
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 09 July 2012
Sec. Cardiac Electrophysiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00252
This article is part of the Research Topic The Effects of Omega-3 Polyunsaturated Fatty Acids on Cardiac Rhythm: Antiarrhythmic, Proarrhythmic, Both or Neither? View all 17 articles
 Zhenghang Zhao1,2*
Zhenghang Zhao1,2* Hairuo Wen2 Nadezhda Fefelova2 Charelle Allen2 Nancy Guillaume2
Hairuo Wen2 Nadezhda Fefelova2 Charelle Allen2 Nancy Guillaume2 Dandan Xiao1 Chen Huang1
Dandan Xiao1 Chen Huang1 Weijin Zang1 Judith K. Gwathmey3,4
Weijin Zang1 Judith K. Gwathmey3,4 Lai-Hua Xie2*
Lai-Hua Xie2*Accumulating evidence has suggested that ω3-polyunsaturated fatty acids (ω3-PUFAs) may have beneficial effects in the prevention/treatment of cardiovascular diseases, while controversies still remain regarding their anti-arrhythmic potential. It is not clear yet whether ω-3-PUFAs can suppress early afterdepolarizations (EADs) induced by oxidative stress. In the present study, we recorded action potentials using the patch-clamp technique in ventricular myocytes isolated from rabbit hearts. The treatment of myocytes with H2O2 (200 μM) prolonged AP durations and induced EADs, which were significantly suppressed by docosahexaenoic acid (DHA, 10 or 25 μM; n = 8). To reveal the ionic mechanisms, we examined the effects of DHA on L-type calcium currents (ICa.L), late sodium (INa), and transient outward potassium currents (Ito) in ventricular myocytes pretreated with H2O2. H2O2 (200 μM) increased ICa.L by 46.4% from control (−8.4 ± 1.4 pA/pF) to a peak level (−12.3 ± 1.8 pA/pF, n = 6, p < 0.01) after 6 min of H2O2 perfusion. H2O2-enhanced ICa.L was significantly reduced by DHA (25 μM; −7.1 ± 0.9 pA/pF, n = 6, p < 0.01). Similarly, H2O2-increased the late INa (−3.2 ± 0.3 pC) from control level (−0.7 ± 0.1 pC). DHA (25 μM) completely reversed the H2O2-induced increase in late INa (to −0.8 ± 0.2 pC, n = 5). H2O2 also increased the peak amplitude of and the steady state Ito from 8.9 ± 1.0 and 2.16 ± 0.25 pA/pF to 12.8 ± 1.21 and 3.13 ± 0.47 pA/pF respectively (n = 6, p < 0.01, however, treatment with DHA (25 μM) did not produce significant effects on current amplitudes and dynamics of Ito altered by H2O2. In addition, DHA (25 μM) did not affect the increase of intracellular reactive oxygen species (ROS) levels induced by H2O2 in rabbit ventricular myocytes. These findings demonstrate that DHA suppresses exogenous H2O2-induced EADs mainly by modulating membrane ion channel functions, while its direct effect on ROS may play a less prominent role.
Extensive studies on the potential effects of fish oil omega-3 poly unsaturated fatty acids (ω-3 PUFA) on cardiac rhythm have provided controversial results (von Schacky, 2008). While some interventional studies reported either no effect or even promotion of arrhythmias in some subgroups of patients with heart disease (Raitt et al., 2005; Coronel et al., 2007; Den Ruijter et al., 2007; Cheng and Santoni, 2008), other studies have reported beneficial effects of ω-3-PUFAs on cardiac rhythm resulting in a reduction in the incidence of sudden cardiac death or mortality (London et al., 2007; Cheng and Santoni, 2008; Nodari et al., 2009). It seems that fish oil fatty acids may exert either pro- or anti-arrhythmic effects, probably depending on different underlying mechanisms for the arrhythmias. Recent studies have also shown ω-3-PUFAs suppress afterdepolarizations and triggered activities induced by K channel blockers or by β-adrenergic stimulation in failing hearts (Den Ruijter et al., 2006, 2008; Berecki et al., 2007; Smith et al., 2009). However, it is unclear whether ω-3-PUFAs have protective effects on arrhythmias induced by oxidative stress. Reactive oxygen species (ROS) have recently been implicated in the pathogenesis of cardiac arrhythmia during ischemic-reperfusion, aging, and heart failure. Oxidative stress caused by exogenous H2O2 induces early afterdepolarizations (EADs) and delayed afterdepolarizations (DADs) that may in turn trigger lethal arrhythmias. These afterdepolarizations are a result of a net increase in inward current, which is induced by activation of late sodium current (INa) and the L-type calcium current (ICaL) via oxidized Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII; Ward and Giles, 1997; Xie et al., 2009; Zhao et al., 2011). Our most recent study suggested that the transient outward potassium current (Ito) may also facilitate EAD generation by H2O2 (Zhao et al., 2012b). In the present study, we aim to assess the effects of docosahexaenoic acid (DHA, one of ω-3-PUFAs) on exogenous H2O2-induced EADs, and to further reveal potential underlying ionic mechanisms.
