
- 1 Experimental Cardiology Group, Center for Heart Failure Research, Academic Medical Center, Amsterdam, Netherlands
- 2 Department of Medical Physiology, University Medical Center Utrecht, Utrecht, Netherlands
- 3 Department of Cardiology, The Hatter Institute, University College London Hospitals, London, UK
- 4 Department of Anesthetics, The Hatter Institute, University College London Hospitals, London, UK
- 5 Department of Cardiothoracic Surgery, The Hatter Institute, University College London Hospitals, London, UK
Abnormal ventricular wall motion is a strong clinical predictor of sudden, arrhythmic, cardiac death. Dispersion in repolarization is a prerequisite for the initiation of re-entrant arrhythmia. We hypothesize that regionally decreased wall motion is associated with heterogeneity of repolarization. We measured local activation times, activation-recovery intervals (ARIs, surrogate for action potential duration), and repolarization times using a multielectrode grid at nine segments on the left ventricular epicardium in 23 patients undergoing coronary artery surgery. Regional wall motion was simultaneously assessed using intraoperative transesophageal echocardiography. Three groups were discriminated: (1) Patients with normal wall motion (n = 11), (2) Patients with one or more hypokinetic segments (n = 6), (3) Patients with one or more akinetic or dyskinetic segments (n = 6). The average ARI was similar in all groups (251 ± 3.7 ms, ±SEM). Dispersion of ARIs between the nine segments was significantly increased in the hypokinetic (84 ± 7.4 ms, p < 0.005) and akinetic/dyskinetic group (94 ± 3.5 ms, p < 0.0005) compared with the normal group (49 ± 5.1 ms), independent from the presence of myocardial infarction. Repolarization heterogeneity occurred primarily in the normally contracting regions of the hearts with abnormal wall motion. An almost maximal increased dispersion of repolarization was observed when there was only a single hypokinetic segment. We conclude that inhomogeneous wall motion abnormality of even moderate severity is associated with increased repolarization inhomogeneity, independent from the presence of infarction.
Introduction
Abnormal ventricular wall motion abnormality (WMA) is one of the strongest clinical predictors of sudden cardiac death due to arrhythmia in patients with heart disease (Tracy et al., 1987; Trappe et al., 1989; Nath et al., 1993; Kober et al., 1997; Camm and Katritsis, 2000; Kohl et al., 2005). In the Strong Heart Study, a large population-based study in patients without clinically recognizable cardiovascular disease, wall motion abnormalities were also associated with an about 2.5 times higher risk of both cardiovascular events and death during an 8-years follow-up (Cicala et al., 2007). In human atria, both in silico (Kuijpers et al., 2011) and in vivo (Coronel et al., 2010a) dilatation has been shown to underlie conduction abnormalities and/or increased dispersion in refractoriness. Increased wall strain in a setting of acute ischemia leads to an increased occurrence of ventricular arrhythmias (Coronel et al., 2002; Janse et al., 2003). In addition, the involvement of mechano-electrical feedback in the development of cardiac memory (Jeyarai et al., 2007) and in LQT syndrome (Haugaa et al., 2010) has been suggested. It has been reported that the timing of stretch in relation with the phase of the action potential is important for the effect (Zabel et al., 1996).
Increased repolarization inhomogeneity is a prerequisite for re-entrant arrhythmias (Han and Moe, 1964; Kuo et al., 1983; Barr et al., 1994). The relevance of mechano-electrical feedback has been demonstrated both in animals and in man (Lab, 1982; Franz et al., 1989; Taggart and Sutton, 1999). Also in man WMA produces increased QT dispersion (Schneider et al., 1997). Because action potential duration (APD) and thus also the repolarization time [RT, sum of local activation time (AT) and local APD; Franz et al., 1991] are altered by mechanical stretch (Kohl et al., 1999), we hypothesized that dispersion of those parameters (APD and RT) is increased in patients with wall motion abnormalities.
We measured dispersion of an index of local APD (activation-recovery intervals, ARIs; Haws and Lux, 1990) and of RT at multiple sites on the epicardium in patients with and without myocardial infarction (MI) during coronary bypass grafting. Local wall abnormalities were scored by simultaneous intraoperative transesophageal echocardiography. We conclude that WMA is associated with increased dispersion of both ARIs and RTs thereby enhancing the electrophysiological substrate for arrhythmias, irrespective of the presence of MI.
Materials and Methods
Patients
The study was approved by the hospital Ethical Committee and written informed consent was obtained from all patients. Twenty-three patients, aged 61 ± 2.2 (mean ± SEM), were studied who were undergoing routine coronary artery surgery at The Middlesex Hospital. Patients were selected randomly from the waiting list from those with (n = 12) and without (n = 11) prior MI. Patients with atrial fibrillation or taking class I or class III antiarrhythmic medication were excluded. Individual patient details are shown in Table 1.
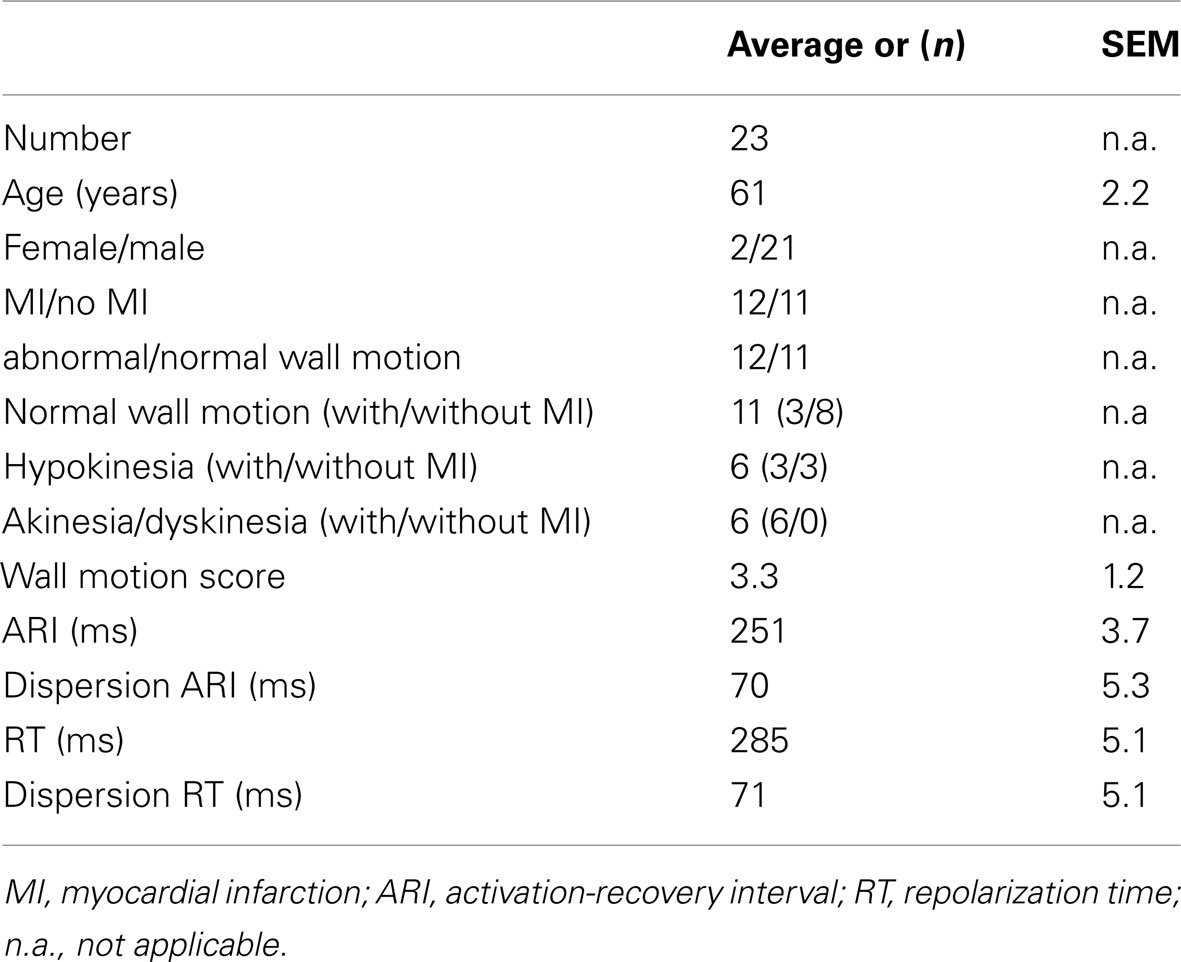
Table 1. Data in the whole patient group (n = 23).
