
 Anders G. Holst1,2
Anders G. Holst1,2

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 15 July 2013
Sec. Cardiac Electrophysiology
Volume 4 - 2013 | https://doi.org/10.3389/fphys.2013.00179
This article is part of the Research Topic Sudden arrhythmic death: from basic science to clinical practice View all 13 articles
Brugada syndrome (BrS) is a clinical entity first described in 1992. BrS is characterized by ST-segment elevations in the right precordial leads and susceptibility to ventricular arrhythmias and sudden cardiac death. It affects young subjects, predominantly males, with structurally normal hearts. The prevalence varies with ethnicity ranging from 1:2,000 to 1:100,000 in different parts of the world. Today, hundreds of variants in 17 genes have been associated with BrS of which mutations in SCN5A, coding for the cardiac voltage-gated sodium channel, accounts for the vast majority. Despite this, approximately 70% of BrS cases cannot be explained genetically with the current knowledge. Moreover, the monogenic role of some of the variants previously described as being associated with BrS has been questioned by their occurrence in about 4% (1:23) of the general population as found in NHLBI GO Exome Sequencing Project (ESP) currently including approximately 6500 individuals. If we add the variants described in the five newest identified genes associated with BrS, they appear at an even higher prevalence in the ESP (1:21). The current standard treatment of BrS is an implantable cardioverter-defibrillator (ICD). The risk stratification and indications for ICD treatment are based on the ECG and on the clinical and family history. In this review we discuss the genetic basis of BrS.
The Brugada syndrome (BrS) was first described as a clinical entity in 1992 (Brugada and Brugada, 1992). It is inherited in an autosomal dominant manner (Antzelevitch et al., 2005). BrS has traditionally been viewed as a consequence of comprised electrical function without structural abnormalities, although the latter has been reported (Coronel et al., 2005; Frustaci et al., 2005; Nademanee et al., 2011; Duthoit et al., 2012).
BrS is characterized by an ST-segment elevation in the right precordial ECG leads V1–V3. The most descriptive ECG changes have been described at consensus conferences, endorsed by Heart Rhythm Society (HRS) and European Heart Rhythm Association (EHRA) over the last decade (Antzelevitch et al., 2005). In a consensus report from 2012, two specific ECG patterns are found to be descriptive (Bayés de Luna et al., 2012).
The BrS ECG pattern is characterized by a coved type ST-segment elevation ≥2 mm followed by a negative T wave in at least one of the right precordial leads (V1–V3) in the presence or absence of a sodium channel-blocking agent (type 1 ECG). BrS is diagnosed when this is seen in conjunction with one of the following: ventricular tachycardia/fibrillation (VF/VT), a family history of sudden cardiac death (SCD) <45 years old, coved-type ECGs in family members, inducibility of VT with programmed electrical stimulation (PES), syncope or nocturnal agonal respiration (Antzelevitch et al., 2005).
Patients with BrS have an increased risk of SCD secondary to VT/VF (Antzelevitch et al., 2005). Different cohorts have reported different risk of developing VT/VF (Brugada et al., 2002; Priori et al., 2002; Eckardt et al., 2005). In the most updated and largest BrS population so far, the cardiac event rate per year was 0.5% in asymptomatic patients, 1.9% in patients with syncope and 7.7% in patients with aborted SCD. The median age of diagnosis was 45 ± 10 years (Probst et al., 2010). In general, men are affected 8–10 fold more often than women, probably due to gender differences in the expression of certain cardiac ion channels (Antzelevitch, 2006). Approximately 20% of Brugada patients also develop supraventricular arrhythmias with atrial fibrillation accounting for most of the cases (Antzelevitch et al., 2005).
The syndrome is estimated to be responsible for 4% of all sudden deaths (SD) and 20% of SD's among patients with structurally normal hearts. The prevalence is ranging between 1:2,000 and 1:100,000 (Hermida et al., 2000; Letsas et al., 2007; Gallagher et al., 2008; Sinner et al., 2009; Holst et al., 2012a) in different countries, and it is the most common cause of death, besides accidents, in men under 40 years in some parts of the world, e.g., in Thailand (Antzelevitch et al., 2005). The syndrome is probably underestimated due to the fact that the characteristic ECG-pattern often is dynamic and concealed and by the fact, that there are several differential diagnoses associated with elevated ST-segments in right precordial leads (Brugada et al., 2009). The characteristic ECG pattern can in some cases be unmasked by administration of sodium channel-blockers, by febrile state or by vasotonic agents. Indeed sodium channel blockers such as flecainide are used in the diagnosis of BrS (Antzelevitch et al., 2005).
Experimental studies have provided some understanding of the pathophysiological basis of the two main clinical characteristics; elevated coved ST-segment in V1–V3 and the increased risk of VT/VF (Yan and Antzelevitch, 1999). However, a consensus concerning the exact mechanism has not been established and there is an ongoing dispute as to whether BrS is a repolarization disorder, a depolarization disorder, or maybe both (Meregalli et al., 2005; Hoogendijk et al., 2010; Wilde et al., 2010). The central mechanism underlying the ECG pattern and arrhythmias, according to the repolarization hypothesis, is a more prominent transmural voltage-gradient in the early repolarization phase due to a more prominent Ito current in the epicardium compared to the endocardium of the right ventricle (Meregalli et al., 2005). Thus, in the epicardium, in the face of reduced sodium current, increased potassium current or reduced calcium current, complete loss of the phase 2 dome can occur. If this appears at some epicardial sites but not at others and a further heterogeneity between the epicardium and the endocardium occur, the result is an epicardial and transmural dispersion of repolarisation, respectively. This could lead to the development of a re-entry loop and premature beats already in the phase 2 of the on-going action potential (AP) that could further trigger VT/VF (Meregalli et al., 2005).
The reason why ST-segment elevation occurs in right precordial leads, and not left, has been suggested to be due to a more prominent Ito in the right ventricle than in the left (Di Diego et al., 1996). Accordingly, this current is believed to have a central role in BrS pathogenesis. This hypothesis has been tested by Calloe et al. (2009), who demonstrated that an Ito activator recapitulated the electrographic and arrhythmic manifestations of BrS.
The depolarization theory states that the substrate for the ECG changes and susceptibility of VT/VF is a slowing of conduction caused both by fibrosis in right ventricular outflow tract (RVOT) and a decreased INa. This decrease in conduction velocity is more prominent in RVOT compared to the rest of the right ventricle which gives rise to the substrate for ECG changes and re-entry arrhythmias (Meregalli et al., 2005).
Recently Hoogendijk et al. stated that none of the proposed mechanism so far described has been irrefutably demonstrated in BrS patients. Therefore they suggested a unifying explanation, the so-called current-to-load mismatch. This hypothesis states that current-to-load mismatch caused by structural and functional abnormality could explain the ST-segment elevation and susceptibility to arrhythmias (Hoogendijk et al., 2010).
To date, 17 genes have been associated with BrS or BrS ECG phenotype (Table 1). SCN5A was the first gene to be associated with BrS and still represents the major gene in BrS pathogenesis. The individual genes associated with BrS are described in detail in the following.
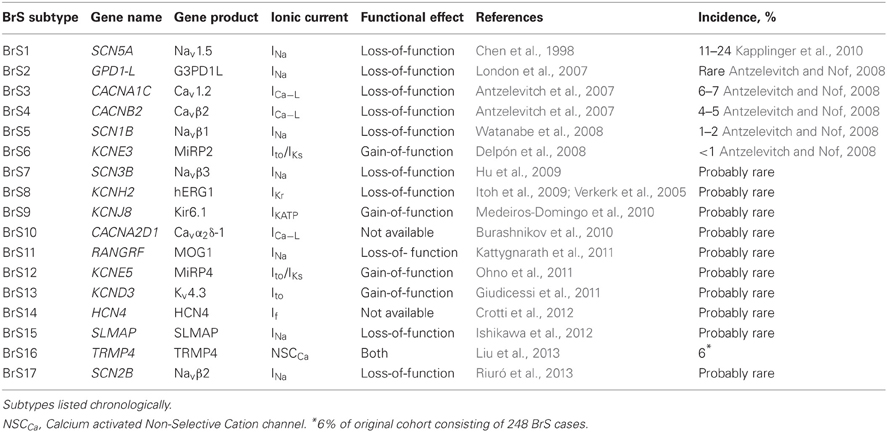
Table 1. Mutations in genes associated with Brugada syndrome.
