
 Lucia Biasutto2
Lucia Biasutto2
- 1Department of Biology, University of Padova, Padova, Italy
- 2CNR Institute of Neuroscience and Department of Biomedical Sciences of the University of Padova, Padova, Italy
- 3Department of Molecular Biology, University of Duisburg-Essen, Essen, Germany
Several types of channels play a role in the maintenance of ion homeostasis in subcellular organelles including endoplasmatic reticulum, nucleus, lysosome, endosome, and mitochondria. Here we give a brief overview of the contribution of various mitochondrial and other organellar channels to cancer cell proliferation or death. Much attention is focused on channels involved in intracellular calcium signaling and on ion fluxes in the ATP-producing organelle mitochondria. Mitochondrial K+ channels (Ca2+-dependent BKCa and IKCa, ATP-dependent KATP, Kv1.3, two-pore TWIK-related Acid-Sensitive K+ channel-3 (TASK-3)), Ca2+ uniporter MCU, Mg2+-permeable Mrs2, anion channels (voltage-dependent chloride channel VDAC, intracellular chloride channel CLIC) and the Permeability Transition Pore (MPTP) contribute importantly to the regulation of function in this organelle. Since mitochondria play a central role in apoptosis, modulation of their ion channels by pharmacological means may lead to death of cancer cells. The nuclear potassium channel Kv10.1 and the nuclear chloride channel CLIC4 as well as the endoplasmatic reticulum (ER)-located inositol 1,4,5-trisphosphate (IP3) receptor, the ER-located Ca2+ depletion sensor STIM1 (stromal interaction molecule 1), a component of the store-operated Ca2+ channel and the ER-resident TRPM8 are also mentioned. Furthermore, pharmacological tools affecting organellar channels and modulating cancer cell survival are discussed. The channels described in this review are summarized on Figure 1. Overall, the view is emerging that intracellular ion channels may represent a promising target for cancer treatment.
Mitochondria
The “impermeable” mitochondrial inner membrane (IMM) allows the formation of an electrochemical proton gradient which drives the aerobic synthesis of ATP. The “semipermeable” outer membrane (OMM) encloses a periplasmic space where proteins with fundamental roles in cell death are stored until a sufficiently strong pro-apoptotic signal arrives. Mitochondria have assumed a peculiar role in cancer cell physiology (Ralph and Neuzil, 2009). They are crucial for the control of intracellular Ca2+ homeostasis, and produce reactive oxygen species (ROS). ROS are involved in the regulation of physiological processes, but may also be harmful if produced excessively. Mitochondria are the checkpoint of the intrinsic pathway of apoptosis: the release of caspase cofactors, such as cytochrome c (cyt c) and SMAC/Diablo, results in the assembly of the apoptosome and in commitment of the cell to apoptosis. In cancer cells mitochondrial metabolism is deregulated to optimize the production of glycolytic intermediates for anabolic reactions. Much effort has been devoted to discover drugs inducing cancer cell death by targeting tumor-specific alterations of mitochondrial metabolism or by stimulating OMM permeabilization and thus, allowing the release of apoptotic co-factors (Fulda et al., 2010). Mitochondrial ion channels play a role in this process by influencing organellar membrane potential, ROS production, volume, calcium homeostasis, and possibly morphology. The mitochondrial channels characterized over the last two decades include outer membrane-located VDAC and MAC in the IMM, potassium channels mtKATP, mtBKCa, mtIKCa, mtKv1.3, TASK-3, the non-selective permeability transition pore PTP, chloride channels, and the calcium uniporter (e.g., Zoratti et al., 2009; Shoshan-Barmatz et al., 2010; Rizzuto et al., 2012; Szabò et al., 2012).
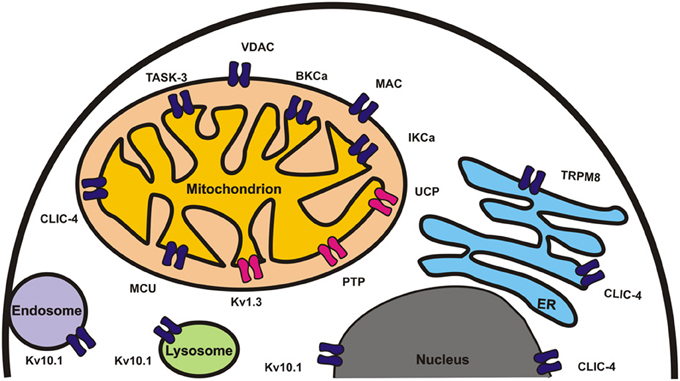
Figure 1. Ion channels involved in regulation of apoptosis and/or tumorigenesis are shown in different organelles. Channels for whose crucial role pharmacological and/or genetic in vivo evidence is available are shown in red.
Channels of the Outer Mitochondrial Membrane Involved in Apoptosis/Cancer
Mitochondrial apoptosis-induced channel (MAC)
OMM permeabilization has been proposed to involve oligomers of pro-apoptotic Bax, which display ion channel activity in phospholipid bilayers (e.g., Tait and Green, 2010). However, the hypothesis that Bax alone is sufficient to induce cyt c release has been challenged, given that a single point mutant of Bax did not mediate cell death in Bax/Bak-less mouse embryonic fibroblasts despite forming channels with properties similar to WT Bax (Brustovetsky et al., 2010; Szabò et al., 2011). A pore (mitochondrial apoptosis-induced channel, MAC) with an estimated diameter sufficient to allow the passage of cyt c was detected by patch clamp (Martinez-Caballero et al., 2009). The timing of cyt c release in apoptotic cells correlated with the onset of MAC activity and with the translocation of Bax to mitochondrial membranes. MAC, whose formation requires Bim-induced activation of Bax and a still unidentified protein, is considered as a target for novel cancer therapies (Peixoto et al., 2012) but specific MAC activators are not available yet. The BH3 mimetic ABT-737, an efficient anti-cancer agent in vivo, activates MAC by disrupting Bcl-2/Bax/Bim complexes (Dejean et al., 2010).
