
 Yanping Zhang
Yanping Zhang

- Department of Biomedical Sciences, Center for Craniofacial Research and Diagnosis, Texas A&M University, Baylor College of Dentistry, Dallas, TX, USA
Palatal fusion is a tightly controlled process which comprises multiple cellular events, including cell movement and differentiation. Midline epithelial seam (MES) degradation is essential to palatal fusion. In this study, we analyzed the function of Snail1 during the degradation of the MES. We also analyzed the mechanism regulating the expression of the Snail1 gene in palatal shelves. Palatal explants treated with Snail1 siRNA did not degrade the MES and E-cadherin was not repressed leading to failure of palatal fusion. Transforming growth factor beta 3 (Tgfβ3) regulated Snail1 mRNA, as Snail1 expression decreased in response to Tgfβ3 neutralizing antibody and a PI-3 kinase (PI3K) inhibitor. Twist1, in collaboration with E2A factors, regulated the expression of Snail1. Twist1/E47 dimers bond to the Snail1 promoter to activate expression. Without E47, Twist1 repressed Snail1 expression. These results support the hypothesis that Tgfβ3 may signal through Twist1 and then Snail1 to downregulate E-cadherin expression during palatal fusion.
Introduction
Secondary palatal fusion is a tightly controlled process that has been described in many reviews and research papers (Ferguson, 1988; Jugessur and Murray, 2005; Gritli-Linde, 2007; Nawshad, 2008; Yu et al., 2009). Briefly, the two palatal shelves initiate as outgrowth from the inner wall of the maxillary prominences as early as embryonic day (E) 12 in mice. They first grow lateral to the tongue and later become reoriented rostral to the tongue. At E14, the shelves contact and adhere at the midline, where the epithelium covering the tip of the palatal shelves forms a seam termed the midline epithelial seam (MES). Later, the seam breaks down to achieve mesenchymal confluence. The mechanisms for this midline MES degradation are not clear and great interest has been raised on this developmental event since failure of this process results in cleft palate.
Morphological analyses have demonstrated that, before fusion, medial edge epithelial (MEE) cells within the epithelial seam bulge and form filopodia-like structures (Taya et al., 1999; Ding et al., 2004; Fujiwara et al., 2008). Filopodia are actin-based structures associated with mesenchymal cell migration or interaction with the extracellular matrix (ECM) or other cells (Gupton and Gertler, 2007; Mattila and Lappalainen, 2008). Filopodia in MEE cells help them to correctly align and adhere to their target partner and close the gap between them, a process known as “adhesion zippering” and are used in cell guidance and migration (Taya et al., 1999; Bridgman et al., 2001; Millard and Martin, 2008). In knockout mice with disrupted Transforming growth factor beta 3 (Tgfβ3) or Platelet-derived growth factor C (Pdgfc) signaling, the filopodia on the apical surface of MEE cells are either reduced or completely lost (Taya et al., 1999; Ding et al., 2004). The extension of the filopodia-like structures on the MEE cell's surface before or during fusion indicates that cell migration contributes to the palatal fusion (Martin-Blanco and Knust, 2001). Thus, it has been proposed that the epithelial cells migrate to the oral and nasal surface (Cuervo and Covarrubias, 2004). In addition, anterior-posterior migration of the cells has also been proposed (Jin and Ding, 2006). Epithelial cell migration often involve their transformation into mesenchymal cells (Yang et al., 2006).
Studies with cell tracking dyes demonstrated that during fusion, the MEE are viable and are separating from the seam as mesenchyme cells (Fitchett and Hay, 1989). Some of the labeled epithelial cells were found in the mesenchyme after fusion (Griffith and Hay, 1992), indicating that epithelial-mesenchymal transition (EMT) occurred. Tgfβ3, by activating PI-3 kinase (PI3K) signaling, has been considered a “master gene” in initiating EMT and regulating MEE cell fate (Hay, 1989; Kang and Svoboda, 2002; Nawshad et al., 2004, 2005). Cultured MEE cells treated with Tgfβ3 undergo EMT, migration and apoptosis in that chronological sequence (Ahmed et al., 2007). However, EMT during palate fusion remains controversial (Vaziri Sani et al., 2005; Dudas et al., 2006; Jin and Ding, 2006; Xu et al., 2006). While some did not find evidence of EMT during secondary palate fusion (Dudas et al., 2006), a MEE cell fate mapping study by Jin and Ding (2006) revealed its presence. However, most of the cells that underwent the transformation eventually died after migrating away from the seam and only a few persisted, probably explaining the contradictory results. The sequential events observed in Jin and Ding study are in agreement with the in vitro study by Ahmed and colleagues (2007). The transformation may be necessary to maintain the fusion suture patency (Jin and Ding, 2006).
Previously, we demonstrated that the basic-helix-loop-helix (bHLH) transcription factor, Twist1 protein is expressed intensively in the MEE cells right before fusion while also expressed in the mesenchyme (Yu et al., 2008), which was confirmed by another group (Kitase et al., 2011). Down regulation of Twist1 using siRNA in palatal organ culture resulted in blocked fusion (Yu et al., 2008). In addition, Twist1 was increased in Tgfβ3 treated chicken palatal shelves and downregulated when mouse palates were treated with neutralizing antibodies against Tgfβ3 (Yu et al., 2008).
Twist1 has been implicated as an EMT regulator. The Twist1 role in tumor progression notably sustains and enhances this theory (Yang et al., 2004). However, Twist1-null heterozygous mice (Twist1+/−) exhibited phenotypes similar to the dominantly inherited Saethre–Chotzen syndrome in the human population (Bourgeois et al., 1998) with a low penetrance of cleft palate (Stoler et al., 2009), indicating that there are other factors compensating for its function in vivo.
Like Twist1, the Snail1 gene is well-documented for its evolutionarily conserved roles in mesoderm development and has been implicated in several cellular events such as EMT, cell migration, and survival (Cano et al., 2000; Barrallo-Gimeno and Nieto, 2005). Snail genes encode DNA binding zinc-finger proteins that act as transcriptional repressors (Carver et al., 2001). Snail1 is expressed in the palatal and dental mesenchyme adjacent to the epithelium (Rice et al., 2005). In addition, Snail1 mRNA was also found in a small subpopulation of the MEE cells after the seam had formed (Martinez-Alvarez et al., 2004). Transgenic mice have provided insights into function of this gene family in palatogenesis. Conditional deletion of the Snail1 gene in neural crest cells did not cause obvious deformities in the craniofacial region unless the mouse was bred with a Snail2−/− mouse (Murray et al., 2007), suggesting that Snail2 may compensate for the loss of Snail1 function. However, the role of Snail1 in epithelial cells has not been fully investigated.
A hierarchical relation between them was proposed based on the evidence that Twist was required for Snail mRNA expression and Snail was required for the maintenance of Twist expression during Drosophila mesoderm formation (Brouzes et al., 2004). Twist1 dimerizes with E2A-encoded proteins E12 and E47, for successful EMT (Perez-Moreno et al., 2001). The target sequence of these bHLH proteins is the E-box. Interestingly, Snail1 binds to the same consensus sequence on the E-cadherin promoter and acts as a repressor in EMT (Batlle et al., 2000; Cano et al., 2000; Oram and Gridley, 2005). Snail1 may compete directly with bHLH proteins for the same binding sequences (Oram and Gridley, 2005). However, Snail1 also cooperates with Twist1 to inhibit the expression of p21-cip1 induced by E2A-gene products in osteoblast-like cell differentiation (Takahashi et al., 2004). Collectively, the functional networks between Snail1, Twist1, and E2A proteins in cell differentiation and movement remain to be elucidated.