This investigation conforms to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 85–23, Revised 1996). All animal experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Medicine and Dentistry of New Jersey-New Jersey Medical School and by the Ethical Committee of Xi’an Jiaotong University. All experiments were performed at 35–37°C.
Ventricular myocytes were enzymatically isolated from the hearts of New Zealand white rabbits (male, 1.8–2.5 kg) as described previously (Xie et al., 2009). Briefly, after rabbits were anesthetized with intravenous pentobarbital hearts were removed and perfused retrogradely at 37°C in Langendorff fashion with nominally Ca2+-free Tyrode’s solution containing 1.4 mg/mL collagenase (Type II; Worthington) and 0.1 mg/ml protease (type XIV, Sigma) for 25–30 min. The hearts were removed from the perfusion apparatus after washing out the enzyme solution, the left ventricles were gently teased apart with forceps in a Petri dish and the myocytes were filtered through a nylon mesh. The Ca2+ concentration was gradually increased to 1.8 mM, and the cells were stored at room temperature and used within 8 h.
Myocytes were current-or voltage-clamped using the perforated whole-cell patch-clamp technique (240 μg/ml amphotericin B; Rae et al., 1991) for recordings of action potential, or ICa.L, Ito, and late INa. Voltage or current signals were measured with a MultiClamp 700A patch-clamp amplifier controlled by a personal computer using a Digidata 1322 acquisition board driven by pCLAMP 10 software (Molecular Devices, Sunnyvale, CA, USA).
To record action potentials (APs), patch pipettes (resistance 2–4 MΩ) were filled with an internal solution containing (in mM): 110 K-aspartate, 30 KCl, 5 NaCl, 10 HEPES, 0.1 EGTA, 5 MgATP, 5 Na2-phosphocreatine, 0.05 cAMP, pH was adjusted to 7.2 with KOH. The cells were superfused with Tyrode’s solution containing (in mM): 136 NaCl, 4.0 KCl, 0.33 Na2PO4, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH was adjusted to 7.4 with NaOH. APs were elicited with 2 ms, 2 to 4 nA square pulses at a pacing cycle length (PCL) of 6 s.
To record the ICa,L, patch pipettes (2–4 MΩ) were filled with an internal solution containing (in mM): 110 Cs-Aspartate, 30 CsCl, 5 NaCl, 10 HEPES, 0.1 EGTA, 5 MgATP, 5 Na2-phosphocreatine, 0.05 cAMP, pH 7.2 with CsOH, and the cells were perfused with a modified Tyrode’s solution, in which KCl was replaced with CsCl. The myocytes were stimulated at a PCL of 6 s with a double-pulse protocol. Following a 100-ms prepulse to −40 mV from the holding potential of −80 mV (to inactivate Na+ current and T-type Ca2+ current), ICa.L was elicited by a subsequent test depolarization step to 0 mV for 300 ms.
Late INa was measured as described previously (Song et al., 2006). Glass pipettes (1–2 MΩ) were filled with an internal solution containing (in mM): 110 Cs-Aspartate, 30 CsCl, 10 HEPES, 0.5 EGTA, 0.2 Na3-GTP, 5 Na2-phosphocreatine-, 5 MgATP, pH 7.2 was adjusted with CsOH. Myocytes were bathed with a modified Tyrode’s solution in which KCl was replaced with CsCl. Nifedipine (30 μM) was added to the bath solution to block calcium channels. Late INa was elicited by 300 ms voltage-clamp pulses from −90 to −30 mV at a PCL of 6 s from a holding potential of −80 mV.
To record Ito, the pipette and superfusion solutions were the same as those for AP recording. Tetrodotoxin (TTX, 10 μM) and CdCl2 (0.5 mM) were added into the Tyrode’s solution to inhibit INa and ICa,L. Ito was evoked by 400 ms depolarizing pulses to test potentials between −40 and +50 mV (0.1 Hz). The holding potential was set at −80 mV and a 100 ms prepulse was applied to −60 mV to inactivate the INa. Ito recovery from inactivation was investigated using a conventional two-pulse protocol: an inactivating pulse depolarizing to +50 mV for 400 ms (P1) followed by a variable recovery interval and subsequent +50 mV test pulse (P2). The inactivation of Ito and recovery from inactivation were best fit with a double exponential equation. All electrophysiological data were normalized as current densities by dividing measured current amplitude by whole-cell capacitance.
All chemicals were purchased from Sigma-Aldrich unless indicated. Because DHA is very sensitive to oxidation, DHA (Sigma-Aldrich) was dissolved in 100% ethanol under N2 and kept at −20°C in the dark. Immediately before use, the DHA stock solution was diluted in the bath solution to reach the final concentrations needed. The maximum final concentration (0.1%) of ethanol had no effect on membrane currents.
The myocytes were incubated with 5 μM C-DCDHF-DA-AM (Invitrogen) for 30 min. C-DCDHF- DA is oxidized by ROS to dichlorofluorescein (DCF). ROS fluorescence (emission: ∼530 nm) was measured by a 200 ms-exposure (excitation: ∼480 nm) every 30 s using the Andor Ixon charge-coupled device camera. Recordings were started after a stable baseline was achieved.
