statistical significance, from 174 ± 6 pF (n = 44) to 187 ± 5 pF (n = 93), respectively. Length of left ventricular cAVB myocytes increased from 193 ± 2 μm (n = 197) to 225 ± 4 μm (n = 154) (P < 0.05) and of right ventricular cAVB myocytes from 200 ± 3 μm (n = 183) to 244 ± 6 μm (n = 154) (P < 0.05). Width of left ventricular cAVB myocytes increased from 27 ± 0.4 μm (n = 197) to 33 ± 1 μm (n = 154) (P < 0.05) and of right ventricular cAVB myocytes from 26 ± 0.4 μm (n = 183) to 35 ± 1 μm (n = 154) (P < 0.05). Cell volume was estimated from the morphologic data, assuming cylindrical cell shapes. The surface-to-volume ratios of left ventricular cAVB myocytes was reduced by 38%, from 0.16 ± 0.01 to 0.10 ± 0.01 μm−1, and from 0.17 ± 0.02 to 0.09 ± 0.01 μm−1, a decrease of 47%, on the right side. By dividing the cytoplasmic J+H (mM/min) by surface-to-volume ratios we arrived at the sarcolemmal J+H per unit area of cell membrane per minute (amol/μm2/min).
Statistics
Results are expressed as mean ± standard error (SE). We conducted statistical analyses (linear regression model with repeated measurements, ANOVA Student's t-tests) using SPSS® software. Two sets of data were considered significantly different if the P-value of these tests was <0.05. The capital “N” represents the number of hearts used. Lower case “n” represents the number of cells measured.
Results
Identification of Sarcolemmal Acid-Base Transporters in Normal Dog Ventricular Myocytes
Before studying the differences between normal and hypertrophied cAVB myocytes, we first identified what types of cardiac acid-base transporters are present. Acid-extruders and acid-loaders were activated by imposing acute acid and alkaline loads on the myocytes. Protocols are illustrated in Figure 1. In HEPES-buffered solutions, withdrawal of 20 mM NH3/NH+4 caused an acid load, the recovery from which was CO2/HCO−3-independent, but Na+-dependent and sensitive to 10 μM cariporide (Figure 1A). These characteristics are typical of NHE, presumably of the NHE-1 isoform. We cannot exclude a small contribution of the cariporide sensitive NHE-2 isoform, when present in dog cardiomyocytes. Withdrawal of 40 mM HAc/Ac− caused an alkaline load, the recovery from which was CO2/HCO−3- and Na+-independent but Cl−-dependent (Figure 1B). These are hallmarks of the Cl−/OH− exchanger (CHE).
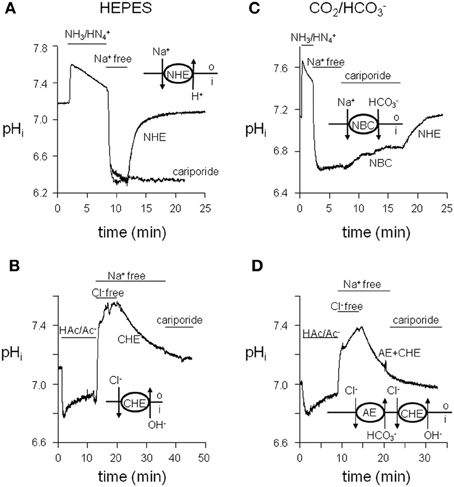
Figure 1. Functional identification of sarcolemmal acid-base transporters in dog ventricular myocytes. (A) pHi recovery after an acute acid load, induced by withdrawal of 20 mM NH3/NH+4, under HEPES-buffered conditions proved extracellular Na+-dependent and cariporide sensitive. (B) pHi recovery after an acute acid load, induced by withdrawal of 20 mM NH3/NH+4, under CO2/HCO−3-buffered conditions proved extracellular Na+-dependent and cariporide insensitive. (C, D) pHi recovery from an acute alkaline load proved extracellular Cl−-dependent and extracellular Na+-independent and was induced by 40 mM HAc/Ac− under HEPES-buffered conditions (C) and by 80 mM HAc/Ac− under CO2/HCO−3-buffered conditions (D).
In CO2/HCO−3-buffered solutions, a Na+-dependent and cariporide-insensitive acid load recovery was observed (Figure 1C). These characteristics are typical of the Na+-HCO−3 cotransporter (NBC). Recovery from an alkaline load upon withdrawal of 80 mM HAc/Ac− was CO2/HCO−3-dependent and Cl−-dependent but Na+-independent (Figure 1D). These are the hallmarks of the Cl−/HCO−3 exchanger (anion exchanger, AE). The recovery from an alkaline load in CO2/HCO−3-buffered solutions is presumably mediated by both CHE and AE.
From these data we conclude that dog cardiac myocytes possess the four classical acid-base transporters NHE-1, CHE, NBC, and AE.
Protocol Used to Study All Acid-Base Transporters in one Myocyte
To limit the time needed for studying all acid-base transporters, we designed a protocol for measuring NHE-1, NBC, AE, and CHE activity in one myocyte. A typical example is shown in Figure 2.
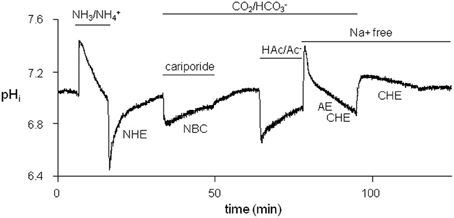
Figure 2. Typical protocol used to successively examine the four sarcolemmal acid-base transporters in each myocyte. Firstly, NHE-1 was studied after an acid-load induced by withdrawal of 20 mM NH+4/NH3. Secondly, NBC was studied, while NHE-1 was inhibited by cariporide, following a second weak acid load induced by replacement of a HEPES-buffered Tyrode's solution by a CO2/HCO−3-buffered Tyrode's solution. Thirdly, AE + CHE was studied in a Na+-free CO2/HCO−3-buffered saline solution after an alkaline load was induced by washout of 80 mM HAc/Ac−. Finally, CHE was studied in Na+-free conditions after an alkaline load was induced by replacing CO2/HCO−3-buffered with HEPES-buffered Tyrode's solutions.
