

- 1 Institute for Cardiovascular Prevention, Ludwig-Maximilians-University Munich, Munich, Germany
- 2 Rudolf Virchow Center, DFG-Research Center for Experimental Medicine, University of Würzburg, Würzburg, Germany
- 3 Department of Vascular Surgery, Klinikum rechts der Isar, Technical University Munich, Munich, Germany
Atherosclerosis, a chronic inflammatory disease of the vessel wall and the underlying cause of cardiovascular disease, is initiated and maintained by innate and adaptive immunity. Accumulating evidence suggests an important contribution of autoimmune responses to this disease. Plasmacytoid dendritic cells (pDCs), a specialized cell type known to produce large amounts of type I interferons (IFNs) in response to bacterial and viral infections, have recently been revealed to play important roles in atherosclerosis. For example, the development of autoimmune complexes consisting of self-DNA and antimicrobial peptides, which trigger chronic type I IFN production by pDCs, promote early atherosclerotic lesion formation. pDCs and pDC-derived type I IFNs can also induce the maturation of conventional DCs and macrophages, and the development of autoreactive B cells and antibody production. These mechanisms, known to play a role in the pathogenesis of other autoimmune diseases such as systemic lupus erythematosus and psoriasis, may also affect the development and progression of atherosclerotic lesion formation. This review discusses emerging evidence showing a contribution of pDCs in the onset and progression of atherosclerosis.
Introduction
Atherosclerosis is the pathophysiological process underlying cardiovascular diseases such as myocardial infarction and stroke, and the leading cause of death in developed countries. Regarded as a chronic inflammatory disease of the vessel wall, atherosclerosis is driven by responses of both innate and adaptive immunity (Hansson et al., 2006; Galkina and Ley, 2009; Weber and Noels, 2011). Pathomechanistically, atherosclerotic plaque development is initiated by local endothelial dysfunction under conditions of hypercholesterolemia, resulting in lipid depositions in the vessel wall. This triggers the infiltration of blood-borne immune cells and an asymmetrical thickening of the intima. The continuous influx of cells sustains plaque progression, resulting in complex mature plaques consisting of a necrotic core, surrounded by foam cells (lipid laden macrophages) and other immune cells (e.g., T cells, dendritic cells), and covered by a cap of smooth muscle cells and a collagen-rich matrix. Eventually, the release of proinflammatory cytokines and proteases may reduce collagen production and cause thinning of the fibrous cap, which can lead to plaque rupture and trigger an acute thrombotic occlusion of the vessel wall (Ross, 1993; Libby, 2002; Hansson et al., 2006; Weber et al., 2008; Galkina and Ley, 2009; Weber and Noels, 2011).
Among the plaque infiltrating leukocytes, monocytes/macrophages, T cells, neutrophils (Zernecke et al., 2008; Drechsler et al., 2010) but also dendritic cells (DCs; Manthey and Zernecke, 2011; Weber et al., 2011), play an important role in atherosclerosis. DCs are professional antigen-presenting cells equipped with high phagocytic activity, that take up, process and present antigen to T cells. DCs are a very heterogeneous cell population with various subtypes differing in location, migratory pathways, and immunological functions (Shortman and Naik, 2007; Geissmann et al., 2010). In mice, DC subtypes include conventional DCs (cDCs) that are further subdivided into migratory and lymphoid-tissue-resident DCs and based on their expression of CD8α and CD4, inflammatory DCs, e.g., GM-CSF-driven monocyte-derived DCs, and plasmacytoid DCs (pDCs; Shortman and Naik, 2007; Naik, 2008; Ju et al., 2010; Miloud et al., 2010). However, DCs are also known as a very plastic cell type, and depending on the local environment, pDCs for instance may also adopt functions of mature DCs that have the ability to stimulate naïve T cells but no longer produce type I interferons, a feature typical for pDCs (Reizis et al., 2011b).