Data are presented as mean ± SEM. Differences were tested for statistical significance by using paired or unpaired Student’s t tests, with p < 0.05 considered significant.
Action potentials were recorded from single ventricular myocytes isolated from rabbit hearts using the perforated whole-cell patch-clamp technique under current-clamp mode. In order to reliably induce EADs, the cells were paced at a PCL of 6 s based on our previous studies (Sato et al., 2009; Xie et al., 2009; Zhao et al., 2012a). The average APD90 of rabbit ventricular myocytes is 266 ± 23 ms (n = 8) at base line. After APD and morphology reached steady state, the cells were perfused with 200 μM H2O2 until EADs consistently appeared. Consecutively, DHA at either 10 or 25 μM was added in the presence of H2O2. The sudden and dramatic increase in APD90 in Figure 1A indicates the incidence of EADs. As shown in Figures 1A,B, EADs were consistently induced by H2O2 at 5 min after perfusion. DHA (25 μM) shortened the APD prolongation from 894 ± 78 ms to 278 ± 52 ms, and significantly suppressed the frequency of EADs induced by H2O2. The incidence of EADs was assessed by counting the number of EADs within 10 APs (from eight cells) in control, after H2O2 (200 μM) and H2O2 (200 μM) + DHA (at 10 or 25 μM). The incidence of EAD was suppressed in all tested cells (n = 8), five of which showed complete abolishment of EADs after 3–5 min of treatment with 25 μM DHA. As summarized in Figure 1C, the incidence of H2O2-induced EADs were significantly reduced by direct perfusion of DHA at both 10 and 25 μM, in a dose-dependent manner (p < 0.05 and p < 0.01, respectively, Fisher’s exact test).
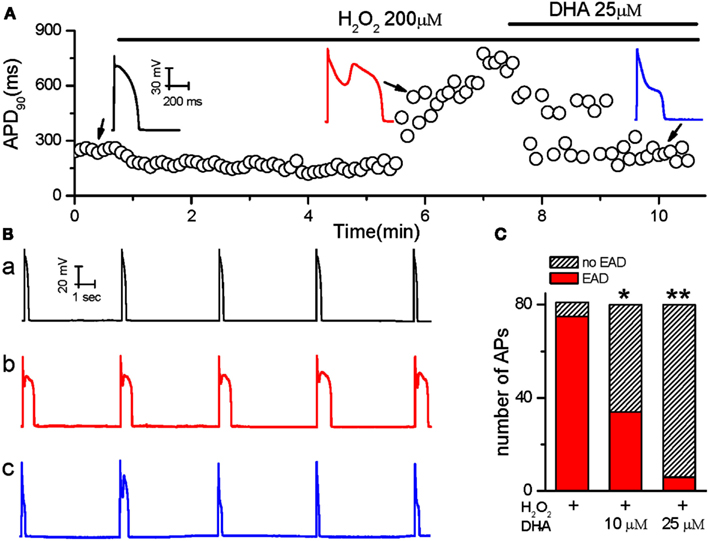
Figure 1. The inhibitory effects of DHA on Early afterdepolarizations (EADs) induced by H2O2. (A) Values of consecutive APD90 are plotted over time. The ventricular myocyte was treated with H2O2 and DHA as indicated by the horizontal bars above the plot. Three representative AP recordings under different conditions are shown in the insets. (B) Five consecutive AP recordings from a cell exposed to control perfusate (a), 200 μM H2O2(b) and 200 μM H2O2 + 25 μM DHA (c). (C) Summarized bar graph showing dose-dependent inhibitory effects of DHA on the incidence of EADs induced by H2O2 (n = 8 cells). *p < 0.05, **p < 0.01; Fisher’s Exact Test vs. H2O2.
Our previous studies have shown that reactivation of ICa.L plays a key role in H2O2-induced EAD in rabbit ventricular myocytes (Xie et al., 2009; Song et al., 2010). Therefore, we first assessed the potential involvement of ICa.L in the inhibitory effect of DHA on H2O2-induced EADs. ICa.L was recorded in rabbit ventricular myocytes using the perforated whole-cell patch-clamp technique under voltage-clamp mode. As shown in Figure 2A, H2O2 (200 μM) gradually increased the amplitude of ICa.L at both peak and late phases (at ∼ 250 ms), which reached the steady state at 5–7 min, consistent with the time course for EAD induction as shown in Figure 1. The I-V relations for the peak current (Figure 2B) showed that the ICa.L amplitude was pronouncedly increased at testing potentials −10 to +40 mV. For example a 46.4% enhancement was caused at 0 mV, i.e., from −8.4 ± 1.4 to −12.3 ± 1.8 pA/pF (n = 6, p < 0.01). DHA (25 μM) significantly suppressed/reversed the elevation of the ICa,L amplitude (e.g., to −7.1 ± 0.9 pA/pF at 0 mV; n = 6, p < 0.01 compared to H2O2-induced effect). In order to test the DHA effect on ICa.L under normal membrane potential conditions, we also performed AP-clamp experiments. As shown in Figures 2C,D, DHA markedly decreased both the peak and the late phase of ICa.L, which were enhanced by H2O2, under AP-clamp conditions.
