 Nishkantha Arulkumaran1,2* Clare M. Turner1 Marije L. Sixma2 Mervyn Singer2 Robert Unwin3 and Frederick W. K. Tam1
Nishkantha Arulkumaran1,2* Clare M. Turner1 Marije L. Sixma2 Mervyn Singer2 Robert Unwin3 and Frederick W. K. Tam1- 1Imperial College Kidney and Transplant Institute, Imperial College London, Hammersmith Hospital, London, UK
- 2Division of Medicine, Bloomsbury Institute of Intensive Care Medicine, University College London, London, UK
- 3Division of Medicine, UCL Centre for Nephrology, Royal Free Campus and Hospital, University College London, London, UK
Extracellular purines have a role in renal physiology and adaption to inflammation. However, inflammatory renal disease may be mediated by extracellular purines, resulting in renal injury. The role of purinergic signaling is dependent on the concentrations of extracellular purines. Low basal levels of purines are important in normal homeostasis and growth. Concentrations of extracellular purines are significantly elevated during inflammation and mediate either an adaptive role or propagate local inflammation. Adenosine signaling mediates alterations in regional renal blood flow by regulation of the renal microcirculation, tubulo-glomerular feedback, and tubular transport of sodium and water. Increased extracellular ATP and renal P2 receptor-mediated inflammation are associated with various renal diseases, including hypertension, diabetic nephropathy, and glomerulonephritis. Experimental data suggests P2 receptor deficiency or receptor antagonism is associated with amelioration of antibody-mediated nephritis, suggesting a pathogenic (rather than adaptive) role of purinergic signaling. We discuss the role of extracellular nucleotides in adaptation to ischemic renal injury and in the pathogenesis of inflammatory renal disease.
Introduction
Purines are ubiquitous molecules that are synthesized as nucleosides. There are several purines with wide-ranging functions. Adenine and guanine are purines that are essential for the synthesis of DNA and RNA. Adenosine is an endogenous purine nucleoside that comprises a molecule of adenine linked to a ribose sugar molecule. As a component in cyclic adenosine monophosphate (cAMP), adenosine plays a major role in signal transduction. Adenosine also provides energy for several intracellular biologic reactions, with adenosine 5′-triphosphate (ATP) and ADP as the principal energy sources. Other purines include xanthine, hypoxanthine, and uric acid.
Among the endogenous purinergic compounds, adenosine and ATP play a key role in the host response to inflammation via extracellular signaling pathways. Extracellular levels of adenosine and ATP are increased several-fold during inflammation, and act on specific purinergic receptors. Activation of adenosine receptors tends to mediate cytoprotection in response to inflammation, whereas activation of the ATP-sensitive P2X7 receptor triggers pro-inflammatory signaling. In this review, we discuss the physiological and pathological roles of purinergic signaling in inflammatory renal disease, with particular focus on adenosine receptors and ATP-sensitive P2 receptors.
Extracellular Purines in the Kidney
Within the cytosol, ATP is present in millimolar concentrations (~3–5 mM in most cells). Much lower concentrations of ATP are found in the extracellular environment under basal conditions. Various physiological and pathological conditions are associated with elevated levels of extracellular nucleotides.
The relative contribution of different cell types to the basal levels of extracellular nucleotides and the mechanism of purine release is an area of active research. The most widely accepted mechanism of release is via cell lysis- either non-specific cytolysis of healthy cells by mechanical trauma or as a result of necrosis or apoptosis. An alternative mechanism involves exocytotic release of ATP and ADP via granules or vesicles that contain compartmentalized nucleotides. Exocytotic release of ATP via granules is well-described in neurons and neuro- endocrine cells (von Kugelgen et al., 1998; Bodin and Burnstock, 2001a; Fabbro et al., 2004). Non-neuronal cells of exocrine and endocrine origin store large amounts of ATP within cytosolic granules which are released into the extracellular environment (>100 mM) (Coco et al., 2003; Obermuller et al., 2005). Extracellular release of ADP via granules has also been described in activated platelets and immune cells (Eltzschig et al., 2006, 2008; Weissmuller et al., 2008).
The conductive release of ATP via intrinsic plasma membrane channels or pores in the absence of irreversible cytolysis is another proposed mechanism of extracellular ATP release. There are various classes of cell membrane channels associated with conductive release of ATP. Examples include pore forming P2X7 receptors (Ferrari et al., 1996), connexins (Kang et al., 2008) and pannexins (Huang et al., 2007), maxi-anion channels and volume-regulated ion channels (Hisadome et al., 2002; Bell et al., 2003, 2009).
The multiple sources of extracellular ATP have also been described for the kidney. ATP is co-released with acetylcholine or noradrenaline from nerve termini via synaptic vesicles within the kidney (Dowdall et al., 1974; Burnstock, 1995). Electrical field stimulation of the sympathetic nerves in the renal cortex induces release of ATP. However, this accounts for only 25% of the total cortical ATP content, with the remainder coming from non-neuronal sources (Vonend et al., 2002). Under basal conditions, nanomolar concentrations of ATP are released from proximal tubule cell primary cultures, cell lines, and in vivo (Schwiebert, 2001; Praetorius and Leipziger, 2010). Glomeruli have been shown to constitutively release ATP, and the macula densa releases ATP via maxi anion channels (Bell et al., 2003, 2009; Karczewska et al., 2007). Thick ascending limb and collecting duct cells have also been shown to release nanomolar concentrations of ATP (Schwiebert, 2001). Apart from epithelial cells, other potential sources of ATP in the kidney include cells of the vascular network such as endothelial and smooth muscle cells (Pearson and Gordon, 1979; Bodin and Burnstock, 1996, 2001b; Yamamoto et al., 2003), platelets (Detwiler and Feinman, 1973; Hechler and Gachet, 2011), mononuclear cells (Maugeri et al., 1990), and erythrocytes (Bergfeld and Forrester, 1992; Wan et al., 2011). The relative amounts of ATP vary within the kidney, with ATP concentration in the proximal tubules being 4-fold greater than in Bowman's space (Vekaria et al., 2006). Cellular release of ATP can be induced by a range of stimuli, including hypoxia (Bergfeld and Forrester, 1992), acute inflammation (Bodin and Burnstock, 1998), fluid shear stress (Bodin and Burnstock, 2001b), osmotic shock (Jans et al., 2002), and mechanical deformation such as distension of the ureter (Knight et al., 2002). Once released from cells, ATP may influence cells in an autocrine or paracrine fashion. Extracellular ATP and other nucleotides are rapidly hydrolyzed by membrane-bound ecto-enzymes such as ecto-5′-nucleotidase (Dawson et al., 1989). Therefore, P2 receptor signaling in the kidney involves a balance between ATP release and ATP breakdown.
