
- 1 Department of Biology, Manhattan College, Riverdale, New York, NY, USA
- 2 Department of Pharmacology, School of Medicine, University of California Irvine, Irvine, CA, USA
- 3 Department of Physiology and Biophysics, School of Medicine, University of California Irvine, Irvine, CA, USA
Voltage-gated potassium (Kv) channels shape the action potentials of excitable cells and regulate membrane potential and ion homeostasis in excitable and non-excitable cells. With 40 known members in the human genome and a variety of homomeric and heteromeric pore-forming α subunit interactions, post-translational modifications, cellular locations, and expression patterns, the functional repertoire of the Kv α subunit family is monumental. This versatility is amplified by a host of interacting proteins, including the single membrane-spanning KCNE ancillary subunits. Here, examining both the secretory and the endocytic pathways, we review recent findings illustrating the surprising virtuosity of the KCNE proteins in orchestrating not just the function, but also the composition, diaspora and retrieval of channels formed by their Kv α subunit partners.
Introduction
Mirroring the lives of the organisms they compose, the lifespan of each protein molecule is marked by maturation, migration, interactions, performance of tasks that define the protein itself, and ultimately degradation. The Kv channel protein family is the most diverse class of voltage-gated ion channels, boasting 40 known pore-forming α subunit genes in the human genome. With additional contribution from several classes of ancillary (β) subunits (Pongs and Schwarz, 2010), homomeric, and heteromeric α subunit complex formation (MacKinnon et al., 1993; Weiser et al., 1994; Rudy and McBain, 2001), and post-translational modifications including phosphorylation (Perozo and Bezanilla, 1991; Holmes et al., 1996a; Thomas et al., 1999), glycosylation (Freeman et al., 2000; Park et al., 2003; Brooks et al., 2006; Chandrasekhar et al., 2011), sumoylation (Dai et al., 2009), nitrosylation (Asada et al., 2009), and oxidation (Cai and Sesti, 2009; Sesti et al., 2010; Cotella et al., 2012), the number of potential combinations of functional Kv channel complexes, each with different functional properties, is staggering. Excitable and non-excitable cells alike exploit this embarrassment of riches to generate the specific repolarization profiles, regulatory responsiveness, and/or ionic gradients required for their cellular physiology. As excitability is defined by the sum properties of channels at the membrane at a given time, in order to understand ion channel function it is vital to examine the mechanisms by which the composition, surface density, and surface residency of Kv channels are controlled, involving both the anterograde (secretory) and retrograde (endocytic) pathways.
In Greek mythology, as a punishment for his wrongdoings, Sisyphus was banished to the underworld and assigned the task of repeatedly rolling a boulder up to the pinnacle of a steep mountain, only for it to come crashing down each time; this torturous and futile process was to be repeated for eternity (Homer, circa 700 BC). Recent work, reviewed here, has highlighted the role played by the KCNE family of ancillary subunits in an ostensibly similar enterprise: shepherding both the anterograde trafficking of α subunits from the endoplasmic reticulum (ER) to the cell surface, and overseeing their retrograde trafficking as they are subsequently internalized following surface expression (Xu et al., 2009; Kanda et al., 2011a,b,c; Roepke et al., 2011b). However, far from being a Sisyphean task, the part that KCNEs play in regulating the timely ebb and flow of Kv α subunits to and from the cell surface helps establish the sum properties of the population of channels at the cell surface at a given moment, and, in turn, the temporal properties of transmembrane ion flux. Therefore, the mechanisms by which Kv channel surface density and residency are controlled by KCNE subunits and other factors, constituting both the secretory and endocytic pathways, are a crucial facet of channel biology.
Secretory Pathway
Translation and Assembly of α Subunits
The anterograde journey of the Kv channel complex, from its creation to its functional residency at the plasma membrane, begins with protein synthesis, occurring at the ER (Palade, 1975). The mRNA encoding any protein is first sent from the nucleus to the cytoplasm, where it associates with ribosomes. This complex is then targeted to the ER membrane. All proteins that are either membrane-bound or secreted are synthesized on the ribosomes of the rough ER (Hegde and Lingappa, 1997). Membrane proteins often follow the paradigm in which they are targeted to the ER membrane as they are synthesized via an ER signal sequence. Kv channels lack this typical signal and are thought to undergo ER targeting by the S2 transmembrane domain, as found for Kv1.3 (Tu et al., 2000); interestingly, in the same study it was found that the S1 transmembrane domain establishes its topology only after S2 is synthesized. During translocation to the ER membrane, nascent polypeptides may acquire their secondary, tertiary, and quaternary structures. The interior of the ER is considered to be the topological equivalent of the extracellular region, and protein oligomerization and folding are aided by chaperones and protein disulfide isomerases within the ER lumen. Some post-translational modifications begin in the ER, such as core-glycosylation, or the attachment of a precursor oligosaccharide to asparagine residues (Alberts et al., 2008). From the ER, Kv channel complexes continue on their journey to the plasma membrane, passing through cis-, medial-, and trans-Golgi elements (McKeown et al., 2008), where further modification occurs, such as N-linked glycosylation.
The basic functional requirements for a Kv channel are four α subunits, with tetramerization occurring in the ER (Nagaya and Papazian, 1997). The N-terminal T1-recognition domain, also known as the N-terminal A and B box (NAB) domain, allows intra-subfamily tetramerization of Kv1.x, Kv2.x, Kv3.x, and Kv4.x subunits, while the proteins are actively being translated from the ribosome (Xu et al., 1995; Papazian, 1999; Lu et al., 2001). Though not much is known regarding the tetramerization process itself, seminal work by the Deutsch laboratory has shown that in vitro assembly of functional Kv1.3 channels involves dimerization of subunit dimers (Tu and Deutsch, 1999). Further, it appears that T1 assembly and tertiary structure formation are coupled, suggesting that channel assembly occurs prior to membrane insertion (Robinson and Deutsch, 2005). One exception to the T1 rule involves the regulatory (also termed “electrically silent”) Kv α subunits, which are retained in the ER (Salinas et al., 1997; Ottschytsch et al., 2002), likely due to a signal in the S6 transmembrane region (Ottschytsch et al., 2005). These subunits, including members of the Kv5, Kv6, Kv8, and Kv9 subfamilies, do not appear to form functional homomers, but can interact and form complexes with members of the Kv2 and Kv3 subfamilies, even though they are classified as members of different subfamilies (Post et al., 1996; Salinas et al., 1997; Stocker et al., 1999; Zhu et al., 1999; Ottschytsch et al., 2002). This results in suppression or in some cases anterograde trafficking as functional heteromers exhibiting altered functional attributes such as gating kinetics, of the Kv2 and Kv3 channel-containing heteromers (Stocker et al., 1999). Another exception is that KCNQ1 and KCNH2 (hERG), from different subfamilies, appear to heteromultimerize (Ehrlich et al., 2004; Ren et al., 2010). Interestingly, the C-terminus of KCNQ (Kv7.x) channels has been shown to direct tetramerization of this subfamily through coiled-coil interactions (Haitin and Attali, 2008), but the potential import of this vis-à-vis KCNQ1-KCNH2 interaction has not been established.
In vivo, Kv channels are probably always formed from complexes of a variety of protein subunits in addition to α, however, our understanding of the events or mechanisms dictating whether Kv channels form from heteromeric or homomeric α subunit assemblies, never mind α−β combinations, is somewhat limited. The N-terminal T1 or NAB domain allows members of the Kv1, Kv2, Kv3, and Kv4 subfamilies to form homotetrameric and intra-subfamily heterotetrameric complexes (Lu et al., 2001). Evidence from the Deutsch laboratory using human T cells suggested that the degree of temporal overlap and kinetics of expression, in terms of in which specific compartments and when different subunits are found together, affects the proportion of homomeric and heteromeric complexes (Panyi and Deutsch, 1996). Others have also found evidence that gene expression levels modulate the probability of heteromeric formation (Weiser et al., 1994).
Regulation of Kv α Subunit Anterograde Trafficking by Non-KCNE Ancillary Subunits
Voltage-gated potassium channel ancillary subunits are integral parts of native Kv channel complexes, altering biophysical properties and trafficking. These subunits come in two distinct flavors: cytoplasmic and transmembrane. Of the cytosolic proteins, the best known are the Kvβ subunits, binding primarily to the α subunit T1 domain in Kv1 channels, and K+ Channel-Interacting Proteins (KChIPs), binding primarily to Kv4 channels (Rettig et al., 1994; An et al., 2000). Kvβ subunits interact with α subunits very early during biosynthesis, at the cytoplasmic face of the ER, as with Kv1.2 and Kvβ2 (Shi et al., 1996).
As also observed for the KChIPs and K+ Channel-Associated Proteins (KChAPs, also cyotosolic), and the single transmembrane segment, dipeptidyl aminopeptidase-like (DPPX) proteins, Kvβ subunits increase the efficiency of Kv channel complex surface expression (Shi et al., 1996; Campomanes et al., 2002; Nadal et al., 2003; Shibata et al., 2003). Thus, in vitro studies have shown that Kvβ1.2 increases the surface expression of Kv1.2, and Kvβ2 likewise with Kv1.4 (Shi et al., 1996; Accili et al., 1997; Manganas and Trimmer, 2000; Zhu et al., 2003b). In vivo studies show that Kvβ subunits also direct regional trafficking of Kv subunits. Axonal trafficking of Kv1.2 channels requires interaction with KIF3/kinesin II and docking of Kvβ2 with the microtubule plus-end tracking protein EB1, associated with the growing tips of microtubules; axonal trafficking does not occur in the absence of Kvβ2 (Gu et al., 2006).