In this study we used a variety of approaches to determine if Snail1 has a function in EMT and palatal fusion. In the presence of Snail1 siRNA, E-cadherin expressing MEE remained at the palatal fusion site, suggesting Snail1 was responsible for E-cadherin down regulation during MES degradation. Snail1 expression was decreased in response to the Tgfβ3 neutralizing antibody and PI3K inhibitor during palatal fusion. In addition, we used transfected cell cultures with luciferase detection to test if Twist1 cooperates with E proteins to regulate the Snail1 promoter activity. Our results support the hypothesis that Twist1 may regulate MES degradation during palatal fusion partially through Snail1 regulation.
Materials and Methods
Animal Manipulation, Palatal Organ Culture, and Cell Culture
The protocol for the use of animals was approved by the Institutional Animal Care and Use Committee at Baylor College of Dentistry, and the animals were euthanized following NIH guidelines. Timed-pregnant CD1 mice (Harlan Sprague-Dawley, Inc.) and fertile chicken eggs (Texas A&M Poultry Science Department) were used in these studies. Mouse embryos were harvested at day E13.5, in Hanks' balanced saline solution (HBSS; GIBCO). The chicken eggs were incubated for 8 days at 37°C before the embryos (Hamburger-Hamilton stages 27–34) were removed from the eggs and rinsed in HBSS; GIBCO. Palatal shelves were dissected and cultured as previously described (Yu et al., 2008). Tgfβ3 neutralizing antibody (R&D Systems) at 10 μg/ml and PI3K inhibitor LY294002 (Calbiochem) at 1 and 10 μM final concentrations were added to the medium of cultured mouse palates, as previously described (Yu et al., 2008). Tissues were cultured for 24 h and three pairs of whole palatal shelves were processed for RNA extraction or protein analysis by western blotting. Tgfβ3 (50 ng/ml, R&D Systems) was added to the chicken palatal organ culture for 15 min to 48 h.
Madin-Darby Canine Kidney Epithelial (MDCK) cells were grown in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin antibiotics. The YFP-MDCK (control) and E2A-MDCK cells were generated by transfection of the pEYFP (control) and E2A-YFP plasmids. The stable cell lines were selected by addition of 500 ug/ml gentamicin (Sigma) for 4 weeks as described before (Perez-Moreno et al., 2001).
Snail1 siRNA Transfection and Treatments
The siRNA oligonucleotides specific for Snail1 mRNA (NM_011427) were purchased from Ambion. 100 and 200 nM of siRNA in 0.1% Lipofectamine were used to transfect cells, following the manufacturer's instructions (Invitrogen). A 21-nucleotides scrambled sequence siRNA was used as a negative control. Tissues were exposed to siRNA treatment for up to 72 h and then processed for analysis. Culture medium was changed every 24 h.
Histology and Immunohistochemistry Staining
Cultured palatal shelves were collected at 72 h and processed for histological analysis as previously described (Kang and Svoboda, 2002; Yu et al., 2008). The average of 20 sections' scores was calculated as the fusion score of one sample. The mean fusion score (MFS) for each treatment group was calculated. Light microscope images were captured using a Zeiss Axioplan microscope with a color RT-Spot camera.
Deparaffinized and rehydrated sections were used for immunohistochemical analysis of E-cadherin expression following standard methods. After blocking with 10% normal donkey serum/PBS, the tissues were incubated with the polyclonal antibody for E-cadherin (Cell Signaling 3195; 1:100 dilution) overnight at 4°C or 1 h at room temperature. After rinsing, the primary antibody was detected with a secondary antibody conjugated with HRP (Molecular Probes). Signal was developed with the ImmPACT DAB kit (Vector Laboratories). Nuclei were counterstained with Hematoxylin. After mounting, the images were photographed as described above.
RNA Extraction and Real-Time PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen). Obtained RNA was reverse transcribed with SuperScriptII reverse-transcriptase (Invitrogen) and the resulting cDNA used for quantitative real-time PCR. The relative quantification value was calculated by the 2−δCt method. All quantifications were normalized to 18s rRNA (SuperArray) and then standardized with the negative control. Experiments were repeated at least three times. Primers user were for mouse Snail1: 5′AAACCCACTCGGATGTGAAG and 5′GAAGGAGTCCTGGCAGTGAG; for chicken: 5′CCTTTCCCGTGCAGATACAT and 3′TGCACAGGAGCACAGGATAG.
Whole Mount in situ Hybridization
Whole-mount in situ hybridization analysis was performed as previously described (Ruest et al., 2004; Ruest and Clouthier, 2009). Embryos were hybridized with digoxigenin (DIG)-labeled cRNA riboprobes against Snail1, Twist1, and E2A. Stained embryos were photographed in whole-mount on an Olympus SZX16 stereoscope fitted with a digital camera.
Immunoprecipitation
The tips of six palatal shelves pairs were dissected and lysed in buffer (50 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1%Nonidet P-40) containing protease inhibitor cocktails 1 and 2 (Sigma). Total protein extracts were used. The co-immunoprecipitation was carried out using the Catch and Release reversible immunoprecipitation system following manufacturer's instructions (Millipore) with 1 μg of antibody specific for E12/47 proteins. Twist1 (sc-6269) and E12/47 (sc-763) antibodies were purchased from SantaCruz.
Protein Extraction and Western Blotting
Tissues or cells were lysed in the RIPA buffer (Sigma) supplemented with protease inhibitors. Protein quantification was performed using the BCA assay (Pierce). Ten microgram total protein was loaded in each well on a 4–12% NuPage Bis-Tris gel (Invitrogen). Protein was transferred onto PVDF membrane (Millipore). The membrane was incubated with polyclonal primary antibody against E-cadherin (1:1000, Cell Signaling), Snail1 (1:1000, Abcam), Twist1 (1:1000), E12/47 (1:1000) overnight at 4°C. IRDye 680 (1:5000 donkey-anti-rabbit, Licor) or IRDye 800 (1:5000, donkey-anti-goat, Licor) secondary antibodies were used to visualize the protein signals with the Odyssey infrared imaging system (Licor).