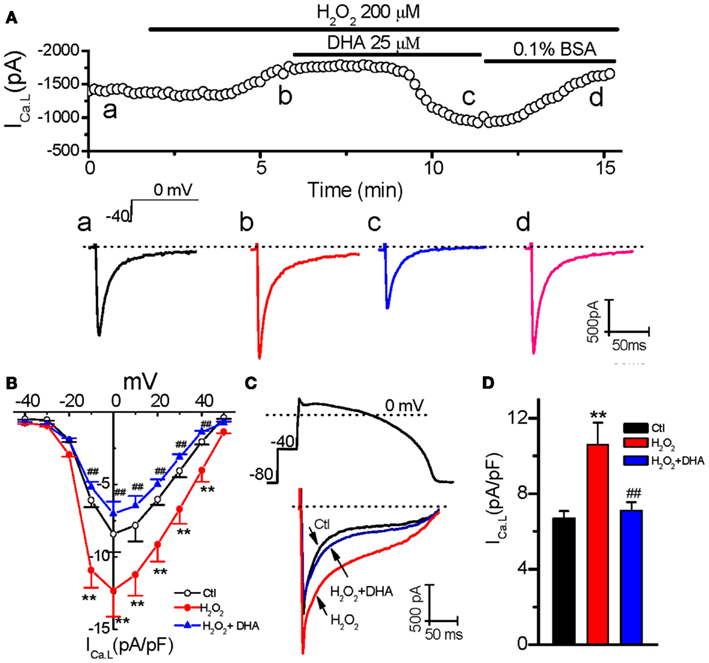
Figure 2. Inhibitory effects of DHA on ICa.L enhanced by H2O2. (A) Time course of peak ICa,L in a myocyte treated with 200 μM H2O2 in the absence and presence of 25 μM DHA, and 0.1% bovine serum albumin (BSA). Representative traces of ICa,L corresponding to points a–d are shown under the plot. (B) The current-voltage relations for peak ICa,L from six cells treated with 200 μM H2O2 in the absence and presence of 25 μM DHA. Test potentials ranged from −40 to +50 mV in 10 mV steps. (C) An AP-clamp waveform (above) and superimposed current traces showing ICa,L under control (Ctl), in the presence of 200 μM H2O2, and DHA(25 μM) + H2O2 are shown respectively. (D) The late phase currents measured at 30-150 ms after the upstroke in (C) were summarized showing an inhibitory effect of DHA on the enhancement of ICa,L by H2O2 (n = 6). **p < 0.01 vs. control; ## p < 0.01 vs. H2O2 group.
Since the activation of late INa also contributes to EAD generation induced by H2O2 (Ward and Giles, 1997; Xie et al., 2009), we next evaluated the effect of DHA on H2O2-enhanced late INa. Late INa was elicited by 300 ms voltage-clamp pulses from −90 to −30 mV at a PCL of 6 s. The magnitude of late INa was evaluated by integration of the area (nA × ms = pC) of the current over the last 50 ms of the −30 mV depolarizing pulse, using the integration (area) feature of the pCLAMP program. As shown in Figure 3, the late current component was significantly enhanced by H2O2 (200 μM) from −0.7 ± 0.1 pC to −3.2 ± 0.3 pC (n = 5, p < 0.01) at 4–6 min after perfusion, when it reaches steady state level. This elevation was completely suppressed by Tetrodotoxin (TTX, 10 μM), a selective INa inhibitor, confirming this late sustained inward current is due to late INa, although we cannot exclude minor contaminations on the baseline current from other currents such as Na-Ca exchange current (INCX), ICa,L or leaky current. H2O2-increased late INa was effectively attenuated by 25 μM DHA (to −0.8 ± 0.2 pC at 2–4 min after DHA application, n = 5, p < 0.01).
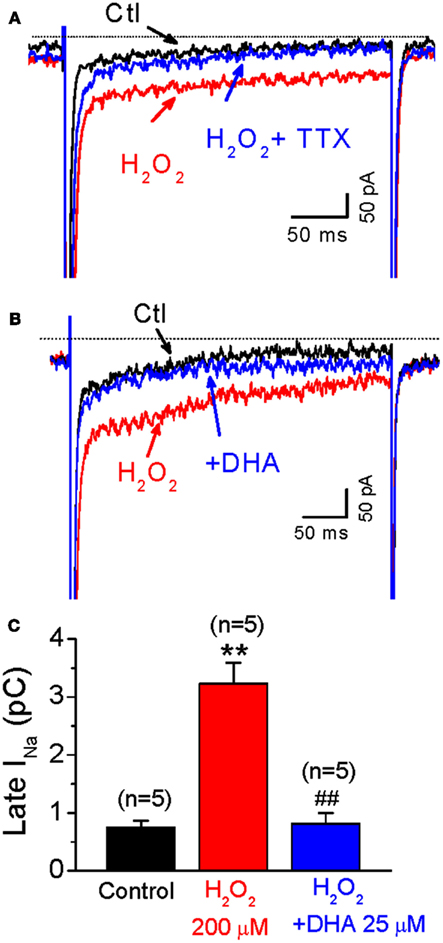
Figure 3. Inhibitory effects of DHA on late INa enhanced by H2O2. (A) Representative INa traces under control condition (Ctl), in the presence of 200 μM H2O, and H2O2 + TTX (10 μM), respectively. (B) Representative INa traces under control condition (Ctl), in the presence of 200 μM H2O, and H2O2 + DHA (25 μM), respectively. (C) A bar graph summarizing 200 μM H2O2-induced increase of late INa, which is significantly suppressed by 25 μM DHA. **p < 0.01 vs. control; ## p < 0.01 vs. H2O2 group.