Surgical Procedure
Following induction of anesthesia an esophageal echo probe was inserted and good quality images established. Patients then underwent routine thoracotomy and right atrial and aortic cannulation in preparation for cardiopulmonary bypass. Transesophageal echocardiographic measurements of ventricular wall motion and epicardial electrophysiological recordings were then made prior to the initiation of cardiopulmonary bypass as described below.
Echocardiography
Multiplane intraoperative transesophageal echocardiography was used to assess cardiac function by scoring left ventricular wall motion in three transgastric short-axis planes. Four segments (left posterior wall, left free wall, left anterior wall, and septum) were scored at the apical, mid-papillary, and basal levels, leading to assessment of wall motion at 12 sites. Each segment was scored as follows: 0, normal wall motion; 1 hypokinesia; 2 akinesia; 3 dyskinesia. Patients with normal wall motion had a total wall motion score zero (n = 11). Hypokinetic patients had a wall motion score 1 in one or more segments (n = 6). Akinetic and/or dyskinetic patients had a wall motion score 2 or 3 in one or more segments (n = 6). Next, the wall motion scores of all 12 sites was summed for each patient. The total WMA score could, theoretically, vary between 1 and 36 (although dyskinesia in all segments is impossible). In this patient group the minimum WMA score was 1 and the maximum was 18. In addition, left ventricular ejection fraction was assessed by measuring fractional area change of the left ventricular cavity in the transgastric mid-papillary short-axis view.
Electrical Recordings
Figure 1 shows the electrode grid of eight terminals in two rows of four with an interelectrode distance of 0.5 cm along rows and 1 cm between rows. Epicardial electrograms were recorded using this multielectrode grid by positioning it in random order at the basal, mid and apical regions of the anterior, lateral, and postero-inferior left ventricular wall (nine regions, septal segments being inaccessible for obvious reason). Signal recording and processing have been described previously (Taggart et al., 2001). During atrial pacing at 600 ms we measured at a maximum of 72 sites (nine regions × eight sites in each) local AT (dV/dtmin of the initial electrogram deflections) and RT (dV/dtmax of the T wave; Coronel et al., 2006). The interval between activation and RT, the ARI, is a surrogate measure of local APD (Haws and Lux, 1990) and local refractoriness (Chinushi et al., 2001). Figure 2 shows typical electrograms in one of the patients. At each of the nine sites we measured the average and the dispersion of AT, ARI, and RT (Figure 1). Dispersion (D) was defined as the difference between maximum and minimum values at each of the nine regions. Local dispersion was determined for the whole left ventricular free wall for AT, ARI, and RT by averaging D (see Figure 1, bottom left). Regional dispersion was defined as the largest difference between the averages of the nine regions (see Figure 1, bottom right).
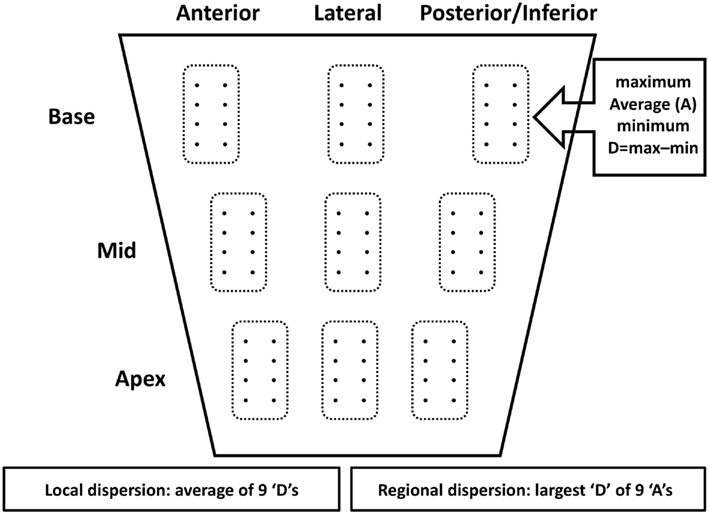
Figure 1. Schematic presentation of location of the multigrid electrode. It consisted of eight (4 × 2) terminals separated 0.5 and 1 cm respectively. In each of nine regions that were successively (and randomly) assessed maximum, average (A), minimum, and dispersion (D: maximum minus minimum) were determined for activation times, activation-recovery intervals and repolarization times. Local dispersion was defined as the average of the 9 “D” values. Regional dispersion was defined as the difference between the largest and smallest “A” (see Materials and Methods for further explanation).
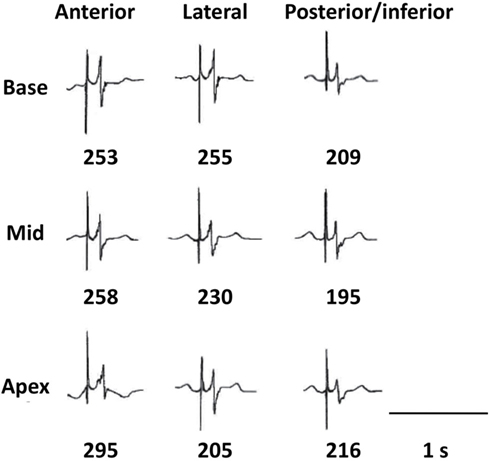
Figure 2. Local electrograms at the nine positions in one of the patients. The first vertical line indicates the stimulus artifact. The number below each of the nine positions is the local activation-recovery-interval (ARI), measured from dV/dtmin of the QRS till dV/dtmax of the T wave (see Materials and Methods).
Statistical Analysis
Data are presented as mean ± SEM. We were dealing with a small group of 23 patients. Differences between patients with normal wall motion and WMA were tested by one-way ANOVA. Further post hoc testing of subgroups was performed by the Student–Newman–Keuls test. Differences were considered statistically significant if p < 0.05.