Chromatin Immunoprecipitation (ChIP) Assay
The Chromatin immunoprecipitation (ChIP) assays were carried out using the EZ ChIP Chromatin Immunoprecipitation Kit following manufacturer's instructions (Upstate Biotechnology). Briefly, tips of E14.5 palatal shelves (six pairs) were fixed with formaldehyde and resuspended in lysis buffer supplemented with protease inhibitors and then processed as indicated with the E12/47 or Twist1 antibody overnight at 4°C with constant agitation. Immunoprecipitated complexes were collected and DNA released using proteinase K. Recovered DNA was used for PCR amplification with primers that were designed to cover the E-boxes present in the Snail1 promoter region. These sites were identified using the MatInspector program from Genomatix. Primers were: E1 5′CCGTTAGGGGCTAAGTCACA and 5′AGGCCTGTTCACAACCTCAC; E2 5′GGGATGAAAGGAAGCCTAGC and 5′TCGTCCCAACGGACAAGT; E3 5′CTGGTCCTTGCTACCTCTGC and 5′TTCCAGGATGAGGTTGGTGT; E4 5′CGGTGCTTCTTCACTTCCTC and 5′ACTACCCAGGGATGCCCTAC; E5 5′TGACCGTACTGTTGGTCACG and 5′ATCATCGCACTTTCTGGCTC. Total DNA extracts were used as input controls for the PCR reactions.
Plasmid Construction
The Snail1 expression plasmid was constructed by inserting the HindIII-BamHI fragment of the mouse Snail1 cDNA into the pEYFP-C1 vector. The mouse Snail1 1.7 kb promoter flanked by the XhoI and HindIII restriction sites was cloned by PCR from genomic DNA (genome sequence NT_039201) of a CD1 mouse using high fidelity DNA polymerase (Pfu turbo, Stratagene) and confirmed by sequencing. The following primers were used:
5′ccgctcgagTGAAAAACCCTAGGTGGCAG (−1683 bp)
5′cccaagcttGCTCGCTATAGTTGGGCTTC (+64 bp).
The fragment was subcloned using the same restriction enzymes into the pGL3 luciferase vector, yielding the 1.7KSnLuc construct. pGL3 basic vector was used as a negative control. The Twist1 plasmid was described previously (Zhang et al., 2012).
Dual Luciferase Assays
Subconfluent cultures of MDCK cells (2 × 104 cells/well in 24-well-plate) were serum starved overnight and transfected with up to 1.2 μg of Twist1 or Snail1 expression plasmids along with 100 ng Snail-promoter luciferase constructs and 10 ng pRL-TK vector (Promega) as internal control. After 24-h incubation, cells were harvested and dual luciferase assay were carried out using the Dual-Glo luciferase assay system according to the manufacturer's recommendations (Promega). All the results were normalized to Renilla luciferase activities (pRL-TK). All assays were performed at least three times in triplicate. The results are mean of different experiments ± standard errors.
Statistical Analysis
Two-tailed Student's t-test analysis or Two-Way ANOVA were used to evaluate the statistical significance of the results. A p < 0.05 was considered significant. The non-parametric Kruskal-Wallis analysis of variance was used to compare MFS between groups. P < 0.05 was also considered statistically significant.
Results
Snail1 Plays a Role in MES Degradation
In order to examine the function of Snail1 during MES degradation, we specifically down regulated Snail1 expression by using siRNA in palatal organ culture. Downregulation efficiency of several Snail1 siRNAs were tested by Western blot (Figure 1A). siRNA1 (si1) suppressed Snail1 protein expression efficiently and was used in all of the following palatal organ culture experiments. A scrambled sequence siRNA (scr) was used as a negative control. The palatal shelves were maintained with or without Snail1 siRNA for 72 h before processing for histological evaluation of palatal fusion (Figure 1B). The immunohistochemical staining of E-cadherin was used to detect epithelial cells in the midline. In control palates and scrambled siRNA groups the palate completely fused without evidence of MES E-cadherin expressing epithelial cells in the mesenchyme (Figure 1B). In palatal shelves treated with 100 nM Snail1 siRNA, the epithelial seam broke down and degraded but triangular clusters of E-cadherin-positive epithelial cells were found primarily on the nasal side (Figure 1B, arrow). In palatal shelves treated with 200 nM Snail1 siRNA, the E-cadherin positive epithelial seam remained mostly intact (Figure 1B, inset 200 nM siRNA). The palatal shelf size between the four treatment groups was similar (Figure 1B, scale bars).
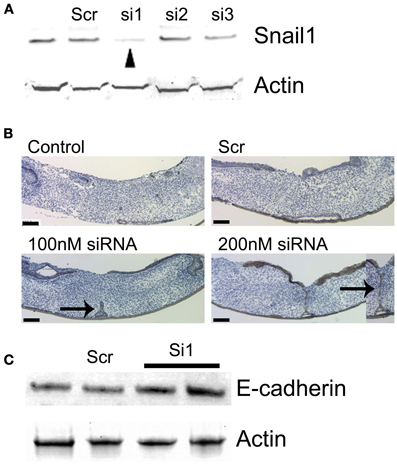
Figure 1. MES degradation was blocked in presence of Snail1 siRNA in mouse palatal organ culture. (A) Snail1 siRNA 1 (si1) decreased Snail1 protein levels in mouse palate as revealed by western blotting (arrowhead). Actin was used a loading control. The other screened siRNAs (si2 and si3) were not efficient. Scrambled sequence siRNA (scr) was used as control. (B) Snail1 siRNA decreased mouse palatal fusion in a dose-dependent manner. Palatal shelves were cultured for 72 h with or without Snail1 siRNA. Sections were stained with E-cadherin antibody (brown) to detect epithelial cells. The tissues were counterstained with hematoxylin (blue). In control and scr control groups, the palates completely fused and no E-cadherin stained cells were found in the midline. In presence of 100 and 200 nM Snail1 siRNA, E-cadherin stained cells were in the epithelial triangle (100 nM siRNA, arrow) and midline seam (arrow in 200 nM siRNA inset). Inset: higher magnification of the midline seam (200 nM siRNA). Scale bars = 100 μm. (C) Western blotting analysis showing that E-cadherin protein levels are elevated in MDCK cells transfected with the Snail1 siRNA.
The degree of palatal fusion was quantified with a scaling system, termed the MFS, as previously described (Yu et al., 2008). Briefly, a score of 5 equals complete palatal fusion. Lower MFS indicate more epithelial cells in the midline seam, indicating that fusion was blocked or decreased (Table 1). In control and scrambled siRNA groups, most sections scored 4–5, indicating complete fusion of the palatal shelves. In presence of Snail1 siRNA at both 100 and 200 nM, the MFS decreased to 3.4, suggesting epithelial islands or seam remnants were found. The non-paprametric Kruskal-Wallis analysis of variance was used to compare MFS between groups. The MFS in 100 and 200 nM Snail1 siRNA group were significantly different from the control and scrambled siRNA control groups, indicating that the degradation of MES was reduced or delayed in presence of Snail1 siRNA in vitro.
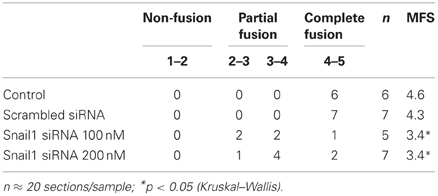
Table 1. Mean fusion score of Snail1 siRNA-treated cultured mouse palates.
Snail1 is a known repressor of E-cadherin expression (Batlle et al., 2000; Cano et al., 2000; Oram and Gridley, 2005). We performed the cognate experiment to confirm (1) the role of Snail1 on E-cadherin abundance and (2) the effect observed in MEE occurs in other epithelial cells. We examined whether reducing Snail1 abundance in cultured MDCK epithelial cells altered E-cadherin protein levels (Figure 1C). In the cells transfected with the Snail1 siRNA, E-cadherin levels increased about two-folds, confirming that Snail1 regulates E-cadherin levels in epithelial cells (Figure 1C).
