
 Deric M. Park2
Deric M. Park2

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Physiol. , 01 October 2013
Sec. Craniofacial Biology and Dental Research
Volume 4 - 2013 | https://doi.org/10.3389/fphys.2013.00273
This article is part of the Research Topic Stem Cell Programming and Reprogramming View all 14 articles
Stem cells, by definition, are able to both self-renew (give rise to more cells of their own kind) and demonstrate multipotential (the ability to differentiate into multiple cell types). To accommodate this unique dual ability, stem cells interpret signal transduction pathways in specialized ways. Notable examples include canonical and non-canonical branches of the Notch signaling pathway, with each controlling different downstream targets (e.g., Hes1 vs. Hes3) and promoting either differentiation or self-renewal. Similarly, stem cells utilize STAT3 signaling uniquely. Most mature cells studied thus far rely on tyrosine phosphorylation (STAT3-Tyr) to promote survival and growth; in contrast, STAT3-Tyr induces the differentiation of neural stem cells (NSCs). NSCs use an alternative phosphorylation site, STAT3-Ser, to regulate survival and growth, a site that is largely redundant for this function in most other cell types. STAT3-Ser regulates Hes3, and together they form a convergence point for several signals, including Notch, Tie2, and insulin receptor activation. Disregulation and manipulation of the STAT3-Ser/Hes3 signaling pathway is important in both tumorigenesis and regenerative medicine, and worthy of extensive study.
Stem cells exhibit the fundamental properties of both self-renewal and differentiation into multiple cell types. The balance between these two must be finely regulated in order to allow stem cells to properly contribute to tissue formation during development and later, in the context of resisting degenerative disease and normal wear-and-tear as we age. Key to this balance is the clear allocation of these two functions to distinct signal transduction pathways. Nature had two options to accomplish this task: (a) to put aside part of the signaling machinery to be used exclusively for self-renewal while have another totally separate set for regulating their multipotential nature, or, (b) to have individual cell types interpret the same signaling pathways differently. In the latter case, a stem cell could, for example, co-opt some typically pro-survival pathways, and utilize them as pro-differentiation signals. Consequently, a stem cell would have fewer pro-survival choices, but it would also be able to encode its multipotential nature without the need for possessing a unique set of signaling molecules. A specific example highlighting the second option is a signal transduction pathway that is emerging as a convergence point for diverse signals and regulates self-renewal vs. differentiation decisions with consequence in regenerative medicine, cancer biology, and drug discovery strategies. At the center of this pathway is the phosphorylation of Signal Transducer and Activator of Transcription 3 (STAT3) on the serine residue at amino acid position 727 (STAT3-Ser) and subsequent induction of the transcription factor Hairy and Enhancer of Split 3 (Hes3), a member of the Hes/Hey gene family of basic helix-loop-helix (bHLH) transcription factors.
The Janus kinase (JAK)-STAT pathway is a major, evolutionarily conserved signaling component in the cells of many organisms, ranging from slime molds to humans. Like other signal transduction cascades, its role is to convey information from the cell's environment all the way to DNA promoters on the genome, resulting in changes in gene expression. Binding of ligands, such as cytokines, hormones and growth factors to surface receptors leads to activation of JAKs. JAK proteins are a family of non-receptor tyrosine kinases and their most prominent targets are STAT proteins. Tyrosine-phosphorylated STATs then enter the nucleus where they regulate gene transcription. The long evolutionary history of the JAK-STAT pathway has incorporated it into many other signaling pathways making it relevant to a vast array of intrinsic and environmental stimuli, cell proliferation, development, hematopoiesis, immunity, metabolic regulation, and cancer (Levy and Darnell, 2002; Shen et al., 2004; Richard and Stephens, 2011; Vera et al., 2011; Wauman and Tavernier, 2011; Camporeale and Poli, 2012; Lafave and Levine, 2012; Mohr et al., 2012; O'shea and Plenge, 2012; Stark and Darnell, 2012; Tibes et al., 2012; Swiatek-Machado and Kaminska, 2013).
In addition to the tyrosine phosphorylation site, some STAT proteins also have a serine phosphorylation site (Decker and Kovarik, 2000; Reich, 2009). STAT-Ser phosphorylation is regulated by different pathways among STAT family members, as demonstrated by pharmacological approaches using receptor ligands and inhibitors of intracellular kinase cascades. In the case of STAT3, serine phosphorylation is induced by EGF, PDGF, insulin, IL-2, BCR, and TCR. In all these cases, STAT3-Ser phosphorylation is reduced by treatment with a MEK1/2 inhibitor (PD98059). In contrast, when STAT3-Ser phosphorylation is induced by treatment with IL-6 or IFN-alpha, phosphorylation is sensitive to a broad spectrum kinase inhibitor, H7 (Chung et al., 1997; Ng and Cantrell, 1997; Stephens et al., 1998; Su et al., 1999; Decker and Kovarik, 2000). Adding to the complexity of STAT-Ser phosphorylation regulation, multiple pathways activated by the same receptor impinge upon different STAT-Ser phosphorylations, adding to the complexity, as well as the utility, of STAT3 in regulating discrete biological processes (Kovarik et al., 1999; Lim and Cao, 1999; Decker and Kovarik, 2000). It will be highly informative to extend such studies using cells from different origins, and address how the regulation of STAT-Ser phosphorylation is affected by cell type, providing an additional level of specificity in terms of pharmacologically manipulating this site.