Basal levels of adenosine are in the range of 300 nM, but may increase up to 4-fold during acute inflammation. Extracellular adenosine is generated from the phosphohydrolysis of extracellular ATP and ADP by constitutive ectonucleoside-triphosphate-diphosphohydrolase-1 (CD39) and ecto-5′-nucleotidase (CD73). CD39 catalyzes the breakdown of extracellular ATP and ADP by rapid conversion to AMP, whereas CD73 is a membrane-bound protein responsible for the final step in generating extracellular adenosine by converting AMP to adenosine. Alternatively, extracellular adenosine may be released by circulating cells such as activated immune cells and platelets (Bauerle et al., 2011).
Purinoceptors in the Kidney
Purinergic signaling occurs via activation of specific receptors called purinergic receptors. Purinergic receptors can be divided into two main families, the P1 receptors that are selective for adenosine and the P2 receptors that are selective for ATP and ADP. Intracellular purines are released into the extracellular space by various mechanisms and act upon extracellular purinergic receptors (Figure 1).
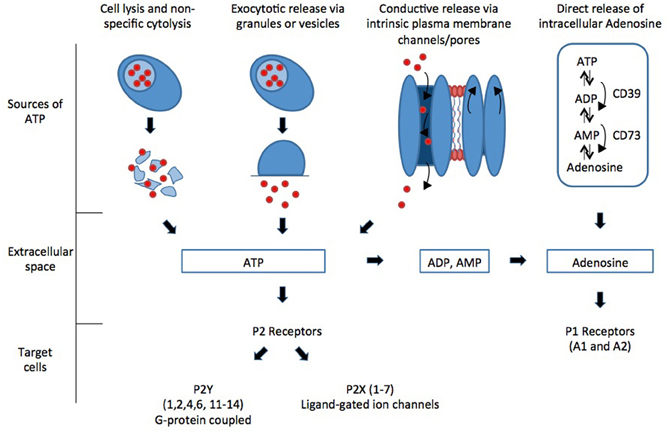
Figure 1. Intracellular purines are released into the extracellular space by various mechanisms and act upon extracellular purinergic receptors.
P1 Receptors
The effects of extracellular adenosine are mediated via G-protein coupled purinergic receptors. These include A1, A2A, A2B, and A3 receptors. Adenosine receptors can be subdivided into those that are activated by physiological levels only of adenosine (10–100 nM) and those that require much higher levels of adenosine (>1 μM), as seen in pathological conditions (Fredholm, 2007). A1, A2A, and A3 receptors require physiological levels of adenosine, whereas A3 receptors require much higher concentrations of adenosine for activation. The A1 (high affinity) and A3 (low affinity) receptors mediate inhibition of the enzyme adenylyl cyclase (AC) and inhibit formation of 3′, 5′-cyclic adenosine monophosphate (cAMP), whereas the A2A (high affinity) and A2B (low affinity) receptors stimulate AC.
The A1AR is widely expressed in the kidney, especially in the afferent arterioles, mesangial cells, proximal convoluted tubules, medullary collecting ducts, and papillary epithelia. The A2AAR receptor is located predominantly in the glomerular epithelium and adjacent vasculature (Bauerle et al., 2011). The A2BAR receptors are found predominantly in the renal vasculature with minimal expression in renal epithelia. The precise location and role of the A3 receptor within the kidney remains unclear. The role of renal adenosine receptors centers primarily on regulation of renal vascular tone and glomerular filtration rate (GFR).
P2 Receptors
The P2 purinergic receptors can in turn be divided into two subcategories: the inotropic P2X (ligand-gated ion channels) and metabotropic P2Y receptors (G-protein-coupled receptors). To date, seven P2X subunits (P2X1-7) and eight P2Y subunits (P2Y1,2,4,6,11–14) have been identified. The effect of extracellular ATP on renal epithelial cells was first reported in 1972, when ATP was shown to increase calcium release from a suspension of cortical tubules (Rorive and Kleinzeller, 1972). ATP is now known to exert its effects via distinct P2 receptors. P2 receptor mRNA and protein are expressed throughout the nephron, vasculature, and interstitial cells (Bailey et al., 2000, 2001; Turner et al., 2003). Differential basolateral and apical cell membrane expression of P2 receptors has been reported (Bailey et al., 2000, 2001; Deetjen et al., 2000; Kishore et al., 2000; Turner et al., 2003). The roles postulated for P2 receptors in the kidney are multiple and diverse, including cell cycle regulation, mediators of hormonal control in the proximal tubule, mediators or moderators of TGF, and regulators of ion transport processes along the nephron. Numerous studies have shown that ATP contributes to many pathophysiological processes, including cell proliferation, apoptosis, vascular remodeling, inflammation and necrosis.
One of the key purinoceptors in promoting inflammation is the P2X7 receptor (P2X7R). Expression of this receptor was originally defined in macrophages and monocytes (Di Virgilio et al., 2001), and has since been characterized in several different cell types, including intrinsic renal cells (Arulkumaran et al., 2011). Unlike other P2X receptors the P2X7 receptor is distinct, because its downstream signaling is coupled to pro- inflammatory cascades (North, 2002). Cell damage and death during active inflammation results in an increase in local concentrations of extracellular ATP. Activation of the P2X7R in vitro requires extracellular concentrations of ATP in the range of 1 mM, in contrast to concentrations <100 μM needed to activate other P2 receptors. Therefore, the P2X7 receptor is thought to act as a “danger signal” in response to damaged or lysed cells. Transient activation of the P2X7R leads to release of the pro-inflammatory cytokines IL-1β and IL-18. Prolonged activation of P2X7R results in irreversible pore formation and allows the non-selective passage of ions and hydrophilic solutes of up to 900 Da, which can result in colloido-osmotic lysis and cell death by apoptosis or necrosis (Ferrari et al., 2006).
