


- 1Physiological Laboratory, Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, UK
- 2Department of Biochemistry, University of Cambridge, Cambridge, UK
Slowed myocardial conduction velocity (θ) is associated with an increased risk of re-entrant excitation, predisposing to cardiac arrhythmia. θ is determined by the ion channel and physical properties of cardiac myocytes and by their interconnections. Thus, θ is closely related to the maximum rate of action potential (AP) depolarization [(dV/dt)max], as determined by the fast Na+ current (INa); the axial resistance (ra) to local circuit current flow between cells; their membrane capacitances (cm); and to the geometrical relationship between successive myocytes within cardiac tissue. These determinants are altered by a wide range of pathophysiological conditions. Firstly, INa is reduced by the impaired Na+ channel function that arises clinically during heart failure, ischemia, tachycardia, and following treatment with class I antiarrhythmic drugs. Such reductions also arise as a consequence of mutations in SCN5A such as those occurring in Lenègre disease, Brugada syndrome (BrS), sick sinus syndrome, and atrial fibrillation (AF). Secondly, ra, may be increased due to gap junction decoupling following ischemia, ventricular hypertrophy, and heart failure, or as a result of mutations in CJA5 found in idiopathic AF and atrial standstill. Finally, either ra or cm could potentially be altered by fibrotic change through the resultant decoupling of myocyte–myocyte connections and coupling of myocytes with fibroblasts. Such changes are observed in myocardial infarction and cardiomyopathy or following mutations in MHC403 and SCN5A resulting in hypertrophic cardiomyopathy (HCM) or Lenègre disease, respectively. This review defines and quantifies the determinants of θ and summarizes experimental evidence that links changes in these determinants with reduced myocardial θ and arrhythmogenesis. It thereby identifies the diverse pathophysiological conditions in which abnormal θ may contribute to arrhythmia.
Introduction
Impaired myocardial action potential (AP) conduction can predispose to arrhythmogenesis through the formation of slow conducting re-entry circuits. Re-entry was first defined by Mines in the early twentieth century as a persisting electrical impulse that reactivates an area of previously activated myocardial tissue that is no longer refractory, resulting in a circus movement of activation (Mines, 1914). Subsequent studies suggest that sustained arrhythmia requires an ectopic AP triggering event to occur within such a substrate capable of generating self-sustaining re-entry processes (Mandapati et al., 2000; Zou et al., 2005). Thus, a triggered electrical impulse must enter a perpetuating electrical circuit containing a unidirectional conduction block along one of its two pathways. Such re-entry within the circuit is more likely to occur following reductions in conduction velocity (θ) and/or the effective refractory period (ERP). As shown in Figure 1, this could result in a reduction in the wavelength of excitation (λ), given by the product of θ and ERP, to values smaller than the dimensions of the available circuits (Mines, 1914; Wiener and Rosenblueth, 1946; Wakili et al., 2011). Alternatively if the site of slowed conduction is such that it prevents propagation of the triggered AP into the circuit, it may discourage re-entry.
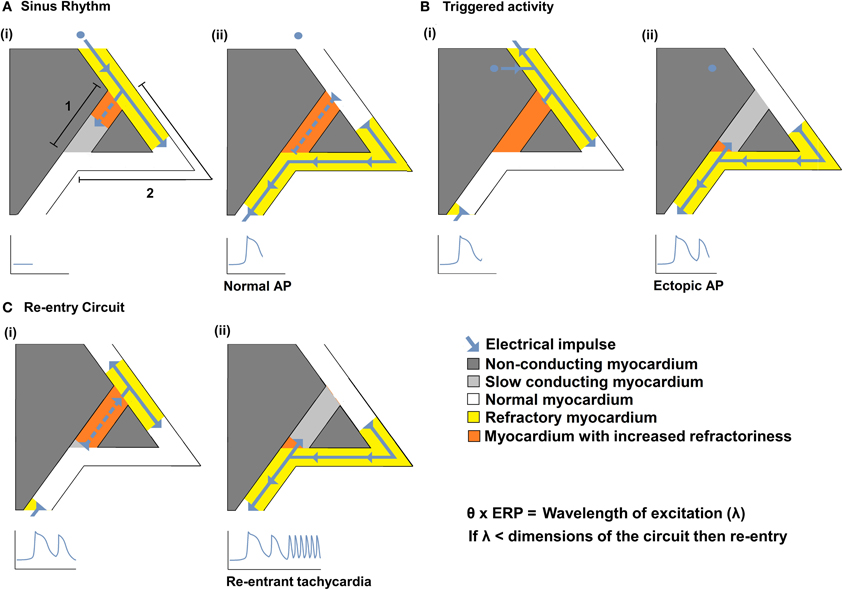
Figure 1. The relationships between θ and re-entrant arrhythmia. A diagram illustrating a typical re-entry circuit, consisting of a pathway of slow conducting myocardium [A(i) path 1, light gray] passing through non-conducting myocardium (dark gray), bordered by a second pathway of normal myocardium [A(i) path 2, white]. (A) An electrical impulse (blue arrow) originating from the SAN, (i) propagates along path 2 (white) and path 1 (light gray) pathways. As the impulse conducts, the myocardium becomes refractory (yellow in path 2 or orange in path 1) (ii) The impulse that travels along path 2 reaches the end of the circuit resulting in a normal AP. The impulse that conducts along 1 cannot exit the circuit as it collides with the refractory tissue of path 2. (B) An abnormal impulse originating from an ectopic focus is triggered immediately following the sinus impulse (i). It cannot conduct down path 1 which remains refractory; it therefore conducts along path 2. (ii) When the impulse reaches the distal end of path 2 it splits, conducting retrogradely along path 1 and orthogradely along path 2. (C) The impulse conducting retrogradely along path 1 then activates the beginning of path 2 (i) without the need of any further stimuli, thereby creating a self-perpetuating re-entrant rhythm (ii). Such re-entry is more likely to occur following reductions in conduction velocity (θ) and/or the effective refractory period (ERP) that reduce the wavelength of excitation (λ), given by the product of the θ and ERP, to values smaller than the dimensions of the available circuits.
This review describes the generation and propagation of the cardiac AP and defines the determinants of θ. It summarizes experimental evidence that links changes in these determinants with reduced myocardial θ and arrhythmogenesis, thereby identifying the diverse pathophysiological conditions in which abnormal θ may contribute to arrhythmogenesis.
The Generation of the Action Potential
Cardiac APs are time-dependent voltage waveforms that propagate along excitable tissues. AP generation arises from specific changes in membrane permeability resulting in a sequence of selective ion fluxes through their contained ion channels down their electrochemical gradients. Cardiac AP waveforms have been divided into five phases. The AP is initiated by transmembrane potential depolarization beyond an “activation threshold” at which inward currents exceed outward currents. This results in a Na+ influx through voltage-sensitive Na+ channels that show a regenerative increase to result in large depolarizing Na+ currents (INa) (of order 400 μA μF−1). These generate the initial rapid (~400 V s−1) phase 0 upstroke of the AP. The maximum rate of AP depolarization [(dV/dt)max] in an isolated cell is thus directly proportional to the total ionic current across the cell membrane (Ii) (Hodgkin and Katz, 1949).
As INa is the major transmembrane current in phase 0, (dV/dt)max is therefore often used as an index of INa. Having reached its peak amplitude, INa quickly inactivates in less than 1 ms, and requires an ERP to elapse before any further excitation. The succeeding brief rapid repolarization (phase 1) is additionally driven by the rapid activation and inactivation of K+ channels carrying the fast and slow transient outward currents, Ito,f, and Ito,s, respectively. This is then followed by a plateau phase (phase 2) during which inward Ca2+ currents (ICaL) through the L-type Ca2+ channels are balanced by INCX, resulting from 3Na+/Ca2+ exchanger (NCX) activity, the ATP-activated K+ current (IKATP), and progressively activating Iks and Ikr,: the slow and rapid components of the delayed outward rectifying K+ current. Finally the outward currents, particularly IKr, terminate the AP during phase 3 rapid repolarization. The resting membrane potential during the final, phase 4, electrical diastole, is primarily set by inward-rectifier currents (IK1), fixing the resting membrane potential close to the K+ equilibrium potential of about −80 to −90 mV.
The resting membrane potential persists until the next appropriately-initiated AP unless there is ectopic activity. Ectopic APs can be triggered by two types of afterdepolarization phenomena: early afterdepolarizations (EADs), or delayed afterdepolarizations (DADs). EADs are repolarization abnormalities characterized by oscillations in the membrane potential during phase 2 or 3 of the AP. They result from abnormalities in INa inactivation, reductions in the outward K+ currents (IK1, IKs, and Ito), or increases in inward ICaL, that then allow reactivation of INa, a persistent late Na+ current, INaL, or both, thereby compromising the net outward current required to repolarize the myocyte. In contrast, DADs are formed during phase 3 or 4 of the AP when Ca2+ released by an abnormal diastolic sarcoplasmic reticulum (SR) Ca2+ discharge is exchanged for extracellular Na+ via the NCX. Since NCX removes only 1 Ca2+ for every 3 Na+ entering, it causes a net inward current and depolarization of the cell. If the resulting afterdepolarization is large enough to displace the membrane voltage beyond the activation threshold, an extrasystole is induced.
Action Potential Propagation and its Determinants
The magnitude of INa also plays a major role in the subsequent propagation of the cardiac impulse to its neighboring cells. In a simple model of AP propagation, an axial current flows along a linear cellular structure, or cable, from one depolarized myocyte to its quiescent neighbor via intercellular channels known as gap junctions (Rohr, 2004). If this axial current is sufficient to depolarize the neighboring cell beyond its activation threshold, voltage sensitive Na+ channels will create transmembrane currents capable of propagating the AP. The axial resistance (ra) to such local circuit currents arises from the resistances of the cytosol and the gap junctions between adjacent cells. Thus, in addition to Na+ channels, gap junctions play a critical role in AP propagation and influence its velocity.
The determinants of θ can be identified using the nonlinear cable equation (Plonsey and Barr, 2007; Keener and Sneyd, 2009). This incorporates circuit elements each made up of a capacitance of unit fiber length, cm, (μF cm−1) in parallel with both a linear membrane resistance of unit fiber length rm (kΩ cm) and nonlinear conductance elements responsible for individual ion channel properties. Together these generate a total current ii in unit fiber length, x, as a function of time, t. Successive circuit elements are connected by elements reflecting cytoplasmic and gap junction resistances intervening between cells. These give rise to the effective intracellular ra of unit fiber length, ra, In classical cable theory, ra is assumed constant. The membrane potential, V, across any given capacitative element depends on the charging of its unit length by currents traversing the local membrane conductance elements, ii, as well as the axial current flow, ia, arising from neighboring regions along the length, x, of the element in question. Thus,
At constant conduction velocity θ = dx/dt, and so:
This simplest version of the cable equation clearly identifies the key determinants of θ as ra, cm, and ii. However, several of its terms are interdependent, as will be discussed below, precluding its analytic solution. Furthermore, it is a stiff equation, requiring a good estimate of θ to be made before numerical solutions may be obtained (Jack et al., 1983) However, it is possible to derive simple relationships between θ and the parameters identified by this equation using a computer model of electrical conduction in a muscle fiber (Fraser et al., 2011). Although this model simulates skeletal rather than cardiac muscle, the insights that it provides into cable properties have general validity for any system in which fast sodium currents dominate the AP upstroke.