The SCN5A gene encodes the α-subunit of the voltage-dependent cardiac sodium channel, Nav1.5 (Gellens et al., 1992). Mutations in this gene in association with BrS were first described in 1998 by Chen et al. (1998). Since then more than 300 mutations in this gene have been associated with BrS.
Functional studies of many different mutations in the gene have been performed and they all lead to a reduction in net sodium current due to one or more of following reasons (Antzelevitch et al., 2005); (1) reduced current density due to failure of the sodium channel to express or defect trafficking of the channel (Baroudi et al., 2001; Valdivia et al., 2004; Pfahnl et al., 2007), (2) a shift in the voltage- and time-dependence of sodium channel current activation, inactivation or reactivation (Keller et al., 2005; Hsueh et al., 2009; Calloe et al., 2013), or (3) entry of the sodium channel into an intermediate state of inactivation from which it recovers relatively slower than normal (Bezzina et al., 1999; Veldkamp et al., 2000; Chiang et al., 2009). A number of knock-out mouse models support the central role of SCN5A in the pathogenesis of BrS (Killeen et al., 2008; Derangeon et al., 2012). Heterozygous knock-out mice have been shown to have compromised conduction velocity, impaired AV conduction and QRS prolongation. Furthermore during programmed ventricular electrical pacing two thirds of the mice developed ventricular tachyarrhythmias (Derangeon et al., 2012).
The arrhythmic potential of mutations in SCN5A is also emphasized by its involvement in other arrhythmic diseases such as LQTS, BrS, SIDS, cardiomyopathy and AF. Various SCN5A mutations are known to present with mixed phenotypes, a presentation known as cardiac sodium channel overlap syndrome (Darbar et al., 2008; Remme et al., 2008; Olesen et al., 2012a). This really emphasises the complexity of SCN5A gene mutations in BrS.
See supplementary Tables S1–S5 for a complete overview of all mutations in SCN5A associated with BrS.
Weiss et al. (2002), linked a locus, close to but distinct from the SCN5A locus, to BrS in a large Italian family (Weiss et al., 2002). London et al. (2007) characterized the locus to be the glycerol-3-phosphat dehydrogenase 1-like, GPD1-L gene. There is 84% homology with the Glycerol-3-phosphate dehydrogenase protein, GPD, a dimer involved in the glycerol phosphate shuttle that transfers electrons from cytosolic NADH to the mitochondrial transport chain. The GPD1-L protein is highly expressed in the heart and is concentrated in the membrane fraction. London et al. found that an A280V mutation from a BrS patient in GPD1-L was linked to a 48% decrease in inward Na+ current and a marked decrease in surface expression of Nav1.5 (London et al., 2007).
A mechanism by which GPD1-L mutations could affect Nav1.5 has been studied since by Liu et al. (2009). They, on the basis of the homology between GPD and GPD1-L, investigated whether the GPD1-L, as GPD, is involved in NAD-dependent energy metabolism and thereby, whether NAD(H) could regulate Nav1.5. Indeed they found that A280V-GPD1-L increased [NADH]i and that this increase in [NADH]i reduced INa. This suggests a link between metabolism and INa.
This gene encodes the α-subunit of the human L-type voltage-gated calcium channel, Cav1.2 (Takimoto et al., 1997). Antzelevitch et al. (2007) identified an association between mutations in CACNA1C and BrS. In a Brugada cohort they found two missense mutations in two probands, G490R and A39V. The ECG of the affected patients revealed short QT interval. Both mutations occurred in highly conserved regions of the Cav1.2 protein and both mutations led to a major loss-of-function in calcium channel activity. The loss-of-function caused by the A39V mutation was found to be caused by a trafficking defect (Antzelevitch et al., 2007).
This gene encodes the β-subunit of Cav1.2, Cavβ2, which is involved in regulation of the gating process of ICa−L, in increasing the ICa−L and in modulation of ICa−L traficking (Cornet et al., 2002; Catterall et al., 2005; Hedley et al., 2009).
Antzelevitch et al. (2007) identified a mutation in CACNB2b (S481L) in one proband with BrS as well as short QT. The S481L mutation was present in all 6 phenotype-positive and absent in all 4 phenotype-negative family members. The ICa−L was found to be reduced markedly. Hedley et al. (2009) suggested that the pathogenic mechanism for this mutation could be interference of the stimulatory role of Cavβ2 on ICa−L, by the fact that the mutation is localized close to the Cav1.2 binding domain.
In 2009, Cordeiro et al. associated a novel mutation in CACNB2b with BrS. They detected a missense mutation in CACNB2b, T11I. They found that the mutation led to an accelerated inactivation of the L-type Ca2+ channel. This change of kinetics resulted in a reduced depolarizing current contributing to the plateau phase of the epicardial AP (Cordeiro et al., 2009). Since, Burashnikov et al. (2010) have revealed additional mutations in CACNB2b (see Table 2).
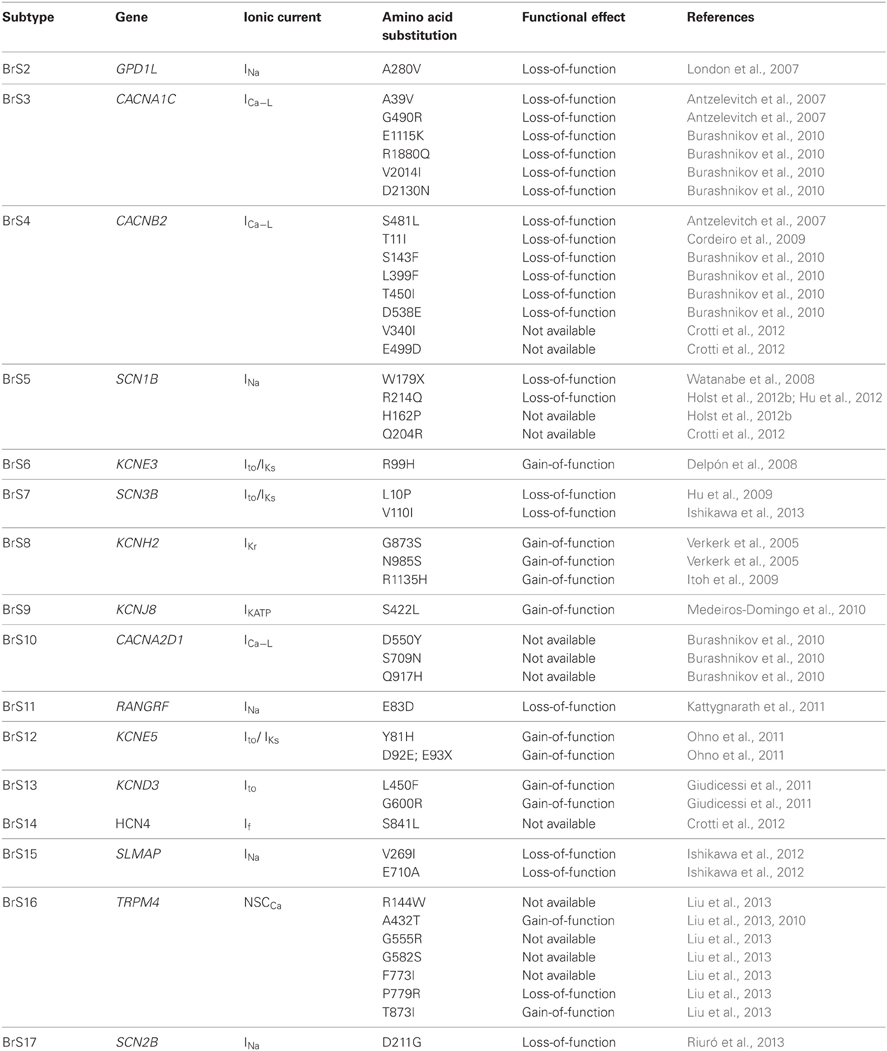
Table 2. Mutations in genes associated with BrS2-BrS17.
The gene encodes the β1-subunit of Nav1.5, Navβ1, which is translated into to isoforms; β1 and β1b. Functions attributed to the β-subunit include an increase in Nav1.5 expression at the cell surface, modulation of channel gating and voltage dependence, and a role in cell adhesion and recruitment of cytosolic proteins such as Ankyrin-G (Isom, 2001; Watanabe et al., 2008).