Mitochondrial voltage dependent anion channel (VDAC)
The major protein of the OMM, porin or VDAC is deeply involved in apoptosis. The role of VDAC1 and of the other isoforms VDAC2 and VDAC3 in cell death is multi-faceted and complex (e.g., McCommis and Baines, 2012; Shoshan-Barmatz and Golan, 2012; Shoshan-Barmatz and Mizrachi, 2012). Formation of a large pore comprising VDAC and Bax/Bak was proposed to account for cyt c release (Tsujimoto and Shimizu, 2000; but see Martinez-Caballero et al., 2009). Alternatively, dimers and higher oligomers of VDAC1 might form the conduit for the efflux of cyt c (Shoshan-Barmatz et al., 2010). Binding of anti-apoptotic Bcl-2 and BclxL to VDAC1 (with resulting inhibition of porin) (Shimizu et al., 2000) has an anti-apoptotic action (e.g., Arbel et al., 2012). In contrast, block of VDAC1 by the phosphorothioate oligonucleotide G3139 (Tan, 2012) or by avicins (plant saponins with anticancer activity) is pro-apoptotic, presumably by reducing flux of metabolites across the OMM (Haridas et al., 2007). VDAC2 inhibits Bak activation and apoptosis (Cheng et al., 2003), and Bak reportedly relocates from the OMM to the ER in the absence of VDAC2 (Raghavan et al., 2012). In contrast, Bax-induced cyt c release from mitochondria isolated from WT or VDAC1−, VDAC3− and VDAC1/VDAC3-null cells was reported to be the same (Baines et al., 2007).
VDAC may inhibit apoptosis and promote tumorigenesis through specific interactions with enzymes favoring glycolysis. It is being examined as a cancer-specific target since tumor cells have elevated glycolysis and increased expression of VDACs (Grills et al., 2011). Overexpression of Hexokinase-2 (HK2) and its association with VDAC are key features of glycolytic cancers (e.g., Wolf et al., 2011). HK2 binding to the conduit channeling ATP out of mitochondria provides a metabolic benefit to cancer cells (Warburg effect) and it antagonizes cell death via inhibition of Bax-induced cyt c release (Pastorino et al., 2002; Gall et al., 2011) and/or inhibition of the Mitochondrial Permeability Transition (MPT) (Chiara et al., 2008). HK detachment seems to favor cell death by disruption of aerobic glycolysis and of the energy balance of the cell, regulation of ROS production, altered interaction of Bcl2 family proteins with mitochondria, facilitation of VDAC oligomer formation (e.g., Shoshan-Barmatz et al., 2010; Shoshan-Barmatz and Golan, 2012). Therefore, a major oncological target is the HK-VDAC complex (e.g., Galluzzi et al., 2008; Simamura et al., 2008; Fulda et al., 2010; Mathupala and Pedersen, 2010). HK2 can be dissociated from mitochondria by peptides interfering with HK-VDAC association, by erastin (Yagoda et al., 2007) and by 3-bromopyruvate (e.g., Cardaci et al., 2012; Ko et al., 2012; Pedersen, 2012; Shoshan, 2012). Antifungal drugs clotrimazole and bifonazole and the plant hormone methyl jasmonate (MJ) are also effective. MJ is particularly promising since it has selective anticancer activity in preclinical studies (Fulda et al., 2010). Finally, the anti-cancer agent furanonaphthoquinone (FNQ) induces caspase-dependent apoptosis via the production of ROS, which is enhanced by VDAC1 overexpression (Simamura et al., 2008). A systematic search for compounds acting at the level of VDAC to antagonize cancer remains to be performed.
Ion Channels of the Inner Mitochondrial Membrane Involved in Apoptosis/Cancer
Permeability transition pore (MPTP)
When the IMM becomes freely permeable to solutes, the consequences for the cell can be catastrophic. Thus, the selective induction of IMM permeabilization in cancer cells is a strategy worth pursuing in oncotherapy. A number of cellular stresses and cytotoxic agents trigger the prime example of such a catastrophe, i.e., the mitochondrial permeability transition (MPT), considered as a final common pathway of cell death (Brenner and Grimm, 2006; Bernardi, 2013). The MPT is caused by the opening of a large Ca2+- and oxidative stress-activated pore [the mitochondrial megachannel, MMC, with a conductance of up to 1.5 nS (Szabó and Zoratti, 1991)] which makes the IMM permeable to ions and solutes up to about 1500 Da MW, leading to matrix swelling.
MPT is considered to bear substantial responsibilities in the tissue damage caused by, e.g., ischemia/reperfusion and oxidative stress. In cancer cells, instead, signaling pathways are activated which desensitize the mitochondria to MPT induction (Rasola et al., 2010; Matassa et al., 2012; Traba et al., 2012), while chemotherapeutic agents causing oxidative stress may activate signals causing death via the MPT (Chiara et al., 2012). Cyclosporin A (CSA), a cyclic endecapeptide, is a powerful inhibitor of the MPTP (Fournier et al., 1987; Crompton et al., 1988; Broekemeier et al., 1989) (and also of calcineurin and thus is a widely used immunosuppressant). CSA inhibits the MPTP via its binding to matrix cyclophilin (CyP) D, a peptidyl-prolyl cis-trans isomerase (PPIase). Patients treated with CSA to prevent transplant rejection have a high incidence of cancer not only because of the drug's immunosuppressive action, but also because CSA inhibits the MPTP (Norman et al., 2010). The molecular nature of the MPTP is being finally delineated: the dimeric form of ATP synthase and CypD as regulator are currently proposed as components (Baines et al., 2005; Bernardi, 2013; Giorgio et al., 2013).
For oncological applications MPT inducers are relevant, despite the likelihood of noxious side-effects, for example on the nervous system. A large number of compounds, often used at relatively high concentrations, have been shown to induce the MPT in cultured cells, often as a consequence of oxidative stress and/or disruption of Ca2+ homeostasis. Some MPTP-targeting molecules such as 4-(N-(S-glutathionylacetyl) amino) phenylarsenoxide are currently being evaluated in clinical trials for cancer treatment of refractory tumors (Brenner and Moulin, 2012; Elliott et al., 2012).
Signaling pathways which modulate occurrence of the MPT have been elucidated, a key component being GSK3α/β whose activation, e.g., by induction of oxidative stress by gold complex AUL12, favors MPTP opening (Chiara and Rasola, 2013). A large portion of MPTP openers are natural compounds like jasmonates (e.g., Raviv et al., 2013), betulinic acid, the synthetic retinoid CD437 (Lena et al., 2009; Javadov et al., 2011), berberine (Pereira et al., 2007, 2008), honokiol (Li et al., 2007), α-bisabolol (Cavalieri et al., 2009) and shikonin (Han et al., 2007), just to name a few. Data on in vivo anti-tumor activities are available for all these compounds (Fulda et al., 2010). Mitochondria-penetrating peptides, such as mastoparan-like sequences, peptides of the innate immunity systems, or the molecules developed by Kelley's group (e.g., Risso et al., 2002; Jones et al., 2008; Horton et al., 2012) also induce MPT. Some MPTP-targeting molecules such as 4-(N-(S-glutathionylacetyl) amino) phenylarsenoxide are currently being evaluated in clinical trials for cancer treatment of refractory tumors (Brenner and Moulin, 2012; Elliott et al., 2012).