Early studies demonstrated that the role of STAT-Ser phosphorylation is largely auxiliary to the function of tyrosine-phosphorylated STAT3, resulting in an increase of its transcriptional activity (Levy and Darnell, 2002). More recent studies have uncovered potentially STAT-Ser—specific effects on cell growth. In this article we will focus on STAT3 because it is extensively studied in terms of its role in regulating the self-renewal and differentiation of neural stem cells (NSCs). In these cells, JAK2 phosphorylates STAT3-Tyr in response to the activation of several receptors, including the CNTF receptor. Tyrosine-phosphorylated STAT3 directly stimulates transcription of genes expressed in astrocytes, most notably glial GFAP, thereby promoting loss of the self-renewal state and differentiation toward the astroglial fate (Bonni et al., 1997; Rajan and McKay, 1998; Panchision and McKay, 2002). STAT3-Tyr phosphorylation is undetectable during self-renewal, showcasing its powerful role in inducing differentiation. This prompted the question of whether STAT3 is irrelevant to NSC growth, or whether NSCs utilized STAT3-Ser for regulating proliferation. Pharmacological and genetic (STAT3 phosphomimetic) approaches demonstrate that STAT3-Ser, in the absence of STAT3-Tyr, is a potent stimulator of NSC survival and growth (Androutsellis-Theotokis et al., 2006).
Many reports today implicate STAT3-Ser in cell growth, proliferation, and survival, in the context of cancer (Plaza-Menacho et al., 2007; Qin et al., 2008; Aggarwal et al., 2009; Lacreusette et al., 2009; Yeh et al., 2009; Banerjee et al., 2010; Sekine et al., 2011; Villalva et al., 2011; Miyakoshi et al., 2012; Tkach et al., 2013; Yang et al., 2013), insulin and neurotrophin signaling (Ng et al., 2006; Kim et al., 2009), neurite outgrowth (Zhou and Too, 2011), and hypertension (Zouein et al., 2013). An extensively studied aspect of STAT3 that is dependent upon serine phosphorylation is its involvement in the regulation of cellular respiration by localizing to the mitochondrion where it regulates reactive oxygen species production and cell death (Zhang et al., 2003; Shulga and Pastorino, 2012).
In this article, we address a particular branch of the signaling pathway involving STAT3-Ser and its downstream mediator, the transcription factor Hes3. This STAT3-Ser/Hes3 Signaling Axis does not operate in parallel to JAK-STAT, nor is it an auxiliary to it. In fact, STAT3-Tyr and STAT3-Ser represent distinct and opposing functions with significant consequences for cellular function and fate determination. Here we discuss its involvement in neuro-regenerative medicine as well as open questions that need to be addressed in order to elevate our understanding of this pathway toward therapeutic benefit.
The Hes/Hey family of bHLH transcription factors is a major target of the Notch signaling pathway (Artavanis-Tsakonas et al., 1999; Imayoshi and Kageyama, 2011; Ueo et al., 2012). They are expressed in NSCs and progenitor cells in vitro and in vivo, where they repress the expression of genes involved in differentiation, making them critical mediators of fate choice (Sasai et al., 1992; Lobe, 1997; Allen and Lobe, 1999; Hirata et al., 2001; Hatakeyama et al., 2004; Androutsellis-Theotokis et al., 2006, 2008a,b, 2009, 2010a,b; Basak and Taylor, 2007, 2009; Rueger et al., 2010; Imayoshi and Kageyama, 2011; Masjkur et al., 2012; Pacioni et al., 2012). Different experimental systems seem to regulate which Hes/Hey family members are more prominently expressed or modulated. For example, Hes1 opposes neuronal differentiation and therefore may support the maintenance of NSCs during mouse development (Hatakeyama et al., 2004; Imayoshi and Kageyama, 2011; Shimojo et al., 2011; Tan et al., 2012). Hes5 is expressed in multipotent progenitors in the embryonic neural tube as well as in neurospheres generated from this region in vitro (Hatakeyama et al., 2004; Basak and Taylor, 2007). In monolayer cultures of fetal and adult mouse NSCs, all family members are expressed (Androutsellis-Theotokis et al., 2006). However, upon Notch activation by treatment of the NSC cultures with a soluble form of the Notch receptor ligands Delta4 or Jagged1, only Hes3 exhibits changes in its expression (mRNA levels increases within 30 min, peaking at 1 h).
It is quite likely, therefore, that several Hes/Hey family members may promote distinct functions in NSCs, or they may cooperate with others. For example, combined deletions of Hes1, Hes3, and Hes5 in mice leads to the depletion of progenitor cells (Hatakeyama et al., 2004; Imayoshi and Kageyama, 2011).