Consistent with our recent finding (Zhao et al., 2012b), H2O2 (200 μM) increased the amplitudes of both peak (from 8.94 ± 1.07 to 12.8 ± 1.21 pA/pF at testing potential of 50 mV, n = 6, p < 0.01) and steady state (late phase at the end of 400 ms pulse; from 2.16 ± 0.25 to 3.13 ± 0.47 pA/pF, n = 6, p < 0.01) component of Ito. Additionally, H2O2 also slowed inactivation (τs, in from 96.6 ± 4.3 to 158.1 ± 5.7 ms; τf, in from 17.4 ± 1.7 to 24.7 ± 14.0 ms, n = 7, p < 0.01). However, DHA at 25 μM, the concentration which dramatically suppressed H2O2-induced EADs, did not show any significant effects on current amplitudes (peak Ito = 12.51 ± 1.47 pA/pF; Ito.ss = 3.34 ± 0.31 pA/pF, n = 6, p > 0.05 compared to H2O2, respectively) or inactivation process of Ito (τs, in: 154.6 ± 6.6 ms and τf, in: 23.9 ± 1.1 ms, n = 7, p > 0.05 compared to H2O2; Figures 4A–C). Furthermore, we found that H2O2 accelerated the recovery from inactivation of Ito mainly by decreasing the fast component (τf. re: from 817.2 ± 79.2 ms to 341.9 ± 26.1 ms, n = 7, p < 0.05), but not by changing the slow component (τs.re: from control 5335.4 ± 504.8 ms to H2O2 4963.2 ± 459.9 ms, p > 0.05). Similarly DHA (25 μM) did not cause any significant alteration in Ito recovery kinetics after H2O2 treatment (Figure 4D).
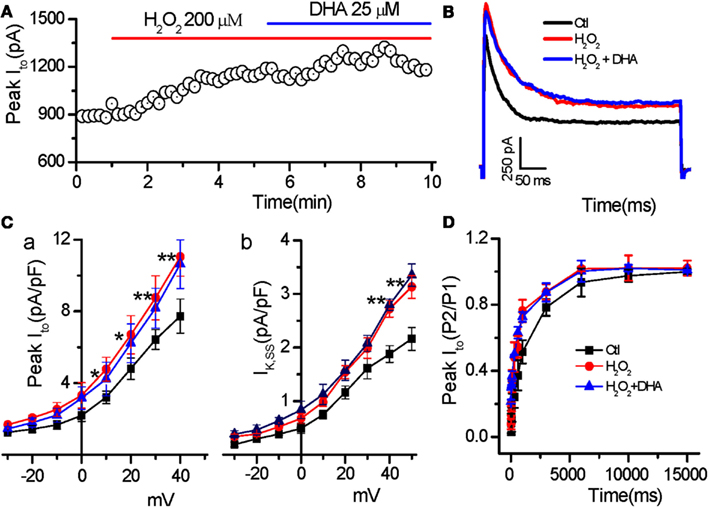
Figure 4. Less effect of DHA on Ito enhanced by H2O2. (A) Time course of peak Ito in a myocyte treated with H2O2 in the absence and presence of DHA. (B) Representative traces of the Ito under control, in the presence of H2O2 (200 μM), and H2O2 + DHA (25 μM), respectively. (C) Current–voltage relations of the peak Ito(C-a) and steady state currents (IK,SS, C-b) showing less effects of DHA on enhancement of peak Ito and IK.ss (n = 6, *p < 0.05, **p < 0.01 vs. control.). Test potentials ranged from −60 to +50 mV in 10 mV steps. (D) Recovery of Ito from inactivation showing no significant effect of DHA (25 μM) on the Ito recovery sped-up by H2O2 (200 μM; p > 0.05, n = 7).
The level of oxidative stress may either increase or decrease in tissues from humans and animals supplemented with fish oil as reported previously (Garrido et al., 1989; Mas et al., 2010; Tsuduki et al., 2011). To determine whether DHA reduces the incidence of EAD via affecting (decreasing) intracellular ROS, the effect of DHA on intracellular ROS levels was measured in isolated ventricular myocytes treated with exogenous H2O2 (200 μM) by monitoring CM-DCF fluorescence intensity. The effect of DHA on intracellular ROS levels in the absence of H2O2 was also measured. As shown in Figure 5, exogenous H2O2 produced a rapid and dramatic increase in DCF fluorescence intensity in the myocytes and the F/F0 of DCF fluorescence intensity reached a steady state value of 2.18 ± 0.24 at 6–10 min after H2O2 treatment. However, DHA (25 μM, either pretreatment or after treatment) showed no significant effect on the fluorescence of CM-DCF either in the absence or presence of H2O2 (2.21 ± 0.33, n = 8, p > 0.05).