Thus, Figure 2 demonstrates the empirical influences of ra, cm, and ii upon θ and AP waveform in a computer model of AP propagation. It confirms that ii is principally determined by INa during the AP upstroke, and demonstrates that iNa(max) α log[PNa(max)] (R2 = 0.9965), where PNa(max) is the maximum permeability of the fast Na+ channels. Several important relationships then emerge that allow measurements of the AP waveform to be used to investigate relative changes in iNa(max) and cm.
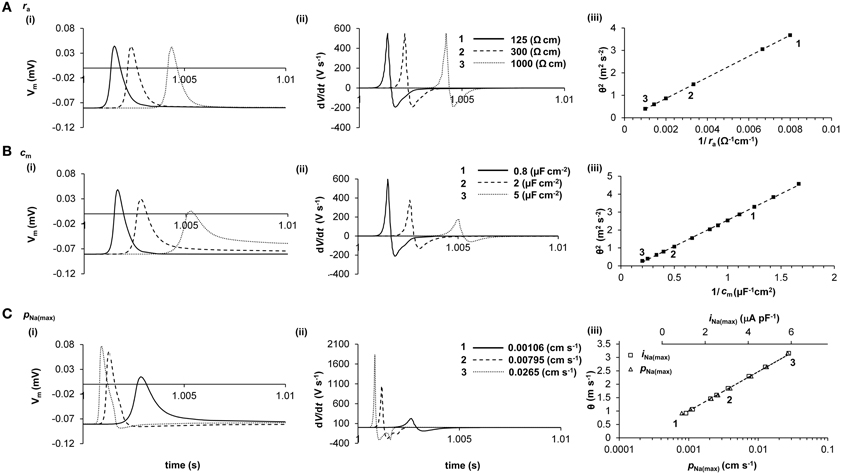
Figure 2. Quantification of the determination of θ in a computer model. The cable equation identifies ra, cm, and ii as the determinants of θ, but does not clearly demonstrate the magnitudes of their influences. Computer modeling of a skeletal muscle fiber (Fraser et al., 2011) shows the influence of ra (panel A), cm (panel B), and iNa(max) (panel C) on AP waveform (i), dV/dt (ii), and θ (iii). In panels i and ii, three representative APs are shown, each stimulated at 1s and recorded 2.5 mm further along the cable, such that increased times to the AP peaks denote slowed conduction. In panel iii, the velocities of these APs are labeled 1, 2, and 3. It will be noted that ra influences θ but not AP waveform, whereas cm and iNa(max) each influence AP waveform and θ. Note that in panel C(iii), θ is plotted against both iNa(max) (upper scale, squares) and PNa(max) (lower scale, triangles). The simple quantitative relationships between these parameters that emerge from this analysis are given in the text.
First, (Figure 2A) ra does not influence the AP waveform [Figure 2A(i)] and thus does not influence dV/dt [Figure 2A(ii)] or d2V/dt2 (not shown). Consequently, θ2 α 1/ra (R2 = 1.0000) [Figure 2A(iii)] as predicted from the cable equation. This simple relationship emerges because ra influences only the AP waveform as a function of distance, not as a function of time. The effect is similar if ra is unevenly distributed, as in cardiac myocytes connected by gap junctions (data not shown). Simulations show that, although uneven distribution of ra produces small increases in AP amplitude and (dV/dt)max immediately before high resistance areas and small decreases in these parameters immediately after, θ and distance-averaged values of AP amplitude and (dV/dt)max are influenced as for evenly-distributed increases in ra.
Second, (Figure 2B) increases in cm influence the AP waveform [Figure 2B(i)], slowing the voltage excursions [Figure 2B(ii)] and producing a reduction in θ. Interestingly, θ2 α 1/cm (R2 = 0.9996) [Figure 2B(iii)], as it appears in the cable equation, despite the influence of cm on dV/dt and d2V/dt2 (not shown). These relationships have good empirical approximations: (dV/dt)max α log(cm) (R2 = 0.9977) and (d2V/dt2)max α 1/cm (R2 = 1.0000).
Finally, (Figure 2C), the relationship between iNa(max) and θ is difficult to derive from the cable equation because of the very large influence of iNa(max) upon AP waveform [Figure 2C(i)] and (dV/dt)max [Figure 2C(ii)] and d2V/dt2 (not shown). Nevertheless, the resultant empirical relationship for the range of values depicted in Figure 2C is straightforward: θ α iNa(max) (R2 = 1.0000) [Figure 2C(iii)]. The AP waveform is influenced by iNa(max) as follows: (dV/dt)max (R2 = 0.9996); and (d2V/dt2)max α iNa(max)3 (R2 = 0.9996).
The cable equation can be extended from geometrically well-defined cylinders to cardiac tissue consisting of a continuous network of electrically-coupled cells. In doing so the analysis above becomes extended to one that determines conduction velocity resulting from the match between current and load (Kucera et al., 1998). Such an approach has been used to describe the macroscopic passive electrical properties of cardiac muscle (Weidmann, 1970; Kléber and Riegger, 1987), the relationship between dV/dt and macroscopic (>1 mm) propagation (Buchanan et al., 1985) and changes in cell to cell coupling.
As summarized in Figure 3, experimental studies have suggested a range of mechanisms through which changes in AP propagation leading to increased arrhythmic tendency can take place. They have been attributed to alterations in Na+ channel and gap junction function, as well as to the consequences of fibrotic change. These could potentially alter the major determinants of θ: transmembrane current (ii), cell to cell coupling (ra), and cell capacitance (cm), outlined in the quantitative analysis above.
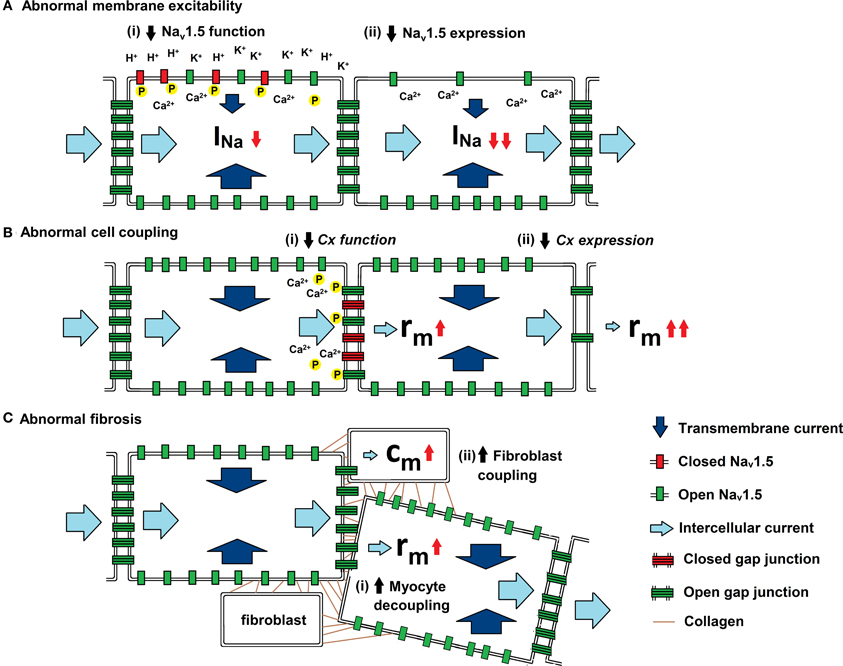
Figure 3. Physiological influences on the determinants of θ. Three diagrams illustrating the mechanisms by which (a) membrane excitability, (b) cell coupling and (c) fibrotic change influence current. Transmembrane current (dark blue arrow) enters through open Nav1.5 (green rectangle) and intercellular current (light blue arrow) passes through open Cxs (green ladder). (A) Abnormal membrane excitability results from reductions in either (i) Nav1.5 function through increases in extracellular [K+] and pH and by increases in [Ca2+]i and phosphorylation, or (ii) Nav1.5 expression by mutations in SCN5A (Brugada syndrome) and through Ca2+ mediated down regulation of the channel. (B) Abnormal cell coupling results from reductions in either (i) Cx function through increases in [Ca2+]i and dephosphorylation or (ii) Cx expression by mutations in either CJA1 or CJA5 (idiopathic AF). (C) Abnormal fibrosis produces either (i) increased myocyte-myocyte decoupling, resulting in increased ra,. or (ii) Cx-mediated myocyte-fibroblast coupling, resulting in increased cm.
The Na+ Channel and its Relationship to INa
Na+ channels are transmembrane proteins responsible for a rapid, voltage-dependent, influx of Na+ ions. They are located within the surface and transverse (t)-tubular membranes (Cohen, 1996) mainly concentrating in the perinexus region near gap junctions (Lin et al., 2011; Rhett et al., 2012). Na+ channels consist of a principal α-subunit composed of four homologous domains each containing six, S1–S6, transmembrane segments. The function of the α-subunit is modulated by one or two associated ancillary β-subunits (Bezzina, 2001).
Several, Nav1.1, Nav1.3, Nav1.5, Nav1.6, α-subunits, are known to be expressed in the mammalian heart. Of these, Nav1.5, encoded by the SCN5A gene is the most abundant. It is a large 260 KDa glycosylated protein that forms the pore component of the channel and has very high selective permeability for Na+ (permeability ratio: Na+:K+ = 100.1) (Gellens et al., 1992; Wang et al., 1996). Na+ influx and the resulting current flow through the open Na+ channel (INa) are responsible for the initial rapid (phase 0) AP depolarization and drives its propagation. It is consequently a key determinant of θ.
Abnormalities in Na+ Channel Function
Abnormalities in conduction can arise from functional mutations in SCN5A that alter INa [Figure 3A(i)]. Of these the SCN5a-1795insD mutation is associated with an overlap syndrome with features of bradycardia, impaired conduction, LQT3, and Brugada syndrome (BrS) (Bezzina et al., 1999). Whilst mice homozygous for the mutation die in utero, the heterozygous Scn5a1798insD/+ mouse shows sinus node dysfunction, conduction slowing, and QT prolongation replicating the phenotype in humans (Remme et al., 2006).
Acquired abnormalities can also lead to changes in Na+ channel function. Abnormalities in AP depolarization were first described by Gelband and Bassett who recorded decreased values of (dV/dt)max and depolarized resting membrane potentials in experimental models of heart failure (Gelband and Bassett, 1973). Subsequent studies have also associated heart failure with reductions in peak INa (Kuryshev et al., 1999; Ufret-Vincenty et al., 2001) through a number of mechanisms including reduced Nav1.5 glycosylation (Ufret-Vincenty et al., 2001). Pathophysiological reductions in Na+ channel availability have been additionally described during the acute phase of ischemia (Downar et al., 1977; Janse et al., 1986; Kleber et al., 1986; Kabell, 1989), tachycardia (Veenstra et al., 1987; Gaspo et al., 1997) and following treatment with class I anti-arrhythmic drugs (Sheldon et al., 1989).