The association of mutations in SCN1B with BrS was first identified by Watanabe et al. (2008). They screened 282 probands with BrS and 44 patients with conduction disease. They identified one mutation (W179X (β1b)) in SCN1B in a patient with BrS ECG phenotype. The mutated form of SCN1B was not able to increase INa as normal. Recently, Holst et al. added further evidence for an association between mutations in SCN1B and BrS. Two mutations in two probands in SCN1Bb (H162P and R214Q) were identified from a cohort of 42 SCN5A-negative BrS patients. However, R214Q was also found in ESP; H162P was not (Holst et al., 2012b; Olesen et al., 2012b). Hu et al. (2012) investigated the functional consequence of the R214Q variant. When co-expressed with WT-SCN5A the mutant SCN1Bb induced a significant decrease in peak sodium current compared to WT-SCN1Bb. Interestingly, when co-expressed with WT-KCND3, the variant induced a greater Ito, suggesting a combined loss of function of sodium channel current and gain of function of transient outward potassium current in BrS pathogenesis.
This gene encodes the protein MiRP2, one of five homologous β-subunits (KCNE1-5) of voltage gated potassium ion channels (Abbott et al., 2001; Hedley et al., 2009). The potassium channel complex, Kv:KCNE, is a heterohexameric structure consisting of four α-subunits and two KCNE peptides. The functional role of KCNE peptides, in general, is modulation of several potassium currents in the heart, for instance Ito and IKs(Delpón et al., 2008). The association of mutations in KCNE3 with BrS was identified by Delpón et al. (2008). They screened 105 probands with BrS and found a R99H missense mutation in one individual who were highly symptomatic with one aborted cardiac arrest and numerous appropriate shocks after ICD implantation. The family of this individual was examined and they found that 4/4 phenotype-positive and 0/3 phenotype-negative family members had the mutation. Co-transfection of R99H-KCNE3 with KCNQ1 produced no alteration in current magnitude or kinetics. Co-expressed with WT Kv4.3, Ito channel, the mutation had a gain-of-function effect leading to an increase in peak current and an accelerated inactivation of Ito. Overall, the mutation led to a significant increase in total charge carried by Ito.
This gene encodes the β3-subunit of the cardiac sodium channel, Navβ3 (Morgan et al., 2000). The functional attribution of Navβ3 is modulation of the channel gating of Nav1.5, similar to the β1-subunit, although with different kinetics (Morgan et al., 2000).
The association of SCN3B with BrS have been identified by Hu et al. (2009). They found a missense mutation (L10P) in an individual with BrS. The mutation led to a decrease in peak sodium current density, accelerated inactivation, and slowed reactivation compared to wild type. The L10P mutation has also been associated with lone atrial fibrillation (AF) suggesting an overlap in phenotypes (Olesen et al., 2011b). Recently, Ishikawa et al. reported another novel SCN3B mutation, V110I, in three of 178 unrelated Japanese BrS patients. (Ishikawa et al., 2013) The mutation was absent in 480 Japanese controls and displayed a loss-of-function effect due to impaired cell surface expression of Nav1.5.
The α-subunit of the rapid delayed rectifier channel (hERG1) is encoded by KCNH2. Verkerk et al. in 2005, identified two mutations (G873S and N985S) in two unrelated SCN5A-negative BrS patients. Functional investigation revealed an increase in the rectifying current, namely an increase in peak current during phase 0 and phase 1 of the ventricular AP. Through computer simulations this gain-of-function in IKr enhanced the susceptibility of loss of AP dome in right ventricular subepicardial myocytes, which is characteristic of BrS. G873S, however, was found in 2 of 500 unrelated Han Chinese controls suggesting that the variant has only a modifying role or is an innocent bystander. Further support for this interpretation is the fact that the glycine at position 873 is not conserved between human, mouse and rat (Verkerk et al., 2005). For this reason, they were not denoted as the first to associate mutations in KCNH2 with BrS. In 2009, Itoh et al., as the “first”, identified a mutation (R1135H) in KCNH2 in a 34-year old man with Brugada-type ECG and short QT interval. The mutation displayed a gain-of-function effect on IKr(Itoh et al., 2009). Subsequently, Wilders and Verkerk, demonstrated, through computer simulations, that R1135H had the same consequence on AP as G873S and N985S (Wilders and Verkerk, 2010).
This gene encodes the cardiac KATP channel, Kir6.1. The Kir6.1 channel facilitates a non-voltage-gated inwardly rectifying potassium current, leading to a shortening of the AP duration under conditions of metabolic stress (Delaney et al., 2012).
Medeiros-Domingo et al. (2010) found a mutation (S422L) in KCNJ8 in a patient with a flecainide induced type 1 ECG pattern. Electrophysiological the mutation displayed a gain-of-function consequence on KATP. Subsequently, Barajas-Martínez et al. (2012) identified the same mutation in three other BrS patients. When KCNJ8-S422L was co-expressed with the wild type regulatory SUR2A, it showed a twofold gain-of-function on IK, ATP. Furthermore, the mutant channel displayed a reduced sensitivity to ATP, pointing to incomplete closing of the channel under normoxic conditions.
CACNA2D1 encodes the α2δ-subunit of the voltage-dependent calcium channel and has been found to share similar functional properties with Cavβ2 (Gurnett et al., 1996; Hobom et al., 2000). Burashnikov et al. identified three different missense mutations in CACNA2D1 (S709N, D550Y and Q917H) in three BrS patients from a cohort consisting of 205 patients with BrS, short QT, idiopathic ventricular fibrillation (IVF) and early repolarisation syndrome. However, in two of the three patients, additional mutations in genes recognized as being associated with BrS were identified. Unfortunately, the authors did not investigate the electrophysiological consequence of the three missense mutations. New mutations in CACNB2b and CACNA1C were also detected in this cohort (see Table 2) (Burashnikov et al., 2010).
Kattygnarath et al. (2011) reported the gene RANGRF, encoding MOG1 (a protein important for the trafficking of SCN5A to the cell membrane), as a new BrS gene. They identified a missense mutation E83D in a BrS patient that dominant-negatively compromised the sodium current. The mutation was not found in 281 control subjects. Olesen et al. identified another MOG1 variant, E61X, in both AF patients and healthy controls indicating that a person may have complete loss of one MOG1 allele without having any signs of disease (Olesen et al., 2011a). Genetic variants that compromise the MOG1 protein are therefore more likely to increase the susceptibility of BrS, rather than to be the major genetic susceptibility variant.
This gene encodes one of the regulatory β-subunits of the Ito/IKs channels mentioned under BrS6. In 2011, Ohno et al. identified two novel variants (Y81H and D92E;E93X) in KCNE5 in a Japanese cohort consisting of 205 patients with BrS or IVF (Ohno et al., 2011). Three probands comprised the Y81H variant, one the D92E;E93X variant. In 300 unrelated healthy Japanese controls, Y81H was identified in 3 women, and D92E; E93X was absent. All four probands were symptomatic and received an ICD. When co-expressed with KCND3 (α-subunit of Ito), the mutant channels significantly increased Ito compared to wild type, displaying a gain-of-function effect. Stimulation study revealed that the two variants induced altered ventricular AP profiles. This could provide the likelihood of a proarrhythmic substrate. Another interesting notion drawn by the authors is that KCNE5 is located on chromosome X. This could in part explain the gender difference seen in prevalence. Indeed, the male phenotype in the study by Ohno et al. was more severe.
KCND3 encodes the α-subunit of Ito, Kv4.3, a voltage-gated potassium channel expressed in heart. In 2011, two novel mutations (L450F and G600R) were identified in two unrelated BrS patients (Giudicessi et al., 2011). Both mutations were absent in 1560 reference alleles. Co-expression of Kv4.3 mutants with KChIP2-WT revealed a significant increase in Ito current density compared with WT-Kv4.3. Moreover, the two mutations induced loss of AP dome in RV epicardial myocytes, demonstrated by computer simulations, providing the arrhythmic substrate for BrS phenotype (Giudicessi et al., 2011).
HCN4 encodes the hyperpolarisation-activated cyclic nucleotide-gated channel 4 which is a pacemaker channel responsible for the funny current (If). Mutations in this gene has formerly been associated with sinus node dysfunction (Ueda et al., 2004). Recently, Crotti et al. identified a mutation (S841L) in HCN4 in one proband of 129 unrelated BrS patients (Crotti et al., 2012). The mutation was absent in ≥1400 ethnicity matched reference alleles and in publicly available databases. The functional effect of this mutation has not been assessed and therefore, before drawing any conclusion in BrS pathogenesis, this gene has to be investigated more thoroughly.