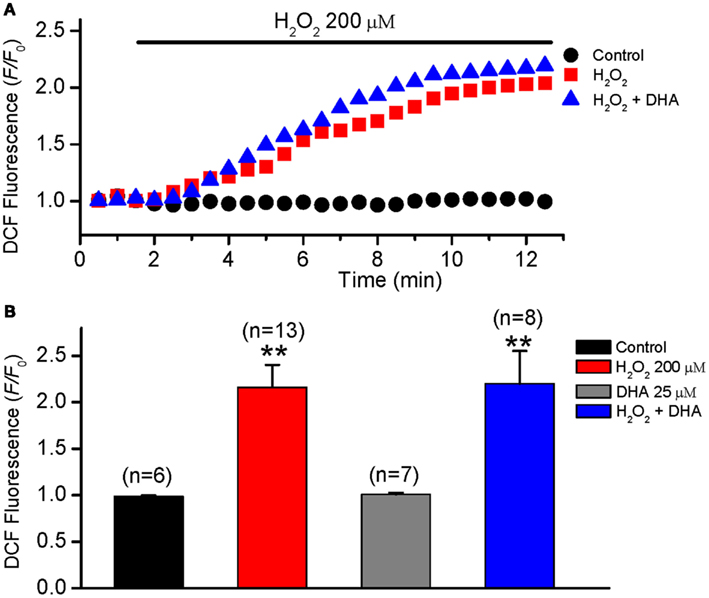
Figure 5. No effect of DHA on ROS levels in isolated rabbit ventricular myocytes. ROS levels were measured by monitoring DCF fluorescence intensity in isolated myocytes every 30 s in control, H2O2 (200 μM) and H2O2 + DHA (25 μM) groups. (A) Time courses of DCF fluorescence intensity (F/F0) in three representative myocytes from the three groups, respectively. (B) Histograms summarizing the DCF intensities for each group measured at 6 min after treatment of H2O2. **p < 0.01 compared to control. Numbers in parentheses indicate the number of cells in each group.
Experimental and clinical studies have obtained controversial results regarding the effects of fish oil or ω-3 PUFA on cardiac rhythm (von Schacky, 2008). Differences in the underlying pathogenic mechanisms for the arrhythmia in differing patient groups or animal models may account for these controversies. We and others have previously shown that both exogenous and endogenous ROS-induced EADs can serve as triggers for arrhythmias. In the present study, we provide the first evidence showing that DHA attenuates EADs induced by H2O2.
The molecular and ionic mechanisms of ion channel modulation by DHA are still not completely understood. A recent review article comprehensively summarized the potential antiarrhythmogenic electrophysiological effect of ω3-PUFAs on the heart (Richardson et al., 2011). Inhibitory effects of DHA on EADs may involve multifactorial mechanisms e.g., (1) via ROS modulation. Although ω3-PUFA may slightly increase levels of oxidative stress due to the susceptibility to oxidation, low to moderate ROS exposure can conversely give rise to up-regulation of antioxidant enzymes and increase antioxidant ability (scavenging ROS) in cardiac tissue (Jahangiri et al., 2006); (2) via direct modulation of ion channels by binding to the channels or affecting cell membrane lipid properties (such as membrane lipid peroxidation). While there is a widespread effect of ω3-PUFA on ion channels and ion pumps, Ca2+ and Na+ currents are most sensitive to ω3-PUFAs (Richardson et al., 2011). Nevertheless, our present data suggest that the ionic mechanisms underlying inhibitory effect of DHA on EADs most likely involve the direct inhibition on the ICa,L and late INa rather than its putative antioxidant ability. This notion was supported by the observation that there was no effect on CM-DCF fluorescence induced by DHA at the same concentration that led to reduction of EADs. In addition, the fast time course for DHA suppression of ICa,L and late INa also supports a mechanism of direct inhibition of ion channels by DHA. Our most recent data showed H2O2 also activates Ito and may facilitate EAD generation (Zhao et al., 2012b). In the present study, however, we showed that DHA did not reverse the Ito activated by H2O2 in rabbit ventricular myocytes, which is inconsistent with previous reports that DHA markedly reduces Ito in human atrial cells and rat ventricular myocytes even at lower concentrations (5–10 μM; Bogdanov et al., 1998; Verkerk et al., 2006; Li et al., 2009). We do not have a ready explanation for this discrepancy, while the molecular subtypes of Ito proteins might be different between rabbits and other species (including humans) or between different locations in the heart (e.g., ventricle vs. atria). In addition, the H2O2-activated Ito seemed to be more resistant to DHA than the Ito at baseline, since we observed the inhibitory effects of 25 μM DHA on Ito (up to ∼50%) in the absence of H2O2.
It has also been reported that n–3-PUFAs are capable of reducing the activity of CaMKII (Zaloga et al., 2006), which may partially account for the inhibitory effect of DHA on EADs. However, since DHA does not alter the ROS level in the presence of H2O2 (Figure 5), the reduction of CaMKII activity, if any, may be mediated by less Ca entry secondarily to ICa,L blockage, rather than by lower oxidation. Further experiments are needed identify the involvement of CaMKII.