Acute ischemia is pro-arrhythmogenic due to changes in intracellular and extracellular ionic concentrations, leading to reduced AP amplitudes, upstroke velocities (Downar et al., 1977; Janse et al., 1986) and conduction delays (Kleber et al., 1986; Kabell, 1989). Ischemic extracellular changes including: increases in [K+], decreases in pH, and hypoxia have been associated with modulation of Na+ channel function (Corr and Yamada, 1995). Microelectrode studies in hypoxic guinea pig papillary muscle demonstrated a decrease in (dV/dt)max and depolarization of the resting membrane potential that was accentuated by increases in [K+] (Kodama et al., 1984). Subsequent studies in canine Purkinje fibers similarly showed a 8% decrease in θ in raised [K+] and a 4% decrease in θ following acidosis (Veenstra et al., 1987).
In addition to the extracellular effects, increases in intracellular cyclic adenosine monophosphate (cAMP) and cytosolic Ca2+ concentrations [(Ca2+)i] have also been reported in acute ischemia. Stimulation of β-adrenergic receptors is thought to produce adenylate cyclase-mediated increases in intracellular cAMP, leading to phosphokinase A (PKA) activation (Bers, 2002). Modulation of Nav1.5 by PKA occurs via phosphorylation at serine 525 and 528 within the DI–DII linker (Murphy et al., 1996). Following PKA activation, Nav1.5 redistributes to the plasma membrane of HEK293 cells (Hallaq et al., 2006). This may explain the increases in INa reported following PKA activation with dibutyryl cAMP in rabbit and canine myocytes (Matsuda et al., 1992; Baba et al., 2004).
However, further experimental studies variously show that treatment with isoproterenol either increases (Matsuda et al., 1992) or decreases (Ono et al., 1989; Schubert et al., 1990) INa. Furthermore, when rabbit myocytes were treated with both a PKA inhibitor and isoproterenol, INa remained elevated suggesting that β-adrenergic stimulation produces an additional, PKA-independent modulation of the Na+ channel. Myocytes treated with a GTP analog and stimulatory G protein subunit showed increased INa implicating involvement of a G protein regulatory pathway (Matsuda et al., 1992).
In contrast to PKA activation, Ca2+-dependent activation of the protein phosphatase calcineurin has also been shown to strongly reduce INa, This has been variously attributed to activation of protein kinase-C (PKC) or modulation of Na+ channel trafficking (Abriel, 2007). PKC also directly modulates Nav1.5 by phosphorylation at serine 1505 in the DIII–DIV inactivation gate, significantly reducing INa (Qu et al., 1996).
Changes in intracellular Ca2+ may also exert direct regulatory effects on the Na+ channel. Indeed the C-terminal region of Na+ channel constructs contain two Ca2+-sensitive regions: a calmodulin binding, IQ, domain, and a Ca2+ binding, EF-hand, motif (Wingo et al., 2004; Chagot et al., 2009). Thus, increases in CaMKII activity have been variously reported to increase (Aiba et al., 2010) or decrease (Wagner et al., 2006) peak INa. Alternatively, intracellular Ca2+ has been shown to directly inhibit INa without affecting channel gating through a permeation block. Indeed, reductions in INa density and (dV/dt)max have been reported following increases in [Ca2+]i brought about by changing the Ca2+ concentration in the pipette solution in patch-clamped WT myocytes (Casini et al., 2009). Furthermore, reductions in (dV/dt)max, θ and increased incidences of arrhythmia have been shown in both homozygous gain of function RyR2-P2328S (RyR2S/S) and caffeine-treated WT hearts that have abnormal diastolic SR Ca2+ release (King et al., 2013b). Both immunohistochemical and biophysical studies subsequently attributed these abnormalities to the effects of Ca2+ homeostasis on INa function (King et al., 2013a).
Finally, treatment with class I anti-arrhythmic drugs modulates Na+ channel function. Thus, lidocaine, mexiletine, tocainide, and aprindine have been shown to block Na+ channels in the inactivated state whilst quinidine and disopyramide block the open channel (Kodama et al., 1986; Sheldon et al., 1989). Furthermore, lidocaine has been associated with impaired conduction and the induction of ventricular tachyarrythmias in experimental models (Anderson et al., 1990).
Abnormalities in Na+ Channel Expression
Abnormalities in AP depolarization could also arise from an alteration in Nav1.5 expression [Figure 3A(ii)]. Knockout mutations in the SCN5A gene decrease INa and are associated with cardiac conduction diseases including Lenègre disease (Schott et al., 1999), BrS (Gussak et al., 1999), sick sinus syndrome (Benson et al., 2003) and atrial fibrillation (AF) (Laitinen-Forsblom et al., 2006; Ellinor et al., 2008).
Of these, BrS is associated with a high incidence of ventricular tachyarrhythmias and sudden cardiac death (Gussak et al., 1999). Although the exact pathophysiological mechanism is not yet known (Hoogendijk et al., 2010), mutations in 17 genes have been associated with BrS of which SCN5A mutations account for a significant proportion (Tan et al., 2001). Furthermore, BrS patients with Na+ channel mutations show significantly longer conduction intervals than those without SCN5A mutations (Smits et al., 2002). In addition, class I anti-arrhythmic drugs have been used to unmask the BrS ECG pattern by exacerbating pre-existing conduction abnormalities (Gasparini et al., 2003).
Conduction alterations in BrS have been studied using a murine model with knock-out mutations in Scn5a (Papadatos et al., 2002). The homozygous embryos die in utero with severe defects in ventricular morphogenesis. Heterozygous mice (Scn5a+/−) haploinsufficient for Nav1.5 show normal survival with several cardiac conduction defects including decreased atrial, ventricular and atrioventricular conduction and increased susceptibility to pacing-induced ventricular arrhythmias (Papadatos et al., 2002).
The expression of Nav1.5 has also been shown to be regulated by changes in intracellular ion concentrations, including Ca2+. Thus, Nav1.5 mRNA and Nav1.5 protein expression increased following treatment with the Ca2+ channel blocker, verapamil, and decreased following treatment with the Ca2+ ionophore calcimycin in rat cardiomyocytes (Offord and Catterall, 1989; Taouis et al., 1991; Duff et al., 1992). Similarly, Na+ current densities increased following elevations of [Ca2+]i brought about by increased extracellular [Ca2+]. It decreased following reductions of [Ca2+]i produced by BAPTA-AM in patch-clamped cultured neonatal rat myocytes (Chiamvimonvat et al., 1995).
Such changes in the expression of Na+ channels have been reported in experimental models of atrial tachycardia that are associated with increased [Ca2+]i (Sun et al., 2001). Thus, atrial tachypacing decreased Nav1.5 mRNA, INa, and θ over several weeks in canine models (Gaspo et al., 1997; Yue et al., 1999). In contrast, the development of AF does not further reduce atrial INa (Yagi et al., 2002) or Nav1.5 mRNA (van der Velden et al., 2000) in canine and goat models, respectively.
Gap Junctions and Their Relationship to ra
Gap junctions are non-selective membrane channels that form low resistance cell-to-cell connections that permit intercellular currents, as well as the transfer of ions, amino acids, and nucleotides. Their distribution within the cell membrane is tissue-specific and helps determine the magnitude and anisotropy of conduction. In general, cardiac cells express gap junctions near Na+ channels and at higher densities toward the ends of cells rather than their lateral margins, resulting in lower ra and hence faster conduction in the longitudinal direction (Kumar and Gilula, 1996).
Gap junction channels are composed of a family of proteins known as connexins (Cx). Adjacent cells each contribute a hemichannel, made up of 6 Cxs, to the junction. There are 15 known Cxs defined by their molecular weight, each with different channel properties and gating mechanisms. Four main variants, Cxs 30.2, 40, 43, and 45, have been described in mammalian cardiac tissue (Davis et al., 1994). The type and distribution of these Cxs determines the properties of passive conduction throughout the heart. Cx43 is the most abundant and is expressed throughout the ventricular and atrial myocardium (Beyer et al., 1987), whilst Cx40 is limited to atrial tissue and the His Purkinje system (Gourdie et al., 1993a,b). Cx30.2 is only found in the atrioventricular node (AVN) (Kreuzberg et al., 2006) and Cx45 is variously reported to be expressed in the specialized conducting system (Coppen et al., 1998, 2001). Cx43 has a moderate conductance of ~110 pS and compared to other cardiac Cx is relatively insensitive to changes in transjunctional voltage (Moreno et al., 1994; Veenstra, 1996). Cx40 conductances are similar to Cx43, ~160 pS, but they have higher sensitivities to transjunctional voltage (Beblo et al., 1995; Bukauskas et al., 1995). In contrast, Cx45 has a much lower conductance at ~30 ps and is extremely sensitive to transjunctional voltage (Veenstra et al., 1994; Veenstra, 1996). These conductances are heavily regulated in healthy and pathophysiological myocardium. The resistance of gap junctions makes up approximately half of the longitudinal resistance in rat atria (Fry et al., 2012). Thus, the conductance of Cxs and therefore gap junctions help determine the magnitude of ra and are an important determinant of θ.
Abnormalities in Gap Junction Function
Mutations in the genes encoding Cxs can change gap junction function [Figure 3B(i)] and thereby reduce cell to cell coupling. Loss-of-function somatic mutations of the CJA5 gene that expresses Cx40 have been shown to result in idiopathic AF (Firouzi et al., 2004) and when combined with a SCN5A mutation, atrial standstill (Groenewegen, 2002). Heterozygous somatic missense mutations and polymorphisms within the gene's regulatory region have also been linked to conduction delays and AF (Gollob et al., 2006; Hauer et al., 2006). Other mutations of the Cx43 gene, such as GJA1, which affects phosphorylation sites, have been associated with cardiac structural abnormalities (Britz-Cunningham et al., 1995; Dasgupta et al., 2001) but without reported conduction abnormalities.
Acquired functional modifications of Cx [Figure 3B(i)] have been shown to arise during both heart failure and myocardial ischemia. Heart failure is associated with increased c-Src tyrosine-mediated tyrosine phosphorylation of Cx43 leading to decreased conductance (Toyofuku et al., 1999), conduction abnormalities and arrhythmia (Laurita et al., 2003).