Snail1 mRNA Expression is Tgfβ3 and PI-3 Kinase (PI3K) Signaling-Dependent
It has been established that Tgfβ3 and PI3K are required for murine palatal fusion (Kaartinen et al., 1995; Proetzel et al., 1995; Kang and Svoboda, 2002). Tgfβ3 signaling is likely mediating PI3K activation in MEE cells. To explore if these connected signaling pathways regulate Snail1 expression during palatal fusion, we used a Tgfβ3 neutralizing antibody and PI3K inhibitor in the palatal organ culture system. In presence of 1 μg/ml Tgfβ3 neutralizing antibody, Snail1 expression did not change (Figure 2A) but in presence of 10 μg/ml Tgfβ3 neutralizing antibody, Snail1 expression was significantly decreased (p = 0.0175). In palates treated with the PI3K inhibitor LY294002, Snail1 expression decreased in a dose-dependent manner (Figure 2B). However, only the higher dose, 10 μM of PI3K inhibitor, produced a significant decrease in Snail1 expression (p = 0.0396). These data were indirectly suggesting that Tgfβ3 was regulating the expression of the gene. To test whether Tgfβ3 was directly regulating Snail1 expression, we used chicken palates. These palates do not normally fuse since Tgfβ3 is not expressed in the MEE, but they fuse when treated in culture with the growth factor (Sun et al., 1998). We used this model to determine if Snail1 expression changed in response to exogenous Tgfβ3. Six hours after Tgfβ3 treatment, the Snail1 expression transiently increased approximately six-folds (p = 0.0008) (Figure 2C). The response appeared temporally limited but the results were indicating that Tgfβ3 can upregulate Snail1 expression.
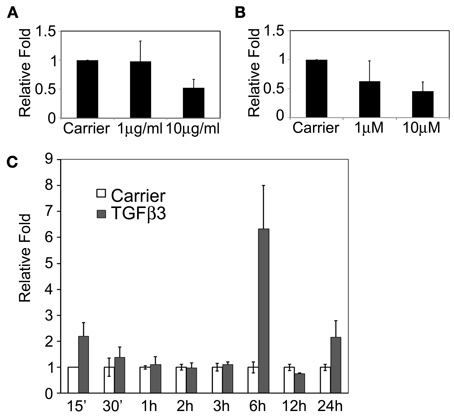
Figure 2. Snail1 expression responses to Tgfβ3 and PI3K signaling in cultured mouse and chicken palatal shelves. (A) Quantitative RT-PCR of Snail1 expression in cultured murine shelves following the blockage of Tgfβ3 signaling with neutralizing antibodies for 24 h (10 μg/ml, p = 0.0175). (B) Quantitative RT-PCR of Snail1 expression in mouse palate following treatment with the PI3K inhibitor LY294001 for 24 h (10 μM, p = 0.0396). Data represented mean ± SE; n = 3. (C) RT-PCR Analysis of Snail1 expression in cultured chicken palatal shelves following treatment with Tgfβ3. Control samples were treated with the carrier and normalized to one. Data represented mean ± SEM; n = 3 (p = 0.0008 at 6 h).
Twist1 Regulates Snail1 Promoter Activity
Although Twist1, Snail1, and E2A genes trigger EMT in different biological contexts independently, evidence supports a differential and hierarchical role for these repressors during the transformation process (Peinado et al., 2004). They may form a complex signaling network to regulate the transition process (Peinado et al., 2007). Tgfβ factors promote the expression of Snai11, Snail2, Zeb1, Zeb2, and Twist1 in cell- or tissue-dependent contexts (Zavadil and Bottinger, 2005; Thuault et al., 2006).
The transcription factor Twist1 plays both positive and negative roles in regulation of embryonic morphogenesis and cell differentiation (O'Rourke and Tam, 2002). Twist1 can form functional homodimers as well as heterodimers with ubiquitously expressed bHLH E protein, such as E2A gene products E12 and E47. In a previous study, we demonstrated that Tgfβ3 regulates the expression of Twist1. Since Snail1 activation by Tgfβ3 was delayed, we investigated whether Twist1 was needed for Snail1 expression. When exploring the Snail1 promoter region, we detected the presence of 7 CANNTG E-boxes upstream of the transcription start site (Figure 3). In a 5′ to 3′ direction, these E-boxes are named E1 (−1550/−1545), E2 (−1289/−1284), E3.1 (−893/−888), E3.2 (−843/−838), E4.1 (−617/−612), E4.2 (−593/−588) and E5 (−120/−115). These E-boxes are presumably E47 and Twist1 binding sites, suggesting that these factors may directly bind the promoter and regulate Snail1 transcription. Thus, we hypothesized that Twist1 cooperates with E47 and acts upstream of Snail1 during palatal EMT.

Figure 3. Mouse Snail1 promoter sequence. The mouse Snail1 promoter was cloned using the oligo primers underlined in the sequence. The ATG transcription start site, located toward the end of the sequence, is bolded. E-boxes are bolded and underlined and numbered in a 5′ to 3′ direction.
We first tested whether Tgfβ3 regulated the Snail1 promoter activity. We cloned the mouse Snail1 promoter into a luciferase reporter vector. The 1.7 Kb promoter-luciferase constructs named 1.7KSnLuc was transfected in the epithelial MDCK cells. These cells were selected based on their similar behavior to palate epithelial cells as described above. The 1.7 Kb promoter showed a significant activation upon Tgfβ3 stimulation (p = 0.0413) (Figure 4A). That response was blocked when the PI3K inhibitor LY294002 was added to the medium (p = 0.030). We then examined the expression of the Snail1, Twist1, and E2A genes in the E13.5 mouse palate by whole mount in situ hybridization. The expression of these three genes overlapped in the palatal shelves (Figure 4B). All three genes were expressed in the palatal shelf along its entire length. A gradient of expression was also observed for Twist1, with lower expression in anterior shelves and higher at the posterior area. E2A expression was lower in the lip/nose pad area, suggesting that functions observed in the developing palate may differ in the developing lip. In addition, we used co-immunoprecipitation to test whether Twist1 and E47 interact in the palatal shelves. We used the tip of the touching E14.0 palatal shelves for protein extractions. E47 antibody was incubated with the protein lysate and Twist1 western blotting was used to detect if the proteins co-immunoprecipitated. Twist1 protein was detected when E47 was immunoprecipitated (Figure 4C). Our results suggested that Twist1 physically interacts with E47 in the palate tissue prior to fusion.