Results
All Patients
Figure 3 shows the minimum and maximum values (connected by vertical lines) for the average of the ARIs in the nine regions in the 23 patients. It, therefore, shows the regional dispersion in ARIs (see Materials and Methods; Figure 1) in each individual patient. Patients 1–12 have wall motion abnormality (wall motion score ≥1) and patients 13–23 have normal wall motion (wall motion score 0). Patients without MI are depicted by thin lines between their minimum and maximum ARIs and patients with MI are marked by bold lines. Without any more sophisticated subgroup analysis, Figure 3 suggests that regional dispersion in ARIs is higher in patients with WMA (patients 1–12), although averaged ARIs were similar in both groups (251 ms; Table 1).

Figure 3. Minimum and maximum activation-recovery intervals (ARIs) in 23 patients (1–12 with WMA, 13–23 with normal wall motion; patient groups separated by dashed line between patients 12 and 13). Thin lines: patients without myocardial infarction, bold lines: patients with myocardial infarction. Whether patients had normal wall motion, or were hypokinetic, akinetic, or dyskinetic can be found in Table 2, where the order of the patients is similar to that in Figure 3.
Wall Motion Abnormality and Dispersion of Activation-Recovery Intervals
Figure 4 shows local (open bars) and regional dispersion of ARIs (black bars) in patients with normal wall motion (two bars at the left) and in patients with WMA (six bars at the right). The local dispersion in ARIs (open bars) was 24 ± 1.7 ms in the normal group and 29 ± 2.4 ms in patients with WMA. The differences between local dispersion in ARIs were not statistically significant. A subdivision of the group with WMA (n = 12) into patients with hypokinesia (n = 6) or a/dyskinesia (n = 6) also yielded similar values for local dispersion (compare all open bars). However, regional dispersion in ARIs (black bars) was 49 ± 5.1 ms in patients with normal wall motion (n = 11), whereas it was 89 ± 4.2 ms (p < 0.005) in patients with WMA (n = 12). In the two subgroups with WMA regional dispersion in ARIs was 84 ± 7.4 ms in patients with hypokinesia (n = 6) and 94 ± 3.5 ms in patients with akinesia or dyskinesia (n = 6). As was the case for the WMA group as a whole, also these two subgroups had larger regional dispersion in ARIs than the group with normal wall motion (ANOVA followed by SNK post hoc test; p < 0.005). The difference between these two subgroups, however, was statistically not significant (SNK post hoc test). Within the group with the most severe WMA, four patients were in the akinetic subgroup and two in the dyskinetic subgroup. The averaged values for regional dispersion in those two subgroups were 93 and 96 ms respectively (data not shown in Figure 4). In the group with normal wall motion 3 out of 11 patients had a history of MI. In the other two groups with WMA these numbers were three out of six (hypokinesia) and six out of six (akinesia/dyskinesia; see below).
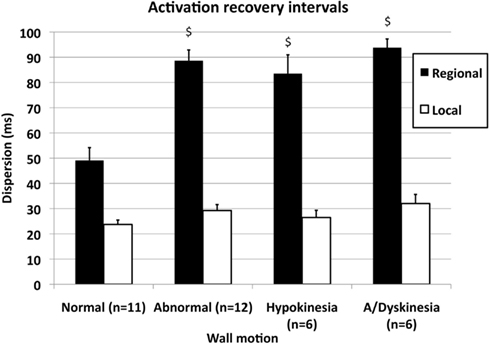
Figure 4. Local (open bars) and regional dispersion (filled bars) of activation-recovery intervals (ARIs) in 11 patients with normal wall motion and in 12 patients with WMA. The patient group with WMA was subdivided into six patients with hypokinesia and six patients with akinesia or dyskinesia. See text for further explanation. $p < 0.005 compared with normal wall motion. The subgroups hypokinesia and a/dyskinesia did not differ from each other, neither for local nor for regional dispersion.
The much larger regional dispersion of ARIs in the group with WMA compared with the group with normal wall motion despite similar averaged ARIs in both groups, suggests that maximum ARIs are increased, whereas minimum ARIs are decreased within the hearts of patients with WMA. Indeed, both the maximum and minimum averaged ARIs at the nine regions differed significantly between the groups (see also Figure 3, 297 ± 4.0 vs. 276 ± 7.3 ms; p < 0.025 for the maximum values in the groups with WMA and normal wall motion and 208 ± 4.3 vs. 227 ± 6.8 ms; p < 0.05 for the minimum values in the same two groups).
The difference in ejection fraction between patients with and without WMA (50% ± 5.1 and 63% ± 4.0, respectively) was not significant.
Myocardial Infarction and Dispersion of Activation-Recovery Intervals
Figure 5 shows the interaction of the effects of WMA and MI on regional dispersion of ARIs. The difference between the five subgroups (there were no patients with akinesia/dyskinesia without MI) was significant (ANOVA; p < 0.0005). However, when we divided the group with normal wall motion (see Figure 4, n = 11) into subgroups with (n = 3) and without MI (n = 8), there were no statistically significant differences (Figure 5, two left bars). Furthermore, when the group with hypokinesia (see Figure 4, n = 6) was subdivided into three patients with and three patients without MI, again no statistically significant differences were found (Figure 5, two middle bars). It should be mentioned, however, that an absence of statistical differences with such small numbers may have little meaning because of the lack of power. The hypokinesia group with infarction and the a/dyskinesia group had significantly larger regional dispersion than both groups with normal wall motion. Although not completely conclusive, Figure 5 suggests that the presence of WMA is more strongly associated with a large regional dispersion in ARIs than the presence of MI.
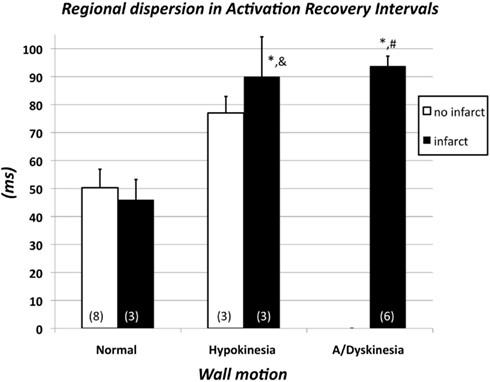
Figure 5. Regional dispersion of activation-recovery intervals in 11 patients with normal wall motion subdivided over subgroups with (n = 3) and without myocardial infarction (n = 8), in six patients with hypokinesia subdivided over subgroups with (n = 3) and without myocardial infarction (n = 3) and in six patients with akinesia or dyskinesia (see also Figure 3). In the latter group all patients had a history of myocardial infarction. The differences between the five groups are significant (ANOVA; p < 0.0005). Subsequently pairwise comparisons were made. *p < 0.01 vs. normal wall motion/no infarct; &p < 0.05 vs. normal wall motion/infarct; #p < 0.01 vs. normal wall motion/infarct.