Dendritic Cells in Atherosclerosis
In general, DC accumulation in regions prone to atherosclerosis suggests that their recruitment accounts for an initial inflammatory or immune activation. The exact localization and origin of vascular DCs, however, is still under debate. CD11c+ DCs have been described in the intima of regions predisposed to atherosclerosis (Jongstra-Bilen et al., 2006; Choi et al., 2009). However, flow cytometric analyses of C57BL/6 as well as Apolipoprotein E-deficient (Apoe−/−) mice revealed the presence of CD11c+ DCs in normal/non-inflamed aortae primarily in the adventitia (Galkina et al., 2006). Moreover, as CD11c expression can also be detected on Ly6Clow monocytes, also these cells may give rise to vascular DCs (Swirski et al., 2007; Tacke et al., 2007; Zhu et al., 2009; Cybulsky and Jongstra-Bilen, 2010). Choi et al. described a CD103+ DC population within the intima, which depends on Flt3L (and thus likely does not originate from monocytes) (Choi et al., 2011). Ccl17+ DCs in turn were described to constitute a subset of mature cDCs in atherosclerotic arteries with pro-atherogenic effects (Weber et al., 2011). All of these DC populations expand during lesion formation, and are able to take up and present antigen. The biggest difference, however, may lay in the fact that CD103+ DCs can already be detected in considerable numbers in the intima during the steady state, whereas Ccl17+ DCs and pDCs primarily seem to enter the vessel wall at sites of inflammation during lesion growth, in line with their “inflammatory” functions.
Lipoproteins deposited in the arterial wall may constitute stimuli or display antigens that participate in early immune activation. oxLDL has been found to induce the upregulation of scavenger receptors on DCs (Nickel et al., 2009), which may contribute to dendritic foam cell formation in the vasculature. Furthermore, short-term depletion of DCs using CD11c-DTR Apoe−/− mice enhanced cholesterolemia implicating that cDCs, possibly via lipoprotein uptake and clearance from the circulation, can control cholesterol homeostasis (Gautier et al., 2009). LDL particles (Hansson and Hermansson, 2011) present within the plaque (or circulation) may affect DC differentiation and function. For example, native LDL and oxLDL can induce the maturation of DCs (HLA-DR and CD86), whereas oxLDL increases the expression of CD40, CD83, CCR7 and interleukin (IL)-6 release and DC-induced T-cell proliferation. Accumulating evidence supports the notion that DCs critically control immune processes in atherosclerosis. The adoptive transfer of OVA-loaded bone marrow derived DCs (BMDCs) induces a profound proliferation of T-cell receptor (TCR)-specific lymphocytes in the adventitia of OTI-Rag-2−/− mice, providing evidence that T cells residing within the aorta can in principle be activated by antigen-presenting cells (Galkina et al., 2006). The antigens recognized in atherosclerosis, however, are still under debate. While modified lipids, e.g., oxidized or malondialdehyde-modified LDL gained most attention due to the early isolation of oxLDL-reactive T cells from human plaque tissue (Stemme et al., 1995), recent studies have in addition implied an important role of native LDL and unmodified ApoB100 as specific antigens in atherosclerotic (Hermansson et al., 2010).
It is well established that T-cell subpopulations with a specific signature of pro- or anti-inflammatory cytokines (Th1, Th2, Th17) control the atherogenic process (Mallat et al., 1999; Binder et al., 2004; Ait-Oufella et al., 2006; Braunersreuther et al., 2007; Kleemann et al., 2008; Taleb et al., 2009; van Es et al., 2009). In particular, also Tregs, which suppress activation of the immune system, have been characterized as inhibitors of atherosclerosis (Gotsman et al., 2006; van Es et al., 2009). Numbers of circulating and lesional Tregs were shown to peak at 4 weeks, but thereafter decrease due to an impaired ability to bind to aortic endothelium after prolonged hypercholesterolemia. Diminished Treg accumulation within lesions is accompanied by an increased accumulation of CD4+ effector T cells, most likely due to the loss of Treg-mediated control functions (Maganto-Garcia et al., 2011). Hence, experimental approaches to expand Tregs may therefore be beneficial. Indeed, adoptive transfer of ApoB100-pulsed myeloid DCs additionally treated with IL-10 resulted in decreased lesion formation in LDL receptor-deficient (Ldlr−/−) mice, accompanied by the induction of regulatory T cells (Tregs) and a reduced lesional T effector cell infiltration (Hermansson et al., 2011). Notably, immunization with apolipoprotein B alone seems to be atheroprotective by stimulating Treg responses in vivo (Wigren et al., 2011).
