 Rakesh K. Kumar1* and Paul S. Foster2
Rakesh K. Kumar1* and Paul S. Foster2- 1Department of Pathology, School of Medical Sciences, University of New South Wales, Sydney, NSW, Australia
- 2Discipline of Microbiology and Immunology, School of Biomedical Sciences and Pharmacy, University of Newcastle and Hunter Medical Research Institute, Newcastle, NSW, Australia
Whether mouse models of chronic asthma can be used to investigate the relationship between airway inflammation/remodeling and airway hyper-responsiveness (AHR) is a vexed question. It raises issues about the extent to which such models replicate key features of the human disease. Here, we review some of the characteristic pathological features of human asthma and their relationship to AHR and examine some limitations of mouse models that are commonly used to investigate these relationships. We compare these conventional models with our mouse model of chronic asthma involving long-term low-level inhalational challenge and review studies of the relationship between inflammation/remodeling and AHR in this model and its derivatives, including models of an acute exacerbation of chronic asthma and of the induction phase of childhood asthma. We conclude that while extrapolating from studies in mouse models to AHR in humans requires cautious interpretation, such experimental work can provide significant insights into the pathogenesis of airway responsiveness and its molecular and cellular regulation.
Introduction
Mouse models can certainly be employed to investigate the relationship between asthmatic inflammation/airway remodeling and airway hyper-responsiveness (AHR): they have been and continue to be used for such research. Whether they should be, i.e., whether the models are sufficiently similar to the human disease to be suitable for the purpose, is a separate question altogether. It raises, in turn, questions about the extent to which such models replicate key features of the human disease and about what exactly is being measured when AHR is assessed in animals. This review focuses on the first of those considerations: technical issues related to the measurement of AHR in mouse models and their relevance to physiological assessment in humans are examined elsewhere in this issue.
Whether mouse models replicate the features of human asthma with sufficient fidelity is a critically important question, which needs to be addressed both by experimentalists and by journal reviewers. Of course all experimental models have limitations, but if the model being investigated is not appropriate to the outcomes being assessed, it may be difficult to extrapolate from the experimental data to the human disease. While this has led some to suggest that mouse models of asthma have yielded little that has been able to be translated into improvements in therapy (Persson, 2002; Boyce and Austen, 2005; Wenzel and Holgate, 2006), many of the carefully performed studies in mouse models have yielded much interesting information about pathogenetic mechanisms of asthma.
To advance discussion about the relevance of mouse models, it would be appropriate to briefly review the characteristic pathological features of human asthma and their relationship to AHR. Some limitations of commonly used mouse models can then be examined, and the merits of a potential approach to overcoming these limitations can be assessed. In drawing comparisons, it is worth emphasizing that available mouse models are largely relevant only to allergic/eosinophilic asthma in humans. The overwhelming majority of children with asthma have evidence of allergy/atopy, but asthma of adult onset is frequently non-eosinophilic (Kumar and Jeffery, 2012). The pathological features described below relate to human allergic asthma.
Airway Inflammation in Asthma
Airway inflammation is a characteristic feature of human asthma. The pathological changes are in general restricted to the conducting airways, mostly involving major bronchi, but also smaller airways in severe disease (James, 2009). Alveolar inflammation has been described in some settings (Kraft et al., 1996; De Magalhaes Simoes et al., 2005), but it should be emphasized that asthma is not primarily a condition involving the lung parenchyma.
Even in the earliest stages of mild clinical asthma, there is accumulation of chronic inflammatory cells in the mucosa of the larger conducting airways (Holgate et al., 1992; Laitinen et al., 1993). These include increased numbers of lymphocytes, plasma cells, mast cells, and macrophages. As has long been emphasized, in allergic asthma the majority of lymphocytes within the airway wall are CD4+ cells which exhibit a T helper type 2 (Th2) profile of cytokine secretion, characterized by production of interleukin (IL)-4, IL-5, and IL-13 (Hamid et al., 1991; Yung et al., 1995). Lymphocytes and mast cells are also prominent deeper in the airway wall, including in the smooth muscle layer (Brightling et al., 2002; Begueret et al., 2007). Superimposed upon this background is an acute inflammatory component, which is typically associated with recruitment of eosinophils into the mucosa (Bradley et al., 1991; Shaver et al., 1997) and in particular into the epithelial layer (Figure 1). Indeed, intraepithelial eosinophils are considered to be a distinctive feature of asthmatic inflammation (Bousquet et al., 1990), whereas recruitment of neutrophils into the airways is uncommon in mild to moderate asthma.
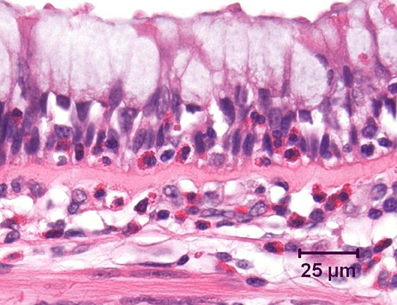
Figure 1. Bronchus from an asthmatic patient, demonstrating numerous eosinophils, both within the airway epithelium and in the underlying lamina propria. Note the marked goblet cell change in the epithelium and the prominent zone of subepithelial fibrosis. Hematoxylin and eosin.
In patients with asthma, there has long been controversy about whether airway inflammation correlates with AHR (Sont et al., 1996; Crimi et al., 1998). This may in part be a consequence of the use of different stimuli to assess airway responses. AHR to indirect stimuli, such as distilled water, hypertonic saline, or mannitol, seems to correlate more closely with airway inflammation than does the response to bronchoconstrictor agents (Chetta et al., 1996; Joos et al., 2003).