We first imposed an acid-load on a myocyte (NH3/NH+4 prepulse technique) under HEPES-buffered conditions. The acid-load was quickly alleviated by means of NHE-1 until pHi, balanced was reached, the pHi value at which an acid-base transporter is exactly balanced by the opposing J+H flux (dpHi/dt equals 0). The myocyte was then subjected to a mild acid-load by replacing HEPES with 5% CO2/22.4 mM HCO−3 (pH 7.4) in the presence of 10 μM cariporide to inhibit NHE-1. The only acid-extruder active under these conditions is the HCO−3-dependent NBC. The NBC-mediated recovery was slow and incomplete, and stopped at the near neutral pHi, balanced value. Washout of cariporide unblocked NHE-1, which led to further restoration of pHi until the original steady-state pHi value under CO2/HCO−3-buffered conditions once again was reached. Thereafter, the myocyte was alkaline loaded (HAc/Ac− prepulse technique). The recovery from this load occurred quickly by means of AE and CHE. By choosing Na+-free condition at this stage of the experiment the Cl−-dependent acid-loaders were not opposed by the Na+-dependent acid extruders at neutral pHi values. In this way their H+ equivalent flux at the steady-state pHi could be determined. Finally, the myocyte was subjected to second and mild alkaline load (replacing CO2/HCO−3 with HEPES). From this alkaline load the myocytes recovered only very slowly by means of CHE and stopped recovering when CHE pHi,balanced value was reached.
Intrinsic Buffering Power (βint) in Normal and Hypertrophied cAVB Myocytes
To compute cytoplasmic H+ equivalent fluxes (J+H) from pHi recovery rates, we determined the pHi-dependence of the intrinsic buffering power (pHi-βint) in left and right ventricular myocytes of normal (left N = 2; n = 7 and right N = 2; n = 7) and cAVB (left N = 2; n = 15 and right N = 2; n = 12) dogs. Myocytes exposed to a series of NH+4/NH3 (20–2.5 mM) solutions showed a stepwise reduction in pHi (Figure 3A). The pHi-βint relationships did not significantly differ between myocytes isolated from the left and right ventricle, or between normal and cAVB hearts (data not shown). For this reason we pooled all data and calculated the average pHi-βint relationship (N = 4; n = 41). The averaged pHi-βint relationship was fitted with a polynomial equation (Figure 3B). Cytoplasmic J+H was calculated at 0.05 pHi intervals. Next, cytoplasmic J+H were divided by the averaged surface-to-volume ratios and plotted as a function of the corresponding pHi to construct sarcolemmal pHi-J+H profiles of NHE-1 (pHi-JNHE-1), NBC (pHi-JNBC), AE + CHE (pHi-JAE + CHE), and CHE (pHi-JCHE).
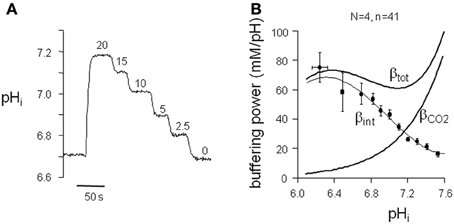
Figure 3. pHi dependence of buffering power in dog ventricular myocytes. (A) Typical pHi trace of a protocol to determine the intrinsic buffering power (βint) of ventricular myocytes. (B) The βint values, within a 0.05 pHi range, were averaged and plotted against the appropriate pHi to construct the pHi-βint relationship. The polynomial fit through the data is used to calculate J+H under HEPES-buffered conditions. For J+H under CO2/HCO−3-buffered conditions the calculated βCO2 was added to βint to arrive at βtot.
Sarcolemmal pHi-J+H Profiles in Normal Left and Right Ventricular Myocytes
In both left (n = 12) and right (n = 9) ventricular myocytes from normal dog hearts (N = 4) sarcolemmal JNHE-1 (i) were equally large at acidic pHi values, (ii) showed a comparable steep pHi-dependency, and (iii) had similar pHi, balanced values (Figure 4A). Likewise, no differences in these three characteristics were found in sarcolemmal JNBC, between left (N = 3, n = 8) and right (N = 4, n = 7) ventricular myocytes (Figure 4B). Compared to NHE-1, (i) NBC fluxes were 77% smaller, (ii) NBC pHi-dependence was 7-fold less, and (iii) NBC pHi, balanced value was ~0.2 pH more acidic. Thus, NHE-1 is the major acid extruder present in the dog myocardium.
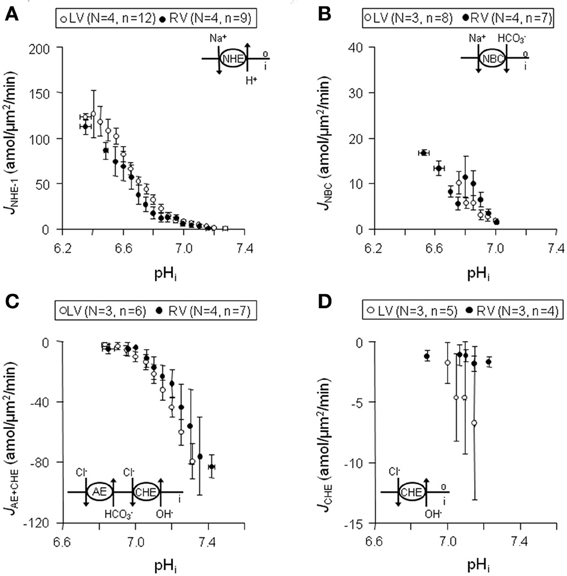
Figure 4. Sarcolemmal J+H through NHE-1, NBC, CHE + AE, and CHE in normal left and right ventricular myocytes. Data obtained from left are shown in blank circles, whereas from right are indicated with filled circles. Panels (A–D) illustrates that sarcolemmal JNHE-1 (A), JNBC (B),JAE + CHE (C), and JCHE (D) are not significantly different throughout the whole pHi range in left compared to right ventricular myocytes.