During ischemia there are pathological decreases in the conductance of gap junctions following increases in [Ca2+]i (Smith et al., 1995; De Groot et al., 2001), intracellular acidification (Yan and Kléber, 1992) and through changes in catecholamine-induced increases in cellular cAMP, which in turn modulate levels of phosphorylation. Acute increases in [Ca2+]i occur in ischemic rabbit models (Dekker et al., 1996) and are associated with gap junctional uncoupling (De Mello, 1975; Smith et al., 1995) and decreased conductance (Kirchhoff et al., 1998; Gutstein et al., 2001). Such changes result in conduction slowing and conduction block (Dekker et al., 1996) that is exacerbated by increases in intracellular pH (Kleber et al., 1986). Myocardial ischemia also causes Cx43 to rapidly dephosphorylate (Huang et al., 1999; Beardslee et al., 2000) leading to its lateralization, transfer from the intercalated disks to intracellular pools and electrical uncoupling (Smith et al., 1991; Matsushita et al., 1999; Beardslee et al., 2000; Lampe et al., 2000). Dephosphorylation of Cxs may also be involved in lateralization of gap junctions resulting in conduction abnormalities in AF (Dobrev et al., 2012). However, other studies have reported that protein kinase C-dependent phosphorylation of Cx43 at serine 368 is associated with decreased gap junctional communication (Lampe et al., 2000) and conductance (Moreno et al., 1994; Kwak et al., 1995).
Abnormalities in Connexin Expression
Abnormalities in AP propagation also arise from changes in the total number of Cxs [Figure 3B(ii)]. Such abnormalities have been extensively modeled experimentally by using genetically modified mice with altered Cx 40, 43, and 45 expression.
Cx45 knockout mice (Kumai et al., 2000) show normal contraction with atrioventricular conduction block and die in utero at day 10, consistent with studies showing Cx45 is uniquely expressed in the atrioventricular canal (Coppen et al., 1999, 2001). The homozygous Cx40 knockout mouse model shows slowed conduction and a partial atrioventricular block (Leaf et al., 2008); however arrhythmia was only observed in one study (Kirchhoff et al., 1998). The homozygote Cx43 knockout mouse (Cx43−/−) dies from neonatal pulmonary outflow obstruction (Reaume et al., 1995) but electrocardiography and optical mapping have been used successfully to measure θ in mice haploid insufficient for Cx43 (Cx43+/−). The first of these studies suggested slowed conduction (Guerrero et al., 1997) but later work contradicted these findings, showing no significant change in θ (Kirchhoff et al., 2000; Thomas et al., 2003). However, ischemic Cx43+/− hearts have shown conduction abnormalities and higher incidences of spontaneous ventricular arrhythmias compared to WT (Lerner et al., 2000).
A cardiac-restricted homozygote knockout of Cx43 model was used to prevent the neonatal lethal structural defects in Cx43−/−. Such mice have a 90% reduction in Cx43 with normal heart structure and contractile function. Epicardial optical mapping showed that both longitudinal and transverse ventricular θ was reduced by 40–50%. In addition, Cx43 conditional knockout mice had high incidences of spontaneous ventricular arrhythmias and sudden cardiac death (Gutstein et al., 2001). These latter findings were supported by recent modeling suggesting that in non-ischemic conditions a 90% reduction in gap junctions is required to decrease θ by 50% (Jongsma and Wilders, 2000; Spach et al., 2000).
Interestingly, recent studies have proposed an additional, non-canonical method of cardiac conduction whereby the intercellular transfer of charge does not only occur by the passive flow of current through gap junctions. These studies suggest a role for extracellular space in modulating θ (Veeraraghavan et al., 2012) and that connexins actively augment current propagation by the accumulation of functional sodium channels at the perinexus of the intercalated disc and by the maintenance of narrow intercellular distances (Agullo-Pascual and Delmar, 2012; Rhett et al., 2013). Such a mechanism is supported by experimental evidence in cardiac restricted Cx43+/− knockout mice that associates reduced Cx43 expression with reduced Nav1.5 expression and INa (Jansen et al., 2012). These findings highlight one of the limitations of using simple cable theory and help explain why decreases in connexin expression that are classically thought to increase ra do not result in proportional decreases in θ.
Acquired changes in Cx expression have also been associated with a number of cardiac conditions including ventricular hypertrophy, heart failure and AF. During the early stages of ventricular hypertrophy there were increases in Cx expression and myocyte coupling. Thus, neonatal rat ventricular myocytes cultured in cAMP (Darrow et al., 1996) or angiotensin II (Dodge et al., 1998), both mediators of hypertrophy, showed increases in Cx43 expression and θ. More recent studies have shown a 100% increase in Cx43 expression and resulting θ in neonatal ventricular myocytes cultured with a mechanical load (Zhuang et al., 2000). θ typically increases in the acute phases of hypertrophy due to both increases in Cx43 expression and cell size that decrease intercellular and longitudinal resistance, respectively (Wiegerinck et al., 2006). Over prolonged periods of hypertrophy θ starts to decrease, as seen in chronic arrhythmic conditions (Cooklin et al., 1997) where Cx43 shows 25–50% downregulation (Kaprielian et al., 1998). The expression of Cx43 is down-regulated in experimental (Akar et al., 2004; Ai and Pogwizd, 2005) and clinical (Dupont et al., 2001; Kostin et al., 2004) studies of heart failure through recruitment of mitogen-activated protein kinase C-Jun NH2- terminal kinase (Petrich et al., 2002). Clinical heart failure studies showed Cx45 (Yamada et al., 2003) and Cx40 (Dupont et al., 2001) are up-regulated, possibly as a compensatory mechanism.
There are inconsistent clinical and experimental reports regarding changes in Cx expression during atrial tachyarrhythmias such as AF. Thus, experimental studies show up regulation of Cx43 in an atrial tachypaced canine model (Elvan et al., 1996; Sakabe et al., 2004), increased heterogeneity of Cx40 expression in tachypaced goat models with no changes in overall Cx expression (van der Velden et al., 1998). Whilst clinical studies support experimental findings of increased Cx expression on lateral cell surfaces (Smith et al., 1991; Polontchouk et al., 2001; Kostin et al., 2002) and increased heterogeneity (Dupont et al., 2001; Kostin et al., 2004) there is no consensus on changes in overall Cx expression. Studies show both increases (Dupont et al., 2001; Polontchouk et al., 2001) and decreases (Kostin et al., 2002; Nao et al., 2003) in Cx40 and Cx43 expression. However, conduction abnormalities are consistently described in experimental (Gaspo et al., 1997) and clinical studies of AF (Konings et al., 1994).
Such findings suggest changes in gap junction conductance do influence θ and arrhythmic susceptibility but only when the majority of gap junctions are impaired. Instead, reductions in membrane excitability and anatomical disruption are likely to have greater roles in most pathologies.
Fibrosis and its Relationship to ra and cm
Fibroblasts make up 75% of the cells in the myocardium (Banerjee et al., 2006). Indeed, sequential contraction of the atria and ventricles is dependent on the annulus fibrosus cordis to electrically insulate the two structures. In normal wound healing they undergo apoptosis after having produced a mixture of cross-linked collagen and other extracellular matrix (ECM) (Gurtner et al., 2008).
However, in certain fibrotic conditions such as myocardial infarction and cardiomyopathy, fibroblasts do not apoptose and instead proliferate, migrate, and differentiate into myofibroblasts (Weber et al., 1994). Fibroblast and myofibroblasts then continue to increase ECM deposition between layers of neighboring cardiomyocytes unabated (Manabe et al., 2002). This results in altered tissue architecture that in turn reduces myocyte-myocyte coupling as well as results in the formation of fibroblast-myocyte couplings (Camelliti et al., 2004). Such changes could in turn alter ra or cm, respectively slowing conduction.
Disruption of Cell-To Cell Coupling and Alterations in ra
When healthy myocardium is disrupted by insulating connective tissue, the current provided by cells preceding the disruption may disperse along alternative branches, giving rise to a local conduction delay. Such current to load mismatches are found in fibrotic myocardium (Spach et al., 1988; De Bakker et al., 1993) and have been shown to impair conduction (Mendez et al., 1970; Spach et al., 1982).
Experimental canine models of fibrosis have shown how the deposition of insulating ECM and the decoupling of myocytes [Figure 3C(i)] lead to the interruption of both longitudinal and transverse cardiomyocyte-bundle continuity, producing zigzag conduction patterns (Spach and Boineau, 1997; Burstein et al., 2009). Such changes lead to conduction delays that predispose to re-entry arrhythmias in both experimental (De Bakker et al., 1993) and clinical studies (Spach, 2007).
Coupling of Cardiomyocytes to Fibroblasts and Alterations in cm
Fibroblasts have been shown to electrically couple with cardiomyocytes through Cx43 and Cx45 binding [Figure 3C(ii)] (Camelliti et al., 2004; Chilton et al., 2007). Fibroblasts lack ion channels associated with excitation. They also have depolarized resting membrane potentials (Kamkin et al., 1999) and would therefore modulate the membrane potential of the myocytes to which they are coupled, potentially inactivating Na+ channels. In addition, the coupling of fibroblasts to myocytes produces a net increase in cell capacitance, cm (Miragoli et al., 2006).
Both these features resulted in a slowing of conduction and re-entrant events in cardiomyocytes co-cultured with fibroblasts (Maleckar et al., 2009; Xie et al., 2009). The magnitude of these effects on conduction appeared to be determined by the number of fibroblasts coupled to each cardiomyocyte, with significant changes observed when 10–30 fibroblasts were coupled per cardiomyocyte (Jacquemet and Henriquez, 2008; Maleckar et al., 2009; Sachse et al., 2009).
Murine Models of Fibrosis
Experimental studies in mice have supported the association between impaired conduction and fibrosis. However, most murine models of fibrosis have concurrent pathological processes that may additionally contribute to abnormal conduction, making interpretation of their findings difficult. Thus, mutations in sarcomere protein genes cause hypertrophic cardiomyopathy (HCM), a congenital condition associated with myocyte enlargement and increased myocardial fibrosis, heart failure, and arrhythmia (Lu et al., 2013). Experimental murine models that carry mutations in the α- myosin heavy chain gene, MHC403, show fibrosis, conduction abnormalities, and increased incidence of inducible arrhythmia (Berul et al., 1998). Mutations in SCN5A are associated with both BrS and Lenègre disease (Schott et al., 1999). The latter condition produces progressive cardiac fibrosis and conduction abnormalities that lead to complete atrioventricular block and episodes of cardiac syncope. Scn5a+/− hearts demonstrate similar age-related fibrosis and deterioration in conduction resembling the clinical phenotype of both Lenègre's disease (Lenegre and Moreau, 1963) and BrS (Coronel et al., 2005). Finally, age-related upregulation of transforming growth factor-β1 (TGF-β1) associated with increased measures of fibrosis have been reported in Scn5a+/− hearts (Hao et al., 2011).
TGF-β1 mediated myocardial fibrosis also directly plays an important role in atrial arrhythmogenesis, including AF (Nattel et al., 2005). Experimental studies suggest that mice with increased expression of TGF-β1 have higher incidences of AF and conduction abnormalities as a result of raised levels of atrial fibrosis (Verheule et al., 2004). Furthermore, atrial fibroblasts appear more sensitive to the actions of TGF-β than their ventricular counterparts (Nakajima et al., 2000). TGF-β1 polymorphisms are also thought to be involved in inducing congenital heart block as a result of fibrosis, leading to a predisposition to AF (Clancy et al., 2003).