Investigating the Relationship between Inflammation and AHR in Mouse Models
Most mouse models of asthma involve short-term challenge of sensitized mice with antigen delivered either by inhalational exposure that is uncontrolled (i.e., with no attempt made to limit the mass concentration of aerosol to which the animals are exposed and no monitoring of the concentration of aerosol in the breathing zone of the animals) or intratracheally. These models are experimentally convenient, but given the nature of the antigenic challenge, they are primarily models of severe acute allergic inflammation. Thus they are very useful for investigating the basis of allergic inflammation, including the role of CD4+ Th2 cells, as well as the various cellular and molecular pathways involved in regulating their activity. However, there are important differences in the pattern of airway inflammation compared to human asthmatics. Notably, inflammation is by no means limited to the conducting airways, but is usually associated with accumulation of large numbers of perivascular inflammatory cells, as well as with marked edema around blood vessels (Figure 2). The severity and extent of peribronchiolar inflammation are variable. There is also a relative over-representation of eosinophils, which comprise 40–80% of the cells in the bronchoalveolar lavage fluid (BALF) of these mice, in contrast to the 1–5% eosinophils in BALF from human asthmatics.
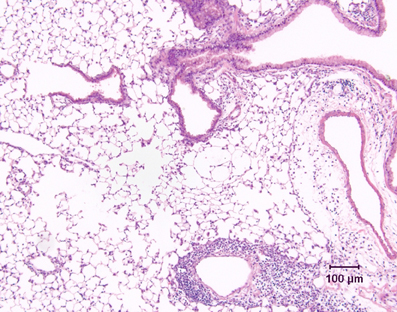
Figure 2. Lung tissue demonstrating the inflammatory response in a conventional mouse model of allergic airway inflammation. Following short-term, uncontrolled inhalational challenge of a sensitized animal with aerosolized ovalbumin, there is marked perivascular inflammation (below, right) and edema (right) but relatively limited peribronchiolar inflammation. Hematoxylin and eosin.
Importantly, chronic inflammation of the airway wall is conspicuously absent in these models, which means that they cannot be used for investigation of the relationship between chronic airway inflammation and AHR. Thus their relevance is likely to be limited to AHR in severe asthma or exacerbations of asthma, subject of course to the problems associated with the lack of comparability of techniques used to assess AHR in mice as compared to humans. There are also questions about the relative contribution of changes in large vs. small airways to the AHR that is able to be demonstrated. Ex vivo studies in these mouse models have provided evidence of altered contractility of bronchial smooth muscle in response to allergen or a non-specific bronchoconstrictor agent (Chiba et al., 2005; Chew et al., 2008) and a component of AHR, measured using the most appropriate available techniques, undoubtedly does originate from the conducting airways. However, it has been suggested that in short-term models, AHR may at least in part be explained on the basis of thickening of the airway mucosa and an increased propensity of small airways to close, without any increase in the degree of airway smooth muscle shortening (Wagers et al., 2004; Lundblad et al., 2007). Notwithstanding this caveat, airway closure in response to a bronchoconstrictor challenge is also a contributory mechanism of AHR in human asthmatics (Berend et al., 2008).
It is also important to realize that the information which may be gained from the use of mouse models to investigate development of AHR is influenced by key experimental variables such as the time point of examination after allergen challenge. For example, we have shown that prolonged AHR (i.e., altered responsiveness detectable at 1 week after allergen challenge, demonstrable as primarily originating from the conducting airways) may largely be driven by an interaction between interferon-γ and pulmonary macrophages rather than involving eosinophilic inflammation (Yang et al., 2010).
Airway Wall Remodeling in Asthma
Pathological remodeling in asthma refers to structural changes in the airway wall. Early on, even in mild or moderate asthma, there is prominent thickening of a fibrillary layer subjacent to the epithelium, which is highly characteristic of the disease (Jeffery, 2001). This is primarily associated with deposition of type III and type V collagens, together with other matrix proteins including laminins (Altraja et al., 1996), tenascin (Laitinen et al., 1997), periostin (Takayama et al., 2006), and a variety of proteoglycans. It is variously referred to as subepithelial fibrosis or thickening of the reticular basement membrane (RBM) and is associated with evidence of Th2-driven allergic inflammation (Woodruff et al., 2009) and the presence of increased numbers of subepithelial myofibroblasts (Brewster et al., 1990). At least in large airways, RBM thickness correlates well with overall thickening of airway wall, so that assessment of the thickness of the subepithelial layer in bronchial biopsies may be a useful indicator of airway remodeling (James et al., 2002). However, its relevance to functional changes in asthma is unclear, and recent investigations suggest that subepithelial thickening may be a consequence of bronchoconstriction rather than a contributor to airflow obstruction (Grainge et al., 2011).
Perhaps the most important feature of asthmatic airway remodeling is thickening of the airway smooth muscle layer (James and Carroll, 2000; James et al., 2012). This includes hypertrophy and hyperplasia of the muscle cells, as well as altered deposition of extracellular matrix (Woodruff et al., 2004; Begueret et al., 2007; Pini et al., 2007; Hassan et al., 2010). Increased smooth muscle mass is highly likely to contribute to AHR because thickening of the airway wall may decrease the degree of airway smooth muscle shortening required to occlude the lumen (James et al., 1989; James and Wenzel, 2007).
Airway mucosal vascularity, especially increased density of newly formed small vessels, is also a feature of remodeling in mild to moderate asthma (Li and Wilson, 1997; Salvato, 2001; Feltis et al., 2006). These vessels may have increased permeability and thus contribute to airway wall edema leading to airflow obstruction (Khor et al., 2009), although the relevance of this to AHR is unknown.