The myocytes showed a rapid alkaline load recovery under CO2/HCO−3-buffered conditions. In left (N = 3, n = 6) and right (N = 4, n = 7) ventricular myocytes sarcolemmal JAE + CHE was (i) equally large at alkaline pHi values, (ii) showed a comparable pHi-dependency, and (iii) had a virtually identical pHi, balanced value of around 6.8 (Figure 4C). Unlike sarcolemmal JAE + CHE, sarcolemmal JCHE proved small and only weakly pHi dependent. Again, no differences were observed between left (N = 3, n = 5) and right (N = 3, n = 4) ventricular myocytes (Figure 4D). Thus, AE is the major acid loader present in the dog myocardium and is largely responsible for alkaline load recoveries under CO2/HCO−3-buffered conditions.
We conclude that no differences exist in sarcolemmal pHi-J+H profiles of NHE-1, NBC, AE, and CHE between myocytes from the left and right ventricle.
Sarcolemmal pHi-J+H Profiles in Normal and Hypertrophied cAVB Myocytes
Next, we examined whether the sarcolemmal pHi-J+H profiles were changed in hypertrophied cAVB myocytes. Like in normal dog hearts, no differences were observed in the sarcolemmal pHi-J+H profiles between left and right hypertrophied ventricular cAVB myocytes (data not shown). For this reason the pooled left and right sarcolemmal pHi-J+H profiles of hypertrophied cAVB myocytes were compared to the pooled left and right sarcolemmal pHi-J+H profiles of normal myocytes (Figure 5).
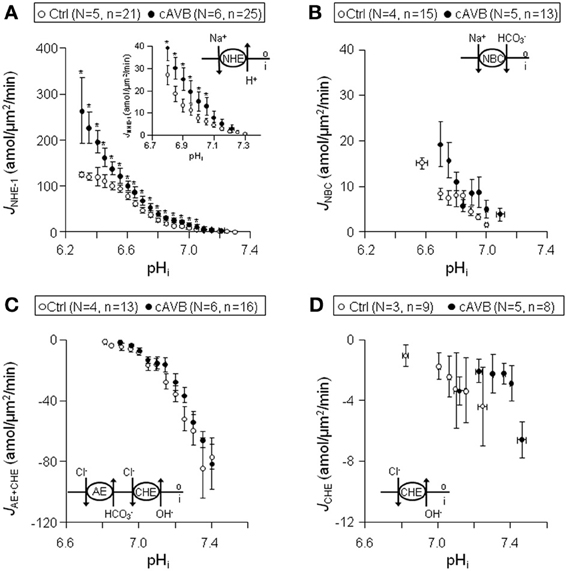
Figure 5. Sarcolemmal J+H through NHE-1, NBC, CHE + AE, and CHE in normal and hypertrophied cAVB ventricular myocytes. Pooled data (LV + RV) obtained from normal (open circles) and cAVB myocytes (filled circles). Panels (A–D) illustrates sarcolemmal pHi-JNHE-1 (A), pHi-JNBC (B), pHi-JAE + CHE (C), and pHi-JCHE (D) profiles. Asterisks “*” indicate statistical significance.
In hypertrophied cAVB myocytes the sarcolemmal JNHE-1 (Figure 5A), (i) was significantly increased at pHi values more acidic than 7.05 (P < 0.05), the increase amounted to 65 ± 6.3% in the pHi interval 6.3–7.2, (ii) showed a 75% steeper pHi-dependency, −4.4 ± 2.2 amol/μm2/min/pH unit (n = 25) vs. −2.2 ± 3.4 amol/μm2/min/pH unit (n = 21) (P < 0.05), and (iii) identical pHi, balanced values of around 7.3. In contrast, no changes were observed in the sarcolemmal pHi-JNBC (Figure 5B), pHi-JAE + CHE (Figure 5C), and pHi-JCHE (Figure 5D) profiles between normal myocytes and hypertrophied cAVB myocytes.
From these data we conclude that sarcolemmal JNHE-1 is increased in hypertrophied cAVB myocytes but otherwise there are no significant differences in sarcolemmal acid-base transport between normal hearts and compensated biventricular hypertrophied hearts.
Sarcolemmal J+H at Resting pHi in Normal and Hypertrophied cAVB Myocytes
When metabolic acid-base production is neglected, resting pHi is defined by the balance of acid loading (JCHE and/or JAE) and acid extrusion (JNHE-1 and/or JNBC). As is shown in Figure 6A, neither under HEPES-buffered conditions nor under CO2/HCO−3-buffered conditions the resting pHi of normal and hypertrophied cAVB myocytes differed significantly. However, in both groups the resting pHi was significantly more alkaline under HEPES-buffered conditions as compared to CO2/HCO−3-buffered conditions (P < 0.05). This difference indicates that in CO2/HCO−3-buffered solutions the acid loading action of AE drives pHi more acidic. In the process NHE-1 and NBC increase their activity. At the new steady-state pHi, acid loading is balanced by acid extrusion, and the cell experiences increased NaCl influx as compared to HEPES-buffered conditions. We estimated the magnitude of the increased sarcolemmal NaCl influx from the sarcolemmal pHi-J+H profiles of the four acid base transporters (Figure 4). In physiological, CO2/HCO−3-buffered solutions the sarcolemmal J+H, at the resting pHi increased from 8.5 ± 1.5 amol/μm2/min in normal myocytes (Figure 6B, left panel) to 15 ± 2.4 amol/μm2/min in hypertrophied cAVB myocytes (Figure 6B, right panel). Thus, the sarcolemmal Na+ influx in CO2/HCO−3-buffered solutions at resting pHi is increased by 76% in hypertrophied cAVB myocytes.