IMM potassium channels Kv1.3, BKca, IKca, and TASK-3 in the regulation of apoptosis/cancer
A functional mitochondrial counterpart of the potassium channel Kv1.3 has been identified in the IMM of several cell types (mtKv1.3) (Szabò et al., 2005; Gulbins et al., 2010). It is expected to participate in regulation of mitochondrial membrane potential, volume, and ROS production. A crucial role of mtKv1.3 in apoptosis became evident since expression of a mitochondria-targeted Kv1.3 construct was sufficient to sensitize apoptosis-resistant CTLL-2 T lymphocytes, which lack Kv channels. MtKv1.3 has been identified as a target of Bax and physical interaction between the two proteins in apoptotic cells has been demonstrated (Szabó et al., 2008; Szabò et al., 2011). Incubating Kv1.3-positive isolated mitochondria with Bax triggered apoptotic events including membrane potential changes (hyperpolarization followed by depolarization due to the opening of MPTP), ROS production and cyt c release, whereas Kv1.3-deficient mitochondria were resistant. Highly conserved Bax lysine 128 protrudes into the intermembrane space (Annis et al., 2005) and mimics a crucial lysine in Kv1.3-blocking peptide toxins. Mutation of Bax at K128 (BaxK128E) abrogated its effects on Kv1.3 and mitochondria, as well as in Bax/Bak-less double knock-out (DKO) mouse embryonic fibroblasts, indicating a toxin-like action of Bax on Kv1.3 to trigger mitochondrial phenomena.
Psora-4, PAP-1 and clofazimine, three membrane-permeant inhibitors of Kv1.3, can induce death by directly targeting the mitochondrial channel, while membrane-impermeant Kv1.3 inhibitors ShK or Margatoxin did not induce apoptosis (Leanza et al., 2012a,b). Importantly, the membrane-permeant drugs killed cells also in the absence of Bax and Bak, in agreement with the above model. Genetic deficiency or siRNA-mediated downregulation of Kv1.3 abrogated the effects of the drugs. Intraperitoneal injection of clofazimine reduced tumor size by 90% in an orthotopic melanoma B16F10 mouse model in vivo, while no adverse effects were observed in several healthy tissues. Similar results were obtained with primary human cancer cells from patients with chronic lymphocytic leukemia (Leanza et al., 2013). The selective action of these drugs on tumor cells is related to a synergistic effect of a higher expression of Kv1.3 and of an altered redox state of cancer cells. The fact that clofazimine is already used in the clinic for the treatment of e.g., leprosis (Ren et al., 2008) and shows an excellent safety profile supports the feasibility of targeting mtKv1.3 for therapy.
The large conductance calcium- and voltage-activated K+ channel BKCa (KCa1.1) has been revealed also in intracellular membranes, including nuclear membrane, ER, Golgi and mitochondria (Xu et al., 2002; O'Rourke, 2007; Singh et al., 2012). Patch clamp experiments with recombinant Bax showed an inhibition of BKCa, which might contribute to opening of the MPTP during cell death (Cheng et al., 2011).
The intermediate conductance potassium channel (IKCa; KCa3.1), selectively inhibited by clotrimazole and TRAM-34, has been recorded from the inner mitochondrial membranes of human cancer cells (De Marchi et al., 2009; Sassi et al., 2010). TRAM-34 used alone did not induce apoptosis (Sassi et al., 2010; Quast et al., 2012), but it synergistically increased sensitivity to the death receptor ligand TRAIL in melanoma cells (Quast et al., 2012). Given that both TRAM-34 and TRAIL have a relatively good safety profile, co-administration of the two drugs might be exploited for melanoma treatment.
Recently TASK-3 (KCNK9), a two-pore potassium channel, was identified in mitochondria of melanoma and keratinocyte (Rusznák et al., 2008) as well as healthy intestinal epithelial cells (Kovács et al., 2005). Reduced expression of TASK-3 resulted in compromised mitochondrial function and cell survival in WM35 melanoma cells (Kosztka et al., 2011). Whether TASK-3 protein gives rise to a functional channel in the IMM and whether it will become an oncological target remain to be determined.
Other IMM Channels Linked to Tumorigenesis: Uncoupling Protein UCP, Mg2+ Channel Mrs-2 and Calcium Uniporter MCU
Uncoupling protein-2 (UCP-2), which mediates proton leak (Cannon and Nedergaard, 2004; Fedorenko et al., 2012), has been proposed to regulate cell survival by decreasing mitochondrial ROS, since a depolarizing proton leak is expected to diminish superoxide production (Baffy et al., 2011). UCP2 over-expression reportedly prevents oxidative injury, thereby possibly contributing to a higher apoptotic threshold assisting survival of cancer cells. Over-expression of UCP2 was found in numerous types of tumors and has been shown to protect cells from oxidative stress (Arsenijevic et al., 2000; Zhang et al., 2007) and even to abolish chemotherapeutic agent- induced apoptosis (Derdak et al., 2008). Ectopic expression of UCP2 in MCF7 breast cancer cells leads to a decreased mitochondrial membrane potential and increased tumorigenic properties as measured by cell migration, in vitro invasion, and anchorage independent growth. Interestingly, UCP2 over-expression has also been proposed to directly contribute to the Warburg phenotype (Samudio et al., 2008) and to development of tumors in an orthotopic model of breast cancer (Ayyasamy et al., 2011). Cisplatin downregulated the expression of UCP2 in colon cancer cells (Santandreu et al., 2010), suggesting that UCP2 over-expression is involved in the development of a variety of cancers. UCP2 can be considered as a promising oncological target.
Mitochondria accumulate Mg2+ via Mrs2, a Mg2+–selective channel of the IMM (Kolisek et al., 2003). An early increase in cytosolic Mg2+ occurs during apoptosis (Chien et al., 1999) and this ion seems to be required for cytochrome c release (Kim et al., 2000). Long-lasting knock-down of Mrs2 caused cell death by inducing loss of respiratory complex I and mitochondrial membrane depolarization (Piskacek et al., 2009). A subtractive hybridization method applied on vincristine or adriamycin resistant and parental human gastric adenocarcinoma cell lines highlighted upregulation of Mrs2 (Chen et al., 2009), suggesting that high expression of Mrs2 may protect against death (Wolf and Trapani, 2009).