Our understanding of the function of different Hes/Hey genes received a great boost through the realization that different branches of the STAT3 signaling pathway regulate and/or involve distinct members of their family. For example, Hes1 and Hes5 form complexes with JAK2, promoting its ability to phosphorylate STAT3-Tyr (Kamakura et al., 2004). In this way, Hes1 and Hes5 are part of the pro-differentiation program in cultured NSCs, resulting in increased astrocyte generation. This is consistent with the in vivo observations showing that Hes1 and Hes5 oppose the acquisition of the neuronal fates. However, the question remains open as to the extent to which the Hes1+/Hes5+ cell population in the brain is undifferentiated (i.e., are these NSCs, or more differentiated intermediates)? Alternatively, the NSC population may be defined as the Hes1+/Hes5+/Hes3+ population. This is consistent with the observations that Hes3 expression in NSC cultures is opposed by JAK activity and supported by various treatments that promote STAT3-Ser phosphorylation (Androutsellis-Theotokis et al., 2006, 2008b, 2009, 2010a,b). In fact, Hes3 can be used as a NSC biomarker in vitro because it is expressed in NSCs and lost upon induction of differentiation. In vivo, Hes3+ cells are found in the established neurogenic zones (subventricular zone, dentate gyrus) as well as many other areas of the adult brain; these can be microdissected and cultures can be established efficiently, demonstrating the NSC properties of Hes3+ cells (Poser and Androutsellis-Theotokis, 2013). Hes3 being downstream of STAT3 is consistent with the observation that, unlike Hes1 and Hes5, Hes3 is not a direct transcriptional target of Notch (Kageyama et al., 2005). Together, these results establish that activation of pathways resulting in STAT3-Ser phosphorylation has functions distinct from those of simply supporting JAK-STAT (Figure 1A).
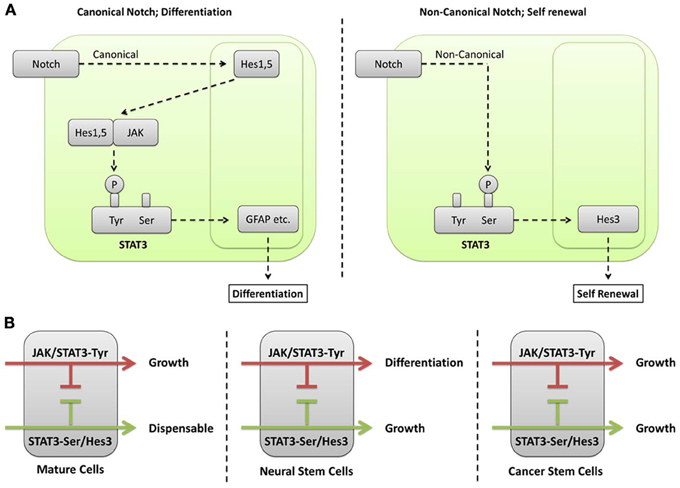
Figure 1. (A) Different phosphorylation sites on STAT3 mediate canonical and non-canonical functions of the Notch signaling pathway. In canonical Notch signaling, the cleaved intracellular domain of the Notch receptor translocates to the nucleus where it regulates transcription of target genes, including Hes1 and Hes5. Hes1 and Hes5 are transcription factors themselves and can therefore regulate additional target genes downstream of Notch activation. However, they also have cytoplasmic functions where they form complexes with JAK2 promoting its ability to phosphorylate STAT3-Tyr (Kamakura et al., 2004). This modification leads to the induction of differentiation in NSCs (Bonni et al., 1997). A non-canonical branch of Notch signaling leads to STAT3-Ser phosphorylation in the absence of detectable STAT3-Tyr phosphorylation (Androutsellis-Theotokis et al., 2006). This is followed by increased transcription of Hes3. Hes3, in turn, promotes growth through poorly understood mechanisms. A clue as to the function of Hes3 comes from observations that force-expressed Hes3 in NSC cultures increases the expression of sonic hedgehog (Androutsellis-Theotokis et al., 2006), a morphogen that also acts as a mitogen for NSCs (Ahn and Joyner, 2005). (B) Different cell types interpret STAT3 and Hes/Hey signaling in specific ways. Many mature cell types (e.g., neurons and astrocytes in the brain), as well as many established cell lines from different tissues utilize the JAK/STAT pathway that leads to STAT3-Tyr phosphorylation for growth. In contrast, STAT3-Ser phosphorylation is only auxiliary to the transcriptional strength of STAT3 and dispensable. NSCs interpret these signals in a different way. The JAK/STAT pathway involving STAT3-Tyr phosphorylation induces their differentiation, preferentially to the astrocytic fate. NSCs can now recruit the STAT3-Ser phosphorylation as a critical regulator of growth. An important mediator of STAT3-Ser action is Hes3. Cancer stem cells (from glioblastoma multiforme) are able to grow utilizing either the JAK-STAT or the STAT3-Ser/Hes3 signaling pathways. The ability of cancer stem cells to grow utilizing distinct signaling pathways blurs the line distinguishing cancer stem cells from more differentiated cancer cells and underlines their ability to evade therapy aimed at disrupting one or a limited set of signaling pathways.
The role of the STAT3-Ser/Hes3 signaling axis in controlling NSC number in vitro and in vivo makes it a potential pharmacological target in neurodegenerative disease therapy (Kittappa et al., 2012). Several diverse treatments that promote the pathway exhibit similar results on cultured cell number, including ligands of the Notch (Delta4, Jagged1) and Tie2 (Angiopoietin 2) receptors, basic FGF, and insulin (Androutsellis-Theotokis et al., 2006, 2008b, 2009, 2010a,b). In vivo, Delta4, Angiopoietin 2, insulin, or a combination of these along with an inhibitor of JAK significantly increase the number of endogenous NSCs, establishing the predictive properties of these culture systems (Androutsellis-Theotokis et al., 2008a; Poser and Androutsellis-Theotokis, 2013). These factors also have known effects on blood vessels with, Angiopoietin 2 promoting angiogenesis whereas Delta4 opposes it (Maisonpierre et al., 1997; Yancopoulos et al., 2000; Noguera-Troise et al., 2006; Hellstrom et al., 2007; Siekmann and Lawson, 2007; Thurston et al., 2007). Together, their antagonistic effects would mitigate any large scale changes in the vasculature that results from their combined therapeutic use, while still yielding a substantial increase in the number of NSCs.