Purines and Cell Survival
P2 purinoceptos have been shown to regulate cell survival and death. The precise functional effect of purinoceptors on cell survival is very much dependent on the local environment. Studies measuring the incorporation of 3H-thymidine into the cellular DNA of mesangial cells after agonist stimulation of P2 receptors have demonstrated that extracellular nucleotides can regulate mesangial cell proliferation in vitro via P2Y receptor activation (Schulze-Lohoff et al., 1992; Huwiler and Pfeilschifter, 1994; Ishikawa et al., 1994; Harada et al., 2000). More recently, ATP-induced proliferation of mesangial cells was shown to be through P2Y receptor activation of the MAPK42/44 signal transduction pathway (Vonend et al., 2003). Extracellular nucleotides may also activate the stress-activated protein kinase (SAPK) and the p38-stress-activated protein kinase (MAPK38) cascades in mesangial cells via P2Y receptors (Huwiler et al., 1997, 2000). These cascades are most often activated by cellular stresses such as chemicals, heat, osmotic shock, and UV irradiation (Paul et al., 1997). The balance between nucleotide-stimulation of the MAPK42/44 pathway (Vonend et al., 2003), the SAPK and MAPK38 pathways, determines whether cells survive or undergo apoptosis (Huwiler et al., 1997, 2000). Cultured mesangial cells have been shown to undergo apoptosis via stimulation of the P2X7R by ATP and inhibited by P2X7 receptor antagonists (Schulze-Lohoff et al., 1998). The trigger for apoptosis has most often been associated with P2X7R activity (Schulze-Lohoff et al., 1998; Bulanova et al., 2005; Turner et al., 2007; Taylor et al., 2009; Kawano et al., 2012) although P2X4R-mediated apoptosis has been reported in cultured human mesangial cells and in a mouse macrophage cell line (Kawano et al., 2012; Solini et al., 2007).
Interestingly a proliferative role has also been suggested for the P2X7 receptor in non-renal cells. P2X7 transfected HEK293 cells exhibited enhanced tumorigenesis when inoculated into immunodeficient BALB/c mice (Adinolfi et al., 2012); moreover, P2X7R expression has been reported to be upregulated in malignant tumor cells (Di Virgilio et al., 2009).
Inflammatory Renal Diseases
Renal Ischemia
During global hypoxia or impaired renal perfusion, ATP consumption exceeds ATP production. This results in elevated adenosine levels (Miller et al., 1978; Beach et al., 1991; Nishiyama et al., 1999). This may be seen in a number of acute clinical scenarios, including hemorrhage and endotoxaemia. Adenosine signaling mediates alterations in regional renal blood flow, preserving bioenergetics and cellular structure (Weinberg and Venkatachalam, 2012).
Tubulo-glomerular feedback (TGF) is a negative feedback mechanism whereby changes in luminal NaCl delivery and reabsorption at the macula densa can adjust the vascular tone of the afferent arteriole to alter and match glomerular filtration (Schnermann et al., 1973). TGF allows each nephron to regulate its GFR in accordance with the capacity of its proximal tubule to reabsorb NaCl. Extracellular adenosine contributes to the regulation of GFR. Renal interstitial adenosine is mainly derived from dephosphorylation of released ATP, AMP, or cAMP by the enzyme ecto-5′-nucleotidase (CD73) (Le Hir and Kaissling, 1993). This enzyme catalyzes the dephosphorylation of 5′-AMP or 5′-IMP to adenosine or inosine, respectively, and is located primarily on the external membranes and mitochondria of proximal tubule cells, but not in distal tubule or collecting duct cells (Miller et al., 1978). ATP consumed in active transport by the macula densa also contributes to the formation of adenosine by 5- nucleotidase (Thomson et al., 2000). Extracellular adenosine activates A1 receptors on vascular afferent arteriolar smooth muscle cells, resulting in vasoconstriction and a reduction in GFR (Schnermann et al., 1990). Renal clearance and micropuncture experiments in CD73 null mice demonstrate a diminished TGF response to increasing proximal tubule perfusion (Huang et al., 2006). These effects of extracellular adenosine are mediated via A1 receptors, since blockade of these receptors completely abrogates TGF, and mice deficient in adenosine A1 receptors also lack a normal TGF response (Vallon et al., 2004). P2X1 receptors autoregulate renal blood flow when juxtamedullary afferent arteriolar pressure is elevated. Activation of P2X1 receptors on preglomerular arteriolar smooth muscle cells results in an increase in intracellular Ca2+ concentration, leading to vasoconstriction. This response is blunted in hypertensive (Angiotensin II treated, high- salt diet) rats, suggesting that P2X1 receptors are important for pressure-mediated autoregulatory responses (Inscho et al., 2011).
Elevated extracellular concentrations of adenosine within the kidney are seen in local ischemia (Miller et al., 1978; Beach et al., 1991; Nishiyama et al., 1999). During ischemia, aerobic metabolism and ATP production are impaired. This limits the ability of the proximal tubule to reabsorb filtered electrolytes and water, a highly energy-dependent process. Elevated adenosine levels may limit GFR via TGF, reducing the need for tubular reabsorption and preserving renal bioenergetics. Experimental work demonstrates that generation of adenosine by renal CD73 during renal ischemia, and selective renal A1 receptor agonists protect against renal ischemia and reperfusion injury (Park et al., 2012; Kim et al., 2013).
Renal concentrations of adenosine are elevated in response to endotoxic shock (Miller et al., 1978; Beach et al., 1991; Nishiyama et al., 1999). In an experimental model of endotoxaemia and shock, reduced mean arterial pressure and renal blood flow were associated with an elevated renal adenosine concentration. Antagonism of the A1 receptor attenuated the reduction in mean arterial pressure and renal blood flow, suggesting that adenosine may preserve renal blood flow during endotoxic shock (Nishiyama et al., 1999).
Selective augmentation of adenosine around afferent arterioles causes persistent vasoconstriction, indicating A1AR dominance. In contrast to the A1AR-receptor, the A2AAR-receptor mediates vasodilatation in deep cortical glomerular vessels, and increases medullary blood flow and oxygenation (Vallon and Osswald, 2009). Global elevation of renal adenosine causes steady-state vasodilatation from A2AR-mediated generation of nitric oxide (Hansen et al., 2005). Thus, global increments in adenosine production and release may increase total blood flow through A2AR generation of NO, while allowing a compensatory reduction in tubular workload by a reduction in A1AR activation. This can maintain renal oxygenation, while at the same time limiting renal workload. Adenosine protects from ischemic acute kidney injury (AKI) in a mouse model of ischemia -reperfusion by preserving peritubular capillary blood flow during reperfusion via activation of A2AR on endothelial cells (Grenz et al., 2012).