The molecular identification of the mitochondrial Ca2+ “uniporter” (MCU), responsible for the low-affinity uptake of calcium into the mitochondrial matrix (Kirichok et al., 2004), has recently been achieved (Baughman et al., 2011; De Stefani et al., 2011). MCU participates in the control of Ca2+ signaling in the whole cell, and may thus be a very useful tool to influence the myriad cellular calcium-dependent processes, including cell death (Rizzuto et al., 2012). Subthreshold apoptotic signals were shown to synergize with cytosolic Ca2+ waves (Pinton et al., 2001) resulting in opening of MPTP. Cells overexpressing MCU underwent more pronounced apoptosis upon challenging with H2O2 and C2-ceramide (De Stefani et al., 2011). Overexpression of an MCU-targeting microRNA, miR-25, in colon cancer cells resulted in MCU downregulation, impaired calcium uptake and increased resistance to apoptosis (Marchi et al., 2013). Thus, MCU seems to be a crucial protein for tumorigenesis and its specific pharmacological activators, if identified, might become useful tools.
Ion Channels in Other Organelles with a Role in Apoptosis/Tumorigenesis
The intracellular chloride channel CLIC4/mtCLIC has both a soluble and a membrane-inserted form and can be localized to the mitochondrial inner membrane (Fernández-Salas et al., 1999), cytoplasm, ER membrane, and the nucleus. CLIC4 overexpression induced apoptosis associated with loss of mitochondrial membrane potential, cytochrome c release, and caspase activation (Fernández-Salas et al., 2002). On the other hand, inhibition of CLIC4 expression triggered mitochondrial apoptosis under starvation and enhanced autophagy in glioma cells (Zhong et al., 2012). Marked changes in expression and subcellular localization of CLIC4 occur early in tumorigenesis. In particular, reduced CLIC4 expression and nuclear localization in cancer cells is associated with the altered redox state and CLIC4 acts as an important suppressor of squamous tumor development and progression (Suh et al., 2012).
A functional “oncogenic” potassium channel, Kv10.1 has been described in the nuclear inner membrane (Chen et al., 2011) where it might participate in setting nuclear [K+] thereby affecting gene expression. The PM Kv10.1 is also rapidly internalized to lysosomes (Kohl et al., 2011), whose patch clamping has been achieved (Wang et al., 2012). The possible influence of these channels on cancer cell survival remains to be determined.
Finally, we should briefly mention other intracellular channels involved in Ca2+ signaling, (Ca2+ permeable channels are discussed in detail by other contributions in this special issue). For example the calcium-permeable ion channel TRPM8, overexpressed in several tumors, has been located to the ER (Zhang and Barritt, 2004), resulting in decreased ER [Ca2+] and increased resistance to apoptosis (Bidaux et al., 2007). Patients suffering of breast cancers with high ER-located STIM1 levels have significantly reduced survival (McAndrew et al., 2011). The PM-located other component, ORAi1 contributes to altered calcium homeostasis as well (Monteith et al., 2012). Expression of ER-resident IP3 receptors acting as Ca2+ store release channels is altered in glioblastoma (Kang et al., 2010). Repression of IP3- mediated Ca2+ elevation by Bcl-2 has been proposed to contribute to the pathophysiology of chronic lymphocytic leukemia (Zhong et al., 2011).
In summary, while considerable further work is required to clarify the mechanisms by which intracellular channels contribute to tumorigenesis and tumor progression, or intervene in cell death, a few in vivo studies targeting these channels underline the importance of pursuing this line of research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Senior post-doc fellowship and Young Investigator grant of the University of Padova to Luigi Leanza is acknowledged. The authors are grateful for funding to the Italian Association for Cancer Research (AIRC) (grant 5118 to Ildikò Szabò), the Progetti di Rilevante Interesse Nazionale (PRIN) program (2010CSJX4F to Ildikò Szabò and 20107Z8XBW to Mario Zoratti), by DFG-grant Gu 335/13–3 (Erich Gulbins), the International Association for Cancer Research (Erich Gulbins); the EMBO Young Investigator Program grant (Ildikò Szabò), the Fondazione Cassa di Risparmio di Padova e Rovigo (to Mario Zoratti); the CNR Project of Special Interest on Aging (Mario Zoratti).
References
Annis, M. G., Soucie, E. L., Dlugosz, P. J., Cruz-Aguado, J. A., Penn, L. Z., Leber, B., et al. (2005). Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 24, 2096–2103. doi: 10.1038/sj.emboj.7600675
Arbel, N., Ben-Hail, D., and Shoshan-Barmatz, V. (2012). Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J. Biol. Chem. 287, 23152–23161. doi: 10.1074/jbc.M112.345918
Arsenijevic, D., Onuma, H., Pecqueur, C., Raimbault, S., Manning, B. S., Miroux, B., et al. (2000). Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 26, 435–439. doi: 10.1038/82565
Ayyasamy, V., Owens, K. M., Desouki, M. M., Liang, P., Bakin, A., Thangaraj, K., et al. (2011). Cellular model of Warburg effect identifies tumor promoting function of UCP2 in breast cancer and its suppression by genipin. PLoS ONE 6:e24792. doi: 10.1371/journal.pone.0024792
Baffy, G., Derdak, Z., and Robson, S. C. (2011). Mitochondrial recoupling: a novel therapeutic strategy for cancer. Br. J. Cancer 105, 469–474. doi: 10.1038/bjc.2011.245
Baines, C. P., Kaiser, R. A., Purcell, N. H., Blair, N. S., Osinska, H., Hambleton, M. A., et al. (2005). Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662. doi: 10.1038/nature03434
Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J., and Molkentin, J. D. (2007). Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555. doi: 10.1038/ncb1575
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Bernardi, P. (2013). The mitochondrial permeability transition pore: a mystery solved. Front. Physiol. 4:95. doi: 10.3389/fphys.2013.00095
Bidaux, G., Flourakis, M., Thebault, S., Zholos, A., Beck, B., Gkika, D., et al. (2007). Prostate cell differentiation status determines transient receptor potential melastatin member 8 channel subcellular localization and function. J. Clin. Invest. 117, 1647–1657. doi: 10.1172/JCI30168
Brenner, C., and Grimm, S. (2006). The permeability transition pore complex in cancer cell death. Oncogene 25, 4744–4756. doi: 10.1038/sj.onc.1209609
Brenner, C., and Moulin, M. (2012). Physiological roles of the permeability transition pore. Circ. Res. 111, 1237–1247. doi: 10.1161/CIRCRESAHA.112.265942
Broekemeier, K. M., Dempsey, M. E., and Pfeiffer, D. R. (1989). Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J. Biol. Chem. 264, 7826–7830.