The observation that distinct phosphorylation sites on STAT3 mediate different outputs (Figure 1B) allows for testable predictions and the generation of new potential treatments based upon an understanding of a given cell's state. In the context of NSCs for example, CNTF, through its activation of both the JAK-STAT and phosphoinositide 3' kinase (PI3K) pathways, leads to STAT3-Tyr phosphorylation as well as STAT3-Ser phosphorylation (via activation of mTOR) (Decker and Kovarik, 2000; Yokogami et al., 2000; Levy and Darnell, 2002). JAK-STAT activation by CNTF is responsible for the powerful differentiation effect of CNTF toward the astrocytic fate. Our understanding of STAT3 signaling allowed us to predict that concomitant treatment of NSCs with CNTF and a JAK inhibitor would suppress STAT3-Tyr phosphorylation while maintaining STAT3-Ser phosphorylation, thus converting the culture conditions from pro-differentiation to pro-self-renewal (Androutsellis-Theotokis et al., 2008b).
The consequences of activating the STAT3-Ser/Hes3 Signaling Axis in vivo revealed new aspects of the regenerative/repair potential of the adult mammalian central nervous system. First, immunohistochemical detection revealed Hes3+ cells throughout the adult rat brain, with the highest densities being found in the subventricular zone lining the lateral ventricles and the dentate gyrus of the hippocampus (Androutsellis-Theotokis et al., 2006, 2009). This is entirely consistent with a putative role of Hes3 as a biomarker of endogenous NSCs/progenitor cells. However, Hes3+ cells were also observed scattered throughout many gray and white matter areas, displaying a characteristic morphology and associating closely with blood vessels. Microdissection of brain and spinal cord areas and subsequent culture at clonal densities demonstrated the growth of Hes3+ cells and their differentiation to neurons, astrocytes, and oligodendrocytes. Robust cultures can be established, taking advantage of the signaling pathway and providing the culture medium with ligands of the Notch and Tie2 receptors (Androutsellis-Theotokis et al., 2008a; Poser and Androutsellis-Theotokis, 2013). Using these cultures, self-renewal and multipotential of Hes3+ cells can be demonstrated (Androutsellis-Theotokis et al., 2009, 2010a,b).
When these factors are injected into the lateral ventricles of rodents, the number of Hes3+ cells in many areas of the brain and spinal cord increases within days, and results in strong behavioral recovery in models of ischemic stroke and Parkinson's disease. (Androutsellis-Theotokis et al., 2006, 2008b, 2009; Masjkur et al., 2012; Pacioni et al., 2012). This benefit occurred in the absence of detectable newly generated and matured neurons, suggesting these cells provide a neuroprotective effect through trophic support rather than outright replacement. Indeed, in the Parkinsonian models, nigrostriatal dopamine neurons that would otherwise have been lost were rescued. These results revealed a mechanism that the adult central nervous system operates for damage repair, coupling vascular signals (e.g., Delta4, Angiopoietin 2) and a widespread multipotent precursor cell to establish a regenerative environment that promotes restoration of function.
As an extension of these observations, carcinogenesis is sometimes viewed as the manifestation of aberrant regenerative mechanisms. The cells that carry the regenerative ability of a tumor, termed cancer stem cells, express genes common to their non-cancerous stem cell counterparts (Hanahan and Weinberg, 2011; Poser et al., 2011). In line with this notion, putative cancer stem cells from the aggressive brain tumor glioblastoma multiforme express Hes3 both in the patient and in culture. Hes3 RNA interference results in increased death in cells isolated from patient biopsies (Park et al., 2013), highlighting another potential therapeutic benefit of targeting the STAT3-Ser/Hes3 Signaling Axis.