We have recently identified the accumulation of a subset of CCL17-expressing cDCs in the aorta of Apoe−/− mice during lesion growth, which mediates the recruitment of T cells to atherosclerotic lesions, but systemically and at sites of plaque growth also constrains Treg-maintenance, thereby driving atherosclerotic lesion formation (Weber et al., 2011). Conversely, the subset of CD103+ DCs identified by Choi et al. (2011) are atheroprotective; their absence results in decreased Treg numbers and increased expression of inflammatory cytokines. These findings imply that different DC subsets may exert distinct and stage-specific functions during plaque evolution. Furthermore, their influence on T-cell immunity, especially with regards to the control of Treg responses is crucial in the development of atherosclerosis.
While functions of DCs in atherosclerosis have recently been reviewed in more detail elsewhere (Bobryshev, 2010; Cybulsky and Jongstra-Bilen, 2010; Niessner and Weyand, 2010; Puddu et al., 2010; Koltsova and Ley, 2011; Manthey and Zernecke, 2011; Moore and Tabas, 2011; Perrins and Bobryshev, 2011), we here focus on the emerging role and current state of knowledge regarding the contribution of pDCs to atherosclerosis.
Plasmacytoid DCs and Type I Interferon Responses
Plasmacytoid DCs are a specialized subpopulation of DCs first described by Lennert and Remmele (1958) as plasmacytoid T cells or plasmacytoid monocytes in humans (Grouard et al., 1997; Facchetti and Vergoni, 2000). Human pDC can now be identified by expression of IL-3 receptor-α chain (CD123; Grouard et al., 1997) and blood DC antigen 2 and 4 (BDCA-2 and BDCA-4; Dzionek et al., 2001), whereas in mice pDCs express sialic-acid-binding immunoglobulin-like lectin H (SiglecH; Blasius et al., 2006a) and bone marrow stromal cell Ag 2 (BST2; Blasius et al., 2006b). pDCs are highly specialized for sensing viral and certain microbial infections, based on their selective expression of TLR7 and TLR9, and their capacity to respond to inactivated viral nucleic acids in the absence of viral replication (Fitzgerald-Bocarsly et al., 2008). Upon activation of TLR7 and TLR9, pDCs have the unique ability to produce large amounts of type I IFNs (Kadowaki et al., 2001). Despite being a relatively rare leukocyte subtype in humans and mice (e.g., in mice ranging at 0.1–0.4% of blood and 1.5% of bone marrow cells, and in humans at up to 0.5% of blood leukocytes), pDCs are capable of producing up to 1000-fold higher levels of type I IFNs compared to other cell types. Hence, they are the main source of type I interferons (IFN) during infection (Siegal et al., 1999; Asselin-Paturel et al., 2003), when pDC numbers increase and become detectable in various tissues, such as lung, vaginal mucosa, and skin (Gilliet et al., 2008; Lande and Gilliet, 2010). The IFN family consists of two main classes of related cytokines: type I and type II IFNs, discriminated by their ability to “interfere” with viral infections. Type I IFNs have potent antiviral and growth-inhibitory activities and comprise a group of cytokines that share structural homologies and bind to one common receptor (IFN receptor I; Pestka et al., 1987). Among all IFNs produced by pDC (IFNα, IFNβ, IFNλ, and IFNω), the type I IFN IFNα is expressed at highest quantities (Ito et al., 2006). This cytokine can enact multiple functions in different cell types during immune responses, such as the induction of the maturation of CD11c+ cDCs (Ito et al., 2001), the skewing of T cells toward the Th1 phenotype (Kadowaki et al., 2000), or the stimulation of B cells to differentiate into antibody-secreting plasma cells (Le Bon et al., 2001).
Plasmacytoid DCs are described to play a proinflammatory role in the context of different autoimmune diseases, e.g., psoriasis (Gerlini et al., 2006), systemic lupus erythematosus (SLE; Blomberg et al., 2001), and multiple sclerosis (Lande et al., 2008). However, pDCs are also equipped to trigger the development of Tregs, thereby inhibiting acute graft-versus host disease (Hadeiba et al., 2008) or mediating tolerance to vascularized grafts (Ochando et al., 2006). pDC-mediated control of local inflammation in part depends on the release of indoleamine 2,3-dioxygenase (IDO), which can be induced by IFNs (type I and II) and requires non-canonical NF-κB signaling. IDO was shown to facilitate Treg differentiation from naïve CD4+ T cells interacting with pDCs by promoting CD40 signaling including the co-receptor systems CTLA4/CD80, CD40/CD40L, and GITR/GITRL. In turn, these signaling events can sustain IKKα-dependent induction of IDO expression by pDCs, triggering and maintaining an immunosuppressive feedback loop (Puccetti and Grohmann, 2007; Johnson et al., 2009). In addition, IDO controls effector T-cell responses by influencing their tryptophan metabolism.