Brustovetsky, T., Li, T., Yang, Y., Zhang, J. T., Antonsson, B., and Brustovetsky, N. (2010). BAX insertion, oligomerization, and outer membrane permeabilization in brain mitochondria: role of permeability transition and SH-redox regulation. Biochim. Biophys. Acta 1797, 1795–1806. doi: 10.1016/j.bbabio.2010.07.006
Cannon, B., and Nedergaard, J. (2004). Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359. doi: 10.1152/physrev.00015.2003
Cardaci, S., Desideri, E., and Ciriolo, M. R. (2012). Targeting aerobic glycolysis: 3-bromopyruvate as a promising anticancer drug. J. Bioenerg. Biomembr. 44, 17–29. doi: 10.1007/s10863-012-9422-7
Cavalieri, E., Bergamini, C., Mariotto, S., Leoni, S., Perbellini, L., Darra, E., et al. (2009). Involvement of mitochondrial permeability transition pore opening in alpha-bisabolol induced apoptosis. FEBS J. 276, 3990–4000. doi: 10.1111/j.1742-4658.2009.07108.x
Chen, Y., Sánchez, A., Rubio, M. E., Kohl, T., Pardo, L. A., and Stühmer, W. (2011). Functional K(v)10.1 channels localize to the inner nuclear membrane. PLoS ONE 6:e19257. doi: 10.1371/journal.pone.0019257
Chen, Y., Wei, X., Yan, P., Han, Y., Sun, S., Wu, K., et al. (2009). Human mitochondrial Mrs2 protein promotes multidrug resistance in gastric cancer cells by regulating p27, cyclin D1 expression and cytochrome C release. Cancer Biol. Ther. 8, 607–614. doi: 10.4161/cbt.8.7.7920
Cheng, E. H., Sheiko, T. V., Fisher, J. K., Craigen, W. J., and Korsmeyer, S. J. (2003). VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301, 513–517. doi: 10.1126/science.1083995
Cheng, Y., Gulbins, E., and Siemen, D. (2011). Activation of the permeability transition pore by Bax via inhibition of the mitochondrial BK channel. Cell. Physiol. Biochem. 27, 191–200. doi: 10.1159/000327944
Chiara, F., Castellaro, D., Marin, O., Petronilli, V., Brusilow, W. S., Juhaszova, M., et al. (2008). Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE 3:e1852. doi: 10.1371/journal.pone.0001852
Chiara, F., Gambalunga, A., Sciacovelli, M., Nicolli, A., Ronconi, L., Fregona, D., et al. (2012). Chemotherapeutic induction of mitochondrial oxidative stress activates GSK-3α/β and Bax, leading to permeability transition pore opening and tumor cell death. Cell Death Dis. 3:e444. doi: 10.1038/cddis.2012.184
Chiara, F., and Rasola, A. (2013). GSK-3 and mitochondria in cancer cells. Front. Oncol. 3:16. doi: 10.3389/fonc.2013.00016
Chien, M. M., Zahradka, K. E., Newell, M. K., and Freed, J. H. (1999). Fas-induced B cell apoptosis requires an increase in free cytosolic magnesium as an early event. J. Biol. Chem. 274, 7059–7066. doi: 10.1074/jbc.274.11.7059
Crompton, M., Ellinger, H., and Costi, A. (1988). Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 255, 357–360.
Dejean, L. M., Ryu, S. Y., Martinez-Caballero, S., Teijido, O., Peixoto, P. M., and Kinnally, K. W. (2010). MAC and Bcl-2 family proteins conspire in a deadly plot. Biochim Biophys Acta 1797, 1231–1238. doi: 10.1016/j.bbabio.2010.01.007
De Marchi, U., Sassi, N., Fioretti, B., Catacuzzeno, L., Cereghetti, G. M., Szabò, I., et al. (2009). Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium 45, 509–516. doi: 10.1016/j.ceca.2009.03.014
Derdak, Z., Mark, N. M., Beldi, G., Robson, S. C., Wands, J. R., and Baffy, G. (2008). The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 68, 2813–2819. doi: 10.1158/0008-5472.CAN-08-0053
De Stefani, D., Raffaello, A., Teardo, E., Szabò, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
Elliott, M. A., Ford, S. J., Prasad, E., Dick, L. J., Farmer, H., Hogg, P. J., et al. (2012). Pharmaceutical development of the novel arsenical based cancer therapeutic GSAO for Phase I clinical trial. Int. J. Pharm. 426, 67–75. doi: 10.1016/j.ijpharm.2012.01.024
Fedorenko, A., Lishko, P. V., and Kirichok, Y. (2012). Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413. doi: 10.1016/j.cell.2012.09.010
Fernández-Salas, E., Sagar, M., Cheng, C., Yuspa, S. H., and Weinberg, W. C. (1999). p53 and tumor necrosis factor alpha regulate the expression of a mitochondrial chloride channel protein. J Biol Chem. 274, 36488–36497. doi: 10.1074/jbc.274.51.36488
Fernández-Salas, E., Suh, K. S., Speransky, V. V., Bowers, W. L., Levy, J. M., Adams, T., et al. (2002). mtCLIC/CLIC4, an organellular chloride channel protein, is increased by DNA damage and participates in the apoptotic response to p53. Mol Cell Biol. 22, 3610–3620. doi: 10.1128/MCB.22.11.3610-3620.2002
Fournier, N., Ducet, G., and Crevat, A. (1987). Action of cyclosporine on mitochondrial calcium fluxes. J. Bioenerg. Biomembr. 19, 297–303. doi: 10.1007/BF00762419
Fulda, S., Galluzzi, L., and Kroemer, G. (2010). Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 9, 447–464. doi: 10.1038/nrd3137
Gall, J. M., Wong, V., Pimental, D. R., Havasi, A., Wang, Z., Pastorino, J. G., et al. (2011). Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress. Kidney Int. 79, 1207–1216. doi: 10.1038/ki.2010.532
Galluzzi, L., Kepp, O., Tajeddine, N., and Kroemer, G. (2008). Disruption of the hexokinase-VDAC complex for tumor therapy. Oncogene 27, 4633–4635. doi: 10.1038/onc.2008.114
Giorgio, V., von Stockum, S., Antoniel, M., Fabbro, A., Fogolari, F., Forte, M., et al. (2013). Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 110, 5887–5892. doi: 10.1073/pnas.1217823110