Nevertheless, our present study suggests fish oil supplements may be effective in preventing/treating arrhythmias under an increased oxidative stress condition and serve as an alternative or complimentary anti-arrhythmic drug. Conditions with elevated oxidative stress level including ischemia/reperfusion, heart failure and aging might benefit from fish oil supplements.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by National Natural Science Foundation of China No. 30930105 (to Weijin Zang) and No. 81170597 (to Zhenghang Zhao), Major International (Regional) Joint Research Project of National Natural Science Foundation of China No. 81120108002 (to Weijin Zang), and NIH/NHLBI R01 HL97979 (to Lai-Hua Xie).
Berecki, G., Den Ruijter, H. M., Verkerk, A. O., Schumacher, C. A., Baartscheer, A., Bakker, D., Boukens, B. J., Van Ginneken, A. C., Fiolet, J. W., Opthof, T., and Coronel, R. (2007). Dietary fish oil reduces the incidence of triggered arrhythmias in pig ventricular myocytes. Heart Rhythm 4, 1452–1460.
Bogdanov, K. Y., Spurgeon, H. A., Vinogradova, T. M., and Lakatta, E. G. (1998). Modulation of the transient outward current in adult rat ventricular myocytes by polyunsaturated fatty acids. Am. J. Physiol. 274, H571–H579.
Cheng, J. W., and Santoni, F. (2008). Omega-3 fatty acid: a role in the management of cardiac arrhythmias? J. Altern. Complement. Med. 14, 965–974.
Coronel, R., Wilms-Schopman, F. J., Den Ruijter, H. M., Belterman, C. N., Schumacher, C. A., Opthof, T., Hovenier, R., Lemmens, A. G., Terpstra, A. H., Katan, M. B., and Zock, P. (2007). Dietary n-3 fatty acids promote arrhythmias during acute regional myocardial ischemia in isolated pig hearts. Cardiovasc. Res. 73, 386–394.
Den Ruijter, H. M., Berecki, G., Opthof, T., Verkerk, A. O., Zock, P. L., and Coronel, R. (2007). Pro- and antiarrhythmic properties of a diet rich in fish oil. Cardiovasc. Res. 73, 316–325.
Den Ruijter, H. M., Berecki, G., Verkerk, A. O., Bakker, D., Baartscheer, A., Schumacher, C. A., Belterman, C. N., De Jonge, N., Fiolet, J. W., Brouwer, I. A., and Coronel, R. (2008). Acute administration of fish oil inhibits triggered activity in isolated myocytes from rabbits and patients with heart failure. Circulation 117, 536–544.
Den Ruijter, H. M., Verkerk, A. O., Berecki, G., Bakker, D., Van Ginneken, A. C., and Coronel, R. (2006). Dietary fish oil reduces the occurrence of early afterdepolarizations in pig ventricular myocytes. J. Mol. Cell. Cardiol. 41, 914–917.
Garrido, A., Garrido, F., Guerra, R., and Valenzuela, A. (1989). Ingestion of high doses of fish oil increases the susceptibility of cellular membranes to the induction of oxidative stress. Lipids 24, 833–835.
Jahangiri, A., Leifert, W. R., Kind, K. L., and McMurchie, E. J. (2006). Dietary fish oil alters cardiomyocyte Ca2+ dynamics and antioxidant status. Free Radic. Biol. Med. 40, 1592–1602.
Li, G. R., Sun, H. Y., Zhang, X. H., Cheng, L. C., Chiu, S. W., Tse, H. F., and Lau, C. P. (2009). Omega-3 polyunsaturated fatty acids inhibit transient outward and ultra-rapid delayed rectifier K+ currents and Na+ current in human atrial myocytes. Cardiovasc. Res. 81, 286–293.
London, B., Albert, C., Anderson, M. E., Giles, W. R., Van Wagoner, D. R., Balk, E., Billman, G. E., Chung, M., Lands, W., Leaf, A., McAnulty, J., Martens, J. R., Costello, R. B., and Lathrop, D. A. (2007). Omega-3 fatty acids and cardiac arrhythmias: prior studies and recommendations for future research: a report from the National Heart, Lung, and Blood Institute and Office Of Dietary Supplements Omega-3 Fatty Acids and their Role in Cardiac Arrhythmogenesis Workshop. Circulation 116, e320–e335.
Mas, E., Woodman, R. J., Burke, V., Puddey, I. B., Beilin, L. J., Durand, T., and Mori, T. A. (2010). The omega-3 fatty acids EPA and DHA decrease plasma F(2)-isoprostanes: results from two placebo-controlled interventions. Free Radic. Res. 44, 983–990.
Nodari, S., Metra, M., Milesi, G., Manerba, A., Cesana, B. M., Gheorghiade, M., and Dei Cas, L. (2009). The role of n-3 PUFAs in preventing the arrhythmic risk in patients with idiopathic dilated cardiomyopathy. Cardiovasc. Drugs Ther. 23, 5–15.
Rae, J., Cooper, K., Gates, P., and Watsky, M. (1991). Low access resistance perforated patch recordings using amphotericin B. J. Neurosci. Methods 37, 15–26.