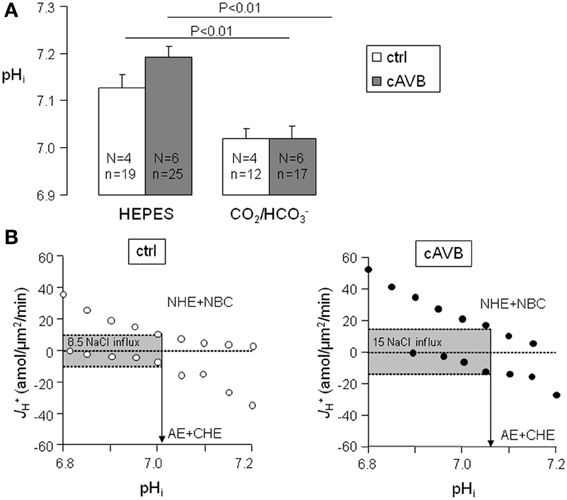
Figure 6. Balanced cytoplasmic J+H and sarcolemmal J+H at steady-state pHi in normal and cAVB myocytes. (A) Steady-state pHi values were obtained in normal (open bars) and cAVB (filled bars) myocytes either under HEPES or CO2/HCO−3-buffered conditions. Significant differences (P < 0.01), the number of animals (N) and observations (n) are indicated. (B) Total sarcolemmal J+H of the acid extruders and the acid loaders are plotted against their pHi in one graph to visualize their overlap at the steady-state pHi. J+H of the acid extruders and acid loaders are balanced in normal myocytes at pHi 7.01 (B, left panel) and in cAVB myocytes at pHi 7.07 (B, right panel), causing respectively a sarcolemmal J+H of 8.5 and 15 amol/μm2/min at these pHi values.
From these data we conclude that at resting pHi there are substantial differences in NaCl loading between normal hearts and compensated biventricular hypertrophied hearts.
Discussion
Overview
We investigated acid-base transport in normal dog hearts, and in hearts from cAVB dogs with compensated hypertrophy. We first identified (Figures 1, 2) and characterized the sarcolemmal acid-base transporters. After determination of the intrinsic cytoplasmic buffering power (Figure 3) we constructed their pHi-J+H profiles (Figure 4). NHE-1 and AE are the most potent acid extruder and acid loader, respectively, whereas the contribution of NBC and CHE to pHi regulation is relatively small (Figure 4). Secondly, neither in normal myocytes nor in hypertrophied cAVB myocytes did we observed left and right differences in acid-base transport (Figure 4). Thirdly, we demonstrated that compensated biventricular hypertrophy is associated with increased sarcolemmal NHE-1 mediated H+ equivalent fluxes but unchanged NBC, AE, or CHE fluxes (Figures 5, 6). Consequently, at resting pHi values, the amount sarcolemmal Na+ influx is significantly increased in hypertrophied cAVB myocytes (Figure 6).
Acid-Base Transport of Normal Dog Ventricular Myocytes
In the past, the role of NHE-1 has been pharmacologically examined in dog hearts with respect to ischemia/reperfusion injury and ischemic preconditioning (Gumina et al., 2000, 2005; Oh et al., 2007), however nothing is known about its activity and pHi-sensitivity. In addition, molecular, pharmacological, and functional data on NBC, AE, and CHE in the dog myocardium are completely lacking. To gain insight in what type of acid-base transporters are present in dog ventricular myocytes we used the experimental approach previously published for guinea-pig and rabbit ventricular myocytes (Leem et al., 1999; van Borren et al., 2006). Except for minor quantitative differences there is good agreement between our data and those published for guinea pig and rabbit. In these species CHE contributes for 15–30% to the acid loading rate (Leem and Vaughan-Jones, 1998), whereas we found a contribution of less than 10% in dog ventricular myocytes. The remaining acid-loading capacity can be attributed to AE. It should be noted that in this study we cannot exclude a possible contribution of residual Cl−/HCO−3 exchange during the alkaline-load recovery under HEPES-buffered conditions.
In dog, NHE-1 is responsible for more than 80% to the total acid extrusion capacity. Similar transport rates were found in rabbit (65%) guinea-pig (Lagadic-Gossmann et al., 1992; Leem et al., 1999), rat (Le Prigent et al., 1997) and sheep purkinje fiber (Dart and Vaughan-Jones, 1992). The other 20% of the acid-extrusion capacity can be attributed to NBC. In this study we did not determine the relative contribution of the different NBC isoforms [electroneutral (NBCn) vs. electrogenic (NBCe)] (Yamamoto et al., 2005). Moreover, the expression levels and molecular identities of the acid-base transporters remain to be determined.
Differences in membrane protein expression levels between left and right ventricular myocardium have been described. For example the current densities of the repolarizing potassium currents, the transient outward potassium current (Ito) and the delayed rectifier (IKs), are larger in right ventricular myocytes (Volders et al., 1999; Di Diego et al., 2002) and may underlie the consequent shorter AP. Also left-right differences in Ca2+i transients and contractions were observed. However, the smaller Ca2+i transients and contractions of right ventricular myocytes were not related to differential expression of Ca2+i handling proteins but to the shorter APs (Kondo et al., 2006).
In this study we demonstrated that steady-state pHi as well as acid-base transport activity of NHE-1, NBC, AE, and CHE do not differ between left and right ventricular myocytes of dogs hearts.
Decreased Surface-to-Volume Ratio in Hypertrophied cAVB Myocytes
Comparison of membrane transport activities, e.g., acid-base transporters, between cells is only allowed when they exhibit an identical cell surface-to-volume ratio value. It has been shown that these values can differ between species and between developmental stages, but are similar between small and large myocytes from the same healthy hearts (Satoh et al., 1996). During cardiac hypertrophy myocyte shape alterations parallel changes in ventricular anatomy (Gerdes, 2002). Indeed, isolated ventricular myocytes of cAVB dogs with eccentric biventricular hypertrophy increased more in length (13–23%) than in width (4–13%). Irrespective whether cells were assumed cylindrical or brick shaped, the cell volume of hypertrophied cAVB myocytes (74–121%) increased more than the surface area (14–26%). Consequently, surface-to-volume ratio decreased in hypertrophied cAVB myocytes by 38–47%. In modest hypertrophy, cardiomyocytes are able to maintain a normal surface-to-volume ratio by increasing T-tubular surface area disproportionally (Gerdes, 2002). Apparently this is not the case in compensated hypertrophied hearts of dogs with cAVB. Changes in surface-to-volume ratios between normal and hypertrophied myocytes have also been found in rat with cardiac hypertrophy (Yamamoto et al., 2007).