SLMAP encodes the sarcolemmal membrane-associated protein, a component of T-tubules and sarcoplasmic reticulum which is involved in excitation-contraction coupling in cardiomyocytes (Ishikawa et al., 2012). Ventricular arrhythmias have previously been linked to mutations in proteins involved in excitation-contraction coupling (Priori et al., 2001). Ishikawa et al. (2012) recently reported two mutations (V269I and E710A) in 190 unrelated BrS patients. In cell lines the two mutations were shown to reduce cell surface expression of Nav1.5 resulting in decreased peak sodium current density. In line with this, the investigators demonstrated that silencing the two SLMAP mutants rescued the decreased surface expression of Nav1.5.
The TRPM4 gene encodes the transient receptor potential melastatin protein number 4 which is a calcium-activated non-selective cation channel (NSCCa) that mediates transport of monovalent cations across membranes, thereby depolarizing the membrane. Mutations in this gene have been associated with cardiac conduction blocks (Kruse et al., 2009; Liu et al., 2010; Stallmeyer et al., 2012) and recently, Liu et al. (2013) associated mutations in TRPM4 with BrS. In 248 BrS cases, the investigators identified 7 mutations absent in approximately 14,000 control alleles. Functional characterization of selected mutations revealed both a decrease in TRPM4 expression (P779R) and an increase in expression (T873I) suggesting that both loss- and gain-of-function mutations in this gene may lead to BrS.
The SCN2B gene encodes the β2-subunit of the cardiac sodium channel. The association of SCN2B with BrS have just recently been identified by Riuró et al. (2013). They found a missense mutation (A211G) in an individual with BrS. The mutation was absent in 500 control alleles and available databases. The mutation led to a significant reduction in sodium current density when co-expressed with Nav1.5 compared to wild type. This reduction was shown to be due to a reduced Nav1.5 cell surface expression (Riuró et al., 2013).
With the recently published exome data from the NHLBI GO Exome Sequencing Project (ESP), knowledge regarding genetic variation in the general population have become available [Exome Variant Server, NHLBI GO (ESP)]. In ESP, next-generation sequencing has been carried out for all protein-coding regions in approximately 6500 persons from different population studies. Risgaard et al. (2013) have, by using these data, found a high genotype prevalence of 1:23 in the ESP of genetic variants in twelve genes (SCN5A, GPD1L, CACNA1C, CACNB2, SCN1B, KCNE3, SCN3B, KCNH2, CACNA2D1, MOG1, KCND3, and KCNJ8) previously associated with BrS. This is a very high prevalence compared to the prevalence of BrS in the general population ranging between 1:2,000 and 1:100,000 (Hermida et al., 2000; Letsas et al., 2007; Gallagher et al., 2008; Sinner et al., 2009; Holst et al., 2012a). Moreover, in a synergistic use of prediction analysis using ≥3 prediction tools, 47% of the variants found in ESP were predicted pathogenic compared to 75% of the variants not found in ESP (p < 0.0001). These data definitely questions the pathogenic role of some of the previously BrS-associated variants. A limitation in the study is that there are no clinical data on the persons in ESP. However, Refsgaard et al. have recently conducted a number of studies that indicate, that the exome database is indeed representative for genetic variation in healthy subject (Refsgaard et al., 2012; Andreasen et al., 2013a,b). Moreover, none of the studies in ESP specifically included patients with channelopathies and at least two studies excluded such patients (Refsgaard et al., 2012).
We investigated the prevalence in ESP of the genes not investigated by Risgaard et al. (2013), BrS subtypes 12 and 14-17. The KCNE5, SCN2B and SLMAP mutations were not found in ESP. The HCN4 mutation (S841L) was found in 3 out of 4289 European American (EA) individuals. The TRPM4 mutation R144W was present in 1 of 2199 Afro-American (AA) individuals, A432T in 9 of 4291 AA, and G582S in 9 of 4291 EA. The rest of the TRPM4 mutations were not present in ESP [Exome Variant Server, NHLBI GO (ESP)]. If we add the variants found in the five newest identified genes associated with BrS, this corresponds to an even higher genotype prevalence of 1:21 (296:6258) in ESP.
ICD is the only widely accepted treatment of BrS thus far (Brugada et al., 1999, 2000). In 2003, a second consensus conference was held which focused on risk stratifications and approaches to therapy (Antzelevitch et al., 2005). This consensus report stated the recommendations for ICD implantation.
Sarkozy et al. (2007) studied the effectiveness of ICD treatment in a retrospective study. 47 high risk Brugada patients (mean age: 44 ± 15 years) with ICD were included. During a mean follow-up of 47.5 months, seven patients had appropriate shocks for potentially life-threatening ventricular arrhythmias. However, seventeen patients received inappropriate shocks due to shocks for sinus tachycardia and atrial arrhythmias, which is common in Brugada patients.
A multicenter study by Sacher et al. (2006) showed the same pattern. In 220 BrS patients with ICD (mean follow up >3 years) 8% experienced appropriate shocks and 20% inappropriate shocks. Overall, complications occurred in 28% of the patients.
In a just published article, Miyazaki et al. (2013) also investigated the prevalence of ICD-related complications. In 41 BrS patients and during a median follow-up of 76 months, 15 patients (37%) experienced adverse effects after ICD implantation. This includes complications in 8 (20%) and inappropriate shocks in 10 (24%). Appropriate shocks were detected in 5 patients (12%), (please keep in mind that some patients experience more than one adverse effect). In a nationwide study by Holst et al. (2012a), 26% experienced appropriate shocks and 8% experienced inappropriate shocks during a median follow-up of 47 months in 35 definite BrS patients. The difference in rate of appropriate and inappropriate shocks compared to the three other studies could be due to a more severe phenotype of patients included and due to difference in ICD discrimination algorithms as suggested by Holst et al. (2012a).
A pharmacological approach with fewer complications is obviously desirable and there is a growing effort to define such a safe and efficient treatment for this specific syndrome.
Loss-of-function mutations are responsible for the vast majority of BrS incidents. This makes it more difficult in regard to a pharmacological therapy as it is difficult to compensate for the missing allele. However some substances may be beneficial. The objective is to rebalance the inward and outward currents during the AP and thereby restoring electrical homogeneity (Brugada et al., 2009).
The prominent Ito in the right ventricle is thought to have a central role in the pathogenesis of BrS, so a drug like quinidine that inhibits Ito (Imaizumi and Giles, 1987) has been suggested to have a therapeutic value in BrS (Yan and Antzelevitch, 1999).
Hermida et al. (2004) observed that hydroquinidine therapy prevented VT/VF inducibility in 22 out of 29 asymptomatic patients with BrS and inducible arrhythmia, as well as VT/VF recurrence in four BrS patients with multiple ICD shocks. Belhassen et al. (2004) reported that quinidine bisulfate prevented VF induction in 22 of 25 BrS patients. All 25 patients had inducible VF before treatment. However, administration of quinidine was associated with a 36% incidence of side-effects that resolved after drug discontinuation. In general, disadvantages of oral quinidine include gastrointestinal side-effects, as observed by Belhassen et al. (2004), and proarrhythmic side-effects (QT prolongation), as observed by Hermida et al. (2004). The latter is probably due to a quinidine block of IKr and IKs, however this side effect is rare (Antzelevitch and Nof, 2008). In a recent study by Márquez et al. (2012) the authors investigate the long-term efficacy of low doses quinidine on malignant arrhythmias in BrS patients. A total of twenty patients, of whom seventeen patients had an ICD, were included. In all but three cases, quinidine effectively suppressed arrhythmic events corresponding to an efficacy of quinidine on 85%. All patients tolerated the medication well.
Taken together the data suggest that preventive treatment by quinidine may be an alternative or complimentary strategy to ICD in BrS patients, both in the short and long term. A more Ito selective compound that does not permeate the blood-brain-barrier would in theory be the optimal treatment. For other possible beneficial agents please see Márquez et al. (2007) and Minoura et al. (2013).