Vascular Remodeling and Hypertension
Immunohistochemistry has shown that P2X1, P2X2, P2X7 and P2Y1 receptors are expressed on the renal vasculature (Lewis and Evans, 2001; Turner et al., 2003) and are responsible for the regulation of renal hemodynamics (reviewed in Guan et al., 2007). Stimulated endothelial cells release endogenous nucleotides and changes in renal arterial pressure result in increased interstitial ATP concentration (Yegutkin et al., 2000; Nishiyama et al., 2001). Studies indicate that alteration in ATP/P2 receptor signaling may result in renal vascular dysfunction and may lead to hypertension. Angiotensin II mediates hypertension by promoting mesangial cell transformation, renal inflammation, and vascular hypertrophy. Mice deficient in P2X1 receptors or treated with the non-specific P2 receptor antagonist PPADS, or a specific P2Y12 receptor antagonist, demonstrate an attenuation of angiotensin II-mediated hypertension (Graciano et al., 2008; Franco et al., 2011). This suggests that angiotensin II mediates hypertension via its action on vascular and glomerular P2 receptors. P2Y2 and P2Y4 receptors regulate ENaC activity in the collecting duct, thereby regulating sodium balance and systemic hypertension (Wildman et al., 2008). Glomerular hypertension results in glomerular capillary wall stretch, endothelial damage, and a rise in protein glomerular filtration, leading to further ATP release (Bodin and Burnstock, 1998; Yegutkin et al., 2000; Nishiyama et al., 2001; Schwiebert, 2001). This autocrine loop of released ATP and cell stimulation exacerbates the inflammatory response by further release of inflammatory cytokines, as supported by a rat model of renin-dependent hypertension in which glomerular P2X7R was up-regulated at 12 weeks (Vonend et al., 2004). The P2X4 receptor has also been shown to regulate vascular remodeling. Local ATP released during increased renal blood flow acts upon endothelial P2X4 receptors, resulting in calcium influx and the production of nitric oxide. This effect is attenuated in P2X4-deficient mice, and, P2X4-deficient mice fail to exhibit adaptive vascular remodeling in response to a chronic decrease in renal blood flow (Yamamoto et al., 2006).
Antibody Mediated Renal Disease
Increased glomerular P2X7R expression has been reported in a mouse model of accelerated nephrotoxic nephritis, with P2X7R expression coinciding with an increase in apoptotic cells (Turner et al., 2007). Similarly, in a rat model of antibody-mediated glomerulonephritis increased P2X7R mRNA expression coincided with the onset of proteinuria and an increase in IL-1β and pro-apoptotic genes (Turner et al., 2007). P2X7R is also upregulated in patients with lupus nephritis. Genetic deletion of P2X7R is associated with amelioration of renal injury in a murine model of antibody-mediated glomerulonephritis, with reduced macrophage infiltration, less fibrin deposition, less glomerular thrombosis, reduced thrombosis and overall protection of renal function. This coincides with a reduction in the chemokine CCL2 in P2X7−/− mice compared with wildtype controls (Taylor et al., 2009). Similarly the P2X7R antagonist A438079 reduces the severity of antibody-mediated glomerulonephritis in rats with reduced proteinuria and renal macrophage infiltration (Taylor et al., 2009). In a model of passive crescentic glomerulonephritis, P2Y1 gene deficiency was also renoprotective, reducing capillary loss, fibrosis, and death by renal failure (Hohenstein et al., 2007). The current evidence are based on models induced by passive injection of glomerular binding antibodies. The roles of purinergic receptors in antigen presentation and development of autoimmune response need to be investigated.
Diabetic Kidney Disease
Significant glomerular expression of P2X7R is reported in rats with steptozotocin-induced diabetes with a corresponding 10-fold increase in P2X7R mRNA expression (Vonend et al., 2004). Electron microscopy demonstrated that P2X7R was located primarily on podocytes and endothelial cells. Both oxygen and glucose deprivation have been shown to increase integration of P2X7R into the cell membrane of HEK 293 cells (Milius et al., 2007). Culturing cells in high glucose-containing media stimulated ATP secretion in rat mesangial cells, but with no change in P2 receptor expression (Solini et al., 2005). High glucose has also been shown to initiate inflammasome activation through P2X4R regulation in HK-2 cells (Chen et al., 2013). In type 2 diabetic patients with nephropathy, tubular P2X4R expression was upregulated and closely related to NLRP3 inflammasome activation and renal interstitial inflammation (Chen et al., 2013). The functional importance of purinergic receptors in diabetic nephropathy still require further investigation in vivo, and in clinical studies.
Ureteric Obstruction
The role of P2X7R and TGF-β in extracellular matrix deposition was demonstrated in a mouse model of unilateral ureteral obstruction (UUO) (Goncalves et al., 2006). Myofibroblast number and collagen deposition were significantly reduced in P2X7R KO mouse compared with wildtype mice in a UUO model. Immunohistochemistry demonstrated significantly less Myofibroblasts and Sirius-red staining in KO mice on day 7 with a reduction in TGF-β. Furthermore, at day 14, there were reduced numbers of infiltrating macrophages associated with less tubular cell apoptosis in kidneys from the P2X7R KO mice. The tubulo-interstitial damage and subsequent fibrosis at this time point were attenuated in P2X7R KO mice, again implicating tubular P2X7R in renal inflammation and fibrosis.
Conclusion
Purines play a key role in maintaining renal physiology during health and mediate adaptation to pathologic conditions, including ischemia and endotoxaemia. P2 purinoreceptors are “danger sensors” and are key players in inflammation. However, in certain circumstances, inflammation mediated via P2 receptors may become pathological, leading to tissue damage and renal injury. The potential role of selective antagonists against purinoreceptors as a therapeutic target in pro-inflammatory renal diseases, including glomerulonephritis and diabetes requires further evaluation.