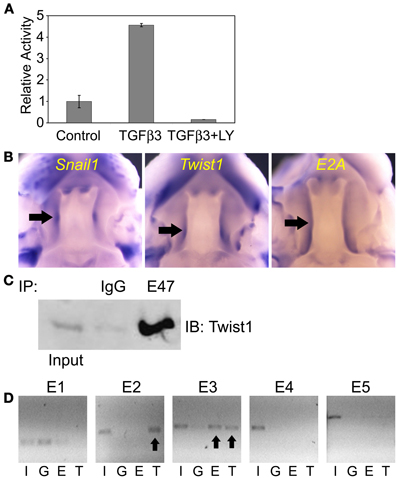
Figure 4. Twist and E2A dimerize and bind to the mouse Snail1 promoter in the developing palate. (A) Luciferase assay results in MDCK cells with the Snail1 promoter showing that after 24 h, Tgfβ3 signaling stimulated the Snail1 promoter (p = 0.0413) but the activation is inhibited by the PI3K inhibitor LY294002 (p = 0.0030). (B) Analysis of Snail1, E2A, and Twist1 expression in palatal shelves (black arrows) in E13.5 mouse embryos using whole mount in situ hybridization. (C) Co-immunoprecipitation results showing that Twist1 and E47 dimerize in pre-fused E14.0 mouse palatal shelves in vivo. Immunoprecipitation (IP) with the E47 antibody and immunoblotting (IB) with the Twist1 antibody demonstrated that the two proteins interact prior to palatal fusion. Unspecific rabbit IgG was used as a negative IP control. (D) ChIP results revealing that Twist1 and E47 bind selective E-boxes of the Snail1 promoter (black arrows) from mouse palatal shelves in vivo. I, input; G, IgG control; E, E47 antibody; T, Twist1 antibody.
In order to test whether E2A (E12/E47) and Twist1 proteins can bind to the Snai11 promoter, we used the ChIP assay on the pre-fusion palatal shelves. We dissected the tip of the mouse palatal shelves where both transcription factors are expressed prior to fusion. Five pairs of primers targeting the different E-boxes on the Snail1 promoter, with no distinction between E3.1 and E3.2 or E4.1 and E4.2 since each pair's E-boxes were close. After immunoprecipitation of the DNA-protein complexes with the Twist1 and Snail1 antibodies, PCR was used to amplify the presumptive targeted E-box regions. Our results show that E47 and Twist1 can both bind to the E3 region (Figure 4D). Only Twist1 bound the E2 region. Twist1 or E47 did not interact with the other E-boxes in the mouse palatal shelves.
However, the ChIP results could not distinguish from Twist1 or E protein response in the epithelial, mesenchymal cells or subpopulations of epithelial cells. Based on our results and those from Yu et al. (2008), the response in the MEE cells is likely triggered by Tgfβ3 inducing Twist1 expression in these cells. To identify how Twist1 regulates the Snail1 promoter activity in epithelial cells, we used the cloned mouse Snail1 promoter into the luciferase reporter vector. The promoter-luciferase constructs 1.7KSnLuc transfected in MDCK cells was significantly repressed in presence of Twist1 (p = 5.73E-05) (Figure 5A). These results are in agreement with others suggesting that often Twist1 acts as a transcriptional repressor (Spicer et al., 1996; Yin et al., 1997). When E1 was removed, the same repression was observed (p = 0.0003). When the E2 site was removed, the repression was abolished but activation was not observed. Only Twist1 bound the E2 site in the ChIP assays. When the E3 region bound by both E47 and Twist1 was removed, a significant decrease in luciferase activity was noted (p = 0.0198). These results indicated the possible inhibitory effects on Snail1 expression exerted by Twist1. Removing the E2 site released the repression, but removing the E3 region possibly blocked the activation of the luciferase expression. To test whether Twist1 and E2A proteins synergistically regulate Snail1 promoter activity, the luciferase vector was transfected along with Twist1 in MDCK cells stably expressing E47 protein. These stably transfected cells allowed testing the different conditions while maintaining a steady level of E47 protein. In these cells, a Twist1 response was observed (p = 0.0048) (Figure 5B). These data suggested that Twist1 and E47 were co-regulating Snail1 expression. The ChIP results indicated that the E3 region was bound by both Twist1 and E47. When the E3 region was removed, the luciferase response significantly decreased (p = 0.02) (Figure 5B), suggesting that the site was essential to regulate Snail1 expression by Twist1 and E proteins.
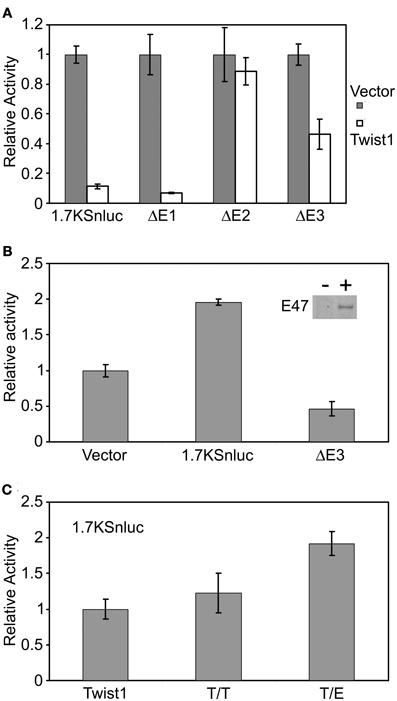
Figure 5. Twist1 regulates the mouse Snail1 promoter activity by dimerizing with E-proteins. (A) Luciferase assay using the Snail1 promoter results revealed that Twist1 alone cannot activate the Snail1 promoter but rather represses the activity of the promoter (p = 5.73E-05). This repression was released when the E2 site was removed (ΔE2). (B) Twist1 increased Snail1 promoter activity in MDCK-E2A cells, indicating that the interaction between Twist1 and E47 was needed to activate the Snail1 promoter. Removal of the E3 region (ΔE3) abolished the activation (p = 0.0048). Inset: western blot analysis of E47 expression in stably transfected cells (10 μg of protein loaded). (C) Luciferase assay results confirming that the interaction between Twist1 and E47 was necessary to activate the Snail1 promoter (p = 0.00096). Forced Twist1/Twist1 (T/T) homodimers and Twist1/E47 (T/E) heterodimers encoding vectors were transfected in the cultured MDCK cells and luciferase assay results compared to the control Twist1 expression vector. Luciferase activity was measured 24 h after transfection. Data represented mean ± SEM; n = 3. An empty expression vector was used as a control for these experiments.
Since Twist1 can form either Twist1/Twist1 (T/T) homodimer or Twist1/E-protein (T/E) heterodimer in different biological events, our results suggested that the regulation of Twist1 on Snail1 promoter activity depends on the ratio of T/T to T/E dimers. To test this hypothesis, we transfected the MDCK cells with plasmids which expressed “forced dimers” of either T/T or T/E. These constructs have been described previously (Connerney et al., 2006). The 1.7KSnLuc response increased two-fold in response to T/E dimers (p = 0.00096) whereas no obvious change was observed in response to the T/T dimer in comparison with cells transfected with Twist1 alone (Figure 5C). These results indicated that Twist1 dimerizes with E-proteins to activate the Snail1 promoter activity.