Presently over 300 mutations in 17 genes have been associated with BrS or BrS ECG phenotype, in contrast to 5 years ago where only mutations in the SCNA5 gene were associated with BrS. The knowledge about BrS associated mutations is therefore rapidly increasing and this could potentially make genetic screening important in future. The intention would be to use this knowledge in risk stratification, as some asymptomatic BrS patients have an appreciable risk of arrhythmia (Probst et al., 2010). The rapidly declining cost of multi-gene screening by Next Generation Sequencing adds to the rationale. A study by Meregalli et al. (2009) reveal an association between the type of SCN5A mutation and the clinical severity. They compared groups having either missense mutations or mutations leading to premature truncation of the protein. They found that the disease phenotype was more severe in the patients with large INa reduction than in those with small INa reduction (truncation versus missense), as evidenced by larger proportions of patients with syncope and SCD. Sommariva et al. (2013) recently demonstrated that SCN5A mutation carriers had a significantly increased risk of major arrhythmic event compared with non-carriers in a BrS cohort. In addition they established an association of five polymorphisms with major arrhythmic event. Crotti et al. (2012) recently conducted a comprehensive mutational analysis of twelve BrS genes in a large BrS cohort. They did not detect any significant difference in mutation yield between those patients with definite BrS and those patients only displaying a type 1 ECG pattern. On this basis, the authors argue that genetic testing should additionally be conducted in patients displaying only type 1 ECG pattern. Secondly, they demonstrated that inclusion of the minor BrS susceptibility genes (genes other than SCN5A) in genetic testing, only minimally affected the sensitivity of the test. Therefore, these genes should only be screened under special circumstances.
These data may show some promise for the use of genetic data in risk stratification regarding clinical outcome in BrS patients. However, the scientific community is increasingly focusing on separating genetic noise from true pathogenic mutations. Risgaard et al. (2013) reported a high prevalence (1:23) of previously BrS-associated variants in ESP. If we added the variants described in the five newest identified genes associated with BrS, they appeared at an even higher prevalence in the ESP (1:21). These data definitely questions the pathogenic role of some of the previously BrS-associated variants. With regard to deletions and insertions in SCN5A (Tables S1–S5) these are more likely susceptibility mutations, as also evidenced by their lack of presence in ESP.
In a HRS/EHRA consensus document, screening is recommended in family members and relatives following the identification of a BrS-causative mutation in an index case (Ackerman et al., 2011). If the clinician should perform risk stratifications based on genetic screening it is important that variants being associated with BrS are truly pathogenic. An index patient with definite BrS but a false-positive variant might have another true pathogenic variant that is not found. This could lead to misdiagnosis of family members with clinical consequences.
Another important motive for identifying the important susceptibility mutations by genetically screening BrS patients could be the tailoring of specific drugs to specific mutations as personal medicine. Presently, the rationale behind the pharmacological approach is to rebalance the inward and outward currents during the AP, regardless of the underlying mutation. Maybe in the future, it could be possible to treat the different BrS types (1–17) differently, according to their mutational consequences, although the disease entity is so small that the drugs will most likely not be developed for this indication in the first case. For instance, in the study done by Liu et al. (2009), they found that the NADH-induced decrease in INa could be antagonized by externally or internally applied NAD+. This result suggests that drugs that increase the availability of NAD+ may be a future treatment strategy for BrS2. Teng et al. (2009) found that a SCN5A non-sense mutation (W822X) associated with BrS effectively could be suppressed by read-through enhancing agents, thereby restoring the expression of normal length sodium channels. This holds promising for all non-sense mutations associated with BrS. Chakrabarti et al. (2013) recently showed that overexpression of MOG1 effectively rescued the trafficking defect and the impaired plasma membrane expression of Nav1.5 caused by the mutations D1275N and G1743, respectively. These data suggests that MOG1-enabled trafficking of Nav1.5 to plasma membrane may serve as a novel therapy for BrS patients with loss-of-function mutations in Nav1.5.
Even though over 300 mutations in 17 genes have been associated with BrS, approximately 70% of BrS incidents cannot be explained genetically at present. The causes may have to be found in epigenetic regulation or in other mechanisms than ion channel mutations. For instance methylation of promoters or mutations in microRNA binding sites as is has been shown for LQTS1 (Amin et al., 2012).
There is a need for an alternative strategy to ICD therapy. Firstly, because ICD treatment is prohibitively expensive in many parts of the world (Brugada et al., 2009). This is the case in Thailand where the prevalence is exceptionally high. Secondly, the high incidence of side-effects/complications associated with ICD (Sacher et al., 2006; Sarkozy et al., 2007; Miyazaki et al., 2013). And thirdly, BrS has been linked to sudden death infant syndrome (Van Norstrand et al., 2007) and ICD therapy in children is challenging in general. The high risk of complications reported in adults is likely to be worse in children (Antzelevitch and Nof, 2008).
The incentive for developing a good pharmacological paradigm is evident. Quinidine is yet the best alternative to ICD and has proven effective in small case series. However, a clear need exists for a large randomized clinical controlled trial to assess the effectiveness of quinidine in BrS patients.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The study was supported by grants from “The John and Birthe Meyer Foundation.”, The Arvid Nilsson Foundation, the Director Ib Henriksens Foundation, The Villadsen Family Foundation and The Stock Broker Henry Hansen and Wife Karla Hansen, born Westergaard, Grant.
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/Cardiac_Electrophysiology_/10.3389/fphys.2013.00179/abstract
Abbott, G. W., Butler, M. H., Bendahhou, S., Dalakas, M. C., Ptacek, L. J., and Goldstein, S. A. (2001). MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 104, 217–231. doi: 10.1016/S0092-8674(01)00207-0
Ackerman, M. J., Priori, S. G., Willems, S., Berul, C., Brugada, R., Calkins, H., et al. (2011). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 8, 1308–1339. doi: 10.1016/j.hrthm.2011.05.020
Amin, A. S., Giudicessi, J. R., Tijsen, A. J., Spanjaart, A. M., Reckman, Y. J., Klemens, C. A., et al. (2012). Variants in the 3' untranslated region of the KCNQ1-encoded Kv7.1 potassium channel modify disease severity in patients with type 1 long QT syndrome in an allele-specific manner. Eur. Heart J. 33, 714–723. doi: 10.1093/eurheartj/ehr473
Andreasen, C., Nielsen, J. B., Refsgaard, L., Holst, A. G., Christensen, A. H., Andreasen, L., et al. (2013a). New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur. J. Hum. Genet. doi: 10.1038/ejhg.2012.283. [Epub ahead of print].
Andreasen, C., Refsgaard, L., Nielsen, J. B., Sajadieh, A., Winkel, B. G., Tfelt-Hansen, J., et al. (2013b). Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can. J. Cardiol. doi: 10.1016/j.cjca.2012.12.002. [Epub ahead of print].
Antzelevitch, C. (2006). Brugada syndrome. Pacing Clin. Electrophysiol. 29, 1130–1159. doi: 10.1111/j.1540-8159.2006.00507.x
Antzelevitch, C., Brugada, P., Borggrefe, M., Brugada, J., Brugada, R., Corrado, D., et al. (2005). Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 111, 659–670. doi: 10.1161/01.CIR.0000152479.54298.51
Antzelevitch, C., and Nof, E. (2008). Brugada syndrome: recent advances and controversies. Curr. Cardiol. Rep. 10, 376–383. doi: 10.1007/s11886-008-0060-y
Antzelevitch, C., Pollevick, G. D., Cordeiro, J. M., Casis, O., Sanguinetti, M. C., Aizawa, Y., et al. (2007). Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442–449. doi: 10.1161/CIRCULATIONAHA.106.668392
Barajas-Martínez, H., Hu, D., Ferrer, T., Onetti, C. G., Wu, Y., Burashnikov, E., et al. (2012). Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm 9, 548–555. doi: 10.1016/j.hrthm.2011.10.035
Baroudi, G., Pouliot, V., Denjoy, I., Guicheney, P., Shrier, A., and Chahine, M. (2001). Novel mechanism for Brugada syndrome: defective surface localization of an SCN5A mutant (R1432G). Circ. Res. 88, E78–E83. doi: 10.1161/hh1201.093270
Bayés de Luna, A., Brugada, J., Baranchuk, A., Borggrefe, M., Breithardt, G., Goldwasser, D., et al. (2012). Current electrocardiographic criteria for diagnosis of Brugada pattern: a consensus report. J. Electrocardiol. 45, 433–442. doi: 10.1016/j.jelectrocard.2012.06.004
Belhassen, B., Glick, A., and Viskin, S. (2004). Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation 110, 1731–1737. doi: 10.1161/01.CIR.0000143159.30585.90
Bezzina, C., Veldkamp, M. W., van Den Berg, M. P., Postma, A. V., Rook, M. B., Viersma, J. W., et al. (1999). A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 85, 1206–1213. doi: 10.1161/01.RES.85.12.1206
Brugada, J., Brugada, R., Antzelevitch, C., Towbin, J., Nademanee, K., and Brugada, P. (2002). Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation 105, 73–78. doi: 10.1161/hc0102.101354
Brugada, J., Brugada, R., and Brugada, P. (2000). Pharmacological and device approach to therapy of inherited cardiac diseases associated with cardiac arrhythmias and sudden death. J. Electrocardiol. 33(Suppl.), 41–47. doi: 10.1054/jelc.2000.20322
Brugada, P., Benito, B., Brugada, R., and Brugada, J. (2009). Brugada syndrome: update 2009. Hellenic J. Cardiol. 50, 352–372.