The Origin of Dispersion in Activation-Recovery Intervals
Figure 6 shows the analysis of the source of dispersion of ARIs in patients with WMA. For this analysis each heart should at least have two normally or two abnormally contracting segments out of a total of nine segments with electrophysiological data. One patient had only abnormally contracting segments and three patients had one abnormally contracting segment. Thus, we selected 8 out of the 12 patients with WMA that fulfilled these criteria. The total regional dispersion was 96 ± 3.6 ms in these eight patients. Next the regional dispersion in ARIs was quantified for the normally contracting segments by subtraction of the minimal from the maximal segmental ARI (within the normally contracting area) of each of these eight hearts. In the same way the regional dispersion in ARIs was quantified for the abnormally contracting segments in each heart. The difference between the average of all ARIs in the normally contracting area and the average of all ARIs in the abnormally contracting area yielded the dispersion between these two areas. These three dispersion parameters were expressed as a percentage of the total regional dispersion in ARIs per individual heart, which was set at 100%. Obviously, the sum of these three parameters could be more than 100%. Figure 6 shows that the regional dispersion of ARIs in patients with WMA arises primarily (72 ± 11.1%) within the normal area of these abnormally contracting hearts. Also, we were able to compare the regional dispersion in ARIs of hearts with normal wall motion (n = 11) with the regional dispersion within the normally contracting area of the hearts with WMA. For this analysis the selection criterion was the presence of at least two normally contracting areas. Because there was only one patient with only abnormally contracting segments, we had to omit only one patient from the WMA group, leading to n = 11 in both the group with normal wall motion and in the group with WMA. Interestingly, the regional dispersion of ARIs within the normal area of abnormally contracting hearts is significantly larger than the regional dispersion of ARIs of hearts with normal wall motion (73 ± 7.7 vs. 49 ± 5.1 ms, ANOVA, p < 0.025, data not shown. Thus, irrespective of the magnitude of regional dispersion between the normally and abnormally contracting areas, the regional dispersion in ARIs within the normal area of hearts with WMA is larger than regional dispersion in ARIs in hearts of patients with normal wall motion.
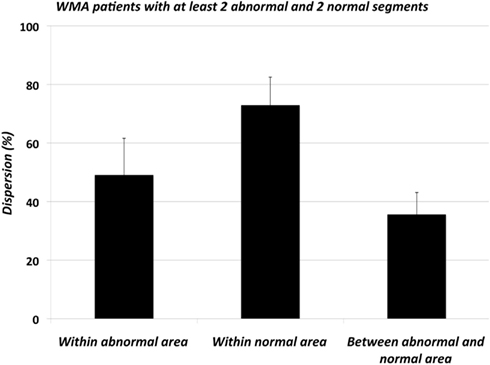
Figure 6. Regional dispersion of activation-recovery intervals (ARIs) in patients with regional wall motion abnormalities expressed as a percentage of the total dispersion in each individual heart (WMA; n = 8, four patients with WMA did not have at least two normally contracting area as well as two abnormally contracting segments and were excluded from this analysis). Dispersion was assessed in the segments within the normal area, within the abnormal area and between the abnormal and normal areas. Regional dispersion was largest within the normally contracting area. Interestingly, the regional dispersion within the normal areas of the hearts with WMA and the hearts with normal wall motion differed significantly (p < 0.05, not shown).
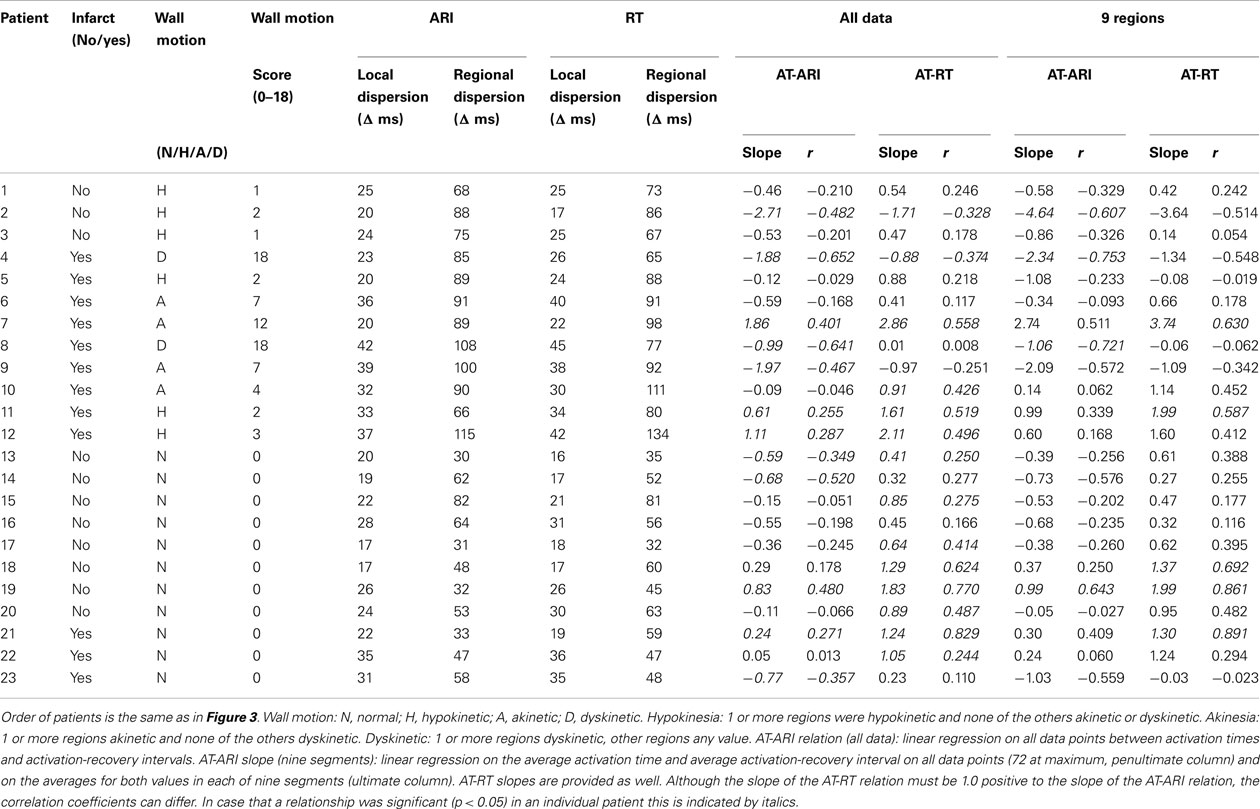
Table 2. Data of individual patients.