The full scope of (p)DC-driven effects in vascular pathologies, however, remains to be elucidated (Bobryshev and Lord, 1995; Galkina and Ley, 2009; Manthey and Zernecke, 2011). We here provide an overview of the functions of pDCs and their signature cytokine IFNα in atherosclerosis.
pDCs in Coronary Artery Disease
Plasmacytoid DCs can be detected in atherosclerotic lesions in humans (Yilmaz et al., 2004) and mice (Jongstra-Bilen et al., 2006), predominantly in the shoulder region of plaques and in clusters with cDCs. As an indirect measure to determine a possible role of pDC in coronary artery disease and atherosclerosis, pDC (and cDC) numbers have been evaluated in the blood of patients with stable and unstable coronary disease, in comparison to controls. van Vre et al. (2006) observed that pDC numbers were decreased in patients with troponin-positive unstable coronary artery syndromes, and that disease severity was inversely correlated with a reduction in pDC numbers (van Brussel et al., 2010). Similarly, Yilmaz et al. (2009) described significantly lower numbers of pDCs (and other DC subsets) in patients with advanced stable coronary artery disease. It was assumed that these findings may relate to an enhanced recruitment of this cell type into plaques. Likewise, pDCs were implicated in myocardial infarction due to their recruitment to the heart post-infarct (Sorrentino et al., 2010). Other studies, however, have also reported unchanged circulating pDC numbers in patients with coronary artery disease (Shi et al., 2007), Kawasaki disease (Yilmaz et al., 2007), and myocardial infarction (Wen et al., 2012). These differences may at least in part originate from the use of different markers to identify pDCs in these studies.
Plasmacytoid DC Phenotype in Atherosclerosis
There is evidence that pDC functions may be impaired in patients suffering from coronary artery disease. Blood-born pDCs from patients with coronary artery disease and cultured in vitro, displayed a reduction in the expression of CD83 and decreased IFNα secretion after stimulation (van Brussel et al., 2011). Functional alterations of pDCs were furthermore observed in patients suffering from both atherosclerosis and type 2 diabetes, with circulating pDCs showing aberrant cytokine secretion (Corrales et al., 2007). Underlying mechanisms for these observations are yet to be determined.
Modified lipoproteins, in particular oxLDL, are key mediators in atherosclerosis. pDCs are also affected by oxLDL; Nickel et al. (2009) reported that oxLDL-stimulation of monocyte-derived DC progenitors promotes the differentiation of BDCA2+ pDCs. We have recently observed that isolated pDCs are able to take up oxLDL ex vivo, and that oxLDL treatment not only upregulates expression of the scavenger receptor CD36, the predominant receptor for oxLDL (Kunjathoor et al., 2002), but also the uptake of other proteins, and to promote pDC-driven antigen-specific T-cell proliferation (Döring et al., 2012; Figure 1). oxLDL may thus enhance pDC-driven immune responses in the context of atherosclerosis.