Another characteristic feature of remodeling in human asthmatics is an increase in the number of mucin-secreting goblet cells in the surface epithelium, due to both hyperplasia and metaplasia (the latter representing altered differentiation and seen mostly in small airways; Ordonez et al., 2001). Again, the relationship between these changes and AHR in patients has not been defined, further emphasizing that the relationship between airway remodeling and AHR in asthmatics remains poorly understood.
Investigating the Relationship between Remodeling and AHR in Mouse Models
As noted above, most commonly used mouse models involve short-term challenge of sensitized animals with large amounts of antigen, triggering severe acute allergic inflammation. Unsurprisingly, such models replicate few features of airway remodeling, although mucous cell change is a very rapidly developing response (Ohkawara et al., 1997) and some studies have yielded interesting information about its potential role in AHR (Nakanishi et al., 2001; Long et al., 2006). In the past dozen years, a number of investigators have sought to develop better animal models based on extending the period of antigenic challenge to 4–8 weeks (Braun et al., 1999; Hogaboam et al., 2000; Tanaka et al., 2001; Henderson et al., 2002; Jain et al., 2002; Leigh et al., 2002; Kenyon et al., 2003; Johnson et al., 2004; Pitchford et al., 2004; Yang et al., 2004; McMillan et al., 2005; Wegmann et al., 2005; Locke et al., 2007; Shinagawa et al., 2007). Many of these do exhibit various features of airway wall remodeling: most reports describe marked goblet cell change and several also demonstrate evidence of increased airway smooth muscle. A single study reported increased airway vascularity in such a model (Su et al., 2008).
However, almost all of these chronic challenge models still have shortcomings. Notably, because of the unregulated exposure to high mass concentrations of inhaled antigen, repeated challenge is frequently associated with severe pulmonary inflammation, sometimes with formation of multinucleated giant cells and even granulomas (Mathew et al., 2001). This response appears to be somewhat variable and possibly dependent on the strain of mice used, because several reports suggest that there may instead be progressive downregulation of airway inflammation and Th2 responses (Irifune et al., 1998; Yiamouyiannis et al., 1999; Sakai et al., 2001; Leigh et al., 2002; Pitchford et al., 2004). Furthermore, the marked peribronchiolar accumulation of collagen that is observed in such models is quite distinct from the limited zone of subepithelial thickening in conducting airways that is characteristic of human asthma. This peribronchiolar scarring, which often extends further including around blood vessels, is not synonymous with subepithelial fibrosis, despite the widespread misuse of the term by investigators, and should instead be described as airway wall fibrosis. Although small airway remodeling has been demonstrated in severe asthmatics (Dolhnikoff et al., 2009), airway fibrosis is not a characteristic lesion of human asthma. Clearly, it would be unwise to draw conclusions about the relationship between airway remodeling and AHR in such a setting.
A More Realistic Mouse Model of Mild Chronic Asthma
We have described a mouse model of chronic asthma that involves long-term challenge of sensitized mice with carefully controlled low mass concentrations of aerosolized ovalbumin (approximately 3 mg/m3 which is some 100–1000 times lower than used in conventional models; Temelkovski et al., 1998). This exhibits changes of chronic asthma that closely resemble the human disease, both in terms of pattern and spatial distribution of cellular responses. Airway disease is established by 4 weeks of inhalational challenge (30 min/day, 3 days/week) with lesions including eosinophil recruitment into the epithelial layer of the conducting airways, chronic inflammation in the airway wall with accumulation of T-lymphocytes and plasma cells, and changes of remodeling such as sub-epithelial fibrosis, epithelial hypertrophy, and goblet cell metaplasia. Importantly, there is minimal parenchymal inflammation, i.e., no perivascular or peribronchiolar inflammation in the lungs (Temelkovski et al., 1998). In terms of pathogenesis, in this model there is clearly a role for both Th2 and Th1 cytokines, as demonstrated in studies using gene-targeted animals and neutralizing antibodies (Kumar et al., 2002, 2004a,c). This model has, therefore, been widely acknowledged to represent a significant improvement in terms of the fidelity with which it reproduces features of human asthma (Shore, 2003; Fulkerson et al., 2005; Nials and Uddin, 2008).
Can this model be used to investigate the pathogenesis of AHR and, in particular, its relationship to airway remodeling? Our studies suggest that, at the very least, the cytokine mechanisms involved in development of AHR following low-level chronic challenge may be different to those demonstrated in short-term models assessed at early time points after challenge, because there appears to be less dependency on Th2 responses and a significant role for interferon-γ (Kumar et al., 2004c). Moreover, although the presence of airway remodeling is often correlated with evidence of AHR, we have clearly demonstrated that these may be dissociated (Foster et al., 2000). We would not suggest that this model provides definitive answers, as we have previously identified some important limitations. Unlike chronic exposure models in which antigenic challenge is uncontrolled, we find no evidence of significant hypertrophy or hyperplasia of airway smooth muscle (Kumar et al., 2004b), which limits comparability to the human disease. In addition, as in other models in mice, there is no evidence of accumulation of mast cells in the airways (Kumar et al., 2000) so the potential contribution of these cells cannot be properly assessed.
Modeling Other Aspects of Asthma
Using our chronic challenge model as a substrate, we have also established a model of an allergen-induced acute exacerbation of chronic asthma, in which following low-level challenge for 4 weeks, animals were exposed for a 30-min period to a single challenge with 30 mg/m3 of aerosolized antigen (i.e., 10-fold higher than usual). This moderate-level challenge is associated with more marked airway inflammation, which extends further distally than in the chronic challenge model (Siegle et al., 2006). Compared to short-term uncontrolled challenge models, however, the inflammatory response is relatively mild and the numbers of eosinophils, lymphocytes and neutrophils in BALF are modest. Table 1 compares some key features of the human disease with short-term, high-level challenge models and the models that we have developed.