Acid-Base Transport in Hypertrophied cAVB Myocytes
Increased NHE-1 activity is observed in human hypertrophied cardiomyocytes (Yokoyama et al., 2000) and in hypertrophied myocytes from various animal models with hypertension or heart failure (Baartscheer, 2006). For instance in rats with monocrotaline-induced right ventricular failure (Chen et al., 2001), in diabetes type-2 rats (Darmellah et al., 2007) and spontaneous hypertensive rats (SHR) (Cingolani et al., 2003; Ennis et al., 2007) with heart failure, and in pressure and volume (Baartscheer et al., 2003; van Borren et al., 2006) and rapid pacing (Aker et al., 2004) induced rabbit models of heart failure NHE-1 activity is markedly enhanced. Both post-translational modulation and increased protein expression have been proposed to cause increased NHE-1 activity (Cingolani et al., 2008). In a number of these animal models chronic NHE-1 inhibition prevented (Kusumoto et al., 2001; Sandmann et al., 2001; Baartscheer et al., 2005) or even reversed the development of hypertrophy or heart failure(Camilion de Hurtado et al., 2002; Cingolani et al., 2003; Baartscheer et al., 2008; Baartscheer and van Borren, 2008). Moreover, transgenic mice with arterial hypertension (lacking the natriuretic peptide receptor type A) (Kilic et al., 2005) or heart failure (beta-adrenergic receptor over-expressing in the heart) (Engelhardt et al., 2002) developed cardiac hypertrophy and exhibit increased NHE-1 activity. Also in these transgenic animal models, NHE-1 inhibition prevented the development of cardiac hypertrophy and heart failure. Recently it became clear that increased cardiac NHE-1 activity alone is sufficient to activate hypertrophic Ca2+i dependent signaling pathways (Nakamura et al., 2008) and to induce dilated hypertrophic cardiomyopathy (Coccaro et al., 2007; Nakamura et al., 2008). Together, these studies underscore the pivotal role of increased NHE-1 activity in the etiology of hypertrophy and heart failure.
In good agreement with to the aforementioned models of cardiac hypertrophy and heart failure, we demonstrated here that also in cAVB dogs with compensated biventricular hypertrophy, sarcolemmal NHE-1 flux is increased. It should be noted that our cAVB dogs are free from heart failure symptoms, hypertension and lack the substantial sustained or progressive increase in the levels of neurohumoral factors (Vos et al., 1998). This suggests that hypertrophy per se is associated with increased sarcolemmal NHE-1 activity. Mechanisms underlying increased sarcolemmal NHE-1 activity in compensated hypertrophied hearts require further investigation, but may include increased wall stretch.
AE, NBC, and CHE, have been studied less extensively than NHE-1. An increased cardiac AE exchange activity has been documented in SHR (Chiappe de Cingolani et al., 2001) and rabbits with heart failure (pressure and volume overload) (van Borren et al., 2006). In our model of compensated cardiac hypertrophy, AE proved unchanged. Perhaps hypertension or heart failure is essential for increased AE activity to occur. Contradicting data exist on NBC. In one study NBC activity proved increased in hypertrophied cardiomyocytes from pressure overloaded rats (Yamamoto et al., 2007), whereas in another NBC activity was unchanged in hypertrophied cardiomyocytes from rabbits with heart failure (pressure and volume overload) (van Borren et al., 2006). Here we add that compensated hypertrophy does not increase cardiac NBC activity. Moreover, like in rabbits with heart failure (van Borren et al., 2006) no significant changes in CHE activity was observed in hypertrophied cAVB myocytes.
Cardiac Acid-Base Transport can Explain Increased Na+ Influx in Hypertrophied cAVB Myocytes
The high Na+i levels observed in hypertrophied cAVB myocytes have been attributed to a reduced affinity of the Na/K pump for Na+i removal and to an increased Na+ influx (Verdonck et al., 2003). The latter was indirectly derived from Na/K pump currents, the major pathway for Na+ efflux that must equal Na+ influx under steady-state conditions in resting myocytes. A recent study revealed that hypertrophied cAVB myocytes exhibit both a reduced peak and late INa, which excludes the sodium channels as a potential source for the enhanced Na+ influx (Antoons et al., 2008). Our data demonstrate that the balanced sarcolemmal J+H flux (JNHE-1 + NBC and JCHE + AE) at steady-state pHi (Figure 6) is increased by 76%. The increased sarcolemmal Na+ influx through balanced acid-base transport needs to be fully compensated by the sarcolemmal Na/K pump. As previously reported by Verdonck et al., the maximal Na/K pump activity (pA/pF, a measure per μm2) remains unchanged and the affinity of the Na/K pump for Na+i is reduced (Verdonck et al., 2003). Therefore, in hypertrophied cAVB myocytes the Na/K pump activity can only be sufficiently increased at higher Na+i concentrations. The higher NHE-1-mediated Na+ influx calculated in this study (76%, Figure 6) closely matches the relative increase of pump current densities in hypertrophied cAVB myocytes (88%) observed by Verdonck et al. (2003). This implies that NHE-1 is the main pathway responsible for increased Na+ influx. Other influx pathways, such as NCX or Na-K-2Cl cotransporter (NKCC, Figure 7) may have a contribution as well. NCX, however, is an unlikely candidate because elevated Na+i would rather promote the reverse mode and load the cell with Ca2+i, in particular in a setting of prolonged APs, as observed in cAVB (Pogwizd et al., 2003). Indeed, increased NCX-mediated Ca2+ loading was observed in hypertrophied cAVB myocytes (Sipido et al., 2000). NKCC may be an additional pathway for increased Na+ influx in cAVB, as documented previously for heart failure (Andersen et al., 2006). We cannot exclude a small contribution of NHE-2, when present in dog cardiomyocytes, since this transporter is also inhibited at the cariporide concentrations we used to identify NHE-1.