Interestingly, Kvβ subunits can also confer N-type inactivation on non-inactivating Kv1 channels, such as Kv1.1 (Rettig et al., 1994). Kvβ1 mediates N-type inactivation by an N-terminal inactivation particle (Rettig et al., 1994). Kvβ2, as we previously mentioned, increases the surface expression of the N-type Kv channel Kv1.4 (Manganas and Trimmer, 2000; Zhu et al., 2003a). Thus, Kvβ subunits play significant roles in modulating N-type inactivation in Kv1 homomeric and heteromeric complexes. Heteromeric complexes formed by the delayed rectifier Kv1.1 and the fast-inactivating “N-type” subunit Kv1.4 display inefficient surface trafficking due to an ER retention determinant in Kv1.1 (Manganas and Trimmer, 2000; Zhu et al., 2003a). The mechanisms behind the effects of Kvβ2 on Kv1.4 are incompletely understood, but it is known that Kvβ2 increases the surface expression of Kv1.2 by masking retention signals (Shi et al., 1996). Another recent report showed that the ER-associated potassium channel regulatory protein KCNRG decreases surface expression of Kv1.1-Kv1.4 complexes by retaining both subunits intracellularly (Usman and Mathew, 2010).
The KCNE Gene Family
The KCNE subunits are single transmembrane domain proteins, varying from 103 to 177 amino acids in length, that do not generate currents themselves, but co-assemble with Kv α subunits to alter their properties (Takumi et al., 1988; Barhanin et al., 1996; Sanguinetti et al., 1996; Abbott et al., 1999, 2001). The founder of this family, KCNE1, also known as MinK (minimal K+ channel protein), or IsK, was first found to co-assemble with the Kv α subunit Kv7.1 (KCNQ1) to form the slowly activating cardiac ventricular repolarization current IKs (Barhanin et al., 1996; Sanguinetti et al., 1996). KCNE1 slows KCNQ1 activation 5- to 10-fold, increases its unitary conductance 4-fold, and eliminates inactivation (Pusch et al., 1998; Sesti and Goldstein, 1998; Seebohm et al., 2003); this complex is the best-understood KCNE-containing channel complex. Four other members of the KCNE family have been identified, termed KCNE2-KCNE5 (Abbott and Goldstein, 1998; Abbott et al., 1999, 2001; Piccini et al., 1999). KCNE2-KCNE5 are also commonly referred to as MiRP1-MiRP4, (MinK-Related Peptides 1–4). Through native and heterologous studies, a variety of functional Kv α-KCNE interactions have been shown. KCNE subunits are quite promiscuous, as each can interact with a multitude of different Kv α subunits (and vice-versa; for a review, see McCrossan and Abbott, 2004).
Regulation of Kv α Subunit Anterograde Trafficking by KCNE Subunits
The importance of KCNE subunits in mammalian physiology is evident from genetic links to ventricular repolarization, gastric acid secretion, thyroid hormone biosynthesis, long QT syndrome (LQTS), deafness, and periodic paralysis (Neyroud et al., 1997; Splawski et al., 1997; Tyson et al., 1997; Abbott et al., 2001; Roepke et al., 2006, 2008, 2009). While KCNEs are most widely recognized for their ability to shape Kv channel kinetics, voltage dependence and conductance (for review see, McCrossan and Abbott, 2004), it is now clear that their influence in channel biology is broader than this. Previous work from the McDonald laboratory showed that KCNE1 could influence KCNH2 current without an apparent change in the classic “biophysical” parameters of this channel (McDonald et al., 1997), hinting at a role for KCNE1 in governing the “cell biology” of KCNH2. This conclusion was bolstered by effects on KCNH2 and KCNQ1 trafficking, of KCNE1 mutations linked to the ventricular repolarization abnormality LQTS (Bianchi et al., 1999).
Currently, there is controversy in the field as to whether Kv α and KCNE subunits associate co-translationally or later on in the secretory pathway. The most-studied KCNE-containing complex, with respect to this and most other facets of KCNE biology, is the KCNQ1-KCNE1 channel that generates the slow component of the human cardiac ventricular repolarization current, IKs. Poulsen and Klaerke (2007) suggest the interaction between KCNQ1 and KCNE1 is transient. Further, KCNE4, which inhibits KCNQ1, was suggested to act on KCNQ1 channels that were already at the surface (Grunnet et al., 2002). A recent study using Xenopus oocytes expressing KCNQ1 and injected with purified KCNE1 protein found that inhibiting the secretory pathway with the blocker brefeldin A inhibited the effect of purified KCNE1 protein on expressed KCNQ1, suggesting KCNQ1-KCNE1 assembly intracellularly; however, in the same study, it was also found that injection of purified KCNE1 protein led to KCNQ1-KCNE1 complex formation, even after inhibition of new KCNQ1 synthesis using cyclohexamide (Vanoye et al., 2010). One potential issue with these studies is that they were performed in Xenopus laevis oocytes and human embryonic kidney (HEK) 293 cells, which contain endogenous Kv subunits (Sanguinetti et al., 1996; Jiang et al., 2002), and Xenopus oocytes also contain endogenous KCNE1, KCNE3, and KCNE4 (Anantharam et al., 2003; Gordon et al., 2006).
The McDonald laboratory also discovered, using LQTS-associated mutant L51H-KCNE1 and an ER-retained version of KCNE1, that both show sequestration of KCNQ1-KCNE1 complexes in the ER, suggesting that the ER is the location of co-assembly of KCNE subunits and Kv α subunits (Krumerman et al., 2004). Work from the Kobertz laboratory also suggests that KCNQ1-KCNE1 complex formation occurs early in the secretory pathway and that when over-expressed alone, most KCNE1 is sequestered intracellularly, not progressing past the Golgi (Chandrasekhar et al., 2006). Furthermore, disrupting glycosylation of KCNE1 can impair KCNQ1-KCNE1 anterograde trafficking (Chandrasekhar et al., 2011).
Work on other KCNE subunits also indicated roles in regulating α subunit forward trafficking. In Caenorhabditis elegans, members of the MPS family of KCNE orthologs are required for stability of α subunits, either because they form complexes early in the secretory pathway and are co-dependent for forward trafficking, or because the proteins are unstable without co-assembly of α and β subunits (Bianchi et al., 2003; Park and Sesti, 2007). KCNE4 has also been shown to suppress Kv1.3 currents by inducing partial ER retention of the complex (Sole et al., 2009). As stated before, the LQTS associated KCNE1 mutation L51H prevents KCNQ1 forward trafficking (Krumerman et al., 2004).
KCNEs Can Regulate the α Subunit Composition of Kv Channels
Adding to the evidence for KCNE input into α subunit cell biology, we recently uncovered roles for KCNE1 and KCNE2 in dictating homomeric versus heteromeric α subunit composition of channels. The surface expression of homomeric “N-type” Kv channels, which generate currents that rapidly decay due to an N-terminal inactivation domain which occludes the pore – preventing K+ ion efflux – was found to be strongly suppressed by KCNE1 and KCNE2, which interact with and trap this family of α subunits early in the secretory pathway (Kanda et al., 2011a) (Figure 1). The question arose, was this suppression the end of the story – merely a means for current inhibition – or did it provide clues of a more interesting regulatory function?