Grills, C., Jithesh, P. V., Blayney, J., Zhang, S. D., and Fennell, D. A. (2011). Gene expression meta-analysis identifies VDAC1 as a predictor of poor outcome in early stage non-small cell lung cancer. PLoS ONE 6:e14635. doi: 10.1371/journal.pone.0014635
Gulbins, E., Sassi, N., Grassmè, H., Zoratti, M., and Szabò, I. (2010). Role of Kv1.3 mitochondrial potassium channel in apoptotic signalling in lymphocytes. Biochim. Biophys. Acta 1797, 1251–1259. doi: 10.1016/j.bbabio.2010.01.018
Han, W., Li, L., Qiu, S., Lu, Q., Pan, Q., Gu, Y., et al. (2007). Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol. Cancer Ther. 6, 1641–1649. doi: 10.1158/1535-7163.MCT-06-0511
Haridas, V., Li, X., Mizumachi, T., Higuchi, M., Lemeshko, V. V., Colombini, M., et al. (2007). Avicins, a novel plant-derived metabolite lowers energy metabolism in tumor cells by targeting the outer mitochondrial membrane. Mitochondrion 7, 234–240. doi: 10.1016/j.mito.2006.12.005
Horton, K. L., Pereira, M. P., Stewart, K. M., Fonseca, S. B., and Kelley, S. O. (2012). Tuning the activity of mitochondria-penetrating peptides for delivery or disruption. Chembiochem. 13, 476–485. doi: 10.1002/cbic.201100415
Javadov, S., Hunter, J. C., Barreto-Torres, G., and Parodi-Rullan, R. (2011). Targeting the mitochondrial permeability transition: cardiac ischemia-reperfusion versus carcinogenesis. Cell Physiol. Biochem. 27, 179–190. doi: 10.1159/000327943
Jones, S., Martel, C., Belzacq-Casagrande, A. S., Brenner, C., and Howl, J. (2008). Mitoparan and target-selective chimeric analogues: membrane translocation and intracellular redistribution induces mitochondrial apoptosis. Biochim. Biophys. Acta 1783, 849–863. doi: 10.1016/j.bbamcr.2008.01.009
Kang, S. S., Han, K. S., Ku, B. M., Lee, Y. K., Hong, J., Shin, H. Y., et al. (2010). Caffeine-mediated inhibition of calcium release channel inositol 1 4, 5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 70, 1173–1183. doi: 10.1158/0008-5472.CAN-09-2886
Kim, T. H., Zhao, Y., Barber, M. J., Kuharsky, D. K., and Yin, X. M. (2000). Bid-induced cytochrome c release is mediated by a pathway independent of mitochondrial permeability transition pore and Bax. J Biol Chem. 275, 39474–39481. doi: 10.1074/jbc.M003370200
Kirichok, Y., Krapivinsky, G., and Clapham, D. E. (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. doi: 10.1038/nature02246
Ko, Y. H., Verhoeven, H. A., Lee, M. J., Corbin, D. J., Vogl, T. J., and Pedersen, P. L. (2012). A translational study “case report” on the small molecule “energy blocke” 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J. Bioenerg. Biomembr. 44, 163–170. doi: 10.1007/s10863-012-9417-4
Kohl, T., Lörinczi, E., Pardo, L. A., and Stühmer, W. (2011). Rapid internalization of the oncogenic K+ channel K(V)10.1. PLoS ONE 6:e26329. doi: 10.1371/journal.pone.0026329
Kolisek, M., Zsurka, G., Samaj, J., Weghuber, J., Schweyen, R. J., and Schweigel, M. (2003). Mrs2p is an essential component of the major electrophoretic Mg2+ influx system in mitochondria. EMBO J. 22, 1235–1244. doi: 10.1093/emboj/cdg122
Kosztka, L., Rusznák, Z., Nagy, D., Nagy, Z., Fodor, J., Szucs, G., et al. (2011). Inhibition of TASK-3 (KCNK9) channel biosynthesis changes cell morphology and decreases both DNA content and mitochondrial function of melanoma cells maintained in cell culture. Melanoma Res. 21, 308–322. doi: 10.1097/CMR.0b013e3283462713
Kovács, I., Pocsai, K., Czifra, G., Sarkadi, L., Szücs, G., Nemes, Z., et al. (2005). TASK-3 immunoreactivity is present but shows differential distribution in the human gastrointestinal tract. Virchows Arch. 446, 402–410. doi: 10.1007/s00428-005-1205-7
Leanza, L., Zoratti, M., Gulbins, E., and Szabò, I. (2012a). Induction of apoptosis in macrophages via Kv1.3 and Kv1.5 potassium channels. Curr. Med. Chem. 19, 5394–5404. doi: 10.2174/092986712803833281
Leanza, L., Henry, B., Sassi, N., Zoratti, M., Chandy, K. G., Gulbins, E., et al. (2012b). Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 4, 577–593. doi: 10.1002/emmm.201200235
Leanza, L., Trentin, L., Becker, K. A., Frezzato, F., Zoratti, M., Semenzato, G., et al. (2013). Clofazimine, Psora-4 and PAP-1, inhibitors of the potassium channel Kv1.3, as a new and selective therapeutic strategy in chronic lymphocytic leukemia. Leukemia 27, 1782–1785. doi: 10.1038/leu.2013.56
Lena, A., Rechichi, M., Salvetti, A., Bartoli, B., Vecchio, D., Scarcelli, V., et al. (2009). Drugs targeting the mitochondrial pore act as cytotoxic and cytostatic agents in temozolomide-resistant glioma cells. J. Transl. Med. 7:13. doi: 10.1186/1479-5876-7-13
Li, L., Han, W., Gu, Y., Qiu, S., Lu, Q., Jin, J., et al. (2007). Honokiol induces a necrotic cell death through the mitochondrial permeability transition pore. Cancer Res. 67, 4894–4903. doi: 10.1158/0008-5472.CAN-06-3818
Marchi, S., Lupini, L., Patergnani, S., Rimessi, A., Missiroli, S., Bonora, M., et al. (2013). Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. 23, 58–63. doi: 10.1016/j.cub.2012.11.026
Martinez-Caballero, S., Dejean, L. M., Kinnally, M. S., Oh, K. J., Mannella, C. A., and Kinnally, K. W. (2009). Assembly of the mitochondrial apoptosis-induced channel, MAC. J. Biol. Chem. 284, 12235–12245. doi: 10.1074/jbc.M806610200
Matassa, D. S., Amoroso, M. R., Maddalena, F., Landriscina, M., and Esposito, F. (2012). New insights into TRAP1 pathway. Am. J. Cancer Res. 2, 235–248.