Conflict of Interest Statement
Nishkantha Arulkumaran, Wellcome Trust pre-doctoral training fellowship. Marije Sixma, Dutch kidney foundation, Kolff student grant. Robert J. Unwin, Consultancy with AstraZeneca. Frederick Tam, Research project grants from Roche Palo Alto, AstraZeneca Limited, Cyclacel Limited and Baxter Biosciences, Consultancy for Roche Palo Alto and Baxter Biosciences. Clare Turner and Mervyn Singer declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adinolfi, E., Raffaghello, L., Giuliani, A. L., Cavazzini, L., Capece, M., Chiozzi, P., et al. (2012). Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 72, 2957–2969. doi: 10.1158/0008-5472.CAN-11-1947
Arulkumaran, N., Unwin, R. J., and Tam, F. W. (2011). A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin. Investig. Drugs 20, 897–915. doi: 10.1517/13543784.2011.578068
Bailey, M. A., Imbert-Teboul, M., Turner, C., Marsy, S., Srai, K., Burnstock, G., et al. (2000). Axial distribution and characterization of basolateral P2Y receptors along the rat renal tubule. Kidney Int. 58, 1893–1901. doi: 10.1111/j.1523-1755.2000.00361.x
Bailey, M. A., Imbert-Teboul, M., Turner, C., Srai, S. K., Burnstock, G., and Unwin, R. J. (2001). Evidence for basolateral P2Y(6) receptors along the rat proximal tubule: functional and molecular characterization. J. Am. Soc. Nephrol. 12, 1640–1647.
Bauerle, J. D., Grenz, A., Kim, J. H., Lee, H. T., and Eltzschig, H. K. (2011). Adenosine generation and signaling during acute kidney injury. J. Am. Soc. Nephrol. 22, 14–20. doi: 10.1681/ASN.2009121217
Beach, R. E., Watts, B. A. 3rd., Good, D. W., Benedict, C. R., and DuBose, T. D. Jr. (1991). Effects of graded oxygen tension on adenosine release by renal medullary and thick ascending limb suspensions. Kidney Int. 39, 836–842. doi: 10.1038/ki.1991.105
Bell, P. D., Komlosi, P., and Zhang, Z. R. (2009). ATP as a mediator of macula densa cell signalling. Purinergic Signal. 5, 461–471. doi: 10.1007/s11302-009-9148-0
Bell, P. D., Lapointe, J. Y., Sabirov, R., Hayashi, S., Peti-Peterdi, J., Manabe, K., et al. (2003). Macula densa cell signaling involves ATP release through a maxi anion channel. Proc. Natl. Acad. Sci. U.S.A. 100, 4322–4327. doi: 10.1073/pnas.0736323100
Bergfeld, G. R., and Forrester, T. (1992). Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc. Res. 26, 40–47. doi: 10.1093/cvr/26.1.40
Bodin, P., and Burnstock, G. (1996). ATP-stimulated release of ATP by human endothelial cells. J. Cardiovasc. Pharmacol. 27, 872–875. doi: 10.1097/00005344-199606000-00015
Bodin, P., and Burnstock, G. (1998). Increased release of ATP from endothelial cells during acute inflammation. Inflamm. Res. 47, 351–354. doi: 10.1007/s000110050341
Bodin, P., and Burnstock, G. (2001a). Purinergic signalling: ATP release. Neurochem. Res. 26, 959–969.
Bodin, P., and Burnstock, G. (2001b). Evidence that release of adenosine triphosphate from endothelial cells during increased shear stress is vesicular. J. Cardiovasc. Pharmacol. 38, 900–908.
Bulanova, E., Budagian, V., Orinska, Z., Hein, M., Petersen, F., Thon, L., et al. (2005). Extracellular ATP induces cytokine expression and apoptosis through P2X7 receptor in murine mast cells. J. Immunol. 174, 3880–3890.
Burnstock, G. (1995). Noradrenaline and ATP: cotransmitters and neuromodulators. J. Physiol. Pharmacol. 46, 365–384.
Chen, K., Zhang, J., Zhang, W., Yang, J., Li, K., and He, Y. (2013). ATP-P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. Int. J. Biochem. Cell Biol. 45, 932–943. doi: 10.1016/j.biocel.2013.02.009
Coco, S., Calegari, F., Pravettoni, E., Pozzi, D., Taverna, E., Rosa, P., et al. (2003). Storage and release of ATP from astrocytes in culture. J. Biol. Chem. 278, 1354–1362. doi: 10.1074/jbc.M209454200
Dawson, T. P., Gandhi, R., Le Hir, M., and Kaissling, B. (1989). Ecto-5'-nucleotidase: localization in rat kidney by light microscopic histochemical and immunohistochemical methods. J. Histochem. Cytochem. 37, 39–47. doi: 10.1177/37.1.2535703
Deetjen, P., Thomas, J., Lehrmann, H., Kim, S. J., and Leipziger, J. (2000). The luminal P2Y receptor in the isolated perfused mouse cortical collecting duct. J. Am. Soc. Nephrol. 11, 1798–1806.
Detwiler, T. C., and Feinman, R. D. (1973). Kinetics of the thrombin-induced release of adenosine triphosphate by platelets. Comparison with release of calcium. Biochemistry 12, 2462–2468. doi: 10.1021/bi00737a015
Di Virgilio, F., Chiozzi, P., Ferrari, D., Falzoni, S., Sanz, J. M., Morelli, A., et al. (2001). Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood 97, 587–600. doi: 10.1182/blood.V97.3.587
Di Virgilio, F., Ferrari, D., and Adinolfi, E. (2009). P2X(7): a growth-promoting receptor-implications for cancer. Purinergic Signal. 5, 251–256. doi: 10.1007/s11302-009-9145-3
Dowdall, M. J., Boyne, A. F., and Whittaker, V. P. (1974). Adenosine triphosphate. A constituent of cholinergic synaptic vesicles. Biochem. J. 140, 1–12.
Eltzschig, H. K., Eckle, T., Mager, A., Kuper, N., Karcher, C., Weissmuller, T., et al. (2006). ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 99, 1100–1108. doi: 10.1161/01.RES.0000250174.31269.70
Eltzschig, H. K., Macmanus, C. F., and Colgan, S. P. (2008). Neutrophils as sources of extracellular nucleotides: functional consequences at the vascular interface. Trends Cardiovasc. Med. 18, 103–107. doi: 10.1016/j.tcm.2008.01.006
Fabbro, A., Skorinkin, A., Grandolfo, M., Nistri, A., and Giniatullin, R. (2004). Quantal release of ATP from clusters of PC12 cells. J. Physiol. 560, 505–517. doi: 10.1113/jphysiol.2004.068924
Ferrari, D., Pizzirani, C., Adinolfi, E., Lemoli, R. M., Curti, A., Idzko, M., et al. (2006). The P2X7 receptor: a key player in IL-1 processing and release. J. Immunol. 176, 3877–3883.