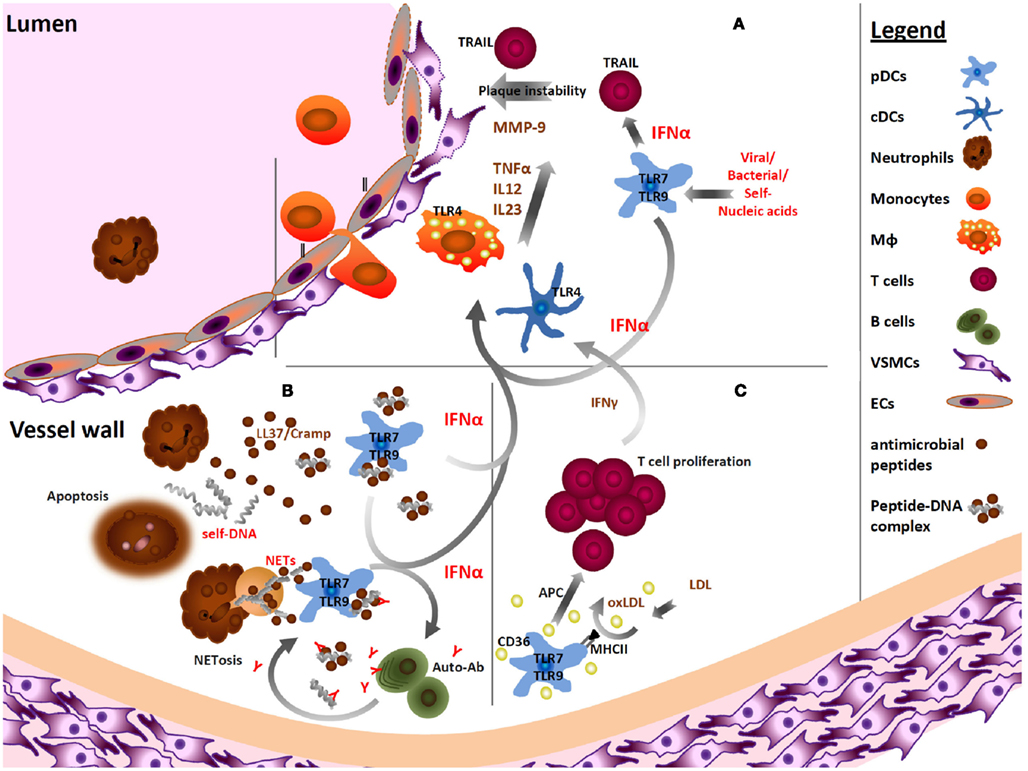
Figure 1. Plasmacytoid DCs in atherosclerosis. (A) pDCs activated by nucleic acids of viral, bacterial or self-origin via TLR7 and TLR9 produce robust amounts of IFNα. This cytokine enhances the sensitivity of other antigen presenting cells by upregulation of TLR4. Furthermore, IFNα upregulates the expression of the pro-apoptotic molecule TRAIL on CD4+ T cells, thereby multiplying their cytotoxic potential. These TRAIL-expressing T cells have the ability to kill plaque-resident cells such as vascular smooth muscle cells (VSMCs) and endothelial cells (ECs). IFNα activates cDCs and macrophages (MΦ) to produce effector molecules such as metalloproteinases (MMP) degrading the extracellular matrix, and proinflammatory cytokines, such as IL-12, TNFα or IL-23 (Niessner and Weyand, 2010). (B) The antimicrobial peptides LL37/Cramp secreted by infiltrating neutrophils bind self-DNA fragments released from dying cells to form aggregates of self-DNA–LL37/Cramp. These complexes activate pDCs and lead to a robust type I interferon production. Furthermore, netting neutrophils release nuclear chromatin structures riddled with antimicrobial granule proteins, and also these complexes may be strong stimulators of IFNα production by pDCs within atherosclerotic lesions. Ultimately, IFNα released by activated pDCs supports humoral immunity, e.g., by enabling B cells to undergo isotype-switching and to mature into antibody-secreting cells. Autoantibodies in turn can bind self-DNA to form autoimmune complexes that in addition trigger IFNα secretion by pDCs (Le Bon et al., 2001). (C) Modified lipids, such as oxLDL, are deposited within atherosclerotic lesions, and may be encountered by pDCs resulting in enhanced CD36 surface expression and antigen uptake by pDCs. Subsequently, pDCs can stimulate increased antigen-specific T-cell proliferation and cytokine secretion by T cells furthermore promoting inflammation. (The order of Roman numbers is not hierarchical, all stimulation scenarios described may happen simultaneously.)
Type I IFNs and pDCs in Lesion Development
Several studies have addressed the principle role of type I IFNs in atherosclerosis. In line with a strong proinflammatory activity of this cytokine, Levy et al. (2003) demonstrated increased atherosclerotic lesion size in LDL-receptor deficient (Ldlr−/−) mice after long-term low-dose treatment with IFNα. However, this was accompanied by increased plasma cholesterol and triglyceride levels, complicating data interpretation. Another more recent study demonstrated that the type I IFN IFNβ aggravates atherosclerotic lesion formation without affecting lipid levels Apoe−/− and Ldlr−/− mice on a high fat diet. Enhanced atherosclerosis was associated with an increased chemokine-dependent macrophage-endothelial cell adhesion and leukocyte attraction to atherosclerosis-prone sites, and an increased macrophage accumulation within plaques (Goossens et al., 2010; Noels and Weber, 2011).