Mathupala, S. P., and Pedersen, P. L. (2010). Voltage dependent anion channel-1 (VDAC-1) as an anti-cancer target. Cancer Biol. Ther. 9, 1053–1056. doi: 10.4161/cbt.9.12.12451
McAndrew, D., Grice, D. M., Peters, A. A., Davis, F. M., Stewart, T., Rice, M., et al. (2011). ORAI1-mediated calcium influx in lactation and in breast cancer. Mol. Cancer Ther. 10, 448–460. doi: 10.1158/1535-7163.MCT-10-0923
McCommis, K. S., and Baines, C. P. (2012). The role of VDAC in cell death: friend or foe. Biochim. Biophys. Acta 1818, 1444–1450. doi: 10.1016/j.bbamem.2011.10.025
Monteith, G. R., Davis, F. M., and Roberts-Thomson, S. J. (2012). Calcium channels and pumps in cancer: changes and consequences. J. Biol. Chem. 287, 31666–31673. doi: 10.1074/jbc.R112.343061
Norman, K. G., Canter, J. A., Shi, M., Milne, G. L., Morrow, J. D., and Sligh, J. E. (2010). Cyclosporine A suppresses keratinocyte cell death through MPTP inhibition in a model for skin cancer in organ transplant recipients. Mitochondrion 10, 94–101. doi: 10.1016/j.mito.2009.10.001
O'Rourke, B. (2007). Mitochondrial ion channels. Annu. Rev. Physiol. 69, 19–49. doi: 10.1146/annurev.physiol.69.031905.163804
Pastorino, J. G., Shulga, N., and Hoek, J. B. (2002). Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 277, 7610–7618. doi: 10.1074/jbc.M109950200
Pedersen, P. L. (2012). 3-Bromopyruvate (3BP) a fast acting, promising, powerful, specific, and effective “small molecule” anti-cancer agent taken from labside to bedside: introduction to a special issue. J. Bioenerg. Biomembr. 44, 1–6. doi: 10.1007/s10863-012-9425-4
Peixoto, P. M., Dejean, L. M., and Kinnally, K. W. (2012). The therapeutic potential of mitochondrial channels in cancer, ischemia-reperfusion injury, and neurodegeneration. Mitochondrion 12, 14–23. doi: 10.1016/j.mito.2011.03.003
Pereira, G. C., Branco, A. F., Matos, J. A., Pereira, S. L., Parke, D., Perkins, E. L., et al. (2007). Mitochondrially targeted effects of berberine [Natural Yellow 18 5,6-dihydro-9,10-dimethoxybenzo(g)-1,3-benzodioxolo(5,6-a) quinolizinium] on K1735-M2 mouse melanoma cells: comparison with direct effects on isolated mitochondrial fractions. J. Pharmacol. Exp. Ther. 323, 636–649. doi: 10.1124/jpet.107.128017
Pereira, C. V., Machado, N. G., and Oliveira, P. J. (2008). Mechanisms of berberine (natural yellow 18)-induced mitochondrial dysfunction: interaction with the adenine nucleotide translocator. Toxicol. Sci. 105, 408–417. doi: 10.1093/toxsci/kfn131
Pinton, P., Ferrari, D., Rapizzi, E., Di Virgilio, F., Pozzan, T., and Rizzuto, R. (2001). The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20, 2690–2701. doi: 10.1093/emboj/20.11.2690
Piskacek, M., Zotova, L., Zsurka, G., and Schweyen, R. J. (2009). Conditional knockdown of hMRS2 results in loss of mitochondrial Mg(2+) uptake and cell death. J. Cell Mol. Med. 13, 693–700. doi: 10.1111/j.1582-4934.2008.00328.x
Quast, S. A., Berger, A., Buttstädt, N., Friebel, K., Schönherr, R., and Eberle, J. (2012). General Sensitization of melanoma cells for TRAIL-induced apoptosis by the potassium channel inhibitor TRAM-34 depends on release of SMAC. PLoS ONE 7:e39290. doi: 10.1371/journal.pone.0039290
Raghavan, A., Sheiko, T., Graham, B. H., and Craigen, W. J. (2012). Voltage-dependant anion channels: novel insights into isoform function through genetic models. Biochim. Biophys. Acta 1818, 1477–1485. doi: 10.1016/j.bbamem.2011.10.019
Ralph, S. J., and Neuzil, J. (2009). Mitochondria as targets for cancer therapy. Mol. Nutr. Food Res. 53, 9–28. doi: 10.1002/mnfr.200800044
Rasola, A., Sciacovelli, M., Pantic, B., and Bernardi, P. (2010). Signal transduction to the permeability transition pore. FEBS Lett. 584, 1989–1996. doi: 10.1016/j.febslet.2010.02.022
Raviv, Z., Cohen, S., and Reischer-Pelech, D. (2013). The anti-cancer activities of jasmonates. Cancer Chemother. Pharmacol. 71, 275–285. doi: 10.1007/s00280-012-2039-z
Ren, Y. R., Pan, F., Parvez, S., Fleig, A., Chong, C. R., Xu, J., et al. (2008). Clofazimine inhibits human Kv1.3 potassium channel by perturbing calcium oscillation in T lymphocytes. PLoS ONE 3:e4009. doi: 10.1371/journal.pone.0004009
Risso, A., Braidot, E., Sordano, M. C., Vianello, A., Macrì, F., Skerlavaj, B., et al. (2002). BMAP-28, an antibiotic peptide of innate immunity, induces cell death through opening of the mitochondrial permeability transition pore. Mol Cell Biol. 22, 1926–1935. doi: 10.1128/MCB.22.6.1926-1935.2002
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Rusznák, Z., Bakondi, G., Kosztka, L., Pocsai, K., Dienes, B., Fodor, J., et al. (2008). Mitochondrial expression of the two-pore domain TASK-3 channels in malignantly transformed and non-malignant human cells. Virchows Arch. 452, 415–426. doi: 10.1007/s00428-007-0545-x
Samudio, I., Fiegl, M., McQueen, T., Clise-Dwyer, K., and Andreeff, M. (2008). The Warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 68, 5198–5205. doi: 10.1158/0008-5472.CAN-08-0555
Santandreu, F. M., Roca, P., and Oliver, J. (2010). Uncoupling protein-2 knockdown mediates the cytotoxic effects of cisplatin. Free Radic. Biol. Med. 49, 658–666. doi: 10.1016/j.freeradbiomed.2010.05.031
Sassi, N., De Marchi, U., Fioretti, B., Biasutto, L., Gulbins, E., Francolini, F., et al. (2010). An investigation of the occurrence and properties of the mitochondrial intermediate-conductance Ca2+-activated K+ channel mtKCa3.1. Biochim. Biophys. Acta 1797, 1260–1267. doi: 10.1016/j.bbabio.2009.12.015
Shimizu, S., Ide, T., Yanagida, T., and Tsujimoto, Y. (2000). Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 275, 12321–12325. doi: 10.1074/jbc.275.16.12321
Shoshan, M. C. (2012). 3-Bromopyruvate: targets and outcomes. J. Bioenerg. Biomembr. 44, 7–15. doi: 10.1007/s10863-012-9419-2
Shoshan-Barmatz, V., Keinan, N., Abu-Hamad, S., Tyomkin, D., and Aram, L. (2010). Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim. Biophys. Acta 1797, 1281–1291. doi: 10.1016/j.bbabio.2010.03.003
Shoshan-Barmatz, V., and Golan, M. (2012). Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 19, 714–735.