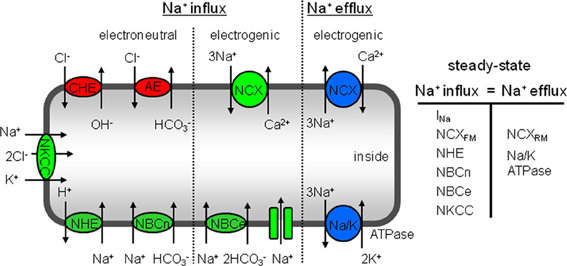
Figure 7. Schematic overview of sarcolemmal transporters that regulate Na+i concentration in cardiac myocytes. At steady-state Na+ influx is exactly balanced by Na+ efflux and consequently Na+i concentration remains unchanged. Na+ enters the cells through electroneutral transporters, such as the NKCC, NHE-1, and NBCn. The Na+-dependent acid extruders are stimulated by a continuous H+ supply via the Cl−-dependent acid loaders CHE and AE. Moreover, the electrogenic pathways through which Na+ enters is by NBCe, forward mode NCX and Na+ channels. The total Na+ influx is balanced by electrogenic Na+ extrusion mainly via the Na/K-pump and a minor part by reverse mode NCX.
Conclusion
Dogs exhibit four cardiac acid-base transporters, namely NHE-1, NBC, CHE, and AE. Their activities do not differ between left and right ventricular myocytes. Compensated hypertrophied hearts from cAVB dogs exhibit increased sarcolemmal JNHE-1 activity that almost fully explains the elevated Na+ influx in these hearts. Whether NHE-1 inhibition can prevent drug-induced Torsade-de-Pointes arrhythmias in cAVB dogs will be subject of future investigations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Drs A. Baartscheer and J.W.T Fiolet for the fruitful discussions and H.D. Beekman for the excellent technical assistance. This work was supported by a NWO grant (916.56.145) to Gudrun Antoons.
References
Aker, S., Snabaitis, A. K., Konietzka, I., Van De Sand, A., Bongler, K., Avkiran, M., et al. (2004). Inhibition of the Na+/H+ exchanger attenuates the deterioration of ventricular function during pacing-induced heart failure in rabbits. Cardiovasc. Res. 63, 273–282. doi: 10.1016/j.cardiores.2004.04.014
Andersen, G. O., Oie, E., Vinge, L. E., Yndestad, A., Attramadal, H., Skomedal, T., et al. (2006). Increased expression and function of the myocardial Na-K-2Cl cotransporter in failing rat hearts. Basic Res. Cardiol. 101, 471–478. doi: 10.1007/s00395-006-0604-5
Antoons, G., Oros, A., Beekman, H., Houtman, M., Engelen, M., Belardinelli, L., et al. (2008). Inhibition of late Na-current by ranolazine reduces Torsade de Pointes arrhythmias in the dog with chronic atrio-ventricular block. Heart Rhythm Abstr. 5, S115.
Antoons, G., Oros, A., Bito, V., Sipido, K. R., and Vos, M. A. (2007). Cellular basis for triggered ventricular arrhythmias that occur in the setting of compensated hypertrophy and heart failure: considerations for diagnosis and treatment. J. Electrocardiol. 40, S8–S14. doi: 10.1016/j.jelectrocard.2007.05.022
Baartscheer, A. (2006). Chronic inhibition of Na+/H+-exchanger in the heart. Curr. Vasc. Pharmacol. 4, 23–29. doi: 10.2174/157016106775203117
Baartscheer, A., and van Borren, M. M. G. J. (2008). Sodium ion transporters as new therapeutic targets in heart failure. Cardiovasc. Hematol. Agents Med. Chem. 6, 229–236. doi: 10.2174/187152508785909546
Baartscheer, A., Hardziyenka, M., Schumacher, C. A., Belterman, C. N., van Borren, M. M. G. J., Verkerk, A. O., et al. (2008). Chronic inhibition of the Na+/H+-exchanger causes regression of hypertrophy, heart failure, and ionic and electrophysiological remodeling. Br. J. Pharmacol. 154, 1266–1275. doi: 10.1038/bjp.2008.189
Baartscheer, A., Schumacher, C. A., van Borren, M. M. G. J., Belterman, C. N., Coronel, R., Opthof, T., et al. (2005). Chronic inhibition of Na+/H+-exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc. Res. 65, 83–92. doi: 10.1016/j.cardiores.2004.09.024
Baartscheer, A., Schumacher, C. A., van Borren, M. M. G. J., Belterman, C. N., Coronel, R., and Fiolet, J. W. (2003). Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc. Res. 57, 1015–1024. doi: 10.1016/S0008-6363(02)00809-X
Bers, D. M., Despa, S., and Bossuyt, J. (2006). Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann. N.Y. Acad. Sci. 1080, 165–77. doi: 10.1196/annals.1380.015
Bikkina, M., Larson, M. G., and Levy, D. (1993). Asymptomatic ventricular arrhythmias and mortality risk in subjects with left ventricular hypertrophy. J. Am. Coll. Cardiol. 22, 1111–1116. doi: 10.1016/0735-1097(93)90424-Y
Camilion de Hurtado, M. C., Portiansky, E. L., Perez, N. G., Rebolledo, O. R., and Cingolani, H. E. (2002). Regression of cardiomyocyte hypertrophy in SHR following chronic inhibition of the Na+/H+ exchanger. Cardiovasc. Res. 53, 862–868. doi: 10.1016/S0008-6363(01)00544-2
Chen, L., Gan, X. T., Haist, J. V., Feng, Q., Lu, X., Chakrabarti, S., et al. (2001). Attenuation of compensatory right ventricular hypertrophy and heart failure following monocrotaline-induced pulmonary vascular injury by the Na+-H+ exchange inhibitor cariporide. J. Pharmacol. Exp. Ther. 298, 469–476.