Discussion
Twist1 Signals Through Snail1 to Downregulate E-cadherin During MEE Cell Migration
The function of Snail genes is best known for their direct repression of E-cadherin expression (Cano et al., 2000; Nieto, 2002). Snail genes have additional cellular functions, such as cell survival, cell adhesion, and migration (Barrallo-Gimeno and Nieto, 2005). Ectopic expression of Snail1 in the MDCK epithelial cell line promotes resistance to apoptosis (Escriva et al., 2008). Previously, Snail1 was localized to a small subgroup of palatal MEE cells (Martinez-Alvarez et al., 2004). However, its role in palatal fusion is not clear. In this study the expression of Snail1 was down regulated with a specific siRNA in palatal organ cultures. In treated cultured shelves, the palates failed to completely fuse and epithelial remnants were present after the 72 h culture period. We used E-cadherin as a marker to establish that the cells retained an epithelial phenotype when Snail1 function was suppressed. Our results suggested that Snail1 was at least required for E-cadherin suppression during MES degradation, in line with other studies (Cano et al., 2000; Medici et al., 2008). Since conditional Snail1 mutant mouse embryos do not develop a cleft palate, our culture results with the siRNA indicate that Snail1 is needed for palatal fusion but fusion is delayed when absent due to compensatory effects by Snail2 (Murray et al., 2007).
In our study, we found increased Snail1 expression in response to Tgfβ3 stimulation and decreased expression when Tgfβ3 signaling was reduced. Our previous study indicated that Tgfβ3 through PI3K activation regulates Twist1 expression (Yu et al., 2008). In this study we show that Twist1 regulates Snail1 expression. The delay observed in the Tgfβ3 response in chicken palate may represent the time needed to activate Twist1 expression. Our results suggest that Snail1 activation by Tgfβ3 may be sequential to Twist1 activation. Reduction of Snail1 expression to blocked Tgfβ3 or PI3K signaling was not as great as expected. This may be due to the fine-tuning feedback mechanism of Snail1 regulation with Snail1 binding its own promoter region to create a negative loop controlling its own expression (Peiro et al., 2006). In addition, signaling pathways other than Tgfβ3 also contribute to Snail1 regulation (Barrallo-Gimeno and Nieto, 2005), many of which play important roles in palatal fusion as well. The activity of the Snail1 promoter during EMT is dependent on Erk2 and Gsk-3β/NFkB pathway activity (Barbera et al., 2004). PI3K activity also contributes to Snail1 transcription and promoter activity (Peinado et al., 2003), possibly acting in the same signal pathway as GSK3β/NFkb. In our study, Snail1 mRNA levels responded to both Tgfβ3 and PI3K, suggesting that Tgfβ3 may signal through the PI3K/Gsk3β route to regulate Snail1 expression levels during MEE transdifferentiation, migration or death.
Twist1 Regulates Snail1 Promoter Activity in Collaboration with E-proteins
We demonstrated that Twist1 binds the Snail1 promoter and regulates its activity by recruiting E-proteins (E12/E47), which are also expressed during palatal fusion. Without the synergy with the E-proteins, Twist1 represses Snail1 expression, probably indicating a spatial or temporal regulatory mechanism (Figure 6). Our in situ results show that Twist1 expression is higher in the posterior half of the developing palate, where fusion occurs later. The E2A gene is more uniformly expressed with a slightly higher expression at the posterior ends of the shelves. The different levels of expression may have an impact on the dimerization of Twist1 and E-proteins and cellular expression and in vivo protein-protein interaction analyses could eventually help resolve this issue. However, based on the respective gene expression patterns, we hypothesize that the higher Twist1 expression in the posterior half of the palate would favor the formation of Twist1/Twist1 dimers instead of Twist1/E-protein dimmers that would be favored more anteriorly. The skewed ratios would favor the repression of Snail1 expression in the posterior palate and promote its expression more anteriorly. Our in situ analysis of Snail1 expression supports this hypothesis since the expression of the gene is higher where Twist1 expression is intermediate. In the anterior palate where Twist1 expression is lower, Snail1 expression is also reduced but not absent, suggesting the presence of other unidentified regulatory mechanisms. The results indicate that the balance between Twist1 and E-proteins regulates the spatial, temporal and expression levels of Snail1 and can explain why both E2 and E3 sites were precipitated from whole palatal shelf protein extracts.
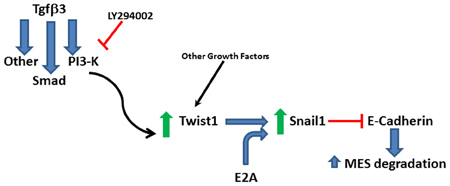
Figure 6. Proposed signaling pathway. Twist1 is upregulated by Tgfβ3 through signaling molecules including PI3-kinase and it dimerizes with E2A proteins to upregulate Snail1 to repress E-cadherin to promote the degradation of the medial edge seam (MES).
Twist1 and Snail proteins are involved in EMT. We think that the regulation of Snail1 expression by Twist1 is tightly regulated based on the short Twist1 protein presence in the MEE (Yu et al., 2008; Kitase et al., 2011). Targeting these factors is likely affecting the EMT associated with palatal fusion and the consequent apoptotic death of the transformed cells (Jin and Ding, 2006; Ahmed et al., 2007), as evidenced by the remaining MES cells in our treated samples. Apoptosis is important for palatal fusion and appears to be regulated by Tgfβ3 signaling (Martinez-Alvarez et al., 2000). In the Tgfβ3 mutant embryos, compensation by Tgfβ1 promotes the expression of Snail1 and 2 in the MEE (Martinez-Alvarez et al., 2004). Elevated Snail gene expression correlates with the resistance to apoptosis in the MEE cells and affects EMT. However, in the Snail1+/−; Snail2−/− mouse embryos, apoptosis resistance was observed (Murray et al., 2007). Our results showing that MES cells remain in the midline are in agreement with these last results indicating that Snail1 function may be needed for apoptosis. It appears that EMT precedes apoptosis (Jin and Ding, 2006; Ahmed et al., 2007), at least in some of the MES cells. This process is also regulated by Tgfβ3 (Martinez-Alvarez et al., 2004). When this growth factor is absent, MEE cells fail to generate lamellipodia and filopodia (Taya et al., 1999), characteristic structures of migratory mesenchymal cells. In palatal fusion, Twist1 may cooperate with E-proteins to activate Snail1 expression and regulate the E-cadherin expression (Figure 6), while using other factors to regulate the cell migratory behavior. Thus, future experiments should explore the theory that Twist1 can modify the filopodia-like structures on the apical surface of the MEE cells through a small GTPase, such as Cdc42.
Several families of transcription factors other than Snail1, such as the ZEB family, independently induce EMT in different contexts (Yang et al., 2004; Liu et al., 2008; Medici et al., 2008). However, the complex and multifaceted process that defines EMT result from a plexus of changes in transcriptional regulation (Peinado et al., 2007). It is very plausible that a number of such EMT-promoting factors may act together as an EMT signaling network (Yang et al., 2006). Snail1 has been implicated in the initial migratory phenotype and considered as an early marker of EMT that sometimes contributes to the induction of other factors. By contrast, Snail2, Zeb1, Zeb2, and/or Twist1 could be responsible for the maintenance of migratory cell behavior (Peinado et al., 2007). During neural crest development in vertebrates, expression of Snail1 and Snail2 occurs at the neural plate border where Twist1 is also expressed, and all three transcription factors play critical roles in neural crest formation (Meulemans and Bronner-Fraser, 2004). However, in Drosophila, Twist induces the expression of the transcription factor Snail to allow invagination and mesoderm differentiation (Furlong et al., 2001). In Twist or Snail mutant Drosophila embryos, the ventral invagination is largely abolished. The double mutant has the strongest phenotype, suggesting that the two genes have both overlapping and distinct functions (Leptin, 1999). A key function of Twist is to collaborate with Dorsal to optimally activate the expression of Snail (Ip and Gridley, 2002). Moreover, genetic rescue experiments demonstrated that forced expression of Snail in the absence of Twist, but not vice versa, can promote ventral-cell invagination. Therefore, it is suggested that although Twist and Snail may each have non-overlapping functions, Snail has a more direct role in regulating downstream events leading to gastrulation (Ip and Gridley, 2002). It is unclear whether similar mechanisms are also involved with Twist1 and Snail1 during palate development.