Ferrari, D., Villalba, M., Chiozzi, P., Falzoni, S., Ricciardi-Castagnoli, P., and Di Virgilio, F. (1996). Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J. Immunol. 156, 1531–1539.
Franco, M., Bautista, R., Tapia, E., Soto, V., Santamaria, J., Osorio, H., et al. (2011). Contribution of renal purinergic receptors to renal vasoconstriction in angiotensin II-induced hypertensive rats. Am. J. Physiol. Renal Physiol. 300, 2.
Fredholm, B. B. (2007). Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 14, 1315–1323. doi: 10.1038/sj.cdd.4402132
Goncalves, R. G., Gabrich, L., Rosario, A. Jr., Takiya, C. M., Ferreira, M. L., Chiarini, L. B., et al. (2006). The role of purinergic P2X7 receptors in the inflammation and fibrosis of unilateral ureteral obstruction in mice. Kidney Int. 70, 1599–1606. doi: 10.1038/sj.ki.5001804
Graciano, M. L., Nishiyama, A., Jackson, K., Seth, D. M., Ortiz, R. M., Prieto-Carrasquero, M. C., et al. (2008). Purinergic receptors contribute to early mesangial cell transformation and renal vessel hypertrophy during angiotensin II-induced hypertension. Am. J. Physiol. Renal Physiol. 294, 7.
Grenz, A., Bauerle, J. D., Dalton, J. H., Ridyard, D., Badulak, A., Tak, E., et al. (2012). Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J. Clin. Invest. 122, 693–710. doi: 10.1172/JCI60214
Guan, Z., Osmond, D. A., and Inscho, E. W. (2007). P2X receptors as regulators of the renal microvasculature. Trends Pharmacol. Sci. 28, 646–652. doi: 10.1016/j.tips.2007.09.010
Hansen, P. B., Hashimoto, S., Oppermann, M., Huang, Y., Briggs, J. P., and Schnermann, J. (2005). Vasoconstrictor and vasodilator effects of adenosine in the mouse kidney due to preferential activation of A1 or A2 adenosine receptors. J. Pharmacol. Exp. Ther. 315, 1150–1157. doi: 10.1124/jpet.105.091017
Harada, H., Chan, C. M., Loesch, A., Unwin, R., and Burnstock, G. (2000). Induction of proliferation and apoptotic cell death via P2Y and P2X receptors, respectively, in rat glomerular mesangial cells. Kidney Int. 57, 949–958. doi: 10.1046/j.1523-1755.2000.00911.x
Hechler, B., and Gachet, C. (2011). P2 receptors and platelet function. Purinergic Signal. 7, 293–303. doi: 10.1007/s11302-011-9247-6
Hisadome, K., Koyama, T., Kimura, C., Droogmans, G., Ito, Y., and Oike, M. (2002). Volume-regulated anion channels serve as an auto/paracrine nucleotide release pathway in aortic endothelial cells. J. Gen. Physiol. 119, 511–520. doi: 10.1085/jgp.20028540
Hohenstein, B., Renk, S., Lang, K., Daniel, C., Freund, M., Leon, C., et al. (2007). P2Y1 gene deficiency protects from renal disease progression and capillary rarefaction during passive crescentic glomerulonephritis. J. Am. Soc. Nephrol. 18, 494–505. doi: 10.1681/ASN.2006050439
Huang, D. Y., Vallon, V., Zimmermann, H., Koszalka, P., Schrader, J., and Osswald, H. (2006). Ecto-5′-nucleotidase (cd73)-dependent and -independent generation of adenosine participates in the mediation of tubuloglomerular feedback in vivo. Am. J. Physiol. Renal Physiol. 291, 8.
Huang, Y. J., Maruyama, Y., Dvoryanchikov, G., Pereira, E., Chaudhari, N., and Roper, S. D. (2007). The role of pannexin 1 hemichannels in ATP release and cell-cell communication in mouse taste buds. Proc. Natl. Acad. Sci. U.S.A. 104, 6436–6441. doi: 10.1073/pnas.0611280104
Huwiler, A., and Pfeilschifter, J. (1994). Stimulation by extracellular ATP and UTP of the mitogen-activated protein kinase cascade and proliferation of rat renal mesangial cells. Br. J. Pharmacol. 113, 1455–1463. doi: 10.1111/j.1476-5381.1994.tb17160.x
Huwiler, A., van Rossum, G., Wartmann, M., and Pfeilschifter, J. (1997). Stimulation by extracellular ATP and UTP of the stress-activated protein kinase cascade in rat renal mesangial cells. Br. J. Pharmacol. 120, 807–812. doi: 10.1038/sj.bjp.0700979
Huwiler, A., Wartmann, M., van den Bosch, H., and Pfeilschifter, J. (2000). Extracellular nucleotides activate the p38-stress-activated protein kinase cascade in glomerular mesangial cells. Br. J. Pharmacol. 129, 612–618. doi: 10.1038/sj.bjp.0703077
Inscho, E. W., Cook, A. K., Clarke, A., Zhang, S., and Guan, Z. (2011). P2X1 receptor-mediated vasoconstriction of afferent arterioles in angiotensin II-infused hypertensive rats fed a high-salt diet. Hypertension 57, 780–787. doi: 10.1161/HYPERTENSIONAHA.110.168955
Ishikawa, S., Kawasumi, M., Kusaka, I., Komatsu, N., Iwao, N., and Saito, T. (1994). Extracellular ATP promotes cellular growth of glomerular mesangial cells mediated via phospholipase C. Biochem. Biophys. Res. Commun. 202, 234–240. doi: 10.1006/bbrc.1994.1917
Jans, D., Srinivas, S. P., Waelkens, E., Segal, A., Lariviere, E., Simaels, J., et al. (2002). Hypotonic treatment evokes biphasic ATP release across the basolateral membrane of cultured renal epithelia (A6). J. Physiol. 545, 543–555. doi: 10.1113/jphysiol.2002.026641
Kang, J., Kang, N., Lovatt, D., Torres, A., Zhao, Z., Lin, J., et al. (2008). Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 28, 4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008
Karczewska, J., Martyniec, L., Dzierzko, G., Stepinski, J., and Angielski, S. (2007). The relationship between constitutive ATP release and its extracellular metabolism in isolated rat kidney glomeruli. J. Physiol. Pharmacol. 58, 321–333.