Chiappe de Cingolani, G., Morgan, P., Mundina-Weilenmann, C., Casey, J., Fujinaga, J., Camilion de Hurtado, M., et al. (2001). Hyperactivity and altered mRNA isoform expression of the Cl−/HCO3− anion-exchanger in the hypertrophied myocardium. Cardiovasc. Res. 51, 71–79. doi: 10.1016/S0008-6363(01)00276-0
Cingolani, H. E., Perez, N. G., Aiello, E. A., Ennis, I. L., Garciarena, C. D., Villa-Abrille, M. C., et al. (2008). Early signals after stretch leading to cardiac hypertrophy. Key role of NHE-1. Front. Biosci. 13, 7096–7114. doi: 10.2741/3213
Cingolani, H. E., Rebolledo, O. R., Portiansky, E. L., Perez, N. G., and Camilion de Hurtado, M. C. (2003). Regression of hypertensive myocardial fibrosis by Na+/H+ exchange inhibition. Hypertension 41, 373–377. doi: 10.1161/01.HYP.0000051502.93374.1C
Coccaro, E., Mraiche, F., Malo, M., Vandertol-Vanier, H., Bullis, B., Robertson, M., et al. (2007). Expression and characterization of the Na+/H+ exchanger in the mammalian myocardium. Mol. Cell. Biochem. 302, 145–155. doi: 10.1007/s11010-007-9436-3
Darmellah, A., Baetz, D., Prunier, F., Tamareille, S., Rucker-Martin, C., and Feuvray, D. (2007). Enhanced activity of the myocardial Na+/H+ exchanger contributes to left ventricular hypertrophy in the Goto-Kakizaki rat model of type 2 diabetes: critical role of Akt. Diabetologia 50, 1335–1344. doi: 10.1007/s00125-007-0628-x
Dart, C., and Vaughan-Jones, R. D. (1992). Na+-HCO3− symport in the sheep cardiac Purkinje fibre. J. Physiol. 451, 365–385.
Di Diego, J. M., Cordeiro, J. M., Goodrow, R. J., Fish, J. M., Zygmunt, A. C., Perez, G. J., et al. (2002). Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation 106, 2004–2011. doi: 10.1161/01.CIR.0000032002.22105.7A
Engelhardt, S., Hein, L., Keller, U., Klambt, K., and Lohse, M. J. (2002). Inhibition of Na+-H+ exchange prevents hypertrophy, fibrosis, and heart failure in beta(1)-adrenergic receptor transgenic mice. Circ. Res. 90, 814–819. doi: 10.1161/01.RES.0000014966.97486.C0
Ennis, I. L., Garciarena, C. D., Escudero, E. M. J., Perez, N. G., Dulce, R. A., Camilion de Hurtado, M. C., et al. (2007). Normalization of the calcineurin pathway underlies the regression of hypertensive hypertrophy induced by Na+/H+ exchanger-1 (NHE-1) inhibition. Can. J. Physiol. Pharmacol. 85, 301–310. doi: 10.1139/y06-072
Gerdes, A. M. (2002). Cardiac myocyte remodeling in hypertrophy and progression to failure. J. Card. Fail. 8, S264–S268. doi: 10.1054/jcaf.2002.129280
Gray, R. P., McIntyre, H., Sheridan, D. S., and Fry, C. H. (2001). Intracellular sodium and contractile function in hypertrophied human and guinea-pig myocardium. Pflügers Arch. 442, 117–123. doi: 10.1007/s004240000512
Gumina, R. J., Auchampach, J., Wang, R., Buerger, E., Eickmeier, C., Moore, J., et al. (2000). Na+/H+ exchange inhibition-induced cardioprotection in dogs: effects on neutrophils versus cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 279, H1563–H1570.
Gumina, R. J., El Schultz, J., Moore, J., Beier, N., Schelling, P., and Gross, G. J. (2005). Cardioprotective-mimetics reduce myocardial infarct size in animals resistant to ischemic preconditioning. Cardiovasc. Drugs Ther. 19, 315–322. doi: 10.1007/s10557-005-3693-8
Kilic, A., Velic, A., De Windt, L. J., Fabritz, L., Voss, M., Mitko, D., et al. (2005). Enhanced activity of the myocardial Na+/H+ exchanger NHE-1 contributes to cardiac remodeling in atrial natriuretic peptide receptor-deficient mice. Circulation 112, 2307–2317. doi: 10.1161/CIRCULATIONAHA.105.542209
Kondo, R. P., Dederko, D. A., Teutsch, C., Chrast, J., Catalucci, D., Chien, K. R., et al. (2006). Comparison of contraction and calcium handling between right and left ventricular myocytes from adult mouse heart: a role for repolarization waveform. J. Physiol. 571, 131–146. doi: 10.1113/jphysiol.2005.101428
Kusumoto, K., Haist, J. V., and Karmazyn, M. (2001). Na+/H+ exchange inhibition reduces hypertrophy and heart failure after myocardial infarction in rats. Am. J. Physiol. Heart Circ. Physiol. 280, H738–H745.
Lagadic-Gossmann, D., Buckler, K. J., and Vaughan-Jones, R. D. (1992). Role of bicarbonate in pH recovery from intracellular acidosis in the guinea-pig ventricular myocyte. J. Physiol. 458, 361–384.
Le Prigent, K., Lagadic-Gossmann, D., Mongodin, E., and Feuvray, D. (1997). HCO−3-dependent alkalinizing transporter in adult rat ventricular myocytes: characterization and modulation. Am. J. Physiol. 273, H2596–H2603.