Summary and Conclusions
This review defines the determinants of θ as INa; ra and cm and summarizes the mechanistic evidence that links changes in these determinants with reduced myocardial θ and arrhythmogenesis. Thus, firstly, (dV/dt)max is reduced by impaired Na+ channel function that arises clinically during heart failure, ischemia, tachycardia, and following treatment with class I anti-arrhythmic drugs. Such reductions also arise as a consequence of mutations in SCN5A such as those in Lenègre disease, BrS, sick sinus syndrome and AF. Secondly, ra may be increased due to decreased gap junction coupling following ischemia, ventricular hypertrophy, and heart failure, or as a result of mutations in CJA5 found in idiopathic AF and atrial standstill. Finally, either ra or cm could potentially be altered by fibrotic change through its effects upon decoupling myocyte-myocyte connections and through myocyte-fibroblast coupling. Such changes are observed in myocardial infarction and cardiomyopathy or following mutations in MHC403 and SCN5A resulting in HCM or Lenègre disease, respectively. The review thereby identifies the diverse pathophysiological conditions in which abnormal θ may contribute to arrhythmogenesis. Such findings provide insight into the mechanisms of arrhythmogenesis in common arrhythmias not usually attributed to impaired conduction such as those associated with abnormal Ca2+ homeostasis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Biotechnology and Biological Sciences Research Council (BBSRC), Medical Research Council (MRC) and the Raymond and Beverly Sackler foundation for their generous financial support. James A. Fraser holds a David Phillips Fellowship from the BBSRC.
References
Abriel, H. (2007). Roles and regulation of the cardiac sodium channel Na v 1.5: recent insights from experimental studies. Cardiovasc. Res. 76, 381–389. doi: 10.1016/j.cardiores.2007.07.019
Agullo-Pascual, E., and Delmar, M. (2012). The noncanonical functions of Cx43 in the heart. J. Membr. Biol. 245, 477–482. doi: 10.1007/s00232-012-9466-y
Ai, X., and Pogwizd, S. M. (2005). Connexin 43 downregulation and dephosphorylation in non-ischemic heart failure is associated with enhanced colocalized protein phosphatase type 2A. Circ. Res. 96, 54–63. doi: 10.1161/01.RES.0000152325.07495.5a
Aiba, T., Hesketh, G. G., Liu, T., Carlisle, R., Villa-Abrille, M. C., O'Rourke, B., et al. (2010). Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc. Res. 85, 454–463. doi: 10.1093/cvr/cvp324
Akar, F. G., Spragg, D. D., Tunin, R. S., Kass, D. A., and Tomaselli, G. F. (2004). Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ. Res. 95, 717–725. doi: 10.1161/01.RES.0000144125.61927.1c
Anderson, K. P., Walker, R., Lux, R. L., Ershler, P. R., Menlove, R., Williams, M. R., et al. (1990). Conduction velocity depression and drug-induced ventricular tachyarrhythmias. Effects of lidocaine in the intact canine heart. Circulation 81, 1024–1038. doi: 10.1161/01.CIR.81.3.1024
Baba, S., Dun, W., and Boyden, P. A. (2004). Can PKA activators rescue Na+ channel function in epicardial border zone cells that survive in the infarcted canine heart. Cardiovasc. Res. 64, 260–267. doi: 10.1016/j.cardiores.2004.06.021
Banerjee, I., Yekkala, K., Borg, T. K., and Baudino, T. A. (2006). Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann. N.Y. Acad. Sci. 1080, 76–84. doi: 10.1196/annals.1380.007
Beardslee, M. A., Lerner, D. L., Tadros, P. N., Laing, J. G., Beyer, E. C., Yamada, K. A., et al. (2000). Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ. Res. 87, 656–662. doi: 10.1161/01.RES.87.8.656
Beblo, D. A., Wang, H. Z., Beyer, E. C., Westphale, E. M., and Veenstra, R. D. (1995). Unique conductance, gating, and selective permeability properties of gap junction channels formed by connexin40. Circ. Res. 77, 813–822. doi: 10.1161/01.RES.77.4.813
Benson, D. W., Wang, D. W., Dyment, M., Knilans, T. K., Fish, F. A., Strieper, M. J., et al. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028.
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Berul, C. I., Christe, M. E., Aronovitz, M. J., Maguire, C. T., Seidman, C. E., Seidman, J. G., et al. (1998). Familial hypertrophic cardiomyopathy mice display gender differences in electrophysiological abnormalities. J. Interv. Card. Electrophysiol. 2, 7–14. doi: 10.1023/A:1009700404218
Beyer, E. C., Paul, D. L., and Goodenough, D. A. (1987). Connexin43: a protein from rat heart homologous to a gap junction protein from liver. J. Cell Biol. 105, 2621–2629. doi: 10.1083/jcb.105.6.2621
Bezzina, C. (2001). Cardiac sodium channel and inherited arrhythmia syndromes. Cardiovasc. Res. 49, 257–271.
Bezzina, C., Veldkamp, M. W., Van den Berg, M. P., Postma, A. V., Rook, M. B., Viersma, J.-W., et al. (1999). A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 85, 1206–1213.
Britz-Cunningham, S. H., Shah, M. M., Zuppan, C. W., and Fletcher, W. H. (1995). Mutations of the Connexin43 gap-junction gene in patients with heart malformations and defects of laterality. N. Engl. J. Med. 332, 1323–1329. doi: 10.1056/NEJM199505183322002
Buchanan, J. W., Saito, T., and Gettes, L. S. (1985). The effects of antiarrhythmic drugs, stimulation frequency, and potassium-induced resting membrane potential changes on conduction velocity and dV/dtmax in guinea pig myocardium. Circ. Res. 56, 696–703.
Bukauskas, F. F., Elfgang, C., Willecke, K., and Weingart, R. (1995). Biophysical properties of gap junction channels formed by mouse connexin40 in induced pairs of transfected human HeLa cells. Biophys. J. 68, 2289–2298. doi: 10.1016/S0006-3495(95)80411-X
Burstein, B., Comtois, P., Michael, G., Nishida, K., Villeneuve, L., Yeh, Y.-H., et al. (2009). Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ. Res. 105, 1213–1222. doi: 10.1161/CIRCRESAHA.108.183400
Camelliti, P., Green, C. R., LeGrice, I., and Kohl, P. (2004). Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 94, 828–835. doi: 10.1161/01.RES.0000122382.19400.14
Casini, S., Verkerk, A. O., Van Borren, M. M. G. J., Van Ginneken, A. C. G., Veldkamp, M. W., De Bakker, J. M. T., et al. (2009). Intracellular calcium modulation of voltage-gated sodium channels in ventricular myocytes. Cardiovasc. Res. 81, 72–81. doi: 10.1093/cvr/cvn274
Chagot, B., Potet, F., Balser, J. R., and Chazin, W. J. (2009). Solution NMR structure of the C-terminal EF-hand domain of human cardiac sodium channel NaV1.5. J. Biol. Chem. 284, 6436–6445. doi: 10.1074/jbc.M807747200
Chiamvimonvat, N., Kargacin, M. E., Clark, R. B., and Duff, H. J. (1995). Effects of intracellular calcium on sodium current density in cultured neonatal rat cardiac myocytes. J. Physiol. 483(Pt 2), 307–318.
Chilton, L., Giles, W. R., and Smith, G. L. (2007). Evidence of intercellular coupling between co-cultured adult rabbit ventricular myocytes and myofibroblasts. J. Physiol. 583, 225–236. doi: 10.1113/jphysiol.2007.135038
Clancy, R. M., Backer, C. B., Yin, X., Kapur, R. P., Molad, Y., and Buyon, J. P. (2003). Cytokine polymorphisms and histologic expression in autopsy studies: contribution of TNF-alpha and TGF-beta 1 to the pathogenesis of autoimmune-associated congenital heart block. J. Immunol. 171, 3253–3261.
Cohen, S. A. (1996). Immunocyto-chemical localization of rH1 sodium channel in adult rat heart atria and ventricle: presence in terminal intercalated disks. Circulation 94, 3083–3086. doi: 10.1161/01.CIR.94.12.3083
Cooklin, M., Wallis, W. R., Sheridan, D. J., and Fry, C. H. (1997). Changes in cell-to-cell electrical coupling associated with left ventricular hypertrophy. Circ. Res. 80, 765–771. doi: 10.1161/01.RES.80.6.765
Coppen, S. R., Dupont, E., Rothery, S., and Severs, N. J. (1998). Connexin45 expression is preferentially associated with the ventricular conduction system in mouse and rat heart. Circ. Res. 82, 232–243. doi: 10.1161/01.RES.82.2.232
Coppen, S. R., Gourdie, R. G., and Severs, N. J. (2001). Connexin45 is the first connexin to be expressed in the central conduction system of the mouse heart. Exp. Clin. Cardiol. 6, 17–23.
Coppen, S. R., Severs, N. J., and Gourdie, R. G. (1999). Connexin45 (alpha 6) expression delineates an extended conduction system in the embryonic and mature rodent heart. Dev. Genet. 24, 82–90.
Coronel, R., Casini, S., Koopmann, T. T., Wilms-Schopman, F. J. G., Verkerk, A. O., De Groot, J. R., et al. (2005). Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112, 2769–2777. doi: 10.1161/CIRCULATIONAHA.105.532614
Corr, P. B., and Yamada, K. A. (1995). Selected metabolic alterations in the ischemic heart and their contributions to arrhythmogenesis. Herz 20, 156–168.
Darrow, B. J., Fast, V. G., Kléber, A. G., Beyer, E. C., and Saffitz, J. E. (1996). Functional and structural assessment of intercellular communication. Increased conduction velocity and enhanced connexin expression in dibutyryl cAMP-treated cultured cardiac myocytes. Circ. Res. 79, 174–183. doi: 10.1161/01.RES.79.2.174
Dasgupta, C., Martinez, A. M., Zuppan, C. W., Shah, M. M., Bailey, L. L., and Fletcher, W. H. (2001). Identification of connexin43 (alpha1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (DGGE). Mutat. Res. 479, 173–186. doi: 10.1016/S0027-5107(01)00160-9
Davis, L. M., Kanter, H. L., Beyer, E. C., and Saffitz, J. E. (1994). Distinct gap junction protein phenotypes in cardiac tissues with disparate conduction properties. J. Am. Coll. Cardiol. 24, 1124–1132. doi: 10.1016/0735-1097(94)90879-6
De Bakker, J. M., Van Capelle, F. J., Janse, M. J., Tasseron, S., Vermeulen, J. T., De Jonge, N., et al. (1993). Slow conduction in the infarcted human heart. “Zigzag” course of activation. Circulation 88, 915–926. doi: 10.1161/01.CIR.88.3.915
De Groot, J. R., Wilms-Schopman, F. J., Opthof, T., Remme, C. A., and Coronel, R. (2001). Late ventricular arrhythmias during acute regional ischemia in the isolated blood perfused pig heart. Role of electrical cellular coupling. Cardiovasc. Res. 50, 362–372. doi: 10.1016/S0008-6363(01)00222-X
Dekker, L. R. C., Fiolet, J. W. T., VanBavel, E., Coronel, R., Opthof, T., Spaan, J. A. E., et al. (1996). Intracellular Ca2+, intercellular electrical coupling, and mechanical activity in ischemic rabbit papillary muscle: effects of preconditioning and metabolic blockade. Circ. Res. 79, 237–246.