Relation between Activation Time and Activation-Recovery Times (AT-ARI Relationship)
Re-entrant arrhythmias primarily depend on dispersion in RTs rather than on dispersion in APD. Therefore, the relation between AT and ARI – as a substitute for APD – is of interest. Under normal physiological conditions the AT-ARI relationship is negative at least along the epicardium of human hearts (Cowan et al., 1988; Franz et al., 1991). A negative relationship leads to a smaller dispersion in RT than in ARI and a positive relationship causes a larger dispersion in RT than in ARI. Figure 7A shows the negative AT-ARI relationship in patient 13 (see also Figure 3; Table 2). This patient had no MI and normal wall motion. The patient had a negative AT-ARI relationship with slope −0.59 (Table 2). Figure 7B shows the same data, but now from a patient with an anterior MI (patient 12 in Figure 3; Table 2). We found a positive AT-ARI relationship with slope 1.11 (Table 2). Both correlations were significant (see Table 2 for further data and explanation). The “normal” patient with slope −0.59 for the AT-ARI relationship had, by definition, a slope of +0.41 for the AT-RT relationship. Also, this positive relation was significant. With such weak correlations the dispersion in ARIs will not be very much different from the dispersion in RTs. In the other “abnormal” patient, the positive AT-ARI relationship with slope 1.11 will lead to an AT-RT relationship of 2.11. Now, the dispersion in repolarization, caused by the activation order adds up to the dispersion in ARIs. Figure 8A shows the same data points as in Figure 7A, but now attributed to the nine regions. This patient with normal wall motion had low values both for local dispersion in ARI (20 ms) and RT (16 ms), and also for regional dispersion in ARI (30 ms) and RT (35 ms; Table 2, patient #13). The local dispersion in each region both in AT (horizontal) and in ARI (vertical) has been indicated by thin lines through the nine data points which indicate the average AT and ARI in each region. The AT relationship through these nine averages has a slope −0.39 (Table 2). Figure 8B shows the same data as in Figure 7B from the patient with an anterior MI again attributed to the nine regions (patient 12 in Figure 3; Table 2). The three posterior/inferior regions, i.e., not the infarcted regions, were all hypokinetic, leading to a wall motion score 3. This patient had slightly higher values for local dispersion in ARI (37 ms) and RT (42 ms) than the “normal” patient, but substantially higher values for regional dispersion in ARI (115 ms) and RT (134 ms; Table 2). Again the thin bars indicate the local dispersion at the nine regions, both for AT (horizontal) and ARI (vertical). The slope of the regression line is still positive (+0.60), explaining why dispersion in RT is higher than dispersion in ARI. Interestingly, the “outlier” in Figure 8B was in the anterior apical position, where the wall motion was normal and the ARI (and RT) was very long (late). The large dispersion in ARI and RT occurred between the posterior-mid region (in the mid of the hypokinetic area) and the apical anterior position, where wall motion was normal. The rough “anterior” localization of the MI prevented a more precise analysis, but it is emphasized that the MI area did not show WMA.
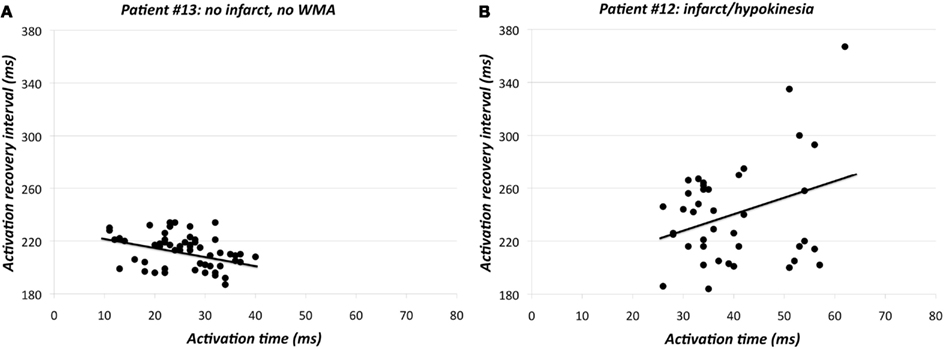
Figure 7. (A) Activation time vs. activation-recovery intervals in patient #13 (see also Figure 3; Table 2). Y = −0.59 X + 228; r = −0.349, p < 0.005. (B) Activation time vs. activation-recovery intervals in patient #12 (see also Figure 3; Table 2). Y = 1.11 X + 196; r = 0.287, p < 0.05.
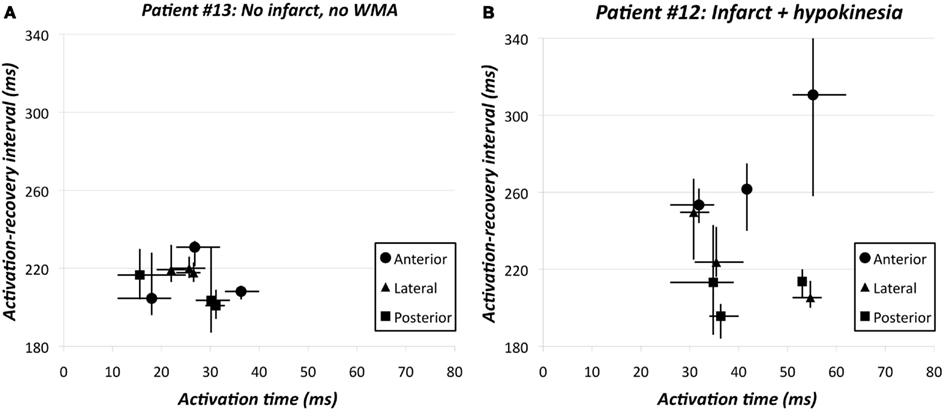
Figure 8. (A) Activation time vs. activation-recovery intervals in the nine regions of patient #13 (see also Figure 3; Table 2). Y = −0.39 X + 224; r = −0.256, ns. Thin lines indicate dispersion in activation times and activation-recovery intervals in each of the nine regions. (B) Activation time vs. activation-recovery intervals in the nine regions of patient #12 (see also Figure 3; Table 2). Y = 0.60 X + 211; r = 0.168, ns. Thin lines indicate dispersion in activation times and activation-recovery intervals in each of the nine regions. The huge dispersion primarily results from the very long activation-recovery intervals in the apical anterior region. The three posterior/inferior regions were all hypokinetic, leading to wall motion score 3 in this patient. The posterior/mid region had the shortest activation-recovery intervals.
We plotted dispersion in RTs as a function of the WMA score. Figure 9 shows dispersion of local (open circles) and regional RT (filled circles) for each individual patient with WMA (see also Table 2). As a reference the stars along the ordinate indicate the local and regional dispersion in RT in the 11 patients with normal wall motion. Although the data as presented in Figures 4 and 5 have already shown that regional dispersion in ARIs (and also RTs) is significantly larger in hearts with WMA, the data in Figure 9 show that this dispersion does not increase with a higher wall motion score. On the contrary, the correlation found between WMA score and regional dispersion of RT (closed circles) was negative (Y = −0.71, X + 93; r = −0.229, n = 12), albeit not significantly. The seven largest dispersion values had a wall motion score of 5 ± 1.4, indicating that WMA in a relatively small area is associated with substantial dispersion in RT. Similar results were obtained for wall motion score vs. ARIs (not shown). Thus, a small abnormality in wall motion (score 1–3) is associated with maximal dispersion in ARIs within the normally contracting area of hearts with WMA.
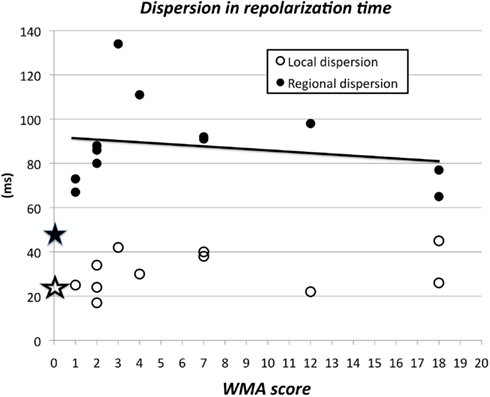
Figure 9. Wall motion abnormality score (index for abnormal wall motion) and regional dispersion in repolarization times. The averaged dispersion in RT of the 11 patients with wall motion score 0 is summarized by the two stars at the ordinate. At WMA score 1 and local dispersion 25 ms there were two patients. A small disturbance of normal wall motion, i.e., hypokinesia in only 1 or 2 out of 12 segments is associated with substantial dispersion in RT. Although the relation between WMA score and regional dispersion in repolarization time (restricted to the 12 patients with WMA) was not significant, the regression line Y = −0.71 × WMA score + 93.0 (r = −0.229, n = 12) had an intercept at the Y-axis of 93, substantially more than the regional dispersion of 53 ± 4.1 ms in the group with normal wall motion (filled asterisk).