In the 1960s, Joseph Altman's experiments demonstrated, through the use of tritiated thymidine, that the adult mammalian brain contains proliferating cells that can become neurons (Altman and Das, 1965). It took decades to begin appreciating the significance of these findings and eventually a vigorous search started for biomarkers to identify this cellular population. The results of this effort uncovered discrete regions that harbor multipotent progenitor cells, the stem cell niches of the subventricular zone and the dentate gyrus. However, the bias of the search toward proliferating (albeit slowly) cells resulted in the field missing the widespread neural stem cell population that may be largely quiescent, possibly not even being able to generate new neurons in their microenvironment in vivo, but nevertheless, are just as critical to the maintenance of the brain as we age, or recover from injury. The reason for the existence of these “parenchymal” stem cells is not clear but it is conceivable that they are the result of an evolutionary adaptation toward brain tissue rigidity, as a means of storing information and lessons learned as the extraordinary regenerative plasticity that lower animals exhibit may be incompatible with long-term memory storage. These cells may have kept some of their progenitor character (e.g., some biomarkers) but have adapted from direct cell replacement to modulating the microenvironment, thus playing a neuroprotective role. In this way, these cells can still contribute to tissue repair without interfering with information storage. This may also be an evolutionary adaptation because as the mammalian brain has grown large, the challenge of new neurons efficiently recapitulating lost neuronal connections has become extremely daunting. This problem is encountered by efforts to replace lost neurons using approaches that involve the grafting of exogenous NSCs or fetal neuronal tissue. Just like placing a bunch of cable bits in a broken computer does not guarantee that the computer will work again, there is still work to be done before we understand how to effectively integrate a graft bolus into the appropriate brain circuitry to facilitate recovery. Until then, boosting natural, innate repair mechanisms, such as those involving the STAT3-Ser/Hes3 Signaling Axis, represents a sound strategy that both seems to work and addresses disease progression.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was funded (in part) by the Helmholtz Alliance ICEMED—Imaging and Curing Environmental Metabolic Diseases, through the Initiative and Network Fund of the Helmholtz Association, and a grant from the Else Kroener-Fresenius Foundation.
Aggarwal, B. B., Kunnumakkara, A. B., Harikumar, K. B., Gupta, S. R., Tharakan, S. T., Koca, C., et al. (2009). Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann. N.Y. Acad. Sci. 1171, 59–76. doi: 10.1111/j.1749-6632.2009.04911.x
Ahn, S., and Joyner, A. L. (2005). In vivo analysis of quiescent adult neural stem cells responding to Sonic hedgehog. Nature 437, 894–897. doi: 10.1038/nature03994
Allen, T., and Lobe, C. G. (1999). A comparison of Notch, Hes and Grg expression during murine embryonic and post-natal development. Cell. Mol. Biol. (Noisy-le-grand) 45, 687–708.
Altman, J., and Das, G. D. (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 124, 319–335. doi: 10.1002/cne.901240303
Androutsellis-Theotokis, A., Leker, R. R., Soldner, F., Hoeppner, D. J., Ravin, R., Poser, S. W., et al. (2006). Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 442, 823–826. doi: 10.1038/nature04940
Androutsellis-Theotokis, A., Murase, S., Boyd, J. D., Park, D. M., Hoeppner, D. J., Ravin, R., et al. (2008a). Generating neurons from stem cells. Methods Mol. Biol. 438, 31–38. doi: 10.1007/978-1-59745-133-8_4
Androutsellis-Theotokis, A., Rueger, M. A., Mkhikian, H., Korb, E., and McKay, R. D. (2008b). Signaling pathways controlling neural stem cells slow progressive brain disease. Cold Spring Harb. Symp. Quant. Biol. 73, 403–410. doi: 10.1101/sqb.2008.73.018
Androutsellis-Theotokis, A., Rueger, M. A., Park, D. M., Mkhikian, H., Korb, E., Poser, S. W., et al. (2009). Targeting neural precursors in the adult brain rescues injured dopamine neurons. Proc. Natl. Acad. Sci. U.S.A. 106, 13570–13575. doi: 10.1073/pnas.0905125106
Androutsellis-Theotokis, A., Rueger, M. A., Park, D. M., Boyd, J. D., Padmanabhan, R., Campanati, L., et al. (2010a). Angiogenic factors stimulate growth of adult neural stem cells. PLoS ONE 5:e9414. doi: 10.1371/journal.pone.0009414
Androutsellis-Theotokis, A., Walbridge, S., Park, D. M., Lonser, R. R., and McKay, R. D. (2010b). Cholera toxin regulates a signaling pathway critical for the expansion of neural stem cell cultures from the fetal and adult rodent brains. PLoS ONE 5:e10841. doi: 10.1371/journal.pone.0010841
Artavanis-Tsakonas, S., Rand, M. D., and Lake, R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science 284, 770–776. doi: 10.1126/science.284.5415.770
Banerjee, S., Byrd, J. N., Gianino, S. M., Harpstrite, S. E., Rodriguez, F. J., Tuskan, R. G., et al. (2010). The neurofibromatosis type 1 tumor suppressor controls cell growth by regulating signal transducer and activator of transcription-3 activity in vitro and in vivo. Cancer Res. 70, 1356–1366. doi: 10.1158/0008-5472.CAN-09-2178
Basak, O., and Taylor, V. (2007). Identification of self-replicating multipotent progenitors in the embryonic nervous system by high Notch activity and Hes5 expression. Eur. J. Neurosci. 25, 1006–1022. doi: 10.1111/j.1460-9568.2007.05370.x
Basak, O., and Taylor, V. (2009). Stem cells of the adult mammalian brain and their niche. Cell. Mol. Life Sci. 66, 1057–1072. doi: 10.1007/s00018-008-8544-x
Bonni, A., Sun, Y., Nadal-Vicens, M., Bhatt, A., Frank, D. A., Rozovsky, I., et al. (1997). Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278, 477–483. doi: 10.1126/science.278.5337.477
Camporeale, A., and Poli, V. (2012). IL-6, IL-17 and STAT3: a holy trinity in auto-immunity? Front. Biosci. (Landmark Ed.) 17, 2306–2326. doi: 10.2741/4054
Chung, J., Uchida, E., Grammer, T. C., and Blenis, J. (1997). STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 17, 6508–6516.