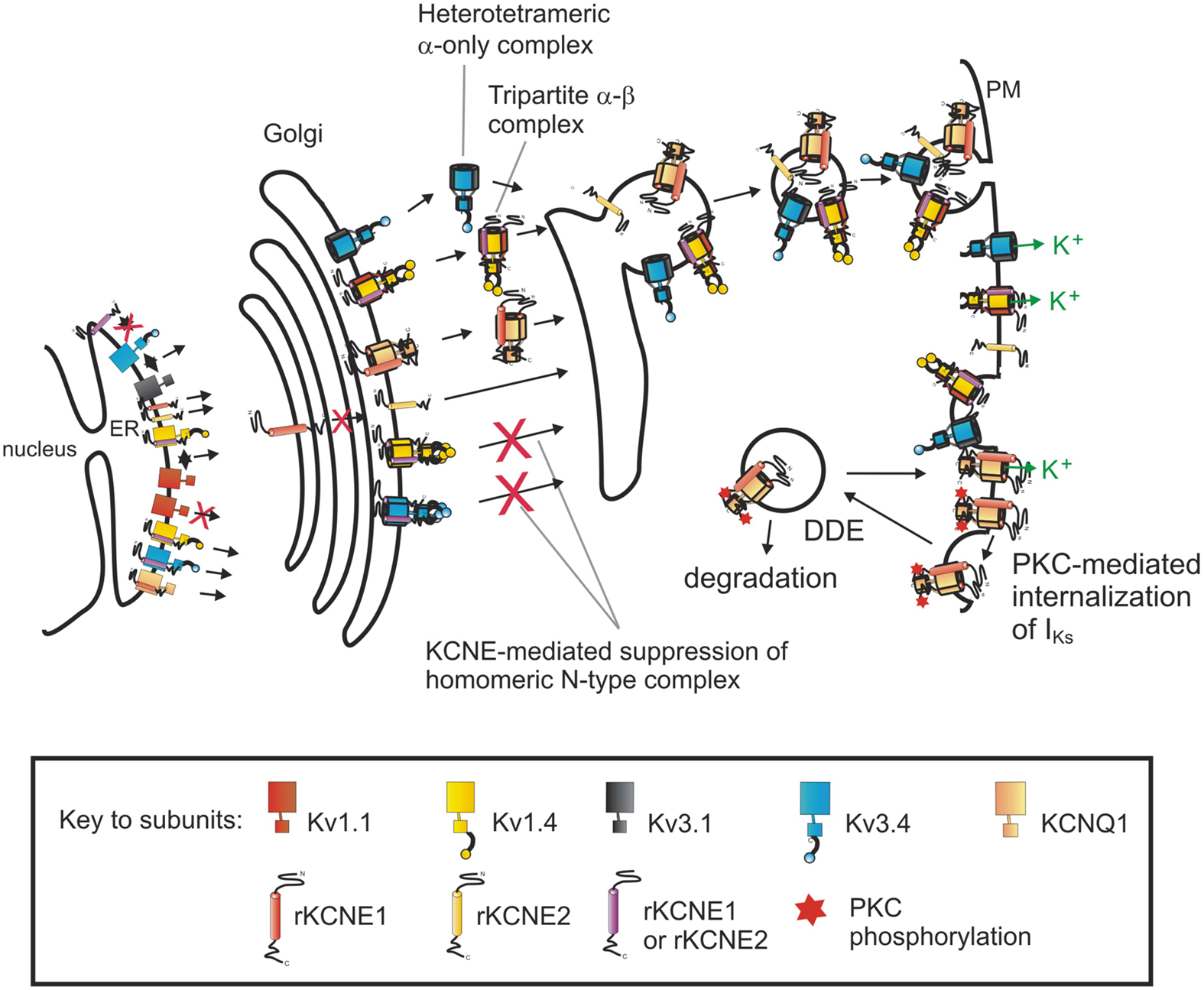
Figure 1. Impact of KCNE subunits on the composition and density of surface-expressed Kv channels. Pore-forming Kv α subunits, as listed in the key, form complexes in the ER that are trafficked through the Golgi to the plasma membrane (PM). KCNE1 and KCNE2 prevent the surface expression of homomeric N-type Kv channel complexes, but ensure heteromeric (mixed-α) complexes, that depending on their α subunit partners, may or may not include KCNEs. In this way, KCNE subunits form an intracellular checkpoint to modulate surface-expressed Kv channel composition. Similarly, KCNE1 dictates the dynamin-dependent endocytosis (DDE) of the KCNQ1-KCNE1 (IKs) complex. This process is further regulated: phosphorylation of KCNE1-S102 by Protein Kinase C (PKC) stimulates KCNQ1-KCNE1 DDE.
N-type subunits are often found in vivo as heteromeric complexes of both N-type and delayed rectifier (slow, C-type inactivating) α subunits (Scott et al., 1994; Wang et al., 1994; Rhodes et al., 1997; Coleman et al., 1999; Baranauskas et al., 2003), most likely as a means to provide a variety of inactivation characteristics and thus refractory periods and firing frequencies. When we examined the effects of additionally expressed delayed rectifier α subunits on KCNE suppression of N-type α subunits, we found the suppression was prevented by co-assembly with intra-subfamily delayed rectifier α subunits (Kanda et al., 2011b) (Figure 1). This rescue resulted in surface-expressed heteromeric N-type complexes, with no homomeric N-type complexes at the surface. Further, extra-subfamily subunits – even those that interact with KCNE1 and KCNE2 – did not rescue N-type subunit surface expression unless engineered to interact with N-type subunits through NAB domain swapping (Kanda et al., 2011b) (Figure 1). Because the fast-inactivating nature of N-type subunits dominates heteromeric or mixed-α complexes (MacKinnon et al., 1993; Weiser et al., 1994; Rudy and McBain, 2001), the ability of KCNE1 and KCNE2 to act as a molecular matchmaker to regulate incorporation of N-type α subunits into channels destined for the cell surface presents a mechanism by which KCNE subunits can determine heteromeric versus homomeric α subunit assembly as a measure to control cellular excitability.
Deutsch and colleagues have suggested that gene expression levels are critical in determining the fate of a channel subunit being complexed either homomerically or heteromerically (Panyi and Deutsch, 1996). We similarly found that heteromeric assembly of the delayed rectifier Kv3.1 and the fast-inactivating N-type subunit Kv3.4 depends on the expression level of each protein (Kanda et al., 2011b). In Chinese hamster ovary (CHO) cells, co-transfecting equal amounts of each α subunit led solely to heteromeric complex formation, as determined by mutating the canonical TxGYG of Kv3.1 to TxSYG, rendering any channel containing this mutant non-functional. Interestingly, with mismatched Kv3.4:Kv3.1 expression levels (10:1) KCNE1 co-expression greatly slowed inactivation compared to without KCNE1, suggesting that only Kv3.1-rescued Kv3.4 heteromeric complexes were expressed at the surface (Kanda et al., 2011b).
Our laboratory has also found roles for KCNE subunits in determining α subunit localization in polarized cells in vivo. In wild-type gastric parietal cells, apical KCNQ1-KCNE2 channels are required for gastric acid secretion because they form a K+ efflux route to provide luminal K+ for the gastric H+K+ATPase to function (Tinel et al., 2000; Dedek and Waldegger, 2001; Waldegger, 2003; Roepke et al., 2006; Heitzmann et al., 2007). In the gastric parietal cells of Kcne2−/− mice, KCNQ1 incorrectly localizes to the basolateral membrane, chaperoned by upregulated KCNE3 (Roepke et al., 2011b). Genetic disruption of both Kcne2 and Kcne3 results in KCNQ1 targeting to the apical side, similar to the situation in wild-type parietal cells – although gastric acid secretion is not restored in the double-knockout Kcne2−/−Kcne3−/− mice because KCNQ1 also requires KCNE2 for reasons other than localization – e.g., stimulation by low pH and constitutive (voltage-independent) activation (Heitzmann et al., 2004). We recently found a similar reversal of the trafficking polarity of both KCNQ1 and Kv1.3 in the choroid plexus epithelium of Kcne2−/− mice (Roepke et al., 2011a). In addition, KCNE2 is required for localization of Kv1.5 to the intercalated disks of mouse ventricular myocytes in vivo (Roepke et al., 2008).
Molecular Determinants of Kv Channel α Subunit Anterograde Trafficking
Some Kv α subunits contain either a primary sequence motif or other determinant conferring forward trafficking, or an ER retention motif. Some of these ER retention motifs act as quality control signals. As with any biological process, Kv channels may not always assemble properly. The ATP-sensitive potassium channel KATP is formed by complexes of four inward rectifier α subunits and four β subunits. An RKR ER retention/retrieval motif in KATP channels causes ER retention of partial complexes bearing fewer than the required eight subunits (Zerangue et al., 1999). The human ether-a-go-go related gene product (hERG; Kv11.1) α subunit contains an RXR (Arg-Xaa-Arg) motif which, when exposed, causes proteasomal degradation and prevents surface trafficking (Delisle et al., 2004). Similarly, the GB1 subunit of the GABAB receptor contains an ER retention motif, preventing cell surface expression; however, heterodimerization with the GB2 subunit masks this retention signal, allowing cell surface expression of the complex (Margeta-Mitrovic et al., 2000). In addition, a hydrophobic region of the N-terminus of Kv4 channels confers ER localization until co-assembly with KChIPs (Shibata et al., 2003). Studies such as these suggest that trafficking efficiency and specific functional properties might result from the same structural changes.
Likewise, the N-type subunit Kv1.4 has a VXXSL motif on its C-terminus which, when altered, prevents surface trafficking and acts to retain the channel in the ER (Li et al., 2000). Mutation of this motif inhibits glycosylation. Alternatively, the pore of Kv1.4 also contains a forward-trafficking determinant, working by dictating the effect of N-glycosylation (Watanabe et al., 2004). The pore of Kv1.1 confers strong ER retention, and the C-terminal confers partial Golgi retention (Zhu et al., 2003a). Heteromeric Kv1.1-Kv1.4 complexes exist and are found in native systems, however Kv1.1 significantly represses the surface expression of the complex (Zhu et al., 2003a). Our recent findings, that KCNE1 and KCNE2 can increase heteromeric Kv1.1-Kv1.4 currents to levels similar to those produced by homomeric Kv1.4 channels, suggests that KCNE subunits might alter structural components of the channel complex to mask ER and Golgi retention signals (Kanda et al., 2011b).