Discussion
In this study we have simultaneously measured ventricular wall motion and local electrograms, allowing us to identify an association between WMA and the electrophysiological substrate for re-entrant arrhythmias. We have shown in the human heart that wall motion abnormalities are associated with increased dispersion of ARIs (an index of APD) and RTs, independent from the presence of MI, and without effect on ATs. Second, within one heart the increased dispersion in repolarization is caused by shortening of ARIs at one or more sites and prolongation at other sites. Third, maximal dispersion in ARIs within the normally contracting area of hearts with WMA is observed when the abnormality in wall motion (score 1–3) is minor. Finally, the increase in dispersion of ARIs is largest in the normally contracting areas of the hearts of patients with WMA. The latter is concordant with the observation in the Strong Heart Study (Cicala et al., 2007) that WMA in patients without recognizable cardiovascular disease is associated with fatal and non-fatal cardiovascular events during follow-up.
Wall Motion Abnormality
The cardiac effects of volume and pressure loading have been known for almost 100 years (Bainbridge, 1915), but mechano-electrical feedback as a consequence of ventricular WMA, has evoked renewed interest by the work of Max Lab and associates in the 1980s (Lab, 1982) and also more recently (Kohl and Ravens, 2003). Myocardial stretch is a strong modulator of APD (Franz et al., 1989; Hansen, 1993; Taggart and Sutton, 1999; Chen et al., 2004). Prolonged altered stretch has been shown to cause changes in repolarizing membrane currents (Jeyaraj and Rosenbaum, 2011). The effect of pulsatile myocardial stretch on repolarization depends on the timing relative to the repolarization process (Reiter et al., 1988; Zabel et al., 1996; Quinn and Kohl, 2011). For example, stretch modulates APD through cation non-selective (SACns) and potassium selective stretch activated channels (SACk). The reversal potential for SACns occurs at about half way between the action potential plateau and resting potential such that stretch occurring before the reversal potential shortens APD and stretch during the later part of the action potential, lengthens APD. Due to the strongly negative reversal potential for SACk, stretch during the action potential tends to shorten APD (Quinn and Kohl, 2011). The effect of regional dyssynchrony in producing regional differences in stretch modulation of APD has been highlighted in regard to electrical remodeling in heart failure patients with asynchronous LV activation and contraction (Jeyaraj and Rosenbaum, 2011). It is therefore possible that in patients with WMA, myocardial stretch occurs at disparate moments during the action potential at different sites in the heart. This may cause divergent effects on APD and RT with a prolongation at one site and a shortening at another. We have documented this in the present study, where the average ARI was unchanged but its dispersion was increased. Dispersion of repolarization constitutes the basis of unidirectional block, which is a prerequisite for the initiation of re-entrant arrhythmias (Janse and Wit, 1989). On the basis of this study we propose that inhomogeneous wall motion may increase inhomogeneity of repolarization and thereby enhance the susceptibility to ventricular arrhythmias. The observation that dispersion in RT is largest in patients with only moderate WMA, suggests a maximal effect of mechano-electrical feedback between sites where a large difference in myocardial stress occurs. The fact that the larger dispersion of ARIs in the normal areas of abnormal hearts is based on measurements in fewer segments than the dispersion of ARIs in hearts with normal wall motion, may add to the potential arrhythmogenic significance of this observation (but see below).
It has been demonstrated in isolated working pig hearts that stretch may evoke premature beats at the interface between contracting and non-contracting myocardium (Coronel et al., 2002). It is conceivable that these premature beats encounter a high degree of repolarization heterogeneity in the normally contracting tissue, and set off a re-entrant arrhythmia. In this manner both substrate and trigger for reentry are present at the same time (Coronel et al., 2002).
Larger Dispersion: More Arrhythogenesis?
We have recently shown that it is too simple to consider large dispersion per se as arrhythmogenic (Coronel et al., 2009). Dispersion in repolarization can be very large, but when it concerns a too small, or a too large area with late repolarization, it will not result in reentry, either because it is impossible to circumvent the area of prolonged refractoriness for spatial or temporal reasons, or because the timing of the retrogradely invading impulse is not suitable to cause reentry in a proximal area with normal refractoriness (Coronel et al., 2010b; Janse et al., 2011). In this study we show that regional dispersion in ARIs and/or RT was in the order of 90 ms when wall motion abnormality was involved vs. about 50 ms when there was normal wall motion (Figures 4 and 9; Table 2). Based on a conduction velocity of about 40 cm/s, it can be estimated that this will require a minimum diameter of the region with the late repolarization of about 2 cm in order to lead to activation at the distal side of that region.
Activation Time-Action Potential Duration Relationship
Normal human myocardium has a negative relation between AT and ARI or other indices of APD (Franz et al., 1987, 1991; Cowan et al., 1988; Yuan et al., 2001; Hanson et al., 2009). In the normal heart this tends to reduce dispersion in RT. An AT vs. APD relation with a slope of −0.50 presents a special case because at that slope the dispersion in APD will equal to the dispersion in RT. With a more negative slope, dispersion in RT will tend to be less than dispersion in APD. At a relation with a slope of −1.0 the decrease in APD along the path of conduction could theoretically lead to the disappearance of dispersion in repolarization and thus to the disappearance of the T wave in any lead of the surface ECG (Conrath and Opthof, 2006; Opthof et al., 2009; Janse et al., 2011). At any slope less negative than −0.50 (including positive slopes) dispersion in RT will exceed dispersion in APD. It should be noted that the relation between AT and ARI was on average less negative (see Table 2) than in previous studies of Franz et al., 1991 and Cowan et al., 1988. Therefore, it can be questioned how “normal” the hearts of our patients with normal wall motion actually were.
It has previously been shown that under pathophysiological conditions a -normal- negative relation between AT and APD is partially or completely lost (Cowan et al., 1988; Franz et al., 1991). Indeed, we observed that in only 10 out of 23 patients the slope between AT and ARIs was more negative than −0.50 (Table 2). It was even positive in seven patients. We did not find differences in slope between patients with normal wall motion and hypo- and/or akinesia or dyskinesia. Probably abnormalities in a single region (as in patient #12, see Figure 8B) are relevant.
Conclusion
In patients, substantial dispersion in repolarization is observed even though there is only minor wall motion abnormality. The presence of MI is not mandatory for this effect.
Limitations
As patients taking antiarrhythmic drugs were excluded from the study, we cannot relate our findings to the presence of arrhythmias in the patients. Therefore, it could be argued that the patient group with the most marked arrhythmogenic substrate was not studied. However, although none of our patients had a history of severe ventricular arrhythmias, patients with significant coronary artery disease constitute a group at risk for arrhythmic cardiac death. Our study shows that one prerequisite for arrhythmogenesis is likely associated with regional depression of wall motion.