In summary, we established that Snail1 expression is needed to repress E-cadherin expression to facilitate the MES degradation. Furthermore, we revealed that Tgfβ3 response requires the Snail1 activation by Twist1, establishing a sequential mechanism leading to MES degradation (Figure 6). We anticipate that this degradation possibly involves the transdifferentiation of palatal epithelial cells into mesenchymal cells before their migration away from the seam or death because of the known functions of Twist1 and Snail1 in EMT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Baylor Oral Health Foundation and March of Dimes Foundation (Kathy K. H. Svoboda, FY06-321), the Cleft Palate Foundation and NIH grant NIH U24 DE16472 (L. Bruno Ruest) for the funds to support this work. We thank Rebecca Kayne and Jo Taylor for technical assistance and Douglas Spicer for the forced dimer clones. We thank Jeanne Santa Cruz for assistance with the manuscript preparation. This work was supported by grant sponsors: Baylor Oral Health Foundation; March of Dimes grant 6-FY06-321 (Kathy K. H. Svoboda); the Cleft Palate Foundation and NIH U24 DE16472 (L. Bruno Ruest).
References
Ahmed, S., Liu, C. C., and Nawshad, A. (2007). Mechanisms of palatal epithelial seam disintegration by transforming growth factor (TGF) beta3. Dev. Biol. 309, 193–207.
Barbera, M. J., Puig, I., Dominguez, D., Julien-Grille, S., Guaita-Esteruelas, S., Peiro, S., et al. (2004). Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene 23, 7345–7354.
Barrallo-Gimeno, A., and Nieto, M. A. (2005). The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 132, 3151–3161.
Batlle, E., Sancho, E., Franci, C., Dominguez, D., Monfar, M., Baulida, J., et al. (2000). The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2, 84–89.
Bourgeois, P., Bolcato-Bellemin, A. L., Danse, J. M., Bloch-Zupan, A., Yoshiba, K., Stoetzel, C., et al. (1998). The variable expressivity and incomplete penetrance of the twist-null heterozygous mouse phenotype resemble those of human Saethre-Chotzen syndrome. Hum. Mol. Genet. 7, 945–957.
Bridgman, P. C., Dave, S., Asnes, C. F., Tullio, A. N., and Adelstein, R. S. (2001). Myosin IIB is required for growth cone motility. J. Neurosci. 21, 6159–6169.
Brouzes, E., Supatto, W., and Farge, E. (2004). Is mechano-sensitive expression of twist involved in mesoderm formation? Biol. Cell 96, 471–477.
Cano, A., Perez-Moreno, M. A., Rodrigo, I., Locascio, A., Blanco, M. J., Del Barrio, M. G., et al. (2000). The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83.
Carver, E. A., Jiang, R., Lan, Y., Oram, K. F., and Gridley, T. (2001). The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell. Biol. 21, 8184–8188.
Connerney, J., Andreeva, V., Leshem, Y., Muentener, C., Mercado, M. A., and Spicer, D. B. (2006). Twist1 dimer selection regulates cranial suture patterning and fusion. Dev. Dyn. 235, 1345–1357.
Cuervo, R., and Covarrubias, L. (2004). Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development 131, 15–24.
Ding, H., Wu, X., Bostrom, H., Kim, I., Wong, N., Tsoi, B., et al. (2004). A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat. Genet. 36, 1111–1116.
Dudas, M., Kim, J., Li, W. Y., Nagy, A., Larsson, J., Karlsson, S., et al. (2006). Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev. Biol. 296, 298–314.
Escriva, M., Peiro, S., Herranz, N., Villagrasa, P., Dave, N., Montserrat-Sentis, B., et al. (2008). Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol. Cell. Biol. 28, 1528–1540.
Fitchett, J. E., and Hay, E. D. (1989). Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev. Biol. 131, 455–474.
Fujiwara, K., Yamada, T., Mishima, K., Imura, H., and Sugahara, T. (2008). Morphological and immunohistochemical studies on cleft palates induced by 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin in mice. Congenit. Anom. (Kyoto) 48, 68–73.
Furlong, E. E., Andersen, E. C., Null, B., White, K. P., and Scott, M. P. (2001). Patterns of gene expression during Drosophila mesoderm development. Science 293, 1629–1633.
Griffith, C. M., and Hay, E. D. (1992). Epithelial-mesenchymal transformation during palatal fusion: carboxyfluorescein traces cells at light and electron microscopic levels. Development 116, 1087–1099.
Gritli-Linde, A. (2007). Molecular control of secondary palate development. Dev. Biol. 301, 309–326.
Gupton, S. L., and Gertler, F. B. (2007). Filopodia: the fingers that do the walking. Sci. STKE 2007, re5.
Hay, E. D. (1989). Extracellular matrix, cell skeletons, and embryonic development. Am. J. Med. Genet. 34, 14–29.
Ip, Y. T., and Gridley, T. (2002). Cell movements during gastrulation: snail dependent and independent pathways. Curr. Opin. Genet. Dev. 12, 423–429.
Jin, J. Z., and Ding, J. (2006). Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development 133, 3341–3347.
Jugessur, A., and Murray, J. C. (2005). Orofacial clefting: recent insights into a complex trait. Curr. Opin. Genet. Dev. 15, 270–278.
Kaartinen, V., Voncken, J. W., Shuler, C., Warburton, D., Bu, D., Heisterkamp, N., et al. (1995). Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 11, 415–421.
Kang, P., and Svoboda, K. K. (2002). PI-3 kinase activity is required for epithelial-mesenchymal transformation during palate fusion. Dev. Dyn. 225, 316–321.
Kitase, Y., Yamashiro, K., Fu, K., Richman, J. M., and Shuler, C. F. (2011). Spatiotemporal localization of periostin and its potential role in epithelial-mesenchymal transition during palatal fusion. Cells Tissues Organs 193, 53–63.
Leptin, M. (1999). Gastrulation in Drosophila: the logic and the cellular mechanisms. EMBO J. 18, 3187–3192.
Liu, Y., El-Naggar, S., Darling, D. S., Higashi, Y., and Dean, D. C. (2008). Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development 135, 579–588.
Martin-Blanco, E., and Knust, E. (2001). Epithelial morphogenesis: filopodia at work. Curr. Biol. 11, R28–R31.
Martinez-Alvarez, C., Blanco, M. J., Perez, R., Rabadan, M. A., Aparicio, M., Resel, E., et al. (2004). Snail family members and cell survival in physiological and pathological cleft palates. Dev. Biol. 265, 207–218.