Post-translational modifications can also play key roles in Kv channel trafficking. Glycosylation is a critical step for the surface expression of some Kv channels. Nagaya and Papazian (1997) have shown that the Drosophila Shaker Kv α subunit is made as a partially glycosylated immature precursor that is converted to a fully glycosylated protein in the Golgi, as only the immature form was sensitive to digestion by endoglycosidase H (Endo H). Endo H solely cleaves N-linked high mannose-type sugars found on immature proteins in the ER, referred to as core-glycosylation (Iyengar and Hildebrandt, 2001). Work from the Thornhill laboratory has shown that Kv1.2 and Kv1.4 both are N-glycosylated, and that this N-linked glycosylation is important for efficient surface expression, as inhibition of N-glycosylation significantly decreases protein stability and induces intracellular retention (Watanabe et al., 2004, 2007). A glycosylation-defective mutant of hERG also shows reduced surface expression (Gong et al., 2002).
We recently found that co-expression of KCNE1 results in a loss of the fully glycosylated or mature Kv3.4 protein (Kanda et al., 2011a). This maturation of proteins occurs in the Golgi, and thus our findings suggest intracellular sequestration and a defect in ER-to-Golgi trafficking of Kv3.4 when KCNE1 is present. We also observed that KCNE1 glycosylation and Kv3.4 glycosylation are both altered when the two partners are co-expressed together. This suggests that the mechanism for N-type intracellular retention by KCNE1 is not as simple as Kv3.4 being unable to chaperone KCNE1 past the ER and to some extent the Golgi, where it is normally trapped when expressed alone. When expressed alone, we found KCNE1 nearly exclusively in the high molecular weight form, while co-expression of Kv3.4 with KCNE1 resulted in a distribution of non-, mono-, and bi-glycosylated KCNE1. These results suggest that KCNE1 is trafficked differently when complexed with Kv3.4 compared to when expressed alone, suggesting that co-assembly of KCNE1 or KCNE2 with N-type Kv α subunits exposes a retention signal that is formed or perhaps hidden under homomeric expression (Kanda et al., 2011a).
Endocytic Pathway
After reaching the cell surface, membrane protein residency can be abbreviated by a variety of different mechanisms of internalization, or endocytosis. Internal membranes are formed from the plasma membrane, with integral membrane proteins and extracellular fluid internalized into the cell (Doherty and McMahon, 2009). The best-understood method of endocytosis is clathrin-mediated endocytosis (CME). CME is mediated by small vesicles with a crystalline coat generated by a complex of proteins associated with clathrin, including coat protein-I (COP-I) and coat protein-II (COP-II). Adaptor proteins promote the recruitment of clathrin and assembly. Clathrin-coated pits are then formed on the inner surface of the plasma membrane where the proteins to be internalized are located; these clathrin-coated pits become clathrin-coated vesicles when fully internalized, with the endocytosed protein still attached in the membrane of the vesicle (Doherty and McMahon, 2009). Once internalized, these clathrin-coated vesicles can then recycle their cargo to the plasma membrane, through endosomes, or send their cargo to lysosomes for degradation. Many proteins are involved in the processes mediating CME, including dynamin, a membrane scission GTPase which, upon GTP hydrolysis, mediates fission of vesicles from the plasma membrane (Robinson, 1994; Sever et al., 2000). Dynamin has also been implicated, at least partially, in caveolae-dependent endocytosis, IL2Rβ-mediated internalization, flotillin-dependent internalization, and in phagocytosis (Doherty and McMahon, 2009).
Though extensively studied for some other protein families, the mechanisms mediating Kv channel internalization are not well-understood. Tyrosine phosphorylation has been shown to down-regulate Kv1.3 (Fadool et al., 1997) and Kv1.2 surface expression (Nesti et al., 2004). For Kv1.2, this tyrosine phosphorylation induced dynamin-dependent internalization of the channel. Kim et al. (2007) have observed in vivo the activity dependent internalization of Kv4.2 channels, where excitatory stimulation led to a clathrin-mediated decrease in the surface expression of Kv4.2 channels. Beyond the KCNE-mediated intracellular suppression of homomeric N-type complexes to prevent surface expression, we also found that Kv3.4 undergoes dynamin-dependent internalization (Kanda et al., 2011a). Interestingly, Kv1.4 is also thought to internalize by a clathrin-dependent mechanism, as it contains a canonical C-terminal di-leucine motif (McKeown et al., 2009). These findings together show the complexity of Kv channel trafficking, where multiple mechanisms can often result in the same overall result: modulation of channel density at the surface.
KCNQ1 and KCNA5 Internalization
The internalization of two Kv channels has been particularly studied, KCNA5 (more commonly referred to as Kv1.5), underlying the atrial specific IKur current, and KCNQ1, which when co-assembled with KCNE1, generates the slow component of the human ventricular repolarization current, IKs (Wang et al., 1993; Barhanin et al., 1996; Sanguinetti et al., 1996). Kv1.5 has been shown to localize with the early endosome marker EEA1 in HEK cells and undergo dynamin-dependent internalization (Choi et al., 2005). Kv1.5 also has a proline-rich Src homology 3 (SH3) binding domain essential for Kv1.5 internalization (Steele et al., 2007), as we found for KCNE1 (Xu et al., 2009). SH3-containing proteins have been shown to regulate intracellular trafficking, by binding to proline-rich domains of proteins involved in endocytic processes, such as dynamin (Marsh and McMahon, 1999). In atrial myocytes, Kv1.5, once internalized, exhibits Rab-GTPase-dependent recycling to the plasma membrane through early endosomes, modulating surface expression (McEwen et al., 2007). Lastly, tyrosine phosphorylation was also shown to down-regulate Kv1.5 (Holmes et al., 1996b).
KCNQ1, independent of KCNE1, can undergo serum- and glucocorticoid-inducible kinase 1 (SGK1)-dependent forward trafficking through Rab 11, and then internalization through Rab5 (Seebohm et al., 2007). Rab5 is involved in organization and maintenance of early endosomes while Rab11 is important in recycling endosomes, and forward trafficking of vesicles from the trans-Golgi network (Zerial and McBride, 2001). One mechanism by which Kv channel complexes are marked for internalization is through ubiquitylation (Staub and Rotin, 2006). The E3 ubiquitin-protein ligase Nedd4.2 down regulates Kv1.3 (Henke et al., 2004), and KCNQ1-5 (Ekberg et al., 2007; Jespersen et al., 2007). Specifically, KCNQ1-KCNE1 complexes can be internalized by Nedd4.2-dependent ubiquitylation, through direct interaction of Nedd4.2 to a PY motif in the C-terminus of KCNQ1 (Jespersen et al., 2007). Recently, it was also found that this Nedd4.2-dependent ubiquitylation was regulated by the serine/threonine kinase, the AMP-activated protein kinase (AMPK; Alesutan et al., 2011).
KCNE Regulation of Kv α Subunit Internalization
The effects of β subunits on Kv channel internalization have remained largely unexplored. We found that three sites on KCNE1 are necessary and sufficient to induce the dynamin-dependent internalization of KCNQ1-KCNE1 complexes in vitro in COS-7 cells and in guinea-pig ventricular myocytes (Xu et al., 2009). These sites include DPFNVY, reminiscent of the YXXΦ motif, which has been shown to mediate binding to AP-2 for the internalization of other proteins into clathrin-coated pits (Trowbridge et al., 1993; Ohno et al., 1995), a consensus SH3-binding domain at the C-terminus, and a consensus Protein Kinase C (PKC) phosphorylation site (S102) previously shown to decrease IKs currents by a then-unknown mechanism (Varnum et al., 1993; Zhang et al., 1994). Dynamin 2 is the ubiquitously expressed isoform that is also enriched in heart and skeletal muscle, whereas dynamin 1 and 3 are neuronal- and testis-specific, respectively (Diatloff-Zito et al., 1995).
In contrast to the ability of KCNE1 to mediate CME of KCNQ1-KCNE1 complexes, we found that homomeric KCNQ1 complexes are not subject to regulation by dynamin 2. These findings provided a novel method by which excitable cells can preferentially internalize IKs complexes (KCNQ1-KCNE1), leaving homomeric KCNQ1 complexes or other KCNQ1-containing complexes at the surface. As KCNE1 is highly influential in a variety of the features of KCNQ1 function (Pusch et al., 1998; Sesti and Goldstein, 1998; Seebohm et al., 2003), which in turn impacts ventricular repolarization, modulating surface expression to favor homomeric KCNQ1 channels could significantly alter cardiac myocyte action potentials and therefore cardiac rhythm.
This being stated, KCNE1 effects could also involve other pathways for internalization, such as caveolae in lipid rafts, which also utilize dynamin, or ubiquitylation. The aspartate residue in the DPFNVY motif, D76, has been found by Seebohm et al. (2008) to be important for Rab 5 mediated internalization of KCNQ1-KCNE1 complexes in Xenopus oocytes. This residue is also the site of a LQTS mutation, D76N, which has a strong dominant-negative effect on IKs currents, significantly decreasing IKs current density (Splawski et al., 1997; Hoppe et al., 2001). D76N converts the DPFNVY motif to simulate an NPXY motif, known to be involved in CME – suggesting the possibility that the D76N mutation decreases IKs density by stimulating CME in addition to effects on channel functional properties (Sesti and Goldstein, 1998).