Although it is known that the effects of mechano-electrical feedback depend on the phase during which mechanical effects impinge on the action potential, our echocardiographic data, which were only qualitative, were not suitable to match these with the electrophysiological observations.
Clinical Implications
Our study not only suggests a mechanism underlying the hitherto unexplained association between WMA and ventricular arrhythmia, but may also have implications for patient management. Patients with moderate or localized WMA may be at potentially greater risk. Correction of WMA, therefore, by improving hemodynamic status and/or revascularization may help in reducing risk of cardiac arrest. Our results underline the importance of incorporating into such studies measures of regional wall motion rather than ejection fraction which may be less specific for arrhythmias based on reentry.
Our results may be relevant to other clinical situations as well. Thus, in patients with severe WMA, positive inotropic medication induces adverse effects in terms of proarrhythmia (Tinker et al., 1976). The background of these adverse effects may be WMA rather than supposed aberrations in calcium homeostasis. Indeed, afterload reduction with nitroprusside results in abolishment of severe ventricular arrhythmias in patients with acute MI (Mukherjee et al., 1976), whereas an increase in volume load has been demonstrated to increase the inducibility of tachyarrhythmias, at least in dogs (Calkins et al., 1989). Also, in some patients with severe cardiac failure resynchronization therapy using biventricular pacing is employed. Our results suggest that in order to protect against arrhythmias resynchronization would need to be complete since even moderately compromised wall motion is associated with substantial dispersion of repolarization (see Figure 9). Indeed, a proarrhythmic potential of resynchronization therapy has been reported (Medina-Ravell et al., 2003).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bainbridge, F. A. (1915). The influence of venous filling upon the rate of the heart. J. Physiol. (Lond.) 50, 65–84.
Barr, C. S., Naas, A., Freeman, M., Lang, C. C., and Struthers, A. D. (1994). QT dispersion and sudden unexpected death in chronic heart failure. Lancet 343, 327–329.
Calkins, H., Maughan, W. L., Weisman, H. F., Sugiura, S., Sagawa, K., and Levine, J. H. (1989). Effect of acute volume load on refractoriness and arrhythmia development in isolated, chronically infarcted canine hearts. Circulation 79, 687–697.
Camm, A. J., and Katritsis, D. G. (2000). “Risk stratification of patients with ventricular arrhythmias,” in Cardiac Electrophysiology: From Cell to Bedside, eds D. P. Zipes and J. Jalife (Philadelphia, PE: WB Saunders), 808–828.
Chen, R. L., Penny, D. J., Greve, G., and Lab, M. J. (2004). Stretch-induced regional mechanoelectric dispersion and arrhythmia in the right ventricle of anaesthetised lambs. Am. J. Physiol. 286, H1008–H1014.
Chinushi, M., Tagawa, M., Kasai, H., Washizuka, T., Abe, A., Furushima, H., and Aizawa, Y. (2001). Correlation between the effective refractory period and activation-recovery interval calculated from the intracardiac unipolar electrogram of humans with and without dl-sotalol treatment. Jpn. Circ. J. 65, 702–706.
Cicala, S., De Simone, G., Roman, M. J., Best, L. G., Lee, E. T., Wang, W., Welty, T. K., Galloway, J. M., Howard, B. V., and Devereux, R. B. (2007). Prevalence and prognostic significance of wall-motion abnormalities in adults without clinically recognized cardiovascular disease. The Strong Heart Study. Circulation 116, 143–150.
Conrath, C. E., and Opthof, T. (2006). Ventricular repolarization. An overview of (patho)physiology, sympathetic effects, and genetic aspects. Prog. Biophys. Mol. Biol. 92, 269–307.
Coronel, R., De Bakker, J. M. T., Wilms-Schopman, F. J., Opthof, T., Linnenbank, A. C., Belterman, C. N., and Janse, M. J. (2006). Monophasic action potentials and activation-recovery-intervals as measures of ventricular action potential duration. Experimental evidence to resolve some controversies. Heart Rhythm 3, 1043–1050.
Coronel, R., Langerveld, J., Boersma, L. V., Wever, E. F., Bon, L., Van Dessel, P. F., Linnenbank, A. C., Van Gilst, W. H., Ernst, S. M., Opthof, T., and Van Hemel, N. M. (2010a). Left atrial pressure reduction for mitral stenosis reverses left atrial direction dependent conduction abnormalities. Cardiovasc. Res. 85, 711–718.
Coronel, R., Wilms-Schopman, F. J. G., and Janse, M. J. (2010b). Anti- or profibrillatory effects of Na+ channel blockade depend on the site of application relative to gradients in repolarization. Front. Physiol. 1:10. doi:10.3389/fphys.2010.00010
Coronel, R., Wilms-Schopman, F. J. G., and De Groot, J. R. (2002). Origin of ischemia-induced phase 1b arrhythmias in pig hearts. J. Am. Coll. Cardiol. 39, 166–176.
Coronel, R., Wilms-Schopman, F. J. G., Opthof, T., and Janse, M. J. (2009). Dispersion of repolarization and arrhythmogenesis. Heart Rhythm 6, 537–543.
Cowan, J. C., Hilton, C. J., Griffiths, C. J., Tansuphaswadikul, S., Bourke, J. P., Murray, A., and Campbell, R. W. (1988). Sequence of epicardial repolarisation and configuration of the T wave. Br. Heart J. 60, 424–433.
Franz, M. R., Bargheer, K., Costard-Jackle, A., Miller, D. C., and Lichtlen, P. R. (1991). Human ventricular repolarization and T wave genesis. Prog. Cardiovasc. Dis. 33, 369–384.
Franz, M. R., Bargheer, K., Rafflenbeul, W., Haverich, A., and Lichtlen, P. R. (1987). Monophasic action potential mapping in human subjects with normal electrocardiograms: direct evidence for the genesis of the T wave. Circulation 75, 379–386.
Franz, M. R., Burkhoff, D., Yue, D. T., and Sagawa, K. (1989). Mechanically induced action potential changes and arrhythmia in isolated and in situ canine hearts. Cardiovasc. Res. 23, 213–223.
Han, J., and Moe, G. K. (1964). Nonuniform recovery of excitability in ventricular muscle. Circ. Res. 14, 44–61.
Hansen, D. E. (1993). Mechanicoelectrical feedback effects of altering preload, afterload and ventricular shortening. Am. J. Physiol. 264, H423–H432.
Hanson, B., Sutton, P., Elameri, N., Gary, M., Critchley, H., Gill, J. S., and Taggart, P. (2009). Interaction of activation-repolarization coupling and restitution properties in humans. Circ. Arrhythm. Electrophysiol. 2, 162–170.
Haugaa, K. H., Amlie, J. P., Berge, K. E., Leren, T. P., Smiseth, O. A., and Edvardsen, T. (2010). Transmural differences in myocardial contraction in long-QT syndrome. Mechanical consequences of ion channel dysfunction. Circulation 122, 1355–1363.
Haws, C. W., and Lux, R. L. (1990). Correlation between in vivo transmembrane action potentials durations and activation-recovery intervals from electrograms. Effects of interventions that alter repolarization time. Circulation 81, 281–288.