Raitt, M. H., Connor, W. E., Morris, C., Kron, J., Halperin, B., Chugh, S. S., McClelland, J., Cook, J., MacMurdy, K., Swenson, R., Connor, S. L., Gerhard, G., Kraemer, D. F., Oseran, D., Marchant, C., Calhoun, D., Shnider, R., and McAnulty, J. (2005). Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: a randomized controlled trial. JAMA 293, 2884–2891.
Richardson, E. S., Iaizzo, P. A., and Xiao, Y. F. (2011). Electrophysiological mechanisms of the anti-arrhythmic effects of omega-3 fatty acids. J. Cardiovasc. Transl. Res. 4, 42–52.
Sato, D., Xie, L. H., Sovari, A. A., Tran, D. X., Morita, N., Xie, F., Karagueuzian, H., Garfinkel, A., Weiss, J. N., and Qu, Z. (2009). Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc. Natl. Acad. Sci. U.S.A. 106, 2983–2988.
Smith, P. J., Blumenthal, J. A., Babyak, M. A., Georgiades, A., Sherwood, A., Sketch, M. H. Jr., and Watkins, L. L. (2009). Association between n-3 fatty acid consumption and ventricular ectopy after myocardial infarction. Am. J. Clin. Nutr. 89, 1315–1320.
Song, Y., Shryock, J. C., Wagner, S., Maier, L. S., and Belardinelli, L. (2006). Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J. Pharmacol. Exp. Ther. 318, 214–222.
Song, Y. H., Cho, H., Ryu, S. Y., Yoon, J. Y., Park, S. H., Noh, C. I., Lee, S. H., and Ho, W. K. (2010). L-type Ca2+ channel facilitation mediated by H2O2-induced activation of CaMKII in rat ventricular myocytes. J. Mol. Cell. Cardiol. 48, 773–780.
Tsuduki, T., Honma, T., Nakagawa, K., Ikeda, I., and Miyazawa, T. (2011). Long-term intake of fish oil increases oxidative stress and decreases lifespan in senescence-accelerated mice. Nutrition 27, 334–337.
Verkerk, A. O., Van Ginneken, A. C., Berecki, G., Den Ruijter, H. M., Schumacher, C. A., Veldkamp, M. W., Baartscheer, A., Casini, S., Opthof, T., Hovenier, R., Fiolet, J. W., Zock, P. L., and Coronel, R. (2006). Incorporated sarcolemmal fish oil fatty acids shorten pig ventricular action potentials. Cardiovasc. Res. 70, 509–520.
von Schacky, C. (2008). Omega-3 fatty acids: antiarrhythmic, proarrhythmic or both? Curr. Opin. Clin. Nutr. Metab. Care 11, 94–99.
Ward, C. A., and Giles, W. R. (1997). Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J. Physiol. (Lond.) 500(Pt 3), 631–642.
Xie, L. H., Chen, F., Karagueuzian, H. S., and Weiss, J. N. (2009). Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 104, 79–86.
Zaloga, G. P., Ruzmetov, N., Harvey, K. A., Terry, C., Patel, N., Stillwell, W., and Siddiqui, R. (2006). (N-3) long-chain polyunsaturated fatty acids prolong survival following myocardial infarction in rats. J. Nutr. 136, 1874–1878.
Zhao, Z., Fefelova, N., Shanmugam, M., Bishara, P., Babu, G. J., and Xie, L. H. (2011). Angiotensin II induces afterdepolarizations via reactive oxygen species and calmodulin kinase II signaling. J. Mol. Cell. Cardiol. 50, 128–136.
Zhao, Z., Wen, H., Fefelova, N., Allen, C., Baba, A., Matsuda, T., and Xie, L. H. (2012a). Revisiting the ionic mechanisms of early afterdepolarizations in cardiomyocytes: predominant by Ca waves or Ca currents? Am. J. Physiol. Heart Circ. Physiol. 302, C1762–C1771.
Keywords: docosahexaenoic acid, H2O2, early afterdepolarizations, reactive oxygen species, L-type calcium channel, sodium channel
Citation: Zhao Z, Wen H, Fefelova N, Allen C, Guillaume N, Xiao D, Huang C, Zang W, Gwathmey JK and Xie L-H (2012) Docosahexaenoic acid reduces the incidence of early afterdepolarizations caused by oxidative stress in rabbit ventricular myocytes. Front. Physio. 3:252. doi: 10.3389/fphys.2012.00252
Received: 01 May 2012; Paper pending published: 15 May 2012;
Accepted: 18 June 2012; Published online: 09 July 2012.
Edited by:
George E. Billman, The Ohio State University, USAReviewed by:
Gudrun Antoons, Medical University of Graz, AustriaCopyright: © 2012 Zhao, Wen, Fefelova, Allen, Guillaume, Xiao, Huang, Zang, Gwathmey and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Zhenghang Zhao, Department of Pharmacology, School of Medicine, Xi’an Jiaotong University, Xi’an 710061, China. e-mail:enpoQG1haWwueGp0dS5lZHUuY24=; Lai-Hua Xie, Department of Cell Biology and Molecular Medicine, UMDNJ-New Jersey Medical School, Newark NJ 07101, USA. e-mail:eGllbGFAdW1kbmouZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.