One of the sites necessary for KCNE1-dependent internalization of IKs complexes is S102-KCNE1, a PKC phosphorylation site. PKC phosphorylation of S102-KCNE1 was previously shown to down-regulate IKs currents through an unknown mechanism (Varnum et al., 1993; Zhang et al., 1994); we recently found it acts by stimulating dynamin-dependent internalization of the KCNQ1-KCNE1 complex (Kanda et al., 2011c) (Figure 1). Thus, the ability of the PKC activator phorbol 12-myristate 13-acetate (PMA) to decrease KCNQ1-KCNE1 currents was abolished by co-expression of dominant-negative mutant K44A dynamin 2; fluorescence microscopy was then used to confirm the integral role of channel internalization in these phenomena (Kanda et al., 2011c). Analogously, it was subsequently discovered that KCNE2 can modulate hERG internalization in a phosphorylation-dependent manner – in this case the site was S98 on KCNE2 (Zhang et al., 2012).
Conclusion
The entire journey of a single Kv channel macromolecular complex, from translation of the constituent subunits, to formation of the channel complex, forward trafficking to the plasma membrane, and internalization from the plasma membrane, has yet to be tracked. We currently only have glimpses of this odyssey, and only for particular travelers. Between the complexities of Kv channels in vivo, multiple players and differential gene expression, these processes remain largely unknown. One particular problem in the field of potassium channel trafficking is that these studies are mostly performed in heterologous systems under conditions where only select Kv subunits are expressed, and under conditions of over-expression. These studies are still quite useful and fruitful, for they push our understanding of these processes, as shown by work reviewed here. Conditions in heterologous systems, however, are not necessarily entirely physiological. Further, issues such as gene expression and translation kinetics are not particularly well-understood.
Consider the multiple Kv channel mRNAs expressed in each cell type. What determines which of these mRNAs are translated and expressed at the surface? At present, channel researchers have essentially only found pieces of this puzzle. For example, it is known that transcription levels of Kv1.5 are modulated by glucocorticoids (Takimoto and Levitan, 1994), that Kv3.4 expression is upregulated by skeletal muscle innervation (Vullhorst et al., 1998), and that PKA phosphorylation at the surface of the ER augments hERG biosynthesis (Chen et al., 2009; Sroubek and McDonald, 2011). Other questions are also raised, such as whether competition occurs between Kv channels for access to these forward and reverse trafficking pathways, what other players are involved in these pathways, and how transcriptional and translational levels of Kv channel subunits are modulated. Beyond modulation of surface expression by trafficking or phosphorylation, ER resident proteins (chaperones), microtubules, scaffolding proteins, anchoring proteins and the actin cytoskeleton are all players in these processes. In regards to scaffolding proteins, we know that membrane-associated guanylate kinases (MAGUKs), which contain PDZ domains, act as scaffolds on which proteins are anchored (Carnegie and Scott, 2003); for instance, the MAGUK PSD-95 binds to Kv1.4 and increases its surface expression (Kim et al., 1995).
We have aimed in this review to highlight the roles played by KCNE subunits in regulating Kv α subunit transit through both the secretory and endocytic pathways, and modulation of Kv channel α subunit composition and surface density. Though cytosolic β subunits have been shown to play roles in the forward trafficking of Kv α subunits, their roles in the endocytosis of Kv complexes have not to our knowledge been examined. It is suggested that KChIPs may be involved in the endocytosis of Kv4 channels (Pongs and Schwarz, 2010), though there are no studies directly examining whether this actually occurs. In terms of the transmembrane β subunits, KCNE1 and KCNE2 are of particular interest – dependent on their α subunit partner, each can play polar opposite roles in trafficking processes. With the family of fast-inactivating N-type Kv subunits, KCNE1 and KCNE2 act to prevent the forward trafficking of homomeric N-type complexes, ensuring heteromeric (mixed-α) complexes, and thereby modulating action potential refractory periods and overall excitability (Kanda et al., 2011a,b) (Figure 1). On the other hand, when complexed with KCNQ1, KCNE1 plays a dominant role in modulating the dynamin-dependent, CME of the IKs complex; likewise KCNE2 can coordinate hERG internalization. Rather than a Sisyphean enterprise, the Herculean task of ensuring Kv α subunits move to the cell surface in a timely fashion and with the correct composition, then after the appropriate surface residency are internalized and either recycled or degraded, is one of several essential roles performed by KCNE subunits in their maintenance of cellular excitability and ion homeostasis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Geoffrey W. Abbott is grateful for financial support from the US NIH National Heart, Lung and Blood Institute HL079275 and HL101190. The authors thank Drs. Geri Kreitzer and Roberto Levi for helpful suggestions.
Abbreviations
Kv channel, voltage-gated potassium channel; MiRP, MinK-related peptide.
References
Abbott, G. W., Butler, M. H., Bendahhou, S., Dalakas, M. C., Ptacek, L. J., and Goldstein, S. A. (2001). MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 104, 217–231.
Abbott, G. W., and Goldstein, S. A. (1998). A super family of small potassium channel subunits: form and function of the MinK-related peptides (MiRPs). Q. Rev. Biophys. 31, 357–398.
Abbott, G. W., Sesti, F., Splawski, I., Buck, M. E., Lehmann, M. H., Timothy, K. W., Keating, M. T., and Goldstein, S. A. (1999). MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 97, 175–187.
Accili, E. A., Kiehn, J., Wible, B. A., and Brown, A. M. (1997). Interactions among inactivating and noninactivating Kvbeta subunits, and Kvalpha1.2, produce potassium currents with intermediate inactivation. J. Biol. Chem. 272, 28232–28236.
Alberts, B., Wilson, J. H., and Hunt, T. (2008). Molecular Biology of the Cell. New York: Garland Science.
Alesutan, I., Foller, M., Sopjani, M., Dermaku-Sopjani, M., Zelenak, C., Frohlich, H., Velic, A., Fraser, S., Kemp, B. E., Seebohm, G., Volkl, H., and Lang, F. (2011). Inhibition of the heterotetrameric K+ channel KCNQ1/KCNE1 by the AMP-activated protein kinase. Mol. Membr. Biol. 28, 79–89.
An, W. F., Bowlby, M. R., Betty, M., Cao, J., Ling, H. P., Mendoza, G., Hinson, J. W., Mattsson, K. I., Strassle, B. W., Trimmer, J. S., and Rhodes, K. J. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556.
Anantharam, A., Lewis, A., Panaghie, G., Gordon, E., Mccrossan, Z. A., Lerner, D. J., and Abbott, G. W. (2003). RNA interference reveals that endogenous Xenopus MinK-related peptides govern mammalian K+ channel function in oocyte expression studies. J. Biol. Chem. 278, 11739–11745.
Asada, K., Kurokawa, J., and Furukawa, T. (2009). Redox- and calmodulin-dependent S-nitrosylation of the KCNQ1 channel. J. Biol. Chem. 284, 6014–6020.
Baranauskas, G., Tkatch, T., Nagata, K., Yeh, J. Z., and Surmeier, D. J. (2003). Kv3.4 subunits enhance the repolarizing efficiency of Kv3.1 channels in fast-spiking neurons. Nat. Neurosci. 6, 258–266.
Barhanin, J., Lesage, F., Guillemare, E., Fink, M., Lazdunski, M., and Romey, G. (1996). K(V)LQT1 and lsK (minK) proteins associate to form the I (Ks) cardiac potassium current. Nature 384, 78–80.
Bianchi, L., Kwok, S. M., Driscoll, M., and Sesti, F. (2003). A potassium channel-MiRP complex controls neurosensory function in Caenorhabditis elegans. J. Biol. Chem. 278, 12415–12424.
Bianchi, L., Shen, Z., Dennis, A. T., Priori, S. G., Napolitano, C., Ronchetti, E., Bryskin, R., Schwartz, P. J., and Brown, A. M. (1999). Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum. Mol. Genet. 8, 1499–1507.
Brooks, N. L., Corey, M. J., and Schwalbe, R. A. (2006). Characterization of N-glycosylation consensus sequences in the Kv3.1 channel. FEBS J. 273, 3287–3300.
Cai, S. Q., and Sesti, F. (2009). Oxidation of a potassium channel causes progressive sensory function loss during aging. Nat. Neurosci. 12, 611–617.
Campomanes, C. R., Carroll, K. I., Manganas, L. N., Hershberger, M. E., Gong, B., Antonucci, D. E., Rhodes, K. J., and Trimmer, J. S. (2002). Kv beta subunit oxidoreductase activity and Kv1 potassium channel trafficking. J. Biol. Chem. 277, 8298–8305.
Carnegie, G. K., and Scott, J. D. (2003). A-kinase anchoring proteins and neuronal signaling mechanisms. Genes Dev. 17, 1557–1568.
Chandrasekhar, K. D., Bas, T., and Kobertz, W. R. (2006). KCNE1 subunits require co-assembly with K+ channels for efficient trafficking and cell surface expression. J. Biol. Chem. 281, 40015–40023.
Chandrasekhar, K. D., Lvov, A., Terrenoire, C., Gao, G. Y., Kass, R. S., and Kobertz, W. R. (2011). O-glycosylation of the cardiac IKs complex. J. Physiol. (Lond.) 589, 3721–3730.