De Mello, W. C. (1975). Effect of intracellular injection of calcium and strontium on cell communication in heart. J. Physiol. 250, 231–245.
Dobrev, D., Carlsson, L., and Nattel, S. (2012). Novel molecular targets for atrial fibrillation therapy. Nat. Rev. Drug Discov. 11, 275–291. doi: 10.1038/nrd3682
Dodge, S. M., Beardslee, M. A., Darrow, B. J., Green, K. G., Beyer, E. C., and Saffitz, J. E. (1998). Effects of angiotensin II on expression of the gap junction channel protein connexin43 in neonatal rat ventricular myocytes. J. Am. Coll. Cardiol. 32, 800–807. doi: 10.1016/S0735-1097(98)00317-9
Downar, E., Janse, M. J., and Durrer, D. (1977). The effect of “ischemic” blood on transmembrane potentials of normal porcine ventricular myocardium. Circulation 55, 455–462. doi: 10.1161/01.CIR.55.3.455
Duff, H. J., Offord, J., West, J., and Catterall, W. A. (1992). Class I and IV antiarrhythmic drugs and cytosolic calcium regulate mRNA encoding the sodium channel alpha subunit in rat cardiac muscle. Mol. Pharmacol. 42, 570–574.
Dupont, E., Matsushita, T., Kaba, R. A., Vozzi, C., Coppen, S. R., Khan, N., et al. (2001). Altered connexin expression in human congestive heart failure. J. Mol. Cell. Cardiol. 33, 359–371. doi: 10.1006/jmcc.2000.1308
Ellinor, P. T., Nam, E. G., Shea, M. A., Milan, D. J., Ruskin, J. N., and MacRae, C. A. (2008). Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm 5, 99–105. doi: 10.1016/j.hrthm.2007.09.015
Elvan, A., Wylie, K., and Zipes, D. P. (1996). Pacing-induced chronic atrial fibrillation impairs sinus node function in dogs. Electrophysiological remodeling. Circulation 94, 2953–2960. doi: 10.1161/01.CIR.94.11.2953
Firouzi, M., Ramanna, H., Kok, B., Jongsma, H. J., Koeleman, B. P. C., Doevendans, et al. (2004). Association of human connexin40 gene polymorphisms with atrial vulnerability as a risk factor for idiopathic atrial fibrillation. Circ. Res. 95, e29–e33. doi: 10.1161/01.RES.0000141134.64811.0a
Fraser, J. A., Huang, C. L.-H., and Pedersen, T. H. (2011). Relationships between resting conductances, excitability, and t-system ionic homeostasis in skeletal muscle. J. Gen. Physiol. 138, 95–116. doi: 10.1085/jgp.201110617
Fry, C. H., Salvage, S. C., Manazza, A., Dupont, E., Labeed, F. H., Hughes, M. P., et al. (2012). Cytoplasm resistivity of mammalian atrial myocardium determined by dielectrophoresis and impedance methods. Biophys. J. 103, 2287–2294. doi: 10.1016/j.bpj.2012.10.023
Gasparini, M., Priori, S. G., Mantica, M., Napolitano, C., Galimberti, P., Ceriotti, C., et al. (2003). Flecainide test in Brugada syndrome: a reproducible but risky tool. Pacing Clin. Electrophysiol. 26, 338–341. doi: 10.1046/j.1460-9592.2003.00045.x
Gaspo, R., Bosch, R. F., Bou-Abboud, E., and Nattel, S. (1997). Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ. Res. 81, 1045–1052. doi: 10.1161/01.RES.81.6.1045
Gelband, H., and Bassett, A. L. (1973). Depressed transmembrane potentials during experimentally induced ventricular failure in cats. Circ. Res. 32, 625–634. doi: 10.1161/01.RES.32.5.625
Gellens, M. E., George, A. L., Chen, L. Q., Chahine, M., Horn, R., Barchi, R. L., et al. (1992). Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. U.S.A. 89, 554–558.
Gollob, M. H., Jones, D. L., Krahn, A. D., Danis, L., Gong, X.-Q., Shao, Q., et al. (2006). Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 354, 2677–2688. doi: 10.1056/NEJMoa052800
Gourdie, R. G., Green, C. R., Severs, N. J., Anderson, R. H., and Thompson, R. P. (1993a). Evidence for a distinct gap-junctional phenotype in ventricular conduction tissues of the developing and mature avian heart. Circ. Res. 72, 278–289.
Gourdie, R. G., Severs, N. J., Green, C. R., Rothery, S., Germroth, P., and Thompson, R. P. (1993b). The spatial distribution and relative abundance of gap-junctional connexin40 and connexin43 correlate to functional properties of components of the cardiac atrioventricular conduction system. J. Cell. Sci. 105(Pt 4), 985–991.
Groenewegen, W. A. (2002). A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ. Res. 92, 14–22.
Guerrero, P. A., Schuessler, R. B., Davis, L. M., Beyer, E. C., Johnson, C. M., Yamada, K. A., et al. (1997). Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J. Clin. Invest. 99, 1991–1998. doi: 10.1172/JCI119367
Gurtner, G. C., Werner, S., Barrandon, Y., and Longaker, M. T. (2008). Wound repair and regeneration. Nature 453, 314–321. doi: 10.1038/nature07039
Gussak, I., Antzelevitch, C., Bjerregaard, P., Towbin, J. A, and Chaitman, B. R. (1999). The Brugada syndrome: clinical, electrophysiologic and genetic aspects. J. Am. Coll. Cardiol. 33, 5–15. doi: 10.1016/S0735-1097(98)00528-2
Gutstein, D. E. E., Morley, G. E. E., Tamaddon, H., Vaidya, D., Schneider, M. D. D., Chen, J. J., et al. (2001). Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ. Res. 88, 333–339. doi: 10.1161/01.RES.88.3.333
Hallaq, H., Yang, Z., Viswanathan, P. C., Fukuda, K., Shen, W., Wang, D. W., et al. (2006). Quantitation of protein kinase A-mediated trafficking of cardiac sodium channels in living cells. Cardiovasc. Res. 72, 250–261. doi: 10.1016/j.cardiores.2006.08.007
Hao, X., Zhang, Y., Zhang, X., Nirmalan, M., Davies, L., Konstantinou, D., et al. (2011). TGF-β1-mediated fibrosis and ion channel remodeling are key mechanisms in producing the sinus node dysfunction associated with SCN5A deficiency and aging. Circ. Arrhythm. Electrophysiol. 4, 397–406. doi: 10.1161/CIRCEP.110.960807
Hauer, R. N. W., Groenewegen, W. A., Firouzi, M., Ramanna, H., and Jongsma, H. J. (2006). Cx40 polymorphism in human atrial fibrillation. Adv. Cardiol. 42, 284–291. doi: 10.1159/000092579
Hodgkin, A. L., and Katz, B. (1949). The effect of sodium ions on the electrical activity of giant axon of the squid. J. Physiol. 108, 37–77.
Hoogendijk, M. G., Opthof, T., Postema, P. G., Wilde, A. A. M., De Bakker, J. M. T., and Coronel, R. (2010). The Brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither. Toward a unifying mechanism of the Brugada syndrome. Circ. Arrhythm. Electrophysiol. 3, 283–290. doi: 10.1161/CIRCEP.110.937029
Huang, X. D., Sandusky, G. E., and Zipes, D. P. (1999). Heterogeneous loss of connexin43 protein in ischemic dog hearts. J. Cardiovasc. Electrophysiol. 10, 79–91. doi: 10.1111/j.1540-8167.1999.tb00645.x
Jack, J., Noble, D., and Tsien, R. (1983). Electric Current Flow in Excitable Cells. Oxford: Oxford University Press.
Jacquemet, V., and Henriquez, C. S. (2008). Loading effect of fibroblast-myocyte coupling on resting potential, impulse propagation, and repolarization: insights from a microstructure model. Am. J. Physiol. Heart Circ. Physiol. 294, H2040–H2052.
Janse, M., Kleber, A., Capucci, A., Coronel, R., and Wilmsschopman, F. (1986). Electrophysiological basis for arrhythmias caused by acute ischemia. Role of the subendocardium. J. Mol. Cell. Cardiol. 18, 339–355.
Jansen, J. A., Noorman, M., Musa, H., Stein, M., De Jong, S., Van der Nagel, R., et al. (2012). Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm 9, 600–607. doi: 10.1016/j.hrthm.2011.11.025
Jongsma, H. J., and Wilders, R. (2000). Gap junctions in cardiovascular disease. Circ. Res. 86, 1193–1197. doi: 10.1161/01.RES.86.12.1193
Kabell, G. (1989). Ischemia-induced conduction delay and ventricular arrhythmias: comparative electropharmacology of bethanidine sulfate and bretylium tosylate. J. Cardiovasc. Pharmacol. 13, 471–482. doi: 10.1097/00005344-198903000-00017
Kamkin, A., Kiseleva, I., Wagner, K. D., Lammerich, A., Bohm, J., Persson, P. B., et al. (1999). Mechanically induced potentials in fibroblasts from human right atrium. Exp. Physiol. 84, 347–356. doi: 10.1017/S0958067099017947
Kaprielian, R. R., Gunning, M., Dupont, E., Sheppard, M. N., Rothery, S. M., Underwood, R., et al. (1998). Downregulation of immunodetectable connexin43 and decreased gap junction size in the pathogenesis of chronic hibernation in the human left ventricle. Circulation 97, 651–660. doi: 10.1161/01.CIR.97.7.651
Keener, J., and Sneyd, J. (2009). Mathematical Physiology: I: Cellular Physiology (Interdisciplinary Applied Mathematics), 2nd Edn. New York, NY: Springer.
King, J. H., Wickramarachchi, C., Kua, K., Du, Y., Jeevaratnam, K., Matthews, H. R., et al. (2013a). Loss of Nav1.5 expression and function in murine atria containing the RyR2-P2328S gain-of-function mutation. Cardiovasc. Res. doi: 10.1093/cvr/cvt141. [Epub ahead of print].
King, J., Zhang, Y., Lei, M., Grace, A., Huang, C.-H., and Fraser, J. (2013b). Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol. 207, 308–323.
Kirchhoff, S., Kim, J. S., Hagendorff, A., Thönnissen, E., Krüger, O., Lamers, W. H., et al. (2000). Abnormal cardiac conduction and morphogenesis in connexin40 and connexin43 double-deficient mice. Circ. Res. 87, 399–405. doi: 10.1161/01.RES.87.5.399
Kirchhoff, S., Nelles, E., Hagendorff, A., Krüger, O., Traub, O., and Willecke, K. (1998). Reduced cardiac conduction velocity and predisposition to arrhythmias in connexin40-deficient mice. Curr. Biol. 8, 299–302. doi: 10.1016/S0960-9822(98)70114-9
Kleber, A. G., Janse, M. J., Wilms-Schopmann, F. J., Wilde, A. A., and Coronel, R. (1986). Changes in conduction velocity during acute ischemia in ventricular myocardium of the isolated porcine heart. Circulation 73, 189–198. doi: 10.1161/01.CIR.73.1.189
Kléber, A. G., and Riegger, C. B. (1987). Electrical constants of arterially perfused rabbit papillary muscle. J. Physiol. 385, 307–324.