Kawano, A., Tsukimoto, M., Noguchi, T., Hotta, N., Harada, H., Takenouchi, T., et al. (2012). Involvement of P2X4 receptor in P2X7 receptor-dependent cell death of mouse macrophages. Biochem. Biophys. Res. Commun. 419, 374–380. doi: 10.1016/j.bbrc.2012.01.156
Kim, M., Ham, A., Kim, Y. K., Brown, K. M., D'Agati, V. D., and Lee, H. T. (2013). The volatile anesthetic isoflurane induces ecto-5′-nucleotidase (CD73) to protect against renal ischemia and reperfusion injury. Kidney Int. 20, 43.
Kishore, B. K., Ginns, S. M., Krane, C. M., Nielsen, S., and Knepper, M. A. (2000). Cellular localization of P2Y(2) purinoceptor in rat renal inner medulla and lung. Am. J. Physiol. Renal Physiol. 278, F43–F51.
Knight, G. E., Bodin, P., De Groat, W. C., and Burnstock, G. (2002). ATP is released from guinea pig ureter epithelium on distension. Am. J. Physiol. Renal Physiol. 282, F281–F288.
Le Hir, M., and Kaissling, B. (1993). Distribution and regulation of renal ecto-5'-nucleotidase: implications for physiological functions of adenosine. Am. J. Physiol.. 264(3 Pt 2), F377–F387.
Lewis, C. J., and Evans, R. J. (2001). P2X receptor immunoreactivity in different arteries from the femoral, pulmonary, cerebral, coronary and renal circulations. J. Vasc. Res. 38, 332–340. doi: 10.1159/000051064
Maugeri, N., Bermejo, E., and Lazzari, M. A. (1990). Adenosine triphosphate released from human mononuclear cells. Thromb. Res. 59, 887–890. doi: 10.1016/0049-3848(90)90403-Y
Milius, D., Groger-Arndt, H., Stanchev, D., Lange-Dohna, C., Rossner, S., Sperlagh, B., et al. (2007). Oxygen/glucose deprivation increases the integration of recombinant P2X7 receptors into the plasma membrane of HEK293 cells. Toxicology 238, 60–69. doi: 10.1016/j.tox.2007.05.028
Miller, W. L., Thomas, R. A., Berne, R. M., and Rubio, R. (1978). Adenosine production in the ischemic kidney. Circ. Res. 43, 390–397. doi: 10.1161/01.RES.43.3.390
Nishiyama, A., Majid, D. S., Walker, M. 3rd., Miyatake, A., and Navar, L. G. (2001). Renal interstitial atp responses to changes in arterial pressure during alterations in tubuloglomerular feedback activity. Hypertension 37, 753–759. doi: 10.1161/01.HYP.37.2.753
Nishiyama, A., Miura, K., Miyatake, A., Fujisawa, Y., Yue, W., Fukui, T., et al. (1999). Renal interstitial concentration of adenosine during endotoxin shock. Eur. J. Pharmacol. 385, 209–216. doi: 10.1016/S0014-2999(99)00716-5
Obermuller, S., Lindqvist, A., Karanauskaite, J., Galvanovskis, J., Rorsman, P., and Barg, S. (2005). Selective nucleotide-release from dense-core granules in insulin-secreting cells. J. Cell. Sci. 118, 4271–4282. doi: 10.1242/jcs.02549
Park, S. W., Kim, J. Y., Ham, A., Brown, K. M., Kim, M., D'Agati, V. D., et al. (2012). A1 adenosine receptor allosteric enhancer PD-81723 protects against renal ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 303, 3.
Paul, A., Wilson, S., Belham, C. M., Robinson, C. J., Scott, P. H., Gould, G. W., et al. (1997). Stress-activated protein kinases: activation, regulation and function. Cell. Signal. 9, 403–410. doi: 10.1016/S0898-6568(97)00042-9
Pearson, J. D., and Gordon, J. L. (1979). Vascular endothelial and smooth muscle cells in culture selectively release adenine nucleotides. Nature 281, 384–386. doi: 10.1038/281384a0
Praetorius, H. A., and Leipziger, J. (2010). Intrarenal purinergic signaling in the control of renal tubular transport. Annu. Rev. Physiol. 72, 377–393. doi: 10.1146/annurev-physiol-021909-135825
Rorive, G., and Kleinzeller, A. (1972). The effect of ATP and Ca 2+ on the cell volume in isolated kidney tubules. Biochim. Biophys. Acta 274, 226–239. doi: 10.1016/0005-2736(72)90296-9
Schnermann, J., Persson, A. E., and Agerup, B. (1973). Tubuloglomerular feedback. Nonlinear relation between glomerular hydrostatic pressure and loop of henle perfusion rate. J. Clin. Invest. 52, 862–869. doi: 10.1172/JCI107250
Schnermann, J., Weihprecht, H., and Briggs, J. P. (1990). Inhibition of tubuloglomerular feedback during adenosine1 receptor blockade. Am. J. Physiol. 258, F553–F561.
Schulze-Lohoff, E., Hugo, C., Rost, S., Arnold, S., Gruber, A., Brune, B., et al. (1998). Extracellular ATP causes apoptosis and necrosis of cultured mesangial cells via P2Z/P2X7 receptors. Am. J. Physiol. 275, F962–F971.
Schulze-Lohoff, E., Zanner, S., Ogilvie, A., and Sterzel, R. B. (1992). Extracellular ATP stimulates proliferation of cultured mesangial cells via P2-purinergic receptors. Am. J. Physiol. 263, F374–F383.