Brugada, P., and Brugada, J. (1992). Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 20, 1391–1396. doi: 10.1016/0735-1097(92)90253-J
Brugada, P., Brugada, R., Brugada, J., and Geelen, P. (1999). Use of the prophylactic implantable cardioverter defibrillator for patients with normal hearts. Am. J. Cardiol. 83, 98D–100D. doi: 10.1016/S0002-9149(98)01009-1
Burashnikov, E., Pfeiffer, R., Barajas-Martinez, H., Delpón, E., Hu, D., Desai, M., et al. (2010). Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872–1882. doi: 10.1016/j.hrthm.2010.08.026
Calloe, K., Cordeiro, J. M., Di Diego, J. M., Hansen, R. S., Grunnet, M., Olesen, S. P., et al. (2009). A transient outward potassium current activator recapitulates the electrocardiographic manifestations of Brugada syndrome. Cardiovasc. Res. 81, 686–694. doi: 10.1093/cvr/cvn339
Calloe, K., Refaat, M. M., Grubb, S., Wojciak, J., Campagna, J., Thomsen, N. M., et al. (2013). Characterization and mechanisms of action of novel NaV1.5 channel mutations associated with Brugada syndrome. Circ. Arrhythm. Electrophysiol. 6, 177–184. doi: 10.1161/CIRCEP.112.974220
Catterall, W. A., Perez-Reyes, E., Snutch, T. P., and Striessnig, J. (2005). International union of pharmacology. Xlviii. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 57, 411–425. doi: 10.1124/pr.57.4.5
Chakrabarti, S., Wu, X., Yang, Z., Wu, L., Yong, S. L., Zhang, C., et al. (2013). MOG1 rescues defective trafficking of Nav1.5 mutations in Brugada syndrome and sick sinus syndrome. Circ. Arrhythm. Electrophysiol. 6, 392–401. doi: 10.1161/CIRCEP.111.000206
Chen, Q., Kirsch, G. E., Zhang, D., Brugada, R., Brugada, J., Brugada, P., et al. (1998). Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296. doi: 10.1038/32675
Chiang, K.-C., Lai, L.-P., and Shieh, R.-C. (2009). Characterization of a novel Nav1.5 channel mutation, A551T, associated with Brugada syndrome. J. Biomed. Sci. 16, 76. doi: 10.1186/1423-0127-16-76
Cordeiro, J. M., Marieb, M., Pfeiffer, R., Calloe, K., Burashnikov, E., and Antzelevitch, C. (2009). Accelerated inactivation of the L-type calcium current due to a mutation in CACNB2b underlies Brugada syndrome. J. Mol. Cell. Cardiol. 46, 695–703. doi: 10.1016/j.yjmcc.2009.01.014
Cornet, V., Bichet, D., Sandoz, G., Marty, I., Brocard, J., Bourinet, E., et al. (2002). Multiple determinants in voltage-dependent P/Q calcium channels control their retention in the endoplasmic reticulum. Eur. J. Neurosci. 16, 883–895. doi: 10.1046/j.1460-9568.2002.02168.x
Coronel, R., Casini, S., Koopmann, T. T., Wilms-Schopman, F. J. G., Verkerk, A. O., de Groot, J. R., et al. (2005). Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112, 2769–2777. doi: 10.1161/CIRCULATIONAHA.105.532614
Crotti, L., Marcou, C. A., Tester, D. J., Castelletti, S., Giudicessi, J. R., Torchio, M., et al. (2012). Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. J. Am. Coll. Cardiol. 60, 1410–1418. doi: 10.1016/j.jacc.2012.04.037
Darbar, D., Kannankeril, P. J., Donahue, B. S., Kucera, G., Stubblefield, T., Haines, J. L., et al. (2008). Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 117, 1927–1935. doi: 10.1161/CIRCULATIONAHA.107.757955
Delaney, J. T., Muhammad, R., Blair, M. A., Kor, K., Fish, F. A., Roden, D. M., et al. (2012). A KCNJ8 mutation associated with early repolarization and atrial fibrillation. Europace 14, 1428–1432. doi: 10.1093/europace/eus150
Delpón, E., Cordeiro, J. M., Núñez, L., Thomsen, P. E. B., Guerchicoff, A., Pollevick, G. D., et al. (2008). Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 1, 209–218. doi: 10.1161/CIRCEP.107.748103
Derangeon, M., Montnach, J., Baró, I., and Charpentier, F. (2012). Mouse models of SCN5A-related cardiac arrhythmias. Front. Physiol. 3:210. doi: 10.3389/fphys.2012.00210
Di Diego, J. M., Sun, Z. Q., and Antzelevitch, C. (1996). I(to) and action potential notch are smaller in left vs. right canine ventricular epicardium. Am. J. Physiol. 271, H548–H561.
Duthoit, G., Fressart, V., Hidden-Lucet, F., Simon, F., Kattygnarath, D., Charron, P., et al. (2012). Brugada ECG pattern: a physiopathological prospective study based on clinical, electrophysiological, angiographic, and genetic findings. Front. Physiol. 3:474. doi: 10.3389/fphys.2012.00474
Eckardt, L., Probst, V., Smits, J. P. P., Bahr, E. S., Wolpert, C., Schimpf, R., et al. (2005). Long-term prognosis of individuals with right precordial ST-segment-elevation Brugada syndrome. Circulation 111, 257–263. doi: 10.1161/01.CIR.0000153267.21278.8D
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP). Seattle, WA. Available online at: http://evs.gs.washington.edu/EVS/ [Accessed February 1, 2013].
Frustaci, A., Priori, S. G., Pieroni, M., Chimenti, C., Napolitano, C., Rivolta, I., et al. (2005). Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 112, 3680–3687. doi: 10.1161/CIRCULATIONAHA.105.520999
Gallagher, M. M., Forleo, G. B., Behr, E. R., Magliano, G., De Luca, L., Morgia, V., et al. (2008). Prevalence and significance of Brugada-type ECG in 12,012 apparently healthy European subjects. Int. J. Cardiol. 130, 44–48. doi: 10.1016/j.ijcard.2007.07.159
Gellens, M. E., George, A. L. Jr., Chen, L. Q., Chahine, M., Horn, R., and Kallen, R. G. (1992). Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. U.S.A. 89, 554–558. doi: 10.1073/pnas.89.2.554
Giudicessi, J. R., Ye, D., Tester, D. J., Crotti, L., Mugione, A., Nesterenko, V. V., et al. (2011). Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 8, 1024–1032. doi: 10.1016/j.hrthm.2011.02.021
Gurnett, C. A., De Waard, M., and Campbell, K. P. (1996). Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron 16, 431–440. doi: 10.1016/S0896-6273(00)80061-6
Hedley, P. L., Jørgensen, P., Schlamowitz, S., Moolman-Smook, J., Kanters, J. K., Corfield, V. A., et al. (2009). The genetic basis of Brugada syndrome: a mutation update. Hum. Mutat. 30, 1256–1266. doi: 10.1002/humu.21066
Hermida, J. S., Lemoine, J. L., Aoun, F. B., Jarry, G., Rey, J. L., and Quiret, J. C. (2000). Prevalence of the brugada syndrome in an apparently healthy population. Am. J. Cardiol. 86, 91–94. doi: 10.1016/S0002-9149(00)00835-3
Hermida, J.-S., Denjoy, I., Clerc, J., Extramiana, F., Jarry, G., Milliez, P., et al. (2004). Hydroquinidine therapy in Brugada syndrome. J. Am. Coll. Cardiol. 43, 1853–1860. doi: 10.1016/j.jacc.2003.12.046
Hobom, M., Dai, S., Marais, E., Lacinova, L., Hofmann, F., and Klugbauer, N. (2000). Neuronal distribution and functional characterization of the calcium channel alpha2delta-2 subunit. Eur. J. Neurosci. 12, 1217–1226. doi: 10.1046/j.1460-9568.2000.01009.x
Holst, A. G., Jensen, H. K., Eschen, O., Henriksen, F. L., Kanters, J., Bundgaard, H., et al. (2012a). Low disease prevalence and inappropriate implantable cardioverter defibrillator shock rate in Brugada syndrome: a nationwide study. Europace 14, 1025–1029. doi: 10.1093/europace/eus002
Holst, A. G., Saber, S., Houshmand, M., Zaklyazminskaya, E. V., Wang, Y., Jensen, H. K., et al. (2012b). Sodium current and potassium transient outward current genes in Brugada syndrome: screening and bioinformatics. Can. J. Cardiol. 28, 196–200. doi: 10.1016/j.cjca.2011.11.011
Hoogendijk, M. G., Opthof, T., Postema, P. G., Wilde, A. A. M., de Bakker, J. M. T., and Coronel, R. (2010). The Brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ. Arrhythm. Electrophysiol. 3, 283–290. doi: 10.1161/CIRCEP.110.937029
Hsueh, C.-H., Chen, W.-P., Lin, J.-L., Tsai, C.-T., Liu, Y.-B., Juang, J.-M., et al. (2009). Distinct functional defect of three novel Brugada syndrome related cardiac sodium channel mutations. J. Biomed. Sci. 16, 23. doi: 10.1186/1423-0127-16-23
Hu, D., Barajas-Martinez, H., Burashnikov, E., Springer, M., Wu, Y., Varro, A., et al. (2009). A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ. Cardiovasc. Genet. 2, 270–278. doi: 10.1161/CIRCGENETICS.108.829192
Hu, D., Barajas-Martínez, H., Medeiros-Domingo, A., Crotti, L., Veltmann, C., Schimpf, R., et al. (2012). A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel currents. Heart Rhythm 9, 760–769. doi: 10.1016/j.hrthm.2011.12.006
Imaizumi, Y., and Giles, W. R. (1987). Quinidine-induced inhibition of transient outward current in cardiac muscle. Am. J. Physiol. 253, H704–H708.