Chen, J., Sroubek, J., Krishnan, Y., Li, Y., Bian, J., and Mcdonald, T. V. (2009). PKA phosphorylation of HERG protein regulates the rate of channel synthesis. Am. J. Physiol. Heart Circ. Physiol. 296, H1244–H1254.
Choi, W. S., Khurana, A., Mathur, R., Viswanathan, V., Steele, D. F., and Fedida, D. (2005). Kv1.5 surface expression is modulated by retrograde trafficking of newly endocytosed channels by the dynein motor. Circ. Res. 97, 363–371.
Coleman, S. K., Newcombe, J., Pryke, J., and Dolly, J. O. (1999). Subunit composition of Kv1 channels in human CNS. J. Neurochem. 73, 849–858.
Cotella, D., Hernandez-Enriquez, B., Wu, X., Li, R., Pan, Z., Leveille, J., Link, C. D., Oddo, S., and Sesti, F. (2012). Toxic role of K+ channel oxidation in Mammalian brain. J. Neurosci. 32, 4133–4144.
Dai, X. Q., Kolic, J., Marchi, P., Sipione, S., and MacDonald, P. E. (2009). SUMOylation regulates Kv2.1 and modulates pancreatic beta-cell excitability. J. Cell Sci. 122, 775–779.
Dedek, K., and Waldegger, S. (2001). Colocalization of KCNQ1/KCNE channel subunits in the mouse gastrointestinal tract. Pflugers Arch. 442, 896–902.
Delisle, B. P., Anson, B. D., Rajamani, S., and January, C. T. (2004). Biology of cardiac arrhythmias: ion channel protein trafficking. Circ. Res. 94, 1418–1428.
Diatloff-Zito, C., Gordon, A. J., Duchaud, E., and Merlin, G. (1995). Isolation of an ubiquitously expressed cDNA encoding human dynamin II, a member of the large GTP-binding protein family. Gene 163, 301–306.
Doherty, G. J., and McMahon, H. T. (2009). Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902.
Ehrlich, J. R., Pourrier, M., Weerapura, M., Ethier, N., Marmabachi, A. M., Hebert, T. E., and Nattel, S. (2004). KvLQT1 modulates the distribution and biophysical properties of HERG. A novel alpha-subunit interaction between delayed rectifier currents. J. Biol. Chem. 279, 1233–1241.
Ekberg, J., Schuetz, F., Boase, N. A., Conroy, S. J., Manning, J., Kumar, S., Poronnik, P., and Adams, D. J. (2007). Regulation of the voltage-gated K(+) channels KCNQ2/3 and KCNQ3/5 by ubiquitination. Novel role for Nedd4-2. J. Biol. Chem. 282, 12135–12142.
Fadool, D. A., Holmes, T. C., Berman, K., Dagan, D., and Levitan, I. B. (1997). Tyrosine phosphorylation modulates current amplitude and kinetics of a neuronal voltage-gated potassium channel. J. Neurophysiol. 78, 1563–1573.
Freeman, L. C., Lippold, J. J., and Mitchell, K. E. (2000). Glycosylation influences gating and pH sensitivity of I(sK). J. Membr. Biol. 177, 65–79.
Gong, Q., Anderson, C. L., January, C. T., and Zhou, Z. (2002). Role of glycosylation in cell surface expression and stability of HERG potassium channels. Am. J. Physiol. Heart Circ. Physiol. 283, H77–H84.
Gordon, E., Roepke, T. K., and Abbott, G. W. (2006). Endogenous KCNE subunits govern Kv2.1 K+ channel activation kinetics in Xenopus oocyte studies. Biophys. J. 90, 1223–1231.
Grunnet, M., Jespersen, T., Rasmussen, H. B., Ljungstrom, T., Jorgensen, N. K., Olesen, S. P., and Klaerke, D. A. (2002). KCNE4 is an inhibitory subunit to the KCNQ1 channel. J. Physiol. (Lond.) 542, 119–130.
Gu, C., Zhou, W., Puthenveedu, M. A., Xu, M., Jan, Y. N., and Jan, L. Y. (2006). The microtubule plus-end tracking protein EB1 is required for Kv1 voltage-gated K+ channel axonal targeting. Neuron 52, 803–816.
Haitin, Y., and Attali, B. (2008). The C-terminus of Kv7 channels: a multifunctional module. J. Physiol. (Lond.) 586, 1803–1810.
Hegde, R. S., and Lingappa, V. R. (1997). Membrane protein biogenesis: regulated complexity at the endoplasmic reticulum. Cell 91, 575–582.
Heitzmann, D., Grahammer, F., Von Hahn, T., Schmitt-Graff, A., Romeo, E., Nitschke, R., Gerlach, U., Lang, H. J., Verrey, F., Barhanin, J., and Warth, R. (2004). Heteromeric KCNE2/KCNQ1 potassium channels in the luminal membrane of gastric parietal cells. J. Physiol. (Lond.) 561, 547–557.
Heitzmann, D., Koren, V., Wagner, M., Sterner, C., Reichold, M., Tegtmeier, I., Volk, T., and Warth, R. (2007). KCNE beta subunits determine pH sensitivity of KCNQ1 potassium channels. Cell. Physiol. Biochem. 19, 21–32.
Henke, G., Maier, G., Wallisch, S., Boehmer, C., and Lang, F. (2004). Regulation of the voltage gated K+ channel Kv1.3 by the ubiquitin ligase Nedd4-2 and the serum and glucocorticoid inducible kinase SGK1. J. Cell Physiol. 199, 194–199.
Holmes, T. C., Fadool, D. A., and Levitan, I. B. (1996a). Tyrosine phosphorylation of the Kv1.3 potassium channel. J. Neurosci. 16, 1581–1590.
Holmes, T. C., Fadool, D. A., Ren, R., and Levitan, I. B. (1996b). Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science 274, 2089–2091.
Hoppe, U. C., Marban, E., and Johns, D. C. (2001). Distinct gene-specific mechanisms of arrhythmia revealed by cardiac gene transfer of two long QT disease genes, HERG and KCNE1. Proc. Natl. Acad. Sci. U.S.A. 98, 5335–5340.
Iyengar, R., and Hildebrandt, J. D. (2001). G Protein Pathways Receptors Part A. San Diego, CA: Academic Press.
Jespersen, T., Membrez, M., Nicolas, C. S., Pitard, B., Staub, O., Olesen, S. P., Baró, I., and Abriel, H. (2007). The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc. Res. 74, 64–74.
Jiang, B., Sun, X., Cao, K., and Wang, R. (2002). Endogenous Kv channels in human embryonic kidney (HEK-293) cells. Mol. Cell. Biochem. 238, 69–79.
Kanda, V. A., Lewis, A., Xu, X., and Abbott, G. W. (2011a). KCNE1 and KCNE2 inhibit forward trafficking of homomeric N-type voltage-gated potassium channels. Biophys. J. 101, 1354–1363.
Kanda, V. A., Lewis, A., Xu, X., and Abbott, G. W. (2011b). KCNE1 and KCNE2 provide a checkpoint governing voltage-gated potassium channel alpha-subunit composition. Biophys. J. 101, 1364–1375.
Kanda, V. A., Purtell, K., and Abbott, G. W. (2011c). Protein kinase C down regulates I(Ks) by stimulating KCNQ1-KCNE1 potassium channel endocytosis. Heart Rhythm 8, 1641–1647.
Kim, E., Niethammer, M., Rothschild, A., Jan, Y. N., and Sheng, M. (1995). Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature 378, 85–88.
Kim, J., Jung, S. C., Clemens, A. M., Petralia, R. S., and Hoffman, D. A. (2007). Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 54, 933–947.
Krumerman, A., Gao, X., Bian, J. S., Melman, Y. F., Kagan, A., and McDonald, T. V. (2004). An LQT mutant minK alters KvLQT1 trafficking. Am. J. Physiol. Cell Physiol. 286, C1453–C1463.
Li, D., Takimoto, K., and Levitan, E. S. (2000). Surface expression of Kv1 channels is governed by a C-terminal motif. J. Biol. Chem. 275, 11597–11602.
Lu, J., Robinson, J. M., Edwards, D., and Deutsch, C. (2001). T1–T1 interactions occur in ER membranes while nascent Kv peptides are still attached to ribosomes. Biochemistry 40, 10934–10946.
MacKinnon, R., Aldrich, R. W., and Lee, A. W. (1993). Functional stoichiometry of shaker potassium channel inactivation. Science 262, 757–759.
Manganas, L. N., and Trimmer, J. S. (2000). Subunit composition determines Kv1 potassium channel surface expression. J. Biol. Chem. 275, 29685–29693.
Margeta-Mitrovic, M., Jan, Y. N., and Jan, L. Y. (2000). A trafficking checkpoint controls GABA(B) receptor heterodimerization. Neuron 27, 97–106.
McCrossan, Z. A., and Abbott, G. W. (2004). The MinK-related peptides. Neuropharmacology 47, 787–821.
McDonald, T. V., Yu, Z., Ming, Z., Palma, E., Meyers, M. B., Wang, K. W., Goldstein, S. A., and Fishman, G. I. (1997). A minK-HERG complex regulates the cardiac potassium current I (Kr). Nature 388, 289–292.