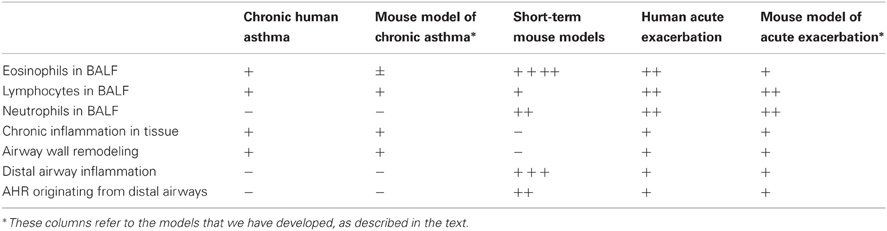
Table 1. Comparison of human asthma and various mouse models.
Does our model permit investigation of the pathogenesis of AHR in patients with an acute exacerbation of chronic asthma? Quite possibly, because these animals demonstrate a pattern of AHR distinct from that seen in the chronic challenge model, presumably reflecting the distal airway involvement (Siegle et al., 2006). Development of AHR appears not to be dependent on eosinophil recruitment and is relatively resistant to treatment with glucocorticosteroids (Ito et al., 2008).
More recently, we have extended the approach of using controlled inhalational challenge to studies seeking to model the induction phase of asthma in childhood, with sensitization early in life via the respiratory tract. Because inhalational sensitization/challenge with an innocuous protein antigen such as ovalbumin does not elicit an allergic immunological response and indeed may lead to tolerance (Gerhold et al., 2003; Wang and McCusker, 2006), early life airway epithelial injury by environmental factors, which promotes sensitization, appears to be essential for the development of features of asthma. We have demonstrated a crucial interaction between neonatal respiratory viral infection and respiratory sensitization/challenge in the pathogenesis of an asthmatic phenotype (Siegle et al., 2010) and have subsequently also investigated the role of early life exposure to environmental particulates in progression to an asthmatic pattern of inflammation (Herbert et al., submitted for publication). These studies have highlighted the likely importance of persistent epithelial injury in the development of AHR in such models.
The Bottom Line
A reasonable answer to the question “Can mouse models be used to investigate the relationship between airway inflammation/remodeling and AHR?” is: up to a point, but it is important to recognize the associated problems and limitations. Both researchers and referees need to be alert to such issues. As an example, smooth muscle changes that appear to be important in the development of AHR in human asthmatics are not reproduced at all well in mouse models except when associated with numerous confounding lesions, which are often ignored. Similarly, the literature contains many reports of studies in which severe peribronchiolar fibrosis is used as an index of remodeling and is then correlated with effects on AHR, yet such fibrosis is not a feature of the airways in patients with asthma. A lack of appreciation of the pathological changes in human asthma can lead to the use of suboptimal animal models and avoidable weaknesses in experimental design.
Given the complexity of asthma, however, it will always be the case that no single animal model—in mice or any other species—will be a perfect system for studies of the pathogenesis of AHR or any other aspect of the human disease. For experimentalists to be able to design/select mouse models that are appropriate for the investigation of the question of interest, this complexity needs to be unraveled. Experimental work focusing on Th2-mediated responses was driven by the recognition that this was a key feature of inflammation in the airways of human asthmatics. However, current approaches to stratifying inflammatory phenotypes of asthma in patients (e.g., based on examination of induced sputum) cannot readily be translated to mouse models. In turn, this means that translation of experimental findings back to the appropriate subset of patients is difficult. Phenotyping of asthma based on pathological changes at the cellular and molecular level, rather than on clinical manifestations and assessment of pulmonary function, is likely to facilitate the design of better studies using appropriate mouse models that shed light on pathogenesis, which in turn can lead to the development of worthwhile therapeutic interventions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in the authors' laboratories is primarily supported by grants from the National Health and Medical Research Council of Australia.
References
Altraja, A., Laitinen, A., Virtanen, I., Kampe, M., Simonsson, B. G., Karlsson, S. E., Hakansson, L., Venge, P., Sillastu, H., and Laitinen, L. A. (1996). Expression of laminins in the airways in various types of asthmatic patients: a morphometric study. Am. J. Respir. Cell Mol. Biol. 15, 482–488.
Begueret, H., Berger, P., Vernejoux, J. M., Dubuisson, L., Marthan, R., and Tunon-De-Lara, J. M. (2007). Inflammation of bronchial smooth muscle in allergic asthma. Thorax 62, 8–15.
Berend, N., Salome, C. M., and King, G. G. (2008). Mechanisms of airway hyperresponsiveness in asthma. Respirology 13, 624–631.
Bousquet, J., Chanez, P., Lacoste, J. Y., Barneon, G., Ghavanian, N., Enander, I., Venge, P., Ahlstedt, S., Simony-Lafontaine, J., Godard, P., and Michel, F. B. (1990). Eosinophilic inflammation in asthma. N. Engl. J. Med. 323, 1033–1039.
Boyce, J. A., and Austen, K. F. (2005). No audible wheezing: nuggets and conundrums from mouse asthma models. J. Exp. Med. 201, 1869–1873.