Decker, T., and Kovarik, P. (2000). Serine phosphorylation of STATs. Oncogene 19, 2628–2637. doi: 10.1038/sj.onc.1203481
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hatakeyama, J., Bessho, Y., Katoh, K., Ookawara, S., Fujioka, M., Guillemot, F., et al. (2004). Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development 131, 5539–5550. doi: 10.1242/dev.01436
Hellstrom, M., Phng, L. K., and Gerhardt, H. (2007). VEGF and Notch signaling: the yin and yang of angiogenic sprouting. Cell Adh. Migr. 1, 133–136. doi: 10.4161/cam.1.3.4978
Hirata, H., Tomita, K., Bessho, Y., and Kageyama, R. (2001). Hes1 and Hes3 regulate maintenance of the isthmic organizer and development of the mid/hindbrain. EMBO J. 20, 4454–4466. doi: 10.1093/emboj/20.16.4454
Imayoshi, I., and Kageyama, R. (2011). The role of Notch signaling in adult neurogenesis. Mol. Neurobiol. 44, 7–12. doi: 10.1007/s12035-011-8186-0
Kageyama, R., Ohtsuka, T., Hatakeyama, J., and Ohsawa, R. (2005). Roles of bHLH genes in neural stem cell differentiation. Exp. Cell Res. 306, 343–348. doi: 10.1016/j.yexcr.2005.03.015
Kamakura, S., Oishi, K., Yoshimatsu, T., Nakafuku, M., Masuyama, N., and Gotoh, Y. (2004). Hes binding to STAT3 mediates crosstalk between Notch and JAK-STAT signalling. Nat. Cell Biol. 6, 547–554. doi: 10.1038/ncb1138
Kim, J. H., Yoon, M. S., and Chen, J. (2009). Signal transducer and activator of transcription 3 (STAT3) mediates amino acid inhibition of insulin signaling through serine 727 phosphorylation. J. Biol. Chem. 284, 35425–35432. doi: 10.1074/jbc.M109.051516
Kittappa, R., Bornstein, S. R., and Androutsellis-Theotokis, A. (2012). The role of eNSCs in neurodegenerative disease. Mol. Neurobiol. 46, 555–562. doi: 10.1007/s12035-012-8303-8
Kovarik, P., Stoiber, D., Eyers, P. A., Menghini, R., Neininger, A., Gaestel, M., et al. (1999). Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 13956–13961. doi: 10.1073/pnas.96.24.13956
Lacreusette, A., Barbieux, I., Nguyen, J. M., Pandolfino, M. C., Dreno, B., Jacques, Y., et al. (2009). Defective activations of STAT3 Ser727 and PKC isoforms lead to oncostatin M resistance in metastatic melanoma cells. J. Pathol. 217, 665–676. doi: 10.1002/path.2490
Lafave, L. M., and Levine, R. L. (2012). JAK2 the future: therapeutic strategies for JAK-dependent malignancies. Trends Pharmacol. Sci. 33, 574–582. doi: 10.1016/j.tips.2012.08.005
Levy, D. E., and Darnell, J. E. Jr. (2002). Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662. doi: 10.1038/nrm909
Lim, C. P., and Cao, X. (1999). Serine phosphorylation and negative regulation of Stat3 by JNK. J. Biol. Chem. 274, 31055–31061. doi: 10.1074/jbc.274.43.31055
Lobe, C. G. (1997). Expression of the helix-loop-helix factor, Hes3, during embryo development suggests a role in early midbrain-hindbrain patterning. Mech. Dev. 62, 227–237. doi: 10.1016/S0925-4773(97)00665-5
Maisonpierre, P. C., Suri, C., Jones, P. F., Bartunkova, S., Wiegand, S. J., Radziejewski, C., et al. (1997). Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277, 55–60. doi: 10.1126/science.277.5322.55
Masjkur, J., Rueger, M. A., Bornstein, S. R., McKay, R., and Androutsellis-Theotokis, A. (2012). Neurovascular signals suggest a propagation mechanism for endogenous stem cell activation along blood vessels. CNS Neurol. Disord. Drug Targets 11, 805–817. doi: 10.2174/1871527311201070805
Miyakoshi, M., Yamamoto, M., Tanaka, H., and Ogawa, K. (2012). Serine 727 phosphorylation of STAT3: an early change in mouse hepatocarcinogenesis induced by neonatal treatment with diethylnitrosamine. Mol. Carcinog. doi: 10.1002/mc.21949. [Epub ahead of print].