Martinez-Alvarez, C., Tudela, C., Perez-Miguelsanz, J., O'Kane, S., Puerta, J., and Ferguson, M. W. (2000). Medial edge epithelial cell fate during palatal fusion. Dev. Biol. 220, 343–357.
Mattila, P. K., and Lappalainen, P. (2008). Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 9, 446–454.
Medici, D., Hay, E. D., and Olsen, B. R. (2008). Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol. Biol. Cell 19, 4875–4887.
Meulemans, D., and Bronner-Fraser, M. (2004). Gene-regulatory interactions in neural crest evolution and development. Dev. Cell 7, 291–299.
Millard, T. H., and Martin, P. (2008). Dynamic analysis of filopodial interactions during the zippering phase of Drosophila dorsal closure. Development 135, 621–626.
Murray, S. A., Oram, K. F., and Gridley, T. (2007). Multiple functions of Snail family genes during palate development in mice. Development 134, 1789–1797.
Nawshad, A. (2008). Palatal seam disintegration: to die or not to die? that is no longer the question. Dev. Dyn. 237, 2643–2656.
Nawshad, A., Lagamba, D., and Hay, E. D. (2004). Transforming growth factor beta (TGFbeta) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch. Oral Biol. 49, 675–689.
Nawshad, A., Lagamba, D., Polad, A., and Hay, E. D. (2005). Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs 179, 11–23.
Nieto, M. A. (2002). The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 3, 155–166.
Oram, K. F., and Gridley, T. (2005). Mutations in snail family genes enhance craniosynostosis of Twist1 haplo-insufficient mice: implications for Saethre-Chotzen Syndrome. Genetics 170, 971–974.
O'Rourke, M. P., and Tam, P. P. (2002). Twist functions in mouse development. Int. J. Dev. Biol. 46, 401–413.
Peinado, H., Olmeda, D., and Cano, A. (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer 7, 415–428.
Peinado, H., Portillo, F., and Cano, A. (2004). Transcriptional regulation of cadherins during development and carcinogenesis. Int. J. Dev. Biol. 48, 365–375.
Peinado, H., Quintanilla, M., and Cano, A. (2003). Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J. Biol. Chem. 278, 21113–21123.
Peiro, S., Escriva, M., Puig, I., Barbera, M. J., Dave, N., Herranz, N., et al. (2006). Snail1 transcriptional repressor binds to its own promoter and controls its expression. Nucleic Acids Res. 34, 2077–2084.
Perez-Moreno, M. A., Locascio, A., Rodrigo, I., Dhondt, G., Portillo, F., Nieto, M. A., et al. (2001). A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J. Biol. Chem. 276, 27424–27431.
Proetzel, G., Pawlowski, S. A., Wiles, M. V., Yin, M., Boivin, G. P., Howles, P. N., et al. (1995). Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 11, 409–414.
Rice, R., Thesleff, I., and Rice, D. P. (2005). Regulation of Twist, Snail, and Id1 is conserved between the developing murine palate and tooth. Dev. Dyn. 234, 28–35.
Ruest, L. B., and Clouthier, D. E. (2009). Elucidating timing and function of endothelin-A receptor signaling during craniofacial development using neural crest cell-specific gene deletion and receptor antagonism. Dev. Biol. 328, 94–108.
Ruest, L. B., Xiang, X., Lim, K. C., Levi, G., and Clouthier, D. E. (2004). Endothelin-A receptor-dependent and -independent signaling pathways in establishing mandibular identity. Development 131, 4413–4423.
Spicer, D. B., Rhee, J., Cheung, W. L., and Lassar, A. B. (1996). Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science 272, 1476–1480.
Stoler, J. M., Rogers, G. F., and Mulliken, J. B. (2009). The frequency of palatal anomalies in Saethre-Chotzen syndrome. Cleft Palate Craniofac. J. 46, 280–284.
Sun, D., Vanderburg, C. R., Odierna, G. S., and Hay, E. D. (1998). TGFbeta3 promotes transformation of chicken palate medial edge epithelium to mesenchyme in vitro. Development 125, 95–105.
Takahashi, E., Funato, N., Higashihori, N., Hata, Y., Gridley, T., and Nakamura, M. (2004). Snail regulates p21(WAF/CIP1) expression in cooperation with E2A and Twist. Biochem. Biophys. Res. Commun. 325, 1136–1144.
Taya, Y., O'Kane, S., and Ferguson, M. W. (1999). Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development 126, 3869–3879.
Thuault, S., Valcourt, U., Petersen, M., Manfioletti, G., Heldin, C. H., and Moustakas, A. (2006). Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 174, 175–183.
Vaziri Sani, F., Hallberg, K., Harfe, B. D., McMahon, A. P., Linde, A., and Gritli-Linde, A. (2005). Fate-mapping of the epithelial seam during palatal fusion rules out epithelial-mesenchymal transformation. Dev. Biol. 285, 490–495.
Xu, X., Han, J., Ito, Y., Bringas, P. Jr., Urata, M. M., and Chai, Y. (2006). Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev. Biol. 297, 238–248.
Yang, J., Mani, S. A., Donaher, J. L., Ramaswamy, S., Itzykson, R. A., Come, C., et al. (2004). Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939.
Yang, J., Mani, S. A., and Weinberg, R. A. (2006). Exploring a new twist on tumor metastasis. Cancer Res. 66, 4549–4552.
Yin, Z., Xu, X. L., and Frasch, M. (1997). Regulation of the twist target gene tinman by modular cis-regulatory elements during early mesoderm development. Development 124, 4971–4982.
Yu, W., Kamara, H., and Svoboda, K. K. (2008). The role of twist during palate development. Dev. Dyn. 237, 2716–2725.
Yu, W., Ruest, L. B., and Svoboda, K. K. (2009). Regulation of epithelial-mesenchymal transition in palatal fusion. Exp. Biol. Med. (Maywood) 234, 483–491.
Zavadil, J., and Bottinger, E. P. (2005). TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774.
Keywords: palatal fusion, Snail1, Twist1, E-cadherin, Tgfβ3, E-proteins, epithelial-mesenchymal transition
Citation: Yu W, Zhang Y, Ruest LB and Svoboda KKH (2013) Analysis of Snail1 function and regulation by Twist1 in palatal fusion. Front. Physio. 4:12. doi: 10.3389/fphys.2013.00012
Received: 28 February 2012; Accepted: 10 January 2013;
Published online: 19 February 2013.
Edited by:
Daniel Graf, University of Zurich, SwitzerlandReviewed by:
David P. Rice, University of Helsinki, FinlandSachiko Iseki, Tokyo Medical and Dental University, Japan
Copyright © 2013 Yu, Zhang, Ruest and Svoboda. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: L. Bruno Ruest and Kathy K. H. Svoboda, Department of Biomedical Sciences, Texas A&M University, Baylor College of Dentistry, 3302 Gaston Ave., Dallas, TX 75246, USA. e-mail:bGJydWVzdEBiY2QudGFtaHNjLmVkdQ==;a3N2b2JvZGFAYmNkLnRhbWhzYy5lZHU=