Bradley, B. L., Azzawi, M., Jacobson, M., Assoufi, B., Collins, J. V., Irani, A. M., Schwartz, L. B., Durham, S. R., Jeffery, P. K., and Kay, A. B. (1991). Eosinophils, T-lymphocytes, mast cells, neutrophils, and macrophages in bronchial biopsy specimens from atopic subjects with asthma: comparison with biopsy specimens from atopic subjects without asthma and normal control subjects and relationship to bronchial hyperresponsiveness. J. Allergy Clin. Immunol. 88, 661–674.
Braun, A., Lommatzsch, M., Mannsfeldt, A., Neuhaus-Steinmetz, U., Fischer, A., Schnoy, N., Lewin, G. R., and Renz, H. (1999). Cellular sources of enhanced brain-derived neurotrophic factor production in a mouse model of allergic inflammation. Am. J. Respir. Cell Mol. Biol. 21, 537–546.
Brewster, C. E. P., Howarth, P. H., Djukanovic, R., Wilson, J., Holgate, S. T., and Roche, W. R. (1990). Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 3, 507–511.
Brightling, C. E., Bradding, P., Symon, F. A., Holgate, S. T., Wardlaw, A. J., and Pavord, I. D. (2002). Mast-cell infiltration of airway smooth muscle in asthma. N. Engl. J. Med. 346, 1699–1705.
Chetta, A., Foresi, A., Del Donno, M., Consigli, G. F., Bertorelli, G., Pesci, A., Barbee, R. A., and Olivieri, D. (1996). Bronchial responsiveness to distilled water and methacholine and its relationship to inflammation and remodeling of the airways in asthma. Am. J. Respir. Crit. Care Med. 153, 910–917.
Chew, A. D., Hirota, J. A., Ellis, R., Wattie, J., Inman, M. D., and Janssen, L. J. (2008). Effects of allergen on airway narrowing dynamics as assessed by lung-slice technique. Eur. Respir. J. 31, 532–538.
Chiba, Y., Ueno, A., Shinozaki, K., Takeyama, H., Nakazawa, S., Sakai, H., and Misawa, M. (2005). Involvement of RhoA-mediated Ca2+ sensitization in antigen-induced bronchial smooth muscle hyperresponsiveness in mice. Respir. Res. 6, 4.
Crimi, E., Spanevello, A., Neri, M., Ind, P. W., Rossi, G. A., and Brusasco, V. (1998). Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am. J. Respir. Crit. Care Med. 157, 4–9.
De Magalhaes Simoes, S., Dos Santos, M. A., Da Silva Oliveira, M., Fontes, E. S., Fernezlian, S., Garippo, A. L., Castro, I., Castro, F. F., De Arruda Martins, M., Saldiva, P. H., Mauad, T., and Dolhnikoff, M. (2005). Inflammatory cell mapping of the respiratory tract in fatal asthma. Clin. Exp. Allergy 35, 602–611.
Dolhnikoff, M., Da Silva, L. F., De Araujo, B. B., Gomes, H. A., Fernezlian, S., Mulder, A., Lindeman, J. H., and Mauad, T. (2009). The outer wall of small airways is a major site of remodeling in fatal asthma. J. Allergy Clin. Immunol. 123, 1090–1097, 1097.e1091.
Feltis, B. N., Wignarajah, D., Zheng, L., Ward, C., Reid, D., Harding, R., and Walters, E. H. (2006). Increased vascular endothelial growth factor and receptors: relationship to angiogenesis in asthma. Am. J. Respir. Crit. Care Med. 173, 1201–1207.
Foster, P. S., Ming, Y., Matthei, K. I., Young, I. G., Temelkovski, J., and Kumar, R. K. (2000). Dissociation of inflammatory and epithelial responses in a murine model of chronic asthma. Lab. Invest. 80, 655–662.
Fulkerson, P. C., Rothenberg, M. E., and Hogan, S. P. (2005). Building a better mouse model: experimental models of chronic asthma. Clin. Exp. Allergy 35, 1251–1253.
Gerhold, K., Bluemchen, K., Franke, A., Stock, P., and Hamelmann, E. (2003). Exposure to endotoxin and allergen in early life and its effect on allergen sensitization in mice. J. Allergy Clin. Immunol. 112, 389–396.
Grainge, C. L., Lau, L. C., Ward, J. A., Dulay, V., Lahiff, G., Wilson, S., Holgate, S., Davies, D. E., and Howarth, P. H. (2011). Effect of bronchoconstriction on airway remodeling in asthma. N. Engl. J. Med. 364, 2006–2015.
Hamid, Q., Azzawi, M., Ying, S., Moqbel, R., Wardlaw, A. J., Corrigan, C. J., Bradley, B., Durham, S. R., Collins, J. V., and Jeffery, P. K. (1991). Expression of mRNA for interleukin-5 in mucosal bronchial biopsies from asthma. J. Clin. Invest. 87, 1541–1546.
Hassan, M., Jo, T., Risse, P. A., Tolloczko, B., Lemiere, C., Olivenstein, R., Hamid, Q., and Martin, J. G. (2010). Airway smooth muscle remodeling is a dynamic process in severe long-standing asthma. J. Allergy Clin. Immunol. 125, 1037–1045.e1033.
Henderson, W. R., Tang, L. O., Chu, S. J., Tsao, S. M., Chiang, G. K., Jones, F., Jonas, M., Pae, C., Wang, H., and Chi, E. Y. (2002). A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am. J. Respir. Crit. Care Med. 165, 108–116.
Hogaboam, C. M., Blease, K., Mehrad, B., Steinhauser, M. L., Standiford, T. J., Kunkel, S. L., and Lukacs, N. W. (2000). Chronic airway hyperreactivity, goblet cell hyperplasia, and peribronchial fibrosis during allergic airway disease induced by Aspergillus fumigatus. Am. J. Pathol. 156, 723–732.