Mohr, A., Chatain, N., Domoszlai, T., Rinis, N., Sommerauer, M., Vogt, M., et al. (2012). Dynamics and non-canonical aspects of JAK/STAT signalling. Eur. J. Cell Biol. 91, 524–532. doi: 10.1016/j.ejcb.2011.09.005
Ng, J., and Cantrell, D. (1997). STAT3 is a serine kinase target in T lymphocytes. Interleukin 2 and T cell antigen receptor signals converge upon serine 727. J. Biol. Chem. 272, 24542–24549. doi: 10.1074/jbc.272.39.24542
Ng, Y. P., Cheung, Z. H., and Ip, N. Y. (2006). STAT3 as a downstream mediator of Trk signaling and functions. J. Biol. Chem. 281, 15636–15644. doi: 10.1074/jbc.M601863200
Noguera-Troise, I., Daly, C., Papadopoulos, N. J., Coetzee, S., Boland, P., Gale, N. W., et al. (2006). Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444, 1032–1037. doi: 10.1038/nature05355
O'shea, J. J., and Plenge, R. (2012). JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36, 542–550. doi: 10.1016/j.immuni.2012.03.014
Pacioni, S., Rueger, M. A., Nistico, G., Bornstein, S. R., Park, D. M., McKay, R. D., et al. (2012). Fast, potent pharmacological expansion of endogenous hes3+/sox2+ cells in the adult mouse and rat hippocampus. PLoS ONE 7:e51630. doi: 10.1371/journal.pone.0051630
Panchision, D. M., and McKay, R. D. (2002). The control of neural stem cells by morphogenic signals. Curr. Opin. Genet. Dev. 12, 478–487. doi: 10.1016/S0959-437X(02)00329-5
Park, D. M., Jung, J., Masjkur, J., Makrogkikas, S., Ebermann, D., Saha, S., et al. (2013). Hes3 regulates cell number in cultures from glioblastoma multiforme with stem cell characteristics. Sci. Rep. 3:1095. doi: 10.1038/srep01095
Plaza-Menacho, I., van der Sluis, T., Hollema, H., Gimm, O., Buys, C. H., Magee, A. I., et al. (2007). Ras/ERK1/2-mediated STAT3 Ser727 phosphorylation by familial medullary thyroid carcinoma-associated RET mutants induces full activation of STAT3 and is required for c-fos promoter activation, cell mitogenicity, and transformation. J. Biol. Chem. 282, 6415–6424. doi: 10.1074/jbc.M608952200
Poser, S. W., Alisky, J., Chung, K.-F., Ebermann, D., Ehrhart-Borsntein, M., Bornstein, S., et al. (2011). “Stem cell growth as a model of carcinogenesis,” in Cancer Stem Cells—The Cutting Edge, ed S. Shostak (Rijeka, Croatia: InTech Europe), 89–110.
Poser, S. W., and Androutsellis-Theotokis, A. (2013). Growing neural stem cells from conventional and non-conventional regions of the adult rodent brain. J. Vis. Exp. (in press).
Qin, H. R., Kim, H. J., Kim, J. Y., Hurt, E. M., Klarmann, G. J., Kawasaki, B. T., et al. (2008). Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 68, 7736–7741. doi: 10.1158/0008-5472.CAN-08-1125
Rajan, P., and McKay, R. D. (1998). Multiple routes to astrocytic differentiation in the CNS. J Neurosci. 18, 3620–3629.
Reich, N. C. (2009). STAT3 revs up the powerhouse. Sci. Signal. 2:pe61. doi: 10.1126/scisignal.290pe61
Richard, A. J., and Stephens, J. M. (2011). Emerging roles of JAK-STAT signaling pathways in adipocytes. Trends Endocrinol. Metab. 22, 325–332. doi: 10.1016/j.tem.2011.03.007
Rueger, M. A., Backes, H., Walberer, M., Neumaier, B., Ullrich, R., Simard, M. L., et al. (2010). Noninvasive imaging of endogenous neural stem cell mobilization in vivo using positron emission tomography. J. Neurosci. 30, 6454–6460. doi: 10.1523/JNEUROSCI.6092-09.2010
Sasai, Y., Kageyama, R., Tagawa, Y., Shigemoto, R., and Nakanishi, S. (1992). Two mammalian helix-loop-helix factors structurally related to Drosophila hairy and Enhancer of split. Genes Dev. 6, 2620–2634. doi: 10.1101/gad.6.12b.2620
Sekine, Y., Suzuki, K., and Remaley, A. T. (2011). HDL and sphingosine-1-phosphate activate stat3 in prostate cancer DU145 cells via ERK1/2 and S1P receptors, and promote cell migration and invasion. Prostate 71, 690–699. doi: 10.1002/pros.21285
Shen, Y., Schlessinger, K., Zhu, X., Meffre, E., Quimby, F., Levy, D. E., et al. (2004). Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol. Cell. Biol. 24, 407–419. doi: 10.1128/MCB.24.1.407-419.2004
Shimojo, H., Ohtsuka, T., and Kageyama, R. (2011). Dynamic expression of notch signaling genes in neural stem/progenitor cells. Front Neurosci 5:78. doi: 10.3389/fnins.2011.00078
Shulga, N., and Pastorino, J. G. (2012). GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J. Cell. Sci. 125, 2995–3003. doi: 10.1242/jcs.103093
Siekmann, A. F., and Lawson, N. D. (2007). Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445, 781–784. doi: 10.1038/nature05577
Stark, G. R., and Darnell, J. E. Jr. (2012). The JAK-STAT pathway at twenty. Immunity 36, 503–514. doi: 10.1016/j.immuni.2012.03.013