Schwiebert, E. M. (2001). ATP release mechanisms, ATP receptors and purinergic signalling along the nephron. Clin. Exp. Pharmacol. Physiol. 28, 340–350. doi: 10.1046/j.1440-1681.2001.03451.x
Solini, A., Iacobini, C., Ricci, C., Chiozzi, P., Amadio, L., Pricci, F., et al. (2005). Purinergic modulation of mesangial extracellular matrix production: role in diabetic and other glomerular diseases. Kidney Int. 67, 875–885. doi: 10.1111/j.1523-1755.2005.00152.x
Solini, A., Santini, E., Chimenti, D., Chiozzi, P., Pratesi, F., Cuccato, S., et al. (2007). Multiple P2X receptors are involved in the modulation of apoptosis in human mesangial cells: evidence for a role of P2X4. Am. J. Physiol. Renal Physiol. 292, 30.
Taylor, S. R., Turner, C. M., Elliott, J. I., McDaid, J., Hewitt, R., Smith, J., et al. (2009). P2X7 deficiency attenuates renal injury in experimental glomerulonephritis. J. Am. Soc. Nephrol. 20, 1275–1281. doi: 10.1681/ASN.2008060559
Thomson, S., Bao, D., Deng, A., and Vallon, V. (2000). Adenosine formed by 5'-nucleotidase mediates tubuloglomerular feedback. J. Clin. Invest. 106, 289–298. doi: 10.1172/JCI8761
Turner, C. M., Tam, F. W., Lai, P. C., Tarzi, R. M., Burnstock, G., Pusey, C. D., et al. (2007). Increased expression of the pro-apoptotic ATP-sensitive P2X7 receptor in experimental and human glomerulonephritis. Nephrol. Dial. Transplant. 22, 386–395. doi: 10.1093/ndt/gfl589
Turner, C. M., Vonend, O., Chan, C., Burnstock, G., and Unwin, R. J. (2003). The pattern of distribution of selected ATP-sensitive P2 receptor subtypes in normal rat kidney: an immunohistological study. Cells Tissues Organs 175, 105–117. doi: 10.1159/000073754
Vallon, V., and Osswald, H. (2009). Adenosine receptors and the kidney. Handb. Exp. Pharmacol. 193, 443–470. doi: 10.1007/978-3-540-89615-9_15
Vallon, V., Richter, K., Huang, D. Y., Rieg, T., and Schnermann, J. (2004). Functional consequences at the single-nephron level of the lack of adenosine A1 receptors and tubuloglomerular feedback in mice. Pflugers Arch. 448, 214–221. doi: 10.1007/s00424-004-1239-8
Vekaria, R. M., Unwin, R. J., and Shirley, D. G. (2006). Intraluminal ATP concentrations in rat renal tubules. J. Am. Soc. Nephrol. 17, 1841–1847. doi: 10.1681/ASN.2005111171
von Kugelgen, I., Goncalves, J., Driessen, B., and Starke, K. (1998). Corelease of noradrenaline and adenosine triphosphate from sympathetic neurones. Adv. Pharmacol. 42, 120–125. doi: 10.1016/S1054-3589(08)60710-3
Vonend, O., Grote, T., Oberhauser, V., Von Kugelgen, I., and Rump, L. C. (2003). P2Y-receptors stimulating the proliferation of human mesangial cells through the MAPK42/44 pathway. Br. J. Pharmacol. 139, 1119–1126. doi: 10.1038/sj.bjp.0705358
Vonend, O., Oberhauser, V., von Kugelgen, I., Apel, T. W., Amann, K., Ritz, E., et al. (2002). ATP release in human kidney cortex and its mitogenic effects in visceral glomerular epithelial cells. Kidney Int. 61, 1617–1626. doi: 10.1046/j.1523-1755.2002.00315.x
Vonend, O., Turner, C. M., Chan, C. M., Loesch, A., Dell'Anna, G. C., Srai, K. S., et al. (2004). Glomerular expression of the ATP-sensitive P2X receptor in diabetic and hypertensive rat models. Kidney Int. 66, 157–166.
Wan, J., Forsyth, A. M., and Stone, H. A. (2011). Red blood cell dynamics: from cell deformation to ATP release. Integr. Biol. 3, 972–981. doi: 10.1039/c1ib00044f
Weinberg, J. M., and Venkatachalam, M. A. (2012). Preserving postischemic reperfusion in the kidney: a role for extracellular adenosine. J. Clin. Invest. 122, 493–496. doi: 10.1172/JCI60957
Weissmuller, T., Campbell, E. L., Rosenberger, P., Scully, M., Beck, P. L., Furuta, G. T., et al. (2008). PMNs facilitate translocation of platelets across human and mouse epithelium and together alter fluid homeostasis via epithelial cell-expressed ecto-NTPDases. J. Clin. Invest. 118, 3682–3692. doi: 10.1172/JCI35874
Wildman, S. S., Marks, J., Turner, C. M., Yew-Booth, L., Peppiatt-Wildman, C. M., King, B. F., et al. (2008). Sodium-dependent regulation of renal amiloride-sensitive currents by apical P2 receptors. J. Am. Soc. Nephrol. 19, 731–742. doi: 10.1681/ASN.2007040443
Yamamoto, K., Sokabe, T., Matsumoto, T., Yoshimura, K., Shibata, M., Ohura, N., et al. (2006). Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat. Med. 12, 133–137. doi: 10.1038/nm1338
Yamamoto, K., Sokabe, T., Ohura, N., Nakatsuka, H., Kamiya, A., and Ando, J. (2003). Endogenously released ATP mediates shear stress-induced Ca2+ influx into pulmonary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 285, 24.
Keywords: purines, inflammation, renal circulation, ischemia, renal disease
Citation: Arulkumaran N, Turner CM, Sixma ML, Singer M, Unwin R and Tam FWK (2013) Purinergic signaling in inflammatory renal disease. Front. Physiol. 4:194. doi: 10.3389/fphys.2013.00194
Received: 21 May 2013; Accepted: 05 July 2013;
Published online: 29 July 2013.
Edited by:
Marcelo D. Carattino, University of Pittsburgh School of Medicine, USAReviewed by:
Shaohu Sheng, University of Pittsburgh, USAPablo D. Cabral, Case Western Reserve University, USA
Copyright © 2013 Arulkumaran, Turner, Sixma, Singer, Unwin and Tam. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Nishkantha Arulkumaran, Division of Medicine, Bloomsbury Institute of Intensive Care Medicine, University College London, Cruciform Building, Gower Street, London, UK e-mail: nish_arul@yahoo.com