Ishikawa, T., Sato, A., Marcou, C. A., Tester, D. J., Ackerman, M. J., Crotti, L., et al. (2012). A novel disease gene for Brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm. Electrophysiol. 5, 1098–1107. doi: 10.1161/CIRCEP.111.969972
Ishikawa, T., Takahashi, N., Ohno, S., Sakurada, H., Nakamura, K., On, Y. K., et al. (2013). Novel SCN3B mutation associated with Brugada syndrome affects intracellular trafficking and function of Nav1.5. Circ. J. 77, 959–967.
Isom, L. L. (2001). Sodium channel beta subunits: anything but auxiliary. Neuroscientist 7, 42–54. doi: 10.1177/107385840100700108
Itoh, H., Sakaguchi, T., Ashihara, T., Ding, W.-G., Nagaoka, I., Oka, Y., et al. (2009). A novel KCNH2 mutation as a modifier for short QT interval. Int. J. Cardiol. 137, 83–85. doi: 10.1016/j.ijcard.2008.05.050
Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M., Brugada, J., et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46. doi: 10.1016/j.hrthm.2009.09.069
Kattygnarath, D., Maugenre, S., Neyroud, N., Balse, E., Ichai, C., Denjoy, I., et al. (2011). MOG1: a new susceptibility gene for Brugada syndrome. Circ. Cardiovasc. Genet. 4, 261–268. doi: 10.1161/CIRCGENETICS.110.959130
Keller, D. I., Rougier, J.-S., Kucera, J. P., Benammar, N., Fressart, V., Guicheney, P., et al. (2005). Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc. Res. 67, 510–519. doi: 10.1016/j.cardiores.2005.03.024
Killeen, M. J., Thomas, G., Sabir, I. N., Grace, A. A., and Huang, C. L.-H. (2008). Mouse models of human arrhythmia syndromes. Acta Physiol. (Oxf). 192, 455–469. doi: 10.1111/j.1748-1716.2007.01822.x
Kruse, M., Schulze-Bahr, E., Corfield, V., Beckmann, A., Stallmeyer, B., Kurtbay, G., et al. (2009). Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Invest. 119, 2737–2744. doi: 10.1172/JCI38292
Letsas, K. P., Gavrielatos, G., Efremidis, M., Kounas, S. P., Filippatos, G. S., Sideris, A., et al. (2007). Prevalence of Brugada sign in a Greek tertiary hospital population. Europace 9, 1077–1080. doi: 10.1093/europace/eum221
Liu, H., Chatel, S., Simard, C., Syam, N., Salle, L., Probst, V., et al. (2013). Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS ONE 8:e54131. doi: 10.1371/journal.pone.0054131
Liu, H., El Zein, L., Kruse, M., Guinamard, R., Beckmann, A., Bozio, A., et al. (2010). Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 3, 374–385. doi: 10.1161/CIRCGENETICS.109.930867
Liu, M., Sanyal, S., Gao, G., Gurung, I. S., Zhu, X., Gaconnet, G., et al. (2009). Cardiac Na+ current regulation by pyridine nucleotides. Circ. Res. 105, 737–745. doi: 10.1161/CIRCRESAHA.109.197277
London, B., Michalec, M., Mehdi, H., Zhu, X., Kerchner, L., Sanyal, S., et al. (2007). Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116, 2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330
Márquez, M. F., Bonny, A., Hernández-Castillo, E., De Sisti, A., Gómez-Flores, J., Nava, S., et al. (2012). Long-term efficacy of low doses of quinidine on malignant arrhythmias in Brugada syndrome with an implantable cardioverter-defibrillator: a case series and literature review. Heart Rhythm 9, 1995–2000. doi: 10.1016/j.hrthm.2012.08.027
Márquez, M. F., Salica, G., Hermosillo, A. G., Pastelín, G., Gómez-Flores, J., Nava, S., et al. (2007). Ionic basis of pharmacological therapy in Brugada syndrome. J. Cardiovasc. Electrophysiol. 18, 234–240. doi: 10.1111/j.1540-8167.2006.00681.x
Medeiros-Domingo, A., Tan, B.-H., Crotti, L., Tester, D. J., Eckhardt, L., Cuoretti, A., et al. (2010). Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm 7, 1466–1471. doi: 10.1016/j.hrthm.2010.06.016
Meregalli, P. G., Tan, H. L., Probst, V., Koopmann, T. T., Tanck, M. W., Bhuiyan, Z. A., et al. (2009). Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 6, 341–348. doi: 10.1016/j.hrthm.2008.11.009
Meregalli, P. G., Wilde, A. A. M., and Tan, H. L. (2005). Pathophysiological mechanisms of Brugada syndrome: depolarization disorder, repolarization disorder, or more? Cardiovasc. Res. 67, 367–378. doi: 10.1016/j.cardiores.2005.03.005
Minoura, Y., Panama, B. K., Nesterenko, V. V., Betzenhauser, M., Barajas-Martínez, H., Hu, D., et al. (2013). Effect of wenxin keli and quinidine to suppress arrhythmogenesis in an experimental model of Brugada syndrome. Heart Rhythm. 10, 1054–1062. doi: 10.1016/j.hrthm.2013.03.011
Miyazaki, S., Uchiyama, T., Komatsu, Y., Taniguchi, H., Kusa, S., Nakamura, H., et al. (2013). Long-term complications of implantable defibrillator therapy in Brugada syndrome. Am. J. Cardiol. 111, 1448–1451. doi: 10.1016/j.amjcard.2013.01.295
Morgan, K., Stevens, E. B., Shah, B., Cox, P. J., Dixon, A. K., Lee, K., et al. (2000). beta 3: an additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc. Natl. Acad. Sci. U.S.A. 97, 2308–2313. doi: 10.1073/pnas.030362197
Nademanee, K., Veerakul, G., Chandanamattha, P., Chaothawee, L., Ariyachaipanich, A., Jirasirirojanakorn, K., et al. (2011). Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 123, 1270–1279. doi: 10.1161/CIRCULATIONAHA.110.972612
Ohno, S., Zankov, D. P., Ding, W.-G., Itoh, H., Makiyama, T., Doi, T., et al. (2011). KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ. Arrhythm. Electrophysiol. 4, 352–361. doi: 10.1161/CIRCEP.110.959619
Olesen, M. S., Jensen, N. F., Holst, A. G., Nielsen, J. B., Tfelt-Hansen, J., Jespersen, T., et al. (2011a). A novel nonsense variant in Nav1.5 cofactor MOG1 eliminates its sodium current increasing effect and may increase the risk of arrhythmias. Can. J. Cardiol. 27, 523.e17–23. doi: 10.1016/j.cjca.2011.01.003
Olesen, M. S., Jespersen, T., Nielsen, J. B., Liang, B., Møller, D. V., Hedley, P., et al. (2011b). Mutations in sodium channel β-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc. Res. 89, 786–793. doi: 10.1093/cvr/cvq348
Olesen, M. S., Yuan, L., Liang, B., Holst, A. G., Nielsen, N., Nielsen, J. B., et al. (2012a). High prevalence of long QT syndrome associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc. Genet. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/22685113 [Accessed August 1, 2012]. 5, 450–459. doi: 10.1161/CIRCGENETICS.111.962597
Olesen, M. S., Holst, A. G., Svendsen, J. H., Haunsø, S., and Tfelt-Hansen, J. (2012b). SCN1Bb R214Q found in 3 patients: 1 with Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm Off. J. Heart Rhythm Soc. 9, 770–773. doi: 10.1016/j.hrthm.2011.12.005