Stephens, J. M., Lumpkin, S. J., and Fishman, J. B. (1998). Activation of signal transducers and activators of transcription 1 and 3 by leukemia inhibitory factor, oncostatin-M, and interferon-gamma in adipocytes. J. Biol. Chem. 273, 31408–31416. doi: 10.1074/jbc.273.47.31408
Su, L., Rickert, R. C., and David, M. (1999). Rapid STAT phosphorylation via the B cell receptor. Modulatory role of CD19. J. Biol. Chem. 274, 31770–31774. doi: 10.1074/jbc.274.45.31770
Swiatek-Machado, K., and Kaminska, B. (2013). STAT signaling in glioma cells. Adv. Exp. Med. Biol. 986, 189–208. doi: 10.1007/978-94-007-4719-7_10
Tan, S. L., Ohtsuka, T., Gonzalez, A., and Kageyama, R. (2012). MicroRNA9 regulates neural stem cell differentiation by controlling Hes1 expression dynamics in the developing brain. Genes Cells 17, 952–961. doi: 10.1111/gtc.12009
Thurston, G., Noguera-Troise, I., and Yancopoulos, G. D. (2007). The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat. Rev. Cancer 7, 327–331. doi: 10.1038/nrc2130
Tibes, R., Bogenberger, J. M., Geyer, H. L., and Mesa, R. A. (2012). JAK2 inhibitors in the treatment of myeloproliferative neoplasms. Expert Opin. Investig. Drugs 21, 1755–1774. doi: 10.1517/13543784.2012.721352
Tkach, M., Rosemblit, C., Rivas, M. A., Proietti, C. J., Diaz Flaque, M. C., Mercogliano, M. F., et al. (2013). p42/p44 MAPK-mediated Stat3Ser727 phosphorylation is required for progestin-induced full activation of Stat3 and breast cancer growth. Endocr. Relat. Cancer 20, 197–212. doi: 10.1530/ERC-12-0194
Ueo, T., Imayoshi, I., Kobayashi, T., Ohtsuka, T., Seno, H., Nakase, H., et al. (2012). The role of Hes genes in intestinal development, homeostasis and tumor formation. Development 139, 1071–1082. doi: 10.1242/dev.069070
Vera, J., Rateitschak, K., Lange, F., Kossow, C., Wolkenhauer, O., and Jaster, R. (2011). Systems biology of JAK-STAT signalling in human malignancies. Prog. Biophys. Mol. Biol. 106, 426–434. doi: 10.1016/j.pbiomolbio.2011.06.013
Villalva, C., Martin-Lanneree, S., Cortes, U., Dkhissi, F., Wager, M., Le Corf, A., et al. (2011). STAT3 is essential for the maintenance of neurosphere-initiating tumor cells in patients with glioblastomas: a potential for targeted therapy? Int. J. Cancer 28, 826–838. doi: 10.1002/ijc.25416
Wauman, J., and Tavernier, J. (2011). Leptin receptor signaling: pathways to leptin resistance. Front. Biosci. (Landmark Ed.) 16, 2771–2793. doi: 10.2741/3885
Yancopoulos, G. D., Davis, S., Gale, N. W., Rudge, J. S., Wiegand, S. J., and Holash, J. (2000). Vascular-specific growth factors and blood vessel formation. Nature 407, 242–248. doi: 10.1038/35025215
Yang, F., Zhang, W., Li, D., and Zhan, Q. (2013). Gadd45a suppresses tumor angiogenesis via inhibition of the mTOR/STAT3 protein pathway. J. Biol. Chem. 288, 6552–6560. doi: 10.1074/jbc.M112.418335
Yeh, H. H., Giri, R., Chang, T. Y., Chou, C. Y., Su, W. C., and Liu, H. S. (2009). Ha-ras oncogene-induced Stat3 phosphorylation enhances oncogenicity of the cell. DNA Cell Biol. 28, 131–139. doi: 10.1089/dna.2008.0762
Yokogami, K., Wakisaka, S., Avruch, J., and Reeves, S. A. (2000). Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 10, 47–50. doi: 10.1016/S0960-9822(99)00268-7
Zhang, J., Yang, J., Roy, S. K., Tininini, S., Hu, J., Bromberg, J. F., et al. (2003). The cell death regulator GRIM-19 is an inhibitor of signal transducer and activator of transcription 3. Proc. Natl. Acad. Sci. U.S.A. 100, 9342–9347. doi: 10.1073/pnas.1633516100
Zhou, L., and Too, H. P. (2011). Mitochondrial localized STAT3 is involved in NGF induced neurite outgrowth. PLoS ONE 6:e21680. doi: 10.1371/journal.pone.0021680
Keywords: stem cells, regenerative medicine, Hes3, STAT3 transcription factor, signal transduction
Citation: Poser SW, Park DM and Androutsellis-Theotokis A (2013) The STAT3-Ser/Hes3 signaling axis: an emerging regulator of endogenous regeneration and cancer growth. Front. Physiol. 4:273. doi: 10.3389/fphys.2013.00273
Received: 23 August 2013; Paper pending published: 02 September 2013;
Accepted: 11 September 2013; Published online: 01 October 2013.
Edited by:
Eumorphia Remboutsika, BSRC “Alexander Fleming,” GreeceReviewed by:
Eumorphia Remboutsika, BSRC “Alexander Fleming,” GreeceCopyright © 2013 Poser, Park and Androutsellis-Theotokis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Androutsellis-Theotokis, Stem Cell Biology Laboratory, Department of Internal Medicine III, University Hospital Carl Gustav Carus at the Technische Universität Dresden, Fetscherstrasse. 74, 01307 Dresden, Germany e-mail:YW5kcmVhcy50aGVvdG9raXNAdW5pa2xpbmlrdW0tZHJlc2Rlbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.