Holgate, S. T., Wilson, J. W., and Howarth, P. H. (1992). New insights into airway inflammation by endobronchial biopsy. Am. Rev. Respir. Dis. 145, S2–S6.
Irifune, K., Yokoyama, A., Kohno, N., Sakai, K., Hirasawa, Y., Kondo, K., and Hiwada, K. (1998). Prolonged exposure to antigen aerosol attenuates eosinophilic lung inflammation in a murine model of asthma. Eur. Respir. J. 12(Suppl. 29), 2s.
Ito, K., Herbert, C., Siegle, J. S., Vuppusetty, C., Hansbro, N., Thomas, P. S., Foster, P. S., Barnes, P. J., and Kumar, R. K. (2008). Steroid-resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. Am. J. Respir. Cell Mol. Biol. 39, 543–550.
Jain, V. V., Kitagaki, K., Businga, T., Hussain, I., George, C., O'Shaughnessy, P., and Kline, J. N. (2002). CpG-oligodeoxynucleotides inhibit airway remodeling in a murine model of chronic asthma. J. Allergy Clin. Immunol. 110, 867–872.
James, A., and Carroll, N. (2000). Airway smooth muscle in health and disease; methods of measurement and relation to function. Eur. Respir. J. 15, 782–789.
James, A. L., Elliot, J. G., Jones, R. L., Carroll, M. L., Mauad, T., Bai, T. R., Abramson, M. J., McKay, K. O., and Green, F. H. (2012). Airway smooth muscle hypertrophy and hyperplasia in asthma. Am. J. Respir. Crit. Care Med. 185, 1058–1064.
James, A. L., Maxwell, P. S., Pearce-Pinto, G., Elliot, J. G., and Carroll, N. G. (2002). The relationship of reticular basement membrane thickness to airway wall remodeling in asthma. Am. J. Respir. Crit. Care Med. 166, 1590–1595.
James, A. L., Pare, P. D., and Hogg, J. C. (1989). The mechanics of airway narrowing in asthma. Am. Rev. Respir. Dis. 139, 242–246.
James, A. L., and Wenzel, S. (2007). Clinical relevance of airway remodelling in airway diseases. Eur. Respir. J. 30, 134–155.
Jeffery, P. K. (2001). Remodelling in asthma and chronic obstructive lung disease. Am. J. Respir. Crit. Care Med. 164, S28–S38.
Johnson, J. R., Wiley, R. E., Fattouh, R., Swirski, F. K., Gajewska, B. U., Coyle, A. J., Gutierrez-Ramos, J. C., Ellis, R., Inman, M. D., and Jordana, M. (2004). Continuous exposure to house dust mite elicits chronic airway inflammation and structural remodeling. Am. J. Respir. Crit. Care Med. 169, 378–385.
Joos, G. F., O'Connor, B., Anderson, S. D., Chung, F., Cockcroft, D. W., Dahlen, B., Dimaria, G., Foresi, A., Hargreave, F. E., Holgate, S. T., Inman, M., Lotvall, J., Magnussen, H., Polosa, R., Postma, D. S., and Riedler, J. (2003). Indirect airway challenges. Eur. Respir. J. 21, 1050–1068.
Kenyon, N. J., Ward, R. W., and Last, J. A. (2003). Airway fibrosis in a mouse model of airway inflammation. Toxicol. Appl. Pharmacol. 186, 90–100.
Khor, Y. H., Teoh, A. K., Lam, S. M., Mo, D. C., Weston, S., Reid, D. W., and Walters, E. H. (2009). Increased vascular permeability precedes cellular inflammation as asthma control deteriorates. Clin. Exp. Allergy 39, 1659–1667.
Kraft, M., Djukanovic, R., Wilson, S., Holgate, S. T., and Martin, R. J. (1996). Alveolar tissue inflammation in asthma. Am. J. Respir. Crit. Care Med. 154, 1505–1510.
Kumar, R. K., Herbert, C., and Foster, P. S. (2004a). Expression of growth factors by airway epithelial cells in a model of chronic asthma: regulation and relationship to subepithelial fibrosis. Clin. Exp. Allergy 34, 567–575.
Kumar, R. K., Herbert, C., and Kasper, M. (2004b). Reversibility of airway inflammation and remodelling following cessation of antigenic challenge in a model of chronic asthma. Clin. Exp. Allergy 34, 1796–1802.
Kumar, R. K., Herbert, C., Webb, D. C., Li, L., and Foster, P. S. (2004c). Effects of anticytokine therapy in a mouse model of chronic asthma. Am. J. Respir. Crit. Care Med. 170, 1043–1048.
Kumar, R. K., Herbert, C., Yang, M., Koskinen, A. M. L., McKenzie, A. N. J., and Foster, P. S. (2002). Role of interleukin-13 in eosinophil accumulation and airway remodelling in a mouse model of chronic asthma. Clin. Exp. Allergy 32, 1104–1111.
Kumar, R. K., and Jeffery, P. K. (2012). “Pathology of asthma,” in Middleton's Allergy, 8th Edn. eds N. F. Adkinson, B. S. Bochner, W. Burks, W. W. Busse, S. T. Holgate, R. F. Lemanske and R. E. O'Hehir (Cambridge: Elsevier) (in press).
Kumar, R. K., Temelkovski, J., McNeil, H. P., and Hunter, N. (2000). Airway inflammation in a murine model of chronic asthma: evidence for a local humoral immune response. Clin. Exp. Allergy 30, 1486–1492.
Laitinen, A., Altraja, A., Kampe, M., Linden, M., Virtanen, I., and Laitinen, L. A. (1997). Tenascin is increased in airway basement membrane of asthmatics and decreased by an inhaled steroid. Am. J. Respir. Crit. Care Med. 156, 951–958.