McEwen, D. P., Schumacher, S. M., Li, Q., Benson, M. D., Iniguez-Lluhi, J. A., Van Genderen, K. M., and Martens, J. R. (2007). Rab-GTPase-dependent endocytic recycling of Kv1.5 in atrial myocytes. J. Biol. Chem. 282, 29612–29620.
McKeown, L., Jones, V. C., and Jones, O. T. (2009). PIN-G reporter for imaging and defining trafficking signals in membrane proteins. Methods Mol. Biol. 574, 235–248.
McKeown, L., Swanton, L., Robinson, P., and Jones, O. T. (2008). Surface expression and distribution of voltage-gated potassium channels in neurons (Review). Mol. Membr. Biol. 25, 332–343.
Nadal, M. S., Ozaita, A., Amarillo, Y., Vega-Saenz De Miera, E., Ma, Y., Mo, W., Goldberg, E. M., Misumi, Y., Ikehara, Y., Neubert, T. A., and Rudy, B. (2003). The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron 37, 449–461.
Nagaya, N., and Papazian, D. M. (1997). Potassium channel alpha and beta subunits assemble in the endoplasmic reticulum. J. Biol. Chem. 272, 3022–3027.
Nesti, E., Everill, B., and Morielli, A. D. (2004). Endocytosis as a mechanism for tyrosine kinase-dependent suppression of a voltage-gated potassium channel. Mol. Biol. Cell 15, 4073–4088.
Neyroud, N., Tesson, F., Denjoy, I., Leibovici, M., Donger, C., Barhanin, J., Faure, S., Gary, F., Coumel, P., Petit, C., Schwartz, K., and Guicheney, P. (1997). A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet. 15, 186–189.
Ohno, H., Stewart, J., Fournier, M. C., Bosshart, H., Rhee, I., Miyatake, S., Saito, T., Gallusser, A., Kirchhausen, T., and Bonifacino, J. S. (1995). Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 269, 1872–1875.
Ottschytsch, N., Raes, A., Van Hoorick, D., and Snyders, D. J. (2002). Obligatory heterotetramerization of three previously uncharacterized Kv channel alpha-subunits identified in the human genome. Proc. Natl. Acad. Sci. U.S.A. 99, 7986–7991.
Ottschytsch, N., Raes, A. L., Timmermans, J. P., and Snyders, D. J. (2005). Domain analysis of Kv6.3, an electrically silent channel. J. Physiol. 568, 737–747.
Panyi, G., and Deutsch, C. (1996). Assembly and suppression of endogenous Kv1.3 channels in human T cells. J. Gen. Physiol. 107, 409–420.
Park, K. H., Kwok, S. M., Sharon, C., Baerga, R., and Sesti, F. (2003). N-Glycosylation-dependent block is a novel mechanism for drug-induced cardiac arrhythmia. FASEB J. 17, 2308–2309.
Park, K. H., and Sesti, F. (2007). An arrhythmia susceptibility gene in Caenorhabditis elegans. J. Biol. Chem. 282, 19799–19807.
Perozo, E., and Bezanilla, F. (1991). Phosphorylation of K+ channels in the squid giant axon. A mechanistic analysis. J. Bioenerg. Biomembr. 23, 599–613.
Piccini, M., Vitelli, F., Seri, M., Galietta, L. J., Moran, O., Bulfone, A., Banfi, S., Pober, B., and Renieri, A. (1999). KCNE1-like gene is deleted in AMME contiguous gene syndrome: identification and characterization of the human and mouse homologs. Genomics 60, 251–257.
Pongs, O., and Schwarz, J. R. (2010). Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 90, 755–796.
Post, M. A., Kirsch, G. E., and Brown, A. M. (1996). Kv2.1 and electrically silent Kv6.1 potassium channel subunits combine and express a novel current. FEBS Lett. 399, 177–182.
Poulsen, A. N., and Klaerke, D. A. (2007). The KCNE1 beta-subunit exerts a transient effect on the KCNQ1 K+ channel. Biochem. Biophys. Res. Commun. 363, 133–139.
Pusch, M., Magrassi, R., Wollnik, B., and Conti, F. (1998). Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75, 785–792.
Ren, X. Q., Liu, G. X., Organ-Darling, L. E., Zheng, R., Roder, K., Jindal, H. K., Centracchio, J., Mcdonald, T. V., and Koren, G. (2010). Pore mutants of HERG and KvLQT1 down regulate the reciprocal currents in stable cell lines. Am. J. Physiol. Heart Circ. Physiol. 299, H1525–H1534.
Rettig, J., Heinemann, S. H., Wunder, F., Lorra, C., Parcej, D. N., Dolly, J. O., and Pongs, O. (1994). Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature 369, 289–294.
Rhodes, K. J., Strassle, B. W., Monaghan, M. M., Bekele-Arcuri, Z., Matos, M. F., and Trimmer, J. S. (1997). Association and colocalization of the Kvbeta1 and Kvbeta2 beta-subunits with Kv1 alpha-subunits in mammalian brain K+ channel complexes. J. Neurosci. 17, 8246–8258.
Robinson, J. M., and Deutsch, C. (2005). Coupled tertiary folding and oligomerization of the T1 domain of Kv channels. Neuron 45, 223–232.
Robinson, M. S. (1994). The role of clathrin, adaptors and dynamin in endocytosis. Curr. Opin. Cell Biol. 6, 538–544.
Roepke, T. K., Anantharam, A., Kirchhoff, P., Busque, S. M., Young, J. B., Geibel, J. P., Lerner, D. J., and Abbott, G. W. (2006). The KCNE2 potassium channel ancillary subunit is essential for gastric acid secretion. J. Biol. Chem. 281, 23740–23747.
Roepke, T. K., Kanda, V. A., Purtell, K., King, E. C., Lerner, D. J., and Abbott, G. W. (2011a). KCNE2 forms potassium channels with KCNA3 and KCNQ1 in the choroid plexus epithelium. FASEB J. 25, 4264–4273.
Roepke, T. K., King, E. C., Purtell, K., Kanda, V. A., Lerner, D. J., and Abbott, G. W. (2011b). Genetic dissection reveals unexpected influence of {beta} subunits on KCNQ1 K+ channel polarized trafficking in vivo. FASEB J. 25, 727–736.
Roepke, T. K., King, E. C., Reyna-Neyra, A., Paroder, M., Purtell, K., Koba, W., Fine, E., Lerner, D. J., Carrasco, N., and Abbott, G. W. (2009). Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat. Med. 15, 1186–1194.
Roepke, T. K., Kontogeorgis, A., Ovanez, C., Xu, X., Young, J. B., Purtell, K., Goldstein, P. A., Christini, D. J., Peters, N. S., Akar, F. G., Gutstein, D. E., Lerner, D. J., and Abbott, G. W. (2008). Targeted deletion of kcne2 impairs ventricular repolarization via disruption of I (K,slow1) and I(to,f). FASEB J. 22, 3648–3660.
Rudy, B., and McBain, C. J. (2001). Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 24, 517–526.
Salinas, M., Duprat, F., Heurteaux, C., Hugnot, J. P., and Lazdunski, M. (1997). New modulatory alpha subunits for mammalian Shab K+ channels. J. Biol. Chem. 272, 24371–24379.
Sanguinetti, M. C., Curran, M. E., Zou, A., Shen, J., Spector, P. S., Atkinson, D. L., and Keating, M. T. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83.
Scott, V. E., Muniz, Z. M., Sewing, S., Lichtinghagen, R., Parcej, D. N., Pongs, O., and Dolly, J. O. (1994). Antibodies specific for distinct Kv subunits unveil a hetero oligomeric basis for subtypes of alpha-dendrotoxin-sensitive K+ channels in bovine brain. Biochemistry 33, 1617–1623.
Seebohm, G., Sanguinetti, M. C., and Pusch, M. (2003). Tight coupling of rubidium conductance and inactivation in human KCNQ1 potassium channels. J. Physiol. (Lond.) 552, 369–378.
Seebohm, G., Strutz-Seebohm, N., Birkin, R., Dell, G., Bucci, C., Spinosa, M. R., Baltaev, R., Mack, A. F., Korniychuk, G., Choudhury, A., Marks, D., Pagano, R. E., Attali, B., Pfeufer, A., Kass, R. S., Sanguinetti, M. C., Tavare, J. M., and Lang, F. (2007). Regulation of endocytic recycling of KCNQ1/KCNE1 potassium channels. Circ. Res. 100, 686–692.
Seebohm, G., Strutz-Seebohm, N., Ureche, O. N., Henrion, U., Baltaev, R., Mack, A. F., Korniychuk, G., Steinke, K., Tapken, D., Pfeufer, A., Kaab, S., Bucci, C., Attali, B., Merot, J., Tavare, J. M., Hoppe, U. C., Sanguinetti, M. C., and Lang, F. (2008). Long QT syndrome-associated mutations in KCNQ1 and KCNE1 subunits disrupt normal endosomal recycling of IKs channels. Circ. Res. 103, 1451–1457.
Sesti, F., and Goldstein, S. A. (1998). Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol. 112, 651–663.