Shoshan-Barmatz, V., and Mizrachi, D. (2012). VDAC1: from structure to cancer therapy. Front. Oncol. 2:164. doi: 10.3389/fonc.2012.00164
Simamura, E., Shimada, H., Hatta, T., and Hirai, K. (2008). Mitochondrial voltage-dependent anion channels (VDACs) as novel pharmacological targets for anti-cancer agents. J. Bioenerg. Biomembr. 40, 213–217. doi: 10.1007/s10863-008-9158-6
Singh, H., Stefani, E., and Toro, L. (2012). Intracellular BKCa (iBKCa) channels. J. Physiol. 590, 5937–5947. doi: 10.1113/jphysiol.2011.215533
Suh, K. S., Malik, M., Shukla, A., Ryscavage, A., Wright, L., Jividen, K., et al. (2012). CLIC4 is a tumor suppressor for cutaneous squamous cell cancer. Carcinogenesis. 33, 986–995. doi: 10.1093/carcin/bgs115
Szabó, I., Bock, J., Grassmé, H., Soddemann, M., Wilker, B., Lang, F., et al. (2008). Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 105, 14861–14866. doi: 10.1073/pnas.0804236105
Szabò, I., Bock, J., Jekle, A., Soddemann, M., Adams, C., Lang, F., et al. (2005). A novel potassium channel in lymphocyte mitochondria. J. Biol. Chem. 280, 12790–12798. doi: 10.1074/jbc.M413548200
Szabò, I., Leanza, L., Gulbins, E., and Zoratti, M. (2012). Physiology of potassium channels in the inner membrane of mitochondria. Pflugers Arch. 463, 231–246. doi: 10.1007/s00424-011-1058-7
Szabò, I., Soddemann, M., Leanza, L., Zoratti, M., and Gulbins, E. (2011). Single-point mutations of a lysine residue change function of Bax and Bcl-xL expressed in Bax- and Bak-less mouse embryonic fibroblasts: novel insights into the molecular mechanisms of Bax-induced apoptosis. Cell Death Differ. 18, 427–438. doi: 10.1038/cdd.2010.112
Szabó, I., and Zoratti, M. (1991). The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J. Biol. Chem. 266, 3376–3379.
Tait, S. W., and Green, D. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632. doi: 10.1038/nrm2952
Tan, W. (2012). VDAC blockage by phosphorothioate oligonucleotides and its implication in apoptosis. Biochim. Biophys. Acta 1818, 1555–1561. doi: 10.1016/j.bbamem.2011.12.032
Traba, J., Del Arco, A., Duchen, M. R., Szabadkai, G., and Satrústegui, J. (2012). SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca(2+) buffering. Cell Death Differ. 19, 650–660. doi: 10.1038/cdd.2011.139
Tsujimoto, Y., and Shimizu, S. (2000). VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 7, 1174–1181. doi: 10.1038/sj.cdd.4400780
Wang, X., Zhang, X., Dong, X. P., Samie, M., Li, X., Cheng, X., et al. (2012). TPC proteins are phosphoinositide- activated sodium-selective ion channels in endosomes and lysosomes. Cell 151, 372–383. doi: 10.1016/j.cell.2012.08.036
Wolf, A., Agnihotri, S., Micallef, J., Mukherjee, J., Sabha, N., Cairns, R., et al. (2011). Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 208, 313–326. doi: 10.1084/jem.20101470
Wolf, F. I., and Trapani, V. (2009). Multidrug resistance phenotypes and MRS2 mitochondrial magnesium channel: two players from one stemness. Cancer Biol. Ther. 8, 615–617. doi: 10.4161/cbt.8.7.8152
Xu, W., Liu, Y., Wang, S., McDonald, T., Van Eyk, J. E., Sidor, A., et al. (2002). Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science 298, 1029–1033. doi: 10.1126/science.1074360
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. doi: 10.1038/nature05859
Zhang, K., Shang, Y., Liao, S., Zhang, W., Nian, H., Liu, Y., et al. (2007). Uncoupling protein 2 protects testicular germ cells from hyperthermia-induced apoptosis. Biochem. Biophys. Res. Commun. 360, 327–332. doi: 10.1016/j.bbrc.2007.06.071
Zhang, L., and Barritt, G. J. (2004). Evidence that TRPM8 is an androgendependent Ca2+ channel required for the survival of prostate cancer cells. Cancer Res. 64, 8365–8373. doi: 10.1158/0008-5472.CAN-04-2146
Zhong, F., Harr, M. W., Bultynck, G., Monaco, G., Parys, J. B., DeSmedt, H., et al. (2011). Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2–IP3 receptor interaction. Blood 117, 2924–2934. doi: 10.1182/blood-2010-09-307405
Zhong, J., Kong, X., Zhang, H., Yu, C., Xu, Y., Kang, J., et al. (2012). Inhibition of CLIC4 enhances autophagy and triggers mitochondrial and ER stress-induced apoptosis in human glioma U251 cells under starvation. PLoS ONE 7:e39378. doi: 10.1371/journal.pone.0039378
Keywords: cancer, organelles, ion channel, apoptosis, pharmacology
Citation: Leanza L, Biasutto L, Managò A, Gulbins E, Zoratti M and Szabò I (2013) Intracellular ion channels and cancer. Front. Physiol. 4:227. doi: 10.3389/fphys.2013.00227
Received: 17 May 2013; Accepted: 05 August 2013;
Published online: 03 September 2013.
Edited by:
Luca Munaron, University of Turin, ItalyReviewed by:
Yong S. Song, Seoul National University College of Medicine, South KoreaFlorian Lang, Eberhard-Karls-University of Tuebingen, Germany
Copyright © 2013 Leanza, Biasutto, Managò, Gulbins, Zoratti and Szabò. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ildikò Szabò, Department of Biology, University of Padova, Viale G. Colombo 3, 35131 Padova, Italy e-mail:aWxkaUBjaXYuYmlvLnVuaXBkLml0