Laitinen, L. A., Laitinen, A., and Haahtela, T. (1993). Airway mucosal inflammation even in patients with newly diagnosed asthma. Am. Rev. Respir. Dis. 147, 697–704.
Leigh, R., Ellis, R., Wattie, J., Southam, D. S., De Hoogh, M., Gauldie, J., O'Byrne, P. M., and Inman, M. D. (2002). Dysfunction and remodeling of the mouse airway persist after resolution of acute allergen-induced airway inflammation. Am. J. Respir. Cell Mol. Biol. 27, 526–535.
Li, X., and Wilson, J. W. (1997). Increased vascularity of the bronchial mucosa in mild asthma. Am. J. Respir. Crit. Care Med. 156, 229–233.
Locke, N. R., Royce, S. G., Wainewright, J. S., Samuel, C. S., and Tang, M. L. (2007). Comparison of airway remodeling in acute, subacute, and chronic models of allergic airways disease. Am. J. Respir. Cell Mol. Biol. 36, 625–632.
Long, A. J., Sypek, J. P., Askew, R., Fish, S. C., Mason, L. E., Williams, C. M., and Goldman, S. J. (2006). Gob-5 contributes to goblet cell hyperplasia and modulates pulmonary tissue inflammation. Am. J. Respir. Cell Mol. Biol. 35, 357–365.
Lundblad, L. K., Thompson-Figueroa, J., Allen, G. B., Rinaldi, L., Norton, R. J., Irvin, C. G., and Bates, J. H. (2007). Airway hyperresponsiveness in allergically inflamed mice: the role of airway closure. Am. J. Respir. Crit. Care Med. 175, 768–774.
Mathew, A., Maclean, J. A., Dehaan, E., Tager, A. M., Green, F. H., and Luster, A. D. (2001). Signal transducer and activator of transcription 6 controls chemokine production and T helper cell type 2 trafficking in allergic pulmonary inflammation. J. Exp. Med. 193, 1087–1096.
McMillan, S. J., Xanthou, G., and Lloyd, C. M. (2005). Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J. Immunol. 174, 5774–5780.
Nakanishi, A., Morita, S., Iwashita, H., Sagiya, Y., Ashida, Y., Shirafuji, H., Fujisawa, Y., Nishimura, O., and Fujino, M. (2001). Role of gob-5 in mucus overproduction and airway hyperresponsiveness in asthma. Proc. Natl. Acad. Sci. U.S.A. 98, 5175–5180.
Nials, A. T., and Uddin, S. (2008). Mouse models of allergic asthma: acute and chronic allergen challenge. Dis. Models. Mech. 1, 213–220.
Ohkawara, Y., Lei, X. F., Stampfli, M. R., Marshall, J. S., Xing, Z., and Jordana, M. (1997). Cytokine and eosinophil responses in the lung, peripheral blood, and bone marrow compartments in a murine model of allergen-induced airways inflammation. Am. J. Respir. Cell Mol. Biol. 16, 510–520.
Ordonez, C. L., Khashayar, R., Wong, H. H., Ferrando, R., Wu, R., Hyde, D. M., Hotchkiss, J. A., Zhang, Y., Novikov, A., Dolganov, G., and Fahy, J. V. (2001). Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am. J. Respir. Crit. Care Med. 163, 517–523.
Persson, C. G. (2002). Con: mice are not a good model of human airway disease. Am. J. Respir. Crit. Care Med. 166, 6–7.
Pini, L., Hamid, Q., Shannon, J., Lemelin, L., Olivenstein, R., Ernst, P., Lemiere, C., Martin, J. G., and Ludwig, M. S. (2007). Differences in proteoglycan deposition in the airways of moderate and severe asthmatics. Eur. Respir. J. 29, 71–77.
Pitchford, S. C., Riffo-Vasquez, Y., Sousa, A., Momi, S., Gresele, P., Spina, D., and Page, C. P. (2004). Platelets are necessary for airway wall remodeling in a murine model of chronic allergic inflammation. Blood 103, 639–647.
Sakai, K., Yokoyama, A., Kohno, N., Hamada, H., and Hiwada, K. (2001). Prolonged antigen exposure ameliorates airway inflammation but not remodeling in a mouse model of bronchial asthma. Int. Arch. Allergy Immunol. 126, 126–134.
Salvato, G. (2001). Quantitative and morphological analysis of the vascular bed in bronchial biopsy specimens from asthmatic and non-asthmatic subjects. Thorax 56, 902–906.
Shaver, J. R., Zangrilli, J. G., Cho, S. K., Cirelli, R. A., Pollice, M., Hastie, A. T., Fish, J. E., and Peters, S. P. (1997). Kinetics of the development and recovery of the lung from IgE-mediated inflammation: dissociation of pulmonary eosinophilia, lung injury, and eosinophil-active cytokines. Am. J. Respir. Crit. Care Med. 155, 442–448.
Shinagawa, K., Martin, J. A., Ploplis, V. A., and Castellino, F. J. (2007). Coagulation factor Xa modulates airway remodeling in a murine model of asthma. Am. J. Respir. Crit. Care Med. 175, 136–143.
Shore, S. A. (2003). Modeling airway remodeling: the winner by a nose? Am. J. Respir. Crit. Care Med. 168, 910–911.
Siegle, J. S., Hansbro, N., Herbert, C., Rosenberg, H. F., Domachowske, J. B., Asquith, K. L., Foster, P. S., and Kumar, R. K. (2010). Early-life viral infection and allergen exposure interact to induce an asthmatic phenotype in mice. Respir. Res. 11, 14.