Sesti, F., Liu, S., and Cai, S. Q. (2010). Oxidation of potassium channels by ROS: a general mechanism of aging and neurodegeneration? Trends Cell Biol. 20, 45–51.
Sever, S., Damke, H., and Schmid, S. L. (2000). Dynamin: GTP controls the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J. Cell Biol. 150, 1137–1148.
Shi, G., Nakahira, K., Hammond, S., Rhodes, K. J., Schechter, L. E., and Trimmer, J. S. (1996). Beta subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron 16, 843–852.
Shibata, R., Misonou, H., Campomanes, C. R., Anderson, A. E., Schrader, L. A., Doliveira, L. C., Carroll, K. I., Sweatt, J. D., Rhodes, K. J., and Trimmer, J. S. (2003). A fundamental role for KChIPs in determining the molecular properties and trafficking of Kv4.2 potassium channels. J. Biol. Chem. 278, 36445–36454.
Sole, L., Roura-Ferrer, M., Perez-Verdaguer, M., Oliveras, A., Calvo, M., Fernandez-Fernandez, J. M., and Felipe, A. (2009). KCNE4 suppresses Kv1.3 currents by modulating trafficking, surface expression and channel gating. J. Cell Sci. 122, 3738–3748.
Splawski, I., Tristani-Firouzi, M., Lehmann, M. H., Sanguinetti, M. C., and Keating, M. T. (1997). Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 17, 338–340.
Sroubek, J., and McDonald, T. V. (2011). Protein kinase A activity at the endoplasmic reticulum surface is responsible for augmentation of human ether-a-go-go-related gene product (HERG). J. Biol. Chem. 286, 21927–21936.
Staub, O., and Rotin, D. (2006). Role of ubiquitylation in cellular membrane transport. Physiol. Rev. 86, 669–707.
Steele, D. F., Eldstrom, J., and Fedida, D. (2007). Mechanisms of cardiac potassium channel trafficking. J. Physiol. (Lond.) 582, 17–26.
Stocker, M., Hellwig, M., and Kerschensteiner, D. (1999). Subunit assembly and domain analysis of electrically silent K+ channel alpha-subunits of the rat Kv9 subfamily. J. Neurochem. 72, 1725–1734.
Takimoto, K., and Levitan, E. S. (1994). Glucocorticoid induction of Kv1.5 K+ channel gene expression in ventricle of rat heart. Circ. Res. 75, 1006–1013.
Takumi, T., Ohkubo, H., and Nakanishi, S. (1988). Cloning of a membrane protein that induces a slow voltage-gated potassium current. Science 242, 1042–1045.
Thomas, D., Zhang, W., Karle, C. A., Kathofer, S., Schols, W., Kubler, W., and Kiehn, J. (1999). Deletion of protein kinase A phosphorylation sites in the HERG potassium channel inhibits activation shift by protein kinase A. J. Biol. Chem. 274, 27457–27462.
Tinel, N., Diochot, S., Borsotto, M., Lazdunski, M., and Barhanin, J. (2000). KCNE2 confers background current characteristics to the cardiac KCNQ1 potassium channel. EMBO J. 19, 6326–6330.
Trowbridge, I. S., Collawn, J. F., and Hopkins, C. R. (1993). Signal-dependent membrane protein trafficking in the endocytic pathway. Annu. Rev. Cell Biol. 9, 129–161.
Tu, L., and Deutsch, C. (1999). Evidence for dimerization of dimers in K+ channel assembly. Biophys. J. 76, 2004–2017.
Tu, L., Wang, J., Helm, A., Skach, W. R., and Deutsch, C. (2000). Transmembrane biogenesis of Kv1.3. Biochemistry 39, 824–836.
Tyson, J., Tranebjaerg, L., Bellman, S., Wren, C., Taylor, J. F., Bathen, J., Aslaksen, B., Sorland, S. J., Lund, O., Malcolm, S., Pembrey, M., Bhattacharya, S., and Bitner-Glindzicz, M. (1997). IsK and KvLQT1: mutation in either of the two subunits of the slow component of the delayed rectifier potassium channel can cause Jervell and Lange–Nielsen syndrome. Hum. Mol. Genet. 6, 2179–2185.
Usman, H., and Mathew, M. K. (2010). Potassium channel regulator KCNRG regulates surface expression of shaker-type potassium channels. Biochem. Biophys. Res. Commun. 391, 1301–1305.
Vanoye, C. G., Welch, R. C., Tian, C., Sanders, C. R., and George, A. L. Jr. (2010). KCNQ1/KCNE1 assembly, co-translation not required. Channels (Austin) 4, 108–114.
Varnum, M. D., Busch, A. E., Bond, C. T., Maylie, J., and Adelman, J. P. (1993). The min K channel underlies the cardiac potassium current IKs and mediates species-specific responses to protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 90, 11528–11532.
Vullhorst, D., Klocke, R., Bartsch, J. W., and Jockusch, H. (1998). Expression of the potassium channel KV3.4 in mouse skeletal muscle parallels fiber type maturation and depends on excitation pattern. FEBS Lett. 421, 259–262.
Waldegger, S. (2003). Heartburn: cardiac potassium channels involved in parietal cell acid secretion. Pflugers Arch. 446, 143–147.
Wang, H., Kunkel, D. D., Schwartzkroin, P. A., and Tempel, B. L. (1994). Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J. Neurosci. 14, 4588–4599.
Wang, Z., Fermini, B., and Nattel, S. (1993). Sustained depolarization-induced outward current in human atrial myocytes. Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ. Res. 73, 1061–1076.
Watanabe, I., Zhu, J., Recio-Pinto, E., and Thornhill, W. B. (2004). Glycosylation affects the protein stability and cell surface expression of Kv1. 4 but Not Kv1.1 potassium channels. A pore region determinant dictates the effect of glycosylation on trafficking. J. Biol. Chem. 279, 8879–8885.
Watanabe, I., Zhu, J., Sutachan, J. J., Gottschalk, A., Recio-Pinto, E., and Thornhill, W. B. (2007). The glycosylation state of Kv1.2 potassium channels affects trafficking, gating, and simulated action potentials. Brain Res. 1144, 1–18.
Weiser, M., Vega-Saenz De Miera, E., Kentros, C., Moreno, H., Franzen, L., Hillman, D., Baker, H., and Rudy, B. (1994). Differential expression of Shaw-related K+ channels in the rat central nervous system. J. Neurosci. 14, 949–972.
Xu, J., Yu, W., Jan, Y. N., Jan, L. Y., and Li, M. (1995). Assembly of voltage-gated potassium channels. Conserved hydrophilic motifs determine subfamily-specific interactions between the alpha-subunits. J. Biol. Chem. 270, 24761–24768.
Xu, X., Kanda, V. A., Choi, E., Panaghie, G., Roepke, T. K., Gaeta, S. A., Christini, D. J., Lerner, D. J., and Abbott, G. W. (2009). MinK-dependent internalization of the IKs potassium channel. Cardiovasc. Res. 82, 430–438.
Zerangue, N., Schwappach, B., Jan, Y. N., and Jan, L. Y. (1999). A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22, 537–548.
Zerial, M., and McBride, H. (2001). Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2, 107–117.
Zhang, M., Wang, Y., Jiang, M., Zankov, D. P., Chowdhury, S., Kasirajan, V., and Tseng, G. N. (2012). KCNE2 protein is more abundant in ventricles than in atria and can accelerate hERG protein degradation in a phosphorylation-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 302, H910–H922.
Zhang, Z. J., Jurkiewicz, N. K., Folander, K., Lazarides, E., Salata, J. J., and Swanson, R. (1994). K+ currents expressed from the guinea pig cardiac IsK protein are enhanced by activators of protein kinase C. Proc. Natl. Acad. Sci. U.S.A. 91, 1766–1770.
Zhu, J., Watanabe, I., Gomez, B., and Thornhill, W. B. (2003a). Heteromeric Kv1 potassium channel expression: amino acid determinants involved in processing and trafficking to the cell surface. J. Biol. Chem. 278, 25558–25567.
Zhu, J., Watanabe, I., Gomez, B., and Thornhill, W. B. (2003b). Trafficking of Kv1.4 potassium channels: interdependence of a pore region determinant and a cytoplasmic C-terminal VXXSL determinant in regulating cell-surface trafficking. Biochem. J. 375, 761–768.
Keywords: MinK-related peptides, trafficking, voltage-gated, endocytosis
Citation: Kanda VA and Abbott GW (2012) KCNE regulation of K+ channel trafficking – a Sisyphean task? Front. Physio. 3:231. doi: 10.3389/fphys.2012.00231
Received: 27 April 2012; Accepted: 08 June 2012;
Published online: 28 June 2012.
Edited by:
M. M. Reddy, University of California, USAReviewed by:
Hai Huang, Oregon Health and Science University, USARoger A. Bannister, University of Colorado Denver – Anschutz Medical Campus, USA
Copyright: © 2012 Kanda and Abbott. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Geoffrey W. Abbott, 356A Med Surge II, Department of Pharmacology, School of Medicine, University of California Irvine, Irvine, CA 92697, USA. e-mail: abbottg@uci.edu