Pfahnl, A. E., Viswanathan, P. C., Weiss, R., Shang, L. L., Sanyal, S., Shusterman, V., et al. (2007). A sodium channel pore mutation causing Brugada syndrome. Heart Rhythm 4, 46–53. doi: 10.1016/j.hrthm.2006.09.031
Priori, S. G., Napolitano, C., Gasparini, M., Pappone, C., Della Bella, P., Giordano, U., et al. (2002). Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 105, 1342–1347. doi: 10.1161/hc1102.105288
Priori, S. G., Napolitano, C., Tiso, N., Memmi, M., Vignati, G., Bloise, R., et al. (2001). Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103, 196–200. doi: 10.1161/01.CIR.103.2.196
Probst, V., Veltmann, C., Eckardt, L., Meregalli, P. G., Gaita, F., Tan, H. L., et al. (2010). Long-term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation 121, 635–643. doi: 10.1161/CIRCULATIONAHA.109.887026
Refsgaard, L., Holst, A. G., Sadjadieh, G., Haunsø, S., Nielsen, J. B., and Olesen, M. S. (2012). High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur. J. Hum. Genet. 20, 905–908. doi: 10.1038/ejhg.2012.23
Remme, C. A., Wilde, A. A. M., and Bezzina, C. R. (2008). Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc. Med. 18, 78–87. doi: 10.1016/j.tcm.2008.01.002
Risgaard, B., Jabbari, R., Refsgaard, L., Holst, A. G., Haunsø, S., Sadjadieh, A., et al. (2013). High prevalence of genetic variants previously associated with Brugada Syndrome in new exome data. Clin. Genet. doi: 10.1111/cge.12126. [Epub ahead of print].
Riuró, H., Beltran-Alvarez, P., Tarradas, A., Selga, E., Campuzano, O., Vergés, M., et al. (2013). A missense mutation in the sodium channel β2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum. Mutat. 34, 961–966. doi: 10.1002/humu.22328
Sacher, F., Probst, V., Iesaka, Y., Jacon, P., Laborderie, J., Mizon-Gérard, F., et al. (2006). Outcome after implantation of a cardioverter-defibrillator in patients with Brugada syndrome: a multicenter study. Circulation 114, 2317–2324. doi: 10.1161/CIRCULATIONAHA.106.628537
Sarkozy, A., Boussy, T., Kourgiannides, G., Chierchia, G.-B., Richter, S., De Potter, T., et al. (2007). Long-term follow-up of primary prophylactic implantable cardioverter-defibrillator therapy in Brugada syndrome. Eur. Heart J. 28, 334–344. doi: 10.1093/eurheartj/ehl450
Sinner, M. F., Pfeufer, A., Perz, S., Schulze-Bahr, E., Mönnig, G., Eckardt, L., et al. (2009). Spontaneous Brugada electrocardiogram patterns are rare in the German general population: results from the KORA study. Europace 11, 1338–1344. doi: 10.1093/europace/eup205
Sommariva, E., Pappone, C., Martinelli Boneschi, F., Di Resta, C., Rosaria Carbone, M., Salvi, E., et al. (2013). Genetics can contribute to the prognosis of Brugada syndrome: a pilot model for risk stratification. Eur. J. Hum. Genet. doi: 10.1038/ejhg.2012.289. [Epub ahead of print].
Stallmeyer, B., Zumhagen, S., Denjoy, I., Duthoit, G., Hébert, J.-L., Ferrer, X., et al. (2012). Mutational spectrum in the Ca(2+)–activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum. Mutat. 33, 109–117. doi: 10.1002/humu.21599
Takimoto, K., Li, D., Nerbonne, J. M., and Levitan, E. S. (1997). Distribution, splicing and glucocorticoid-induced expression of cardiac alpha 1C and alpha 1D voltage-gated Ca2+ channel mRNAs. J. Mol. Cell. Cardiol. 29, 3035–3042. doi: 10.1006/jmcc.1997.0532
Teng, S., Gao, L., Paajanen, V., Pu, J., and Fan, Z. (2009). Readthrough of nonsense mutation W822X in the SCN5A gene can effectively restore expression of cardiac Na+ channels. Cardiovasc. Res. 83, 473–480. doi: 10.1093/cvr/cvp116
Ueda, K., Nakamura, K., Hayashi, T., Inagaki, N., Takahashi, M., Arimura, T., et al. (2004). Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J. Biol. Chem. 279, 27194–27198. doi: 10.1074/jbc.M311953200
Valdivia, C. R., Tester, D. J., Rok, B. A., Porter, C.-B. J., Munger, T. M., Jahangir, A., et al. (2004). A trafficking defective, Brugada syndrome-causing SCN5A mutation rescued by drugs. Cardiovasc. Res. 62, 53–62. doi: 10.1016/j.cardiores.2004.01.022
Van Norstrand, D. W., Valdivia, C. R., Tester, D. J., Ueda, K., London, B., Makielski, J. C., et al. (2007). Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation 116, 2253–2259. doi: 10.1161/CIRCULATIONAHA.107.704627
Veldkamp, M. W., Viswanathan, P. C., Bezzina, C., Baartscheer, A., Wilde, A. A., and Balser, J. R. (2000). Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ. Res. 86, E91–E97. doi: 10.1161/01.RES.86.9.e91
Verkerk, A. O., Wilders, R., Schulze-Bahr, E., Beekman, L., Bhuiyan, Z. A., Bertrand, J., et al. (2005). Role of sequence variations in the human ether-a-go-go-related gene (HERG, KCNH2) in the Brugada syndrome. Cardiovasc. Res. 68, 441–453. doi: 10.1016/j.cardiores.2005.06.027
Watanabe, H., Koopmann, T. T., Le Scouarnec, S., Yang, T., Ingram, C. R., Schott, J.-J., et al. (2008). Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268. doi: 10.1172/JCI33891
Weiss, R., Barmada, M. M., Nguyen, T., Seibel, J. S., Cavlovich, D., Kornblit, C. A., et al. (2002). Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation 105, 707–713. doi: 10.1161/hc0602.103618
Wilde, A. A. M., Postema, P. G., Di Diego, J. M., Viskin, S., Morita, H., Fish, J. M., et al. (2010). The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J. Mol. Cell. Cardiol. 49, 543–553. doi: 10.1016/j.yjmcc.2010.07.012
Wilders, R., and Verkerk, A. O. (2010). Role of the R1135H KCNH2 mutation in Brugada syndrome. Int. J. Cardiol. 144, 149–151. doi: 10.1016/j.ijcard.2008.12.177
Keywords: Brugada syndrome, genetics, Exome Sequencing Project, mutation, treatment
Citation: Nielsen MW, Holst AG, Olesen S-P and Olesen MS (2013) The genetic component of Brugada syndrome. Front. Physiol. 4:179. doi: 10.3389/fphys.2013.00179
Received: 29 April 2013; Accepted: 24 June 2013;
Published online: 15 July 2013.
Edited by:
Ian N. Sabir, King's College London, UKReviewed by:
Ruben Coronel, Academic Medical Center, NetherlandsCopyright © 2013 Nielsen, Holst, Olesen and Olesen. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Morten S. Olesen, Department of Cardiology, Laboratory for Molecular Cardiology, Section 9312, Copenhagen University Hospital, Rigshospitalet, Juliane Mariesvej 20, Copenhagen Ø, 2100, Denmark e-mail:bW9ydGVuLnNhbGxpbmcub2xlc2VuQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.