Leem, C. H., Lagadic-Gossmann, D., and Vaughan-Jones, R. D. (1999). Characterization of intracellular pH regulation in the guinea-pig ventricular myocyte. J. Physiol. 517(Pt 1), 159–180. doi: 10.1111/j.1469-7793.1999.0159z.x
Leem, C. H., and Vaughan-Jones, R. D. (1998). Out-of-equilibrium pH transients in the guinea-pig ventricular myocyte. J. Physiol. 509(Pt 2), 471–485. doi: 10.1111/j.1469-7793.1998.471bn.x
Nakamura, T. Y., Iwata, Y., Arai, Y., Komamura, K., and Wakabayashi, S. (2008). Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ. Res. 103, 891–899. doi: 10.1161/CIRCRESAHA.108.175141
Oh, K. S., Seo, H. W., Yi, K. Y., Lee, S., Yoo, S. E., and Lee, B. H. (2007). Effects of KR-33028, a novel Na+/H+ exchanger-1 inhibitor, on ischemia and reperfusion-induced myocardial infarction in rats and dogs. Fundam. Clin. Pharmacol. 21, 255–263. doi: 10.1111/j.1472-8206.2007.00491.x
Oikarinen, L., Nieminen, M. S., Viitasalo, M., Toivonen, L., Wachtell, K., Papademetriou, V., et al. (2001). Relation of QT interval and QT dispersion to echocardiographic left ventricular hypertrophy and geometric pattern in hypertensive patients. The LIFE study. The Losartan Intervention For Endpoint Reduction. J. Hypertens. 19, 1883–1891. doi: 10.1097/00004872-200110000-00025
Pogwizd, S. M., Sipido, K. R., Verdonck, F., and Bers, D. M. (2003). Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc. Res. 57, 887–896. doi: 10.1016/S0008-6363(02)00735-6
Sandmann, S., Yu, M., Kaschina, E., Blume, A., Bouzinova, E., Aalkjaer, C., et al. (2001). Differential effects of angiotensin AT1 and AT2 receptors on the expression, translation and function of the Na+-H+ exchanger and Na+-HCO−3 symporter in the rat heart after myocardial infarction. J. Am. Coll. Cardiol. 37, 2154–2165. doi: 10.1016/S0735-1097(01)01287-6
Satoh, H., Delbridge, L. M., Blatter, L. A., and Bers, D. M. (1996). Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects. Biophys. J. 70, 1494–1504. doi: 10.1016/S0006-3495(96)79711-4
Schoenmakers, M., Ramakers, C., van Opstal, J. M., Leunissen, J. D., Londono, C., and Vos, M. A. (2003). Asynchronous development of electrical remodeling and cardiac hypertrophy in the complete AV block dog. Cardiovasc. Res. 59, 351–359. doi: 10.1016/S0008-6363(03)00430-9
Sipido, K. R., Volders, P. G., de Groot, S. H., Verdonck, F., Van de Werf, F., Wellens, H. J., et al. (2000). Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation 102, 2137–2144. doi: 10.1161/01.CIR.102.17.2137
Thomsen, M. B., Verduyn, S. C., Stengl, M., Beekman, J. D., de Pater, G., van Opstal, J., et al. (2004). Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation 110, 2453–2459. doi: 10.1161/01.CIR.0000145162.64183.C8
Tomaselli, G. F., and Marban, E. (1999). Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc. Res. 42, 270–283. doi: 10.1016/S0008-6363(99)00017-6
van Borren, M. M. G. J., Baartscheer, A., Wilders, R., and Ravesloot, J. H. (2004). NHE-1 and NBC during pseudo-ischemia/reperfusion in rabbit ventricular myocytes. J. Mol. Cell. Cardiol. 37, 567–577. doi: 10.1016/j.yjmcc.2004.05.017
van Borren, M. M. G. J., Zegers, J., Baartscheer, A., and Ravesloot, J. H. (2006). Contribution of NHE-1 to cell length shortening of normal and failing rabbit cardiac myocytes. J. Mol. Cell. Cardiol. 41, 706–715. doi: 10.1016/j.yjmcc.2006.07.001
van Opstal, J. M., Verduyn, S. C., Leunissen, H. D., de Groot, S. H., Wellens, H. J., and Vos, M. A. (2001). Electrophysiological parameters indicative of sudden cardiac death in the dog with chronic complete AV-block. Cardiovasc. Res. 50, 354–361. doi: 10.1016/S0008-6363(01)00226-7
Verdonck, F., Volders, P. G., Vos, M. A., and Sipido, K. R. (2003). Increased Na+ concentration and altered Na/K pump activity in hypertrophied canine ventricular cells. Cardiovasc. Res. 57, 1035–1043. doi: 10.1016/S0008-6363(02)00734-4
Verduyn, S. C., Vos, M. A., van der Zande, J., Kulcsar, A., and Wellens, H. J. (1997). Further observations to elucidate the role of interventricular dispersion of repolarization and early afterdepolarizations in the genesis of acquired torsade de pointes arrhythmias: a comparison between almokalant and d-sotalol using the dog as its own control. J. Am. Coll. Cardiol. 30, 1575–1584. doi: 10.1016/S0735-1097(97)00333-1
Volders, P. G., Sipido, K. R., Carmeliet, E., Spatjens, R. L., Wellens, H. J., and Vos, M. A. (1999). Repolarizing K+ currents ITO1 and IKs are larger in right than left canine ventricular midmyocardium. Circulation 99, 206–210. doi: 10.1161/01.CIR.99.2.206
Vos, M. A., de Groot, S. H., Verduyn, S. C., van der Zande, J., Leunissen, H. D., Cleutjens, J. P., et al. (1998). Enhanced susceptibility for acquired torsade de pointes arrhythmias in the dog with chronic, complete AV block is related to cardiac hypertrophy and electrical remodeling. Circulation 98, 1125–1135. doi: 10.1161/01.CIR.98.11.1125
Yamamoto, T., Shirayama, T., Sakatani, T., Takahashi, T., Tanaka, H., Takamatsu, T., et al. (2007). Enhanced activity of ventricular Na+-HCO−3 cotransport in pressure overload hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 293, H1254–H1264. doi: 10.1152/ajpheart.00964.2006
Yamamoto, T., Swietach, P., Rossini, A., Loh, S. H., Vaughan-Jones, R. D., and Spitzer, K. W. (2005). Functional diversity of electrogenic Na+-HCO−3 cotransport in ventricular myocytes from rat, rabbit and guinea pig. J. Physiol. 562, 455–475. doi: 10.1113/jphysiol.2004.071068
Keywords: NHE-1, NBC, AE, CHE, compensated cardiac hypertrophy
Citation: van Borren MMGJ, Vos MA, Houtman MJC, Antoons G and Ravesloot JH (2013) Increased sarcolemmal Na+/H+ exchange activity in hypertrophied myocytes from dogs with chronic atrioventricular block. Front. Physiol. 4:322. doi: 10.3389/fphys.2013.00322
Received: 01 August 2013; Paper pending published: 26 August 2013;
Accepted: 16 October 2013; Published online: 26 November 2013.
Edited by:
Mark O. Bevensee, University of Alabama at Birmingham, USAReviewed by:
Ignacio Gimenez, Aragon's Health Sciences Institute, SpainJohn Cuppoletti, University of Cincinnati, USA
Copyright © 2013 van Borren, Vos, Houtman, Antoons and Ravesloot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan H. Ravesloot, Department of Anatomy, Embryology, and Physiology, University of Amsterdam, Academic Medical Center, Meibergdreef 15, 1105 AZ, Amsterdam, Netherlands e-mail:ai5oLnJhdmVzbG9vdEBhbWMudXZhLm5s