Janse, M. J., Coronel, R., and Opthof, T. (2011). M cells do not have a functional role in the ventricular myocardium of the intact heart. Heart Rhythm 8, 934–937.
Janse, M. J., Coronel, R., Wilms-Schopman, F. J. G., and De Groot, J. R. (2003). Mechanical effects on arrhythmogenesis: from pipette to patient. Prog. Biophys. Mol. Biol. 82, 187–195.
Janse, M. J., and Wit, A. L. (1989). Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 69, 1049–1169.
Jeyarai, D., Wilson, L. D., Zhong, J., Flask, C., Saffitz, J. E., Deschênes, I., Yu, X., and Rosenbaum, D. S. (2007). Mechanoelectrical feedback as novel mechanism of electrical remodelling. Circulation 115, 3145–3155.
Jeyaraj, D., and Rosenbaum, D. S. (2011). “Mechanical triggers of long-term ventricular electrical remodelling,” in Cardiac Mechano-Electric Coupling and Arrhythmias, eds P. Kohl, F. Sachs and M. R. Franz (Oxford: Oxford University Press), 187–192.
Kober, L., Torp-Pendersen, C., Elming, H., and Burchhardt, H. (1997). Use of left ventricular ejection fraction or wall-motion score index in predicting arrhythmic death in patients following an acute myocardial infarction. The TRACE Study Group. Pacing Clin. Electrophysiol. 10, 2553–2559.
Kohl, P., Hunter, P., and Noble, D. (1999). Stretch-induced changes in heart rate and rhythm: clinical observations, experiments and mathematical models. Prog. Biophys. Mol. Biol. 71, 91–138.
Kohl, P., and Ravens, U. (2003). Cardiac mechano-electric feedback: past, present, and prospect. Prog. Biophys. Mol. Biol. 82, 3–9.
Kohl, P., Sachs, F., and Franz, M. R. (2005). Cardiac Mechano-Electric Feedback and Arrhythmias: From Pipette to Patient. Philadelphia: Elsevier.
Kuijpers, N. H. L., Potse, M., Van Dam, P. M., Ten Eikelder, H. M. M., Verheule, S., Prinzen, F. W., and Schotten, U. (2011). Mechanoelectrical coupling enhances initiation and affects perpetuation of atrial fibrillation during acute atrial dilatation. Heart Rhythm 8, 429–436.
Kuo, C. S., Munakata, K., Pratap Reddy, C., and Surawicz, B. (1983). Characteristics and possible mechanisms of ventricular arrhythmias dependent on the dispersion of action potential durations. Circulation 67, 1356–1367.
Lab, M. J. (1982). Contraction-excitation feedback in myocardium: physiological basis and clinical relevance. Circ. Res. 50, 757–766.
Medina-Ravell, V. A., Lankipalli, R. S., Yan, G. X., Antzelevitch, C., Medina-Malpica, N. A., Medina-Malpica, O. A., Droogan, C., and Kowey, P. R. (2003). Effect of epicardial or biventricular pacing to prolong QT interval and increase transmural dispersion of repolarisation: does resynchronisation therapy pose a risk for patients predisposed to long QT or torsades de pointes? Circulation 107, 740–746.
Mukherjee, D., Feldman, M. S., and Helfant, R. H. (1976). Nitroprusside therapy. Treatment of hypertensive patients with recurrent chest pain, ST-segment elevation, and ventricular arrhythmias. JAMA 235, 2406–2409.
Nath, S., DeLacey, W. A., Haines, D. E., Barry, V. A., Barber, M. J., Kron, I. L., and DiMarco, J. P. (1993). Use of regional wall motion score to enhance risk stratification of patients receiving an implantable cardioverter-defibrillator. J. Am. Coll. Cardiol. 22, 1093–1099.
Opthof, T., Coronel, R., and Janse, M. J. (2009). Repolarization gradients in the intact heart. Circ. Arrhythm. Electrophysiol. 2, 89–96.
Quinn, T. A., and Kohl, P. (2011). “Mechanical triggers and facilitators of ventricular arrhythmias,” in Cardiac Mechano-Electric Coupling and Arrhythmias, eds P. Kohl, F. Sachs and M. R. Franz (Oxford: Oxford University Press), 161–167.
Reiter, M. J., Synhorst, D. P., and Mann, D. P. (1988). Electrophysiological effects of acute ventricular dilatation in the isolated rabbit heart. Circ. Res. 62, 554–562.
Schneider, C. A., Voth, E., Baer, F. M., Horst, M., Wagner, R., and Sechtem, U. (1997). QT dispersion is determined by the extent of viable myocardium in patients with chronic Q-wave myocardial infarction. Circulation 96, 3913–3920.
Taggart, P., and Sutton, P. M. (1999). Cardiac mechano-electric feedback in man: clinical relevance. Prog. Biophys. Mol. Biol. 71, 139–154.
Taggart, P., Sutton, P. M. I., Opthof, T., Coronel, R., Trimlett, R., Pugsley, W., and Kallis, P. (2001). Transmural repolarisation in the left ventricle in humans during normoxia and ischaemia. Cardiovasc. Res. 50, 454–462.
Tinker, J. H., Tarhan, S., White, R. D., Pluth, J. R., and Barnhorst, D. A. (1976). Dobutamine for inotropic support during emergence from cardiopulmonary bypass. Anesthesiology 44, 281–286.
Tracy, C. M., Winkler, J., Brittain, E., Leon, M. B., Epstein, S. E., and Bonow, R. O. (1987). Determinants of ventricular arrhythmias in mildly symptomatic patients with coronary artery disease and influence of inducible left ventricular dysfunction on arrhythmia frequency. J. Am. Coll. Cardiol. 9, 483–488.
Trappe, H. J., Lichtlen, P. R., Klein, H., Wenzlaff, P., and Hartwig, C. A. (1989). Natural history of single vessel disease. Risk of sudden coronary death in relation to coronary anatomy and arrhythmia profile. Eur. Heart J. 10, 514–524.
Yuan, S. W., Kongstad, O., Hertervig, E., Holm, M., Grins, E., and Olsson, B. (2001). Global repolarization sequence of the ventricular endocardium: monophasic action potential mapping in swine and humans. Pacing Clin. Electrophysiol. 24, 1479–1488.
Keywords: dispersion, activation-recovery interval, repolarization, abnormal wall motion, myocardial infarction
Citation: Opthof T, Sutton P, Coronel R, Wright S, Kallis P and Taggart P (2012) The association of abnormal ventricular wall motion and increased dispersion of repolarization in humans is independent of the presence of myocardial infarction. Front. Physio. 3:235. doi: 10.3389/fphys.2012.00235
Received: 06 February 2011; Accepted: 11 June 2012;
Published online: 03 July 2012.
Edited by:
Sander Verheule, Maastricht University, NetherlandsReviewed by:
T. Alexander Quinn, Imperial College London, UKMichael Franz, Georgetown University, USA
Copyright: © 2012 Opthof, Sutton, Coronel, Wright, Kallis and Taggart. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Tobias Opthof, Experimental Cardiology Group, Center for Heart Failure Research, Academic Medical Center K2-105, Meibergdreef 9, 1105 AZ Amsterdam, Netherlands. e-mail: t.opthof@inter.nl.net