Kodama, I., Toyama, J., and Yamada, K. (1986). Open and inactivated sodium channel block by class-I antiarrhythmic drugs. Jpn. Heart J. 27(Suppl. 1), 83–89.
Kodama, I., Wilde, A., Janse, M. J., Durrer, D., and Yamada, K. (1984). Combined effects of hypoxia, hyperkalemia and acidosis on membrane action potential and excitability of guinea-pig ventricular muscle. J. Mol. Cell. Cardiol. 16, 247–259. doi: 10.1016/S0022-2828(84)80591-X
Konings, K. T., Kirchhof, C. J., Smeets, J. R., Wellens, H. J., Penn, O. C., and Allessie, M. A. (1994). High-density mapping of electrically induced atrial fibrillation in humans. Circulation 89, 1665–1680. doi: 10.1161/01.CIR.89.4.1665
Kostin, S., Dammer, S., Hein, S., Klovekorn, W. P., Bauer, E. P., and Schaper, J. (2004). Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc. Res. 62, 426–436. doi: 10.1016/j.cardiores.2003.12.010
Kostin, S., Klein, G., Szalay, Z., Hein, S., Bauer, E. P., and Schaper, J. (2002). Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 54, 361–379. doi: 10.1016/S0008-6363(02)00273-0
Kreuzberg, M. M., Schrickel, J. W., Ghanem, A., Kim, J.-S., Degen, J., Janssen-Bienhold, U., et al. (2006). Connexin30.2 containing gap junction channels decelerate impulse propagation through the atrioventricular node. Proc. Natl. Acad. Sci. U.S.A. 103, 5959–5964.
Kucera, J. P., Kléber, A. G., and Rohr, S. (1998). Slow conduction in cardiac tissue, II: effects of branching tissue geometry. Circ. Res. 83, 795–805. doi: 10.1161/01.RES.83.8.795
Kumai, M., Nishii, K., Nakamura, K., Takeda, N., Suzuki, M., and Shibata, Y. (2000). Loss of connexin45 causes a cushion defect in early cardiogenesis. Development 127, 3501–3512.
Kumar, N. M., and Gilula, N. B. (1996). The gap junction communication channel. Cell 84, 381–388. doi: 10.1016/S0092-8674(00)81282-9
Kuryshev, Y. A., Brittenham, G. M., Fujioka, H., Kannan, P., Shieh, C.-C., Cohen, S. A., et al. (1999). Decreased sodium and increased transient outward potassium currents in iron-loaded cardiac myocytes: implications for the arrhythmogenesis of human siderotic heart disease. Circulation 100, 675–683. doi: 10.1161/01.CIR.100.6.675
Kwak, B. R., Hermans, M. M., De Jonge, H. R., Lohmann, S. M., Jongsma, H. J., and Chanson, M. (1995). Differential regulation of distinct types of gap junction channels by similar phosphorylating conditions. Mol. Biol. Cell 6, 1707–1719.
Laitinen-Forsblom, P. J., Makynen, P., Makynen, H., Yli-Mayry, S., Virtanen, V., Kontula, K., et al. (2006). SCN5A mutation associated with cardiac conduction defect and atrial arrhythmias. J. Cardiovasc. Electrophysiol. 17, 480–485. doi: 10.1111/j.1540-8167.2006.00411.x
Lampe, P. D., TenBroek, E. M., Burt, J. M., Kurata, W. E., Johnson, R. G., and Lau, A. F. (2000). Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 149, 1503–1512. doi: 10.1083/jcb.149.7.1503
Laurita, K. R., Chuck, E. T., Yang, T., Dong, W.-Q., Kuryshev, Y. A., Brittenham, G. M., et al. (2003). Optical mapping reveals conduction slowing and impulse block in iron-overload cardiomyopathy. J. Lab. Clin. Med. 142, 83–89. doi: 10.1016/S0022-2143(03)00060-X
Leaf, D. E., Feig, J. E., Vasquez, C., Riva, P. L., Yu, C., Lader, J. M., et al. (2008). Connexin40 imparts conduction heterogeneity to atrial tissue. Circ. Res. 103, 1001–1008. doi: 10.1161/CIRCRESAHA.107.168997
Lenegre, J., and Moreau, P. (1963). [Chronic auriculo-ventricular block. Anatomical, clinical and histological study]. Arch. Mal. Coeur Vaiss. 56, 867–888.
Lerner, D. L., Yamada, K. A., Schuessler, R. B., and Saffitz, J. E. (2000). Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation 101, 547–552. doi: 10.1161/01.CIR.101.5.547
Lin, X., Liu, N., Lu, J., Zhang, J., Anumonwo, J. M. B., Isom, L. L., et al. (2011). Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm 8, 1923–1930. doi: 10.1016/j.hrthm.2011.07.016
Lu, Q.-W., Wu, X.-Y., and Morimoto, S. (2013). Inherited cardiomyopathies caused by troponin mutations. J. Geriatr. Cardiol. 10, 91–101.
Maleckar, M. M., Greenstein, J. L., Giles, W. R., and Trayanova, N. A. (2009). Electrotonic coupling between human atrial myocytes and fibroblasts alters myocyte excitability and repolarization. Biophys. J. 97, 2179–2190. doi: 10.1016/j.bpj.2009.07.054
Manabe, I., Shindo, T., and Nagai, R. (2002). Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ. Res. 91, 1103–1113. doi: 10.1161/01.RES.0000046452.67724.B8
Mandapati, R., Skanes, A., Chen, J., Berenfeld, O., and Jalife, J. (2000). Stable microreentrant sources as a mechanism of atrial fibrillation in the isolated sheep heart. Circulation 101, 194–199. doi: 10.1161/01.CIR.101.2.194
Matsuda, J. J., Lee, H., and Shibata, E. F. (1992). Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circ. Res. 70, 199–207.
Matsushita, T., Oyamada, M., Fujimoto, K., Yasuda, Y., Masuda, S., Wada, Y., et al. (1999). Remodeling of cell-cell and cell-extracellular matrix interactions at the border zone of rat myocardial infarcts. Circ. Res. 85, 1046–1055. doi: 10.1161/01.RES.85.11.1046
Mendez, C., Mueller, W. J., and Urguiaga, X. (1970). Propagation of impulses across the Prukinje fiber-muscle junctions in the dog heart. Circ. Res. 26, 135–150. doi: 10.1161/01.RES.26.2.135
Mines, G. R. (1914). On circulating excitations in heart muscles and their possible relation to tachycardia and fibrillation. Trans. R. Soc. Can. 8, 43–52.
Miragoli, M., Gaudesius, G., and Rohr, S. (2006). Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ. Res. 98, 801–810. doi: 10.1161/01.RES.0000214537.44195.a3
Moreno, A. P., Saez, J. C., Fishman, G. I., and Spray, D. C. (1994). Human connexin43 gap junction channels. Regulation of unitary conductances by phosphorylation. Circ. Res. 74, 1050–1057.
Murphy, B. J., Rogers, J., Perdichizzi, A. P., Colvin, A. A., and Catterall, W. A. (1996). cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J. Biol. Chem. 271, 28837–28843.
Nakajima, H., Nakajima, H. O., Salcher, O., Dittie, A. S., Dembowsky, K., Jing, S., et al. (2000). Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor- 1 transgene in the heart. Circ. Res. 86, 571–579.
Nao, T., Ohkusa, T., Hisamatsu, Y., Inoue, N., Matsumoto, T., Yamada, J., et al. (2003). Comparison of expression of connexin in right atrial myocardium in patients with chronic atrial fibrillation versus those in sinus rhythm. Am. J. Cardiol. 91, 678–683. doi: 10.1016/S0002-9149(02)03403-3
Nattel, S., Shiroshita-Takeshita, A., Brundel, B. J., and Rivard, L. (2005). Mechanisms of atrial fibrillation: lessons from animal models. Prog. Cardiovasc. Dis. 48, 9–28. doi: 10.1016/j.pcad.2005.06.002
Offord, J., and Catterall, W. A. (1989). Electrical activity, cAMP, and cytosolic calcium regulate mRNA encoding sodium channel alpha subunits in rat muscle cells. Neuron 2, 1447–1452. doi: 10.1016/0896-6273(89)90190-6
Ono, K., Kiyosue, T., and Arita, M. (1989). Isoproterenol, DBcAMP, and forskolin inhibit cardiac sodium current. Am. J. Physiol. 256, C1131–C1137.
Papadatos, G. A., Wallerstein, P. M. R., Head, C. E. G., Ratcliff, R., Brady, P. A, Benndorf, K., et al. (2002). Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. U.S.A. 99, 6210–6215.
Petrich, B. G., Gong, X., Lerner, D. L., Wang, X., Brown, J. H., Saffitz, J. E., et al. (2002). c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circ. Res. 91, 640–647. doi: 10.1161/01.RES.0000035854.11082.01
Plonsey, R., and Barr, R. (2007). Bioelectricity: A Quantitative Approach, 3rd Edn. New York, NY: Springer.
Polontchouk, L., Haefliger, J. A., Ebelt, B., Schaefer, T., Stuhlmann, D., Mehlhorn, U., et al. (2001). Effects of chronic atrial fibrillation on gap junction distribution in human and rat atria. J. Am. Coll. Cardiol. 38, 883–891. doi: 10.1016/S0735-1097(01)01443-7
Qu, Y., Rogers, J. C., Tanada, T. N., Catterall, W. A, and Scheuer, T. (1996). Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. J. Gen. Physiol. 108, 375–379. doi: 10.1085/jgp.108.5.375
Reaume, A. G., De Sousa, P. A., Kulkarni, S., Langille, B. L., Zhu, D., Davies, T. C., et al. (1995). Cardiac malformation in neonatal mice lacking connexin43. Science 267, 1831–1834.
Remme, C. A., Verkerk, A. O., Nuyens, D., Van Ginneken, A. C. G., Van Brunschot, S., Belterman, C. N. W., et al. (2006). Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A-1795insD. Circulation 114, 2584–2594. doi: 10.1161/CIRCULATIONAHA.106.653949
Rhett, J. M., Ongstad, E. L., Jourdan, J., and Gourdie, R. G. (2012). Cx43 associates with Na(v)1.5 in the cardiomyocyte perinexus. J. Membr. Biol. 245, 411–422. doi: 10.1007/s00232-012-9465-z
Rhett, J. M., Veeraraghavan, R., Poelzing, S., and Gourdie, R. G. (2013). The perinexus: sign-post on the path to a new model of cardiac conduction. Trends Cardiovasc. Med. doi: 10.1016/j.tcm.2012.12.005. [Epub ahead of print].