Siegle, J. S., Hansbro, N., Herbert, C., Yang, M., Foster, P. S., and Kumar, R. K. (2006). Airway hyperreactivity in exacerbation of chronic asthma is independent of eosinophilic inflammation. Am. J. Respir. Cell Mol. Biol. 35, 565–570.
Sont, J. K., Han, J., Van Krieken, J. M., Evertse, C. E., Hooijer, R., Willems, L. N., and Sterk, P. J. (1996). Relationship between the inflammatory infiltrate in bronchial biopsy specimens and clinical severity of asthma in patients treated with inhaled steroids. Thorax 51, 496–502.
Su, X., Taniuchi, N., Jin, E., Fujiwara, M., Zhang, L., Ghazizadeh, M., Tashimo, H., Yamashita, N., Ohta, K., and Kawanami, O. (2008). Spatial and phenotypic characterization of vascular remodeling in a mouse model of asthma. Pathobiology 75, 42–56.
Takayama, G., Arima, K., Kanaji, T., Toda, S., Tanaka, H., Shoji, S., McKenzie, A. N., Nagai, H., Hotokebuchi, T., and Izuhara, K. (2006). Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J. Allergy Clin. Immunol. 118, 98–104.
Tanaka, H., Masuda, T., Tokuoka, S., Komai, M., Takahashi, T., and Nagai, H. (2001). The effect of allergen-induced airway inflammation on airway remodeling in a murine model of allergic asthma. Inflamm. Res. 50, 616–624.
Temelkovski, J., Hogan, S. P., Shepherd, D. P., Foster, P. S., and Kumar, R. K. (1998). An improved murine model of asthma: selective airway inflammation, epithelial lesions and increased methacholine responsiveness following chronic exposure to aerosolised allergen. Thorax 53, 849–856.
Wagers, S., Lundblad, L. K., Ekman, M., Irvin, C. G., and Bates, J. H. (2004). The allergic mouse model of asthma: normal smooth muscle in an abnormal lung? J. Appl. Physiol. 96, 2019–2027.
Wang, Y., and McCusker, C. (2006). Neonatal exposure with LPS and/or allergen prevents experimental allergic airways disease: development of tolerance using environmental antigens. J. Allergy Clin. Immunol. 118, 143–151.
Wegmann, M., Fehrenbach, H., Fehrenbach, A., Held, T., Schramm, C., Garn, H., and Renz, H. (2005). Involvement of distal airways in a chronic model of experimental asthma. Clin. Exp. Allergy 35, 1263–1274.
Wenzel, S., and Holgate, S. T. (2006). The mouse trap: it still yields few answers in asthma. Am. J. Respir. Crit. Care Med. 174, 1173–1176; discussion 1176–1178.
Woodruff, P. G., Dolganov, G. M., Ferrando, R. E., Donnelly, S., Hays, S. R., Solberg, O. D., Carter, R., Wong, H. H., Cadbury, P. S., and Fahy, J. V. (2004). Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am. J. Respir. Crit. Care Med. 169, 1001–1006.
Woodruff, P. G., Modrek, B., Choy, D. F., Jia, G., Abbas, A. R., Ellwanger, A., Koth, L. L., Arron, J. R., and Fahy, J. V. (2009). T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 180, 388–395.
Yang, G., Volk, A., Petley, T., Emmell, E., Giles-Komar, J., Shang, X., Li, J., Das, A. M., Shealy, D., Griswold, D. E., and Li, L. (2004). Anti-IL-13 monoclonal antibody inhibits airway hyperresponsiveness, inflammation and airway remodeling. Cytokine 28, 224–232.
Yang, M., Kumar, R. K., and Foster, P. S. (2010). Interferon-gamma and pulmonary macrophages contribute to the mechanisms underlying prolonged airway hyperresponsiveness. Clin. Exp. Allergy 40, 163–173.
Yiamouyiannis, C. A., Schramm, C. M., Puddington, L., Stengel, P., Baradaran-Hosseini, E., Wolyniec, W. W., Whitely, H. E., and Thrall, R. S. (1999). Shifts in lung lymphocyte profiles correlate with the sequential development of acute allergic and chronic tolerant stages in a murine asthma model. Am. J. Pathol. 154, 1911–1921.
Yung, S., Durham, S. R., Corrigan, C. J., Hamid, Q., and Kay, A. B. (1995). Phenotype of cells expressing mRNA for TH2-type (interleukin 4 and interleukin 5) and TH1-type (interleukin 2 and interferon gamma) cytokines in bronchoalveolar lavage and bronchial biopsies from atopic asthmatic and normal control subjects. Am. J. Respir. Cell Mol. Biol. 12, 477–487.
Keywords: airway hyper-responsiveness, airway inflammation, airway remodeling, animal models, asthma
Citation: Kumar RK and Foster PS (2012) Are mouse models of asthma appropriate for investigating the pathogenesis of airway hyper-responsiveness? Front. Physio. 3:312. doi: 10.3389/fphys.2012.00312
Received: 28 May 2012; Paper pending published: 27 June 2012;
Accepted: 15 July 2012; Published online: 31 July 2012.
Edited by:
Julia K. L. Walker, Duke University Medical Center, USAReviewed by:
Nicola Scichilone, University of Palermo, ItalyAhmed El-Hashim, Kuwait University, Kuwait
Copyright © 2012 Kumar and Foster. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Rakesh K. Kumar, Department of Pathology, School of Medical Sciences, University of New South Wales, Sydney, NSW 2052, Australia. e-mail:ci5rdW1hckB1bnN3LmVkdS5hdQ==