Rohr, S. (2004). Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc. Res. 62, 309–322. doi: 10.1016/j.cardiores.2003.11.035
Sachse, F. B., Moreno, A. P., Seemann, G., and Abildskov, J. A. (2009). A model of electrical conduction in cardiac tissue including fibroblasts. Ann. Biomed. Eng. 37, 874–389. doi: 10.1007/s10439-009-9667-4
Sakabe, M., Fujiki, A., Nishida, K., Sugao, M., Nagasawa, H., Tsuneda, T., et al. (2004). Enalapril prevents perpetuation of atrial fibrillation by suppressing atrial fibrosis and over-expression of connexin43 in a canine model of atrial pacing-induced left ventricular dysfunction. J. Cardiovasc. Pharmacol. 43, 851–859. doi: 10.1097/00005344-200406000-00015
Schott, J. J., Alshinawi, C., Kyndt, F., Probst, V., Hoorntje, T. M., Hulsbeek, M., et al. (1999). Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 23, 20–21. doi: 10.1038/12618
Schubert, B., Vandongen, A. M., Kirsch, G. E., and Brown, A. M. (1990). Inhibition of cardiac Na+ currents by isoproterenol. Am. J. Physiol. 258, H977–H982.
Sheldon, R. S., Hill, R. J., and Duff, H. J. (1989). Antiarrhythmic drugs and the cardiac sodium channel: current models. Clin. Chem. 35, 748–754.
Smith, J. H., Green, C. R., Peters, N. S., Rothery, S., and Severs, N. J. (1991). Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am. J. Pathol. 139, 801–821.
Smith, W. T., Fleet, W. F., Johnson, T. A., Engle, C. L., and Cascio, W. E. (1995). The Ib phase of ventricular arrhythmias in ischemic in situ porcine heart is related to changes in cell-to-cell electrical coupling. Experimental Cardiology Group, University of North Carolina. Circulation 92, 3051–3060. doi: 10.1161/01.CIR.92.10.3051
Smits, J. P. P., Eckardt, L., Probst, V., Bezzina, C. R., Schott, J. J., Remme, C. A., et al. (2002). Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non-SCN5A-related patients. J. Am. Coll. Cardiol. 40, 350–356. doi: 10.1016/S0735-1097(02)01962-9
Spach, M. S. (2007). Mounting evidence that fibrosis generates a major mechanism for atrial fibrillation. Circ. Res. 101, 743–745. doi: 10.1161/CIRCRESAHA.107.163956
Spach, M. S., and Boineau, J. P. (1997). Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: a major mechanism of structural heart disease arrhythmias. Pacing Clin. Electrophysiol. 20, 397–413. doi: 10.1111/j.1540-8159.1997.tb06199.x
Spach, M. S., Dolber, P. C., and Heidlage, J. F. (1988). Influence of the passive anisotropic properties on directional differences in propagation following modification of the sodium conductance in human atrial muscle. A model of reentry based on anisotropic discontinuous propagation. Circ. Res. 62, 811–832. doi: 10.1161/01.RES.62.4.811
Spach, M. S., Heidlage, J. F., Dolber, P. C., and Barr, R. C. (2000). Electrophysiological effects of remodeling cardiac gap junctions and cell size: experimental and model studies of normal cardiac growth. Circ. Res. 86, 302–311. doi: 10.1161/01.RES.86.3.302
Spach, M. S., Miller, W. T., Dolber, P. C., Kootsey, J. M., Sommer, J. R., and Mosher, C. E. (1982). The functional role of structural complexities in the propagation of depolarization in the atrium of the dog. Cardiac conduction disturbances due to discontinuities of effective axial resistivity. Circ. Res. 50, 175–191. doi: 10.1161/01.RES.50.2.175
Sun, H., Chartier, D., Leblanc, N., and Nattel, S. (2001). Intracellular calcium changes and tachycardia-induced contractile dysfunction in canine atrial myocytes. Cardiovasc. Res. 49, 751–761. doi: 10.1016/S0008-6363(00)00294-7
Tan, H. L., Bink-Boelkens, M. T., Bezzina, C. R., Viswanathan, P. C., Beaufort-Krol, G. C., Van Tintelen, P. J., et al. (2001). A sodium-channel mutation causes isolated cardiac conduction disease. Nature 409, 1043–1047. doi: 10.1038/35059090
Taouis, M., Sheldon, R. S., and Duff, H. J. (1991). Upregulation of the rat cardiac sodium channel by in vivo treatment with a class I antiarrhythmic drug. J. Clin. Invest. 88, 375–378. doi: 10.1172/JCI115313
Thomas, S. P., Kucera, J. P., Bircher-Lehmann, L., Rudy, Y., Saffitz, J. E., and Kléber, A. G. (2003). Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ. Res. 92, 1209–1216. doi: 10.1161/01.RES.0000074916.41221.EA
Toyofuku, T., Yabuki, M., Otsu, K., Kuzuya, T., Tada, M., and Hori, M. (1999). Functional role of c-Src in gap junctions of the cardiomyopathic heart. Circ. Res. 85, 672–681. doi: 10.1161/01.RES.85.8.672
Ufret-Vincenty, C. A., Baro, D. J., Lederer, W. J., Rockman, H. A, Quinones, L. E., and Santana, L. F. (2001). Role of sodium channel deglycosylation in the genesis of cardiac arrhythmias in heart failure. J. Biol. Chem. 276, 28197–28203. doi: 10.1074/jbc.M102548200
van der Velden, H., van der Zee, L., Wijffels, M. C., van Leuven, C., Dorland, R., Vos, M. A., et al. (2000). Atrial fibrillation in the goat induces changes in monophasic action potential and mRNA expression of ion channels involved in repolarization. J. Cardiovasc. Electrophysiol. 11, 1262–1269. doi: 10.1046/j.1540-8167.2000.01262.x
van der Velden, H. M., Van Kempen, M. J., Wijffels, M. C., Van Zijverden, M., Groenewegen, W. A., Allessie, M. A., et al. (1998). Altered pattern of connexin40 distribution in persistent atrial fibrillation in the goat. J. Cardiovasc. Electrophysiol. 9, 596–607. doi: 10.1111/j.1540-8167.1998.tb00940.x
Veenstra, R. D. (1996). Size and selectivity of gap junction channels formed from different connexins. J. Bioenerg. Biomembr. 28, 327–337. doi: 10.1007/BF02110109
Veenstra, R. D., Joyner, R. W., Wiedmann, R. T., Young, M. L., and Tan, R. C. (1987). Effects of hypoxia, hyperkalemia, and metabolic acidosis on canine subendocardial action potential conduction. Circ. Res. 60, 93–101. doi: 10.1161/01.RES.60.1.93
Veenstra, R. D., Wang, H. Z., Beyer, E. C., and Brink, P. R. (1994). Selective dye and ionic permeability of gap junction channels formed by connexin45. Circ. Res. 75, 483–490.
Veeraraghavan, R., Salama, M. E., and Poelzing, S. (2012). Interstitial volume modulates the conduction velocity-gap junction relationship. Am. J. Physiol. Heart Circ. Physiol. 302, H278–H286.
Verheule, S., Sato, T., Everett, T., Engle, S. K., Otten, D., Rubart-von der Lohe, M., et al. (2004). Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ. Res. 94, 1458–1465. doi: 10.1161/01.RES.0000129579.59664.9d
Wagner, S., Dybkova, N., Rasenack, E. C. L., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi: 10.1172/JCI26620
Wakili, R., Voigt, N., Kääb, S., Dobrev, D., and Nattel, S. (2011). Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 121, 2955–2968. doi: 10.1172/JCI46315
Wang, Q., Li, Z., Shen, J., and Keating, M. T. (1996). Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics 34, 9–16. doi: 10.1006/geno.1996.0236
Weber, K. T., Sun, Y., Tyagi, S. C., and Cleutjens, J. P. (1994). Collagen network of the myocardium: function, structural remodeling and regulatory mechanisms. J. Mol. Cell. Cardiol. 26, 279–292. doi: 10.1006/jmcc.1994.1036
Weidmann, S. (1970). Electrical constants of trabecular muscle from mammalian heart. J. Physiol. 210, 1041–1054.
Wiegerinck, R. F., Verkerk, A. O., Belterman, C. N., Van Veen, T. A. B., Baartscheer, A., Opthof, T., et al. (2006). Larger cell size in rabbits with heart failure increases myocardial conduction velocity and QRS duration. Circulation 113, 806–813. doi: 10.1161/CIRCULATIONAHA.105.565804
Wiener, N., and Rosenblueth, A. (1946). The mathematical formulation of the problem of conduction of impulses in a network of connected excitable elements, specifically in cardiac muscle. Arch. Instit. Cardiol. Méx. 16, 205–265.
Wingo, T. L., Shah, V. N., Anderson, M. E., Lybrand, T. P., Chazin, W. J., and Balser, J. R. (2004). An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat. Struct. Mol. Biol. 11, 219–225. doi: 10.1038/nsmb737
Xie, Y., Garfinkel, A., Camelliti, P., Kohl, P., Weiss, J. N., and Qu, Z. (2009). Effects of fibroblast-myocyte coupling on cardiac conduction and vulnerability to reentry: a computational study. Heart Rhythm 6, 1641–1649.
Yagi, T., Pu, J., Chandra, P., Hara, M., Danilo, P., Rosen, M. R., et al. (2002). Density and function of inward currents in right atrial cells from chronically fibrillating canine atria. Cardiovasc. Res. 54, 405–415. doi: 10.1016/S0008-6363(02)00279-1
Yamada, K. A., Rogers, J. G., Sundset, R., Steinberg, T. H., and Saffitz, J. E. (2003). Up-regulation of connexin45 in heart failure. J. Cardiovasc. Electrophysiol. 14, 1205–1212. doi: 10.1046/j.1540-8167.2003.03276.x
Yan, G. X., and Kléber, A. G. (1992). Changes in extracellular and intracellular pH in ischemic rabbit papillary muscle. Circ. Res. 71, 460–470. doi: 10.1161/01.RES.71.2.460
Yue, L., Melnyk, P., Gaspo, R., Wang, Z., and Nattel, S. (1999). Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ. Res. 84, 776–784. doi: 10.1161/01.RES.84.7.776
Zhuang, J., Yamada, K. A., Saffitz, J. E., and Kléber, A. G. (2000). Pulsatile stretch remodels cell-to-cell communication in cultured myocytes. Circ. Res. 87, 316–322. doi: 10.1161/01.RES.87.4.316
Keywords: conduction velocity, arrhythmia, sodium channel, gap junction, fibrosis
Citation: King JH, Huang CL-H and Fraser JA (2013) Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front. Physiol. 4:154. doi: 10.3389/fphys.2013.00154
Received: 30 April 2013; Accepted: 10 June 2013;
Published online: 28 June 2013.
Edited by:
Ian N. Sabir, King's College, London, UKReviewed by:
Ruben Coronel, Academic Medical Center, NetherlandsRob Gourdie, Medical University of South Carolina, USA
Copyright © 2013 King, Huang and Fraser. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: James A. Fraser, Physiological Laboratory, University of Cambridge, Downing Street, CB2 3EG, Cambridge, UK e-mail:amFmMjFAY2FtLmFjLnVr