In human plaques, secretion of IFNα was shown to correlate with plaque instability. IFNα stimulation triggered the production of IFNγ and TRAIL by CD4+ T cells (TNF-related apoptosis inducing ligand) in an antigen-independent manner, resulting in the killing of vascular smooth muscle cells (Niessner et al., 2006; Figure 1). In addition, IFNα induced the upregulation of TLR4 on cDCs, leading to their sensitization toward TLR4 ligands. In addition, an increased production of proinflammatory cytokines, such as TNFα, IL-12, and IL-23 was observed in cDCs in response to IFNα (Niessner et al., 2007; Figure 1). These findings imply that pDCs and their index cytokine IFNα function as inflammatory amplifiers in vascular disease.
Importantly, patients that suffer from SLE-display chronically increased titers of IFNα and succumb to an increased risk of atherosclerosis (Ronnblom, 2011). Accumulating evidence suggests that type I IFNs (and in particular IFNα) alter the phenotype and function of endothelial progenitor cells and can thereby hamper vascular repair, which may predispose to an increased cardiovascular risk and premature atherosclerosis in SLE (Denny et al., 2007; Lee et al., 2007; Thacker et al., 2010). In addition, IFNα was shown to trigger an increased expression of class A macrophage scavenger receptor (SR-A) expression in peripheral blood mononuclear cells (predominantly in monocytes/macrophages) in SLE patients (Li et al., 2011). An enhanced SR-A-mediated lipid uptake may in consequence support accelerated foam cell formation within plaques, thereby contributing to increased atherosclerosis in patients suffering from SLE.
Recent studies in mice have now probed the direct in vivo role of pDCs in atherosclerosis. Daissormont et al. (2011) reported that pDC depletion with a specific antibody against bone marrow stromal cell antigen 2 (BST2) in Ldlr−/− mice fed a high fat diet, aggravated atherosclerotic lesion development in the carotid artery after collar placement and in the aortic root. Mechanistically, the authors observed that increased plaque formation in the absence of pDCs may be attributable to a loss of IDO-dependent restraint in T-cell proliferation (Daissormont et al., 2011). These findings may be in line with observations by Nakajima et al. (2011) and functions of IDO in DCs in general, demonstrating that the oral administration of eicosapentaenoic acid to Ldlr−/− mice induced an increased IDO expression and immature phenotype in cDCs, which was associated with diminished T effector cell numbers and a substantial regression of atherosclerotic lesions. In contrast, we have recently observed that pDC depletion using a different antibody against BST2 significantly decreased diet-induced early lesion formation in the aortic root and aorta, whereas the specific stimulation of pDCs with type A CpG oligonucleotides (ODNs) lead to a clear increase in plaque burden in high fat diet-fed Apoe−/− mice. Furthermore, administration of IFNα to Apoe−/− mice promoted increased lesion formation, corroborating previous observations by Levy et al. (2003) and Goossens et al. (2010). Notably, in vivo approaches employed by Döring et al. and Daissormont et al. to define the role of pDCs in lesion development significantly differ in a number of ways. Both groups have used an antibody which recognizes the antigen BST2; Daissormont et al. used the 120G8 antibody from Bioceros BV, we have used the commercially available PDCA1 antibody from Miltenyi. While Daissormont et al. reported a repopulation of blood pDCs at 72 h after antibody depletion, and therefore have applied repetitive 120G8 antibody injections four times a week throughout the study period, we have injected the PDCA1 antibody twice within 7 days at the beginning of our study and have observed restoration of pDCs numbers in spleen and bone marrow to require more than 14 days after the last injection. Daissormont et al. were able to detect sufficient numbers of pDCs in blood to monitor antibody depletion and reconstitution; we found it very difficult to trace blood pDC numbers with baseline frequencies ranging at around ∼0.1% of CD45+ blood leukocytes. This may point at important differences in the mouse models employed, with Ldlr−/− mice used by Daissormont et al., and Apoe−/− animals in our study. Moreover, the composition of the high fat diet was different in these studies, and higher cholesterol content was used for Ldlr−/− mice. Daissormont et al. studied atherosclerotic lesion formation in the carotid artery after collar placement, representing a different vascular bed and model system, mostly employed for studying advanced lesion formation, compared to primary atherosclerosis in the aortic root and aorta, as addressed in our study. However, Daissormont et al. in addition analyzed lesion formation in the aortic root, similarly showing accelerated atherosclerosis in pDC-depleted mice. Of note, the same animals that had received a collar were subjected to subsequent plaque analysis in the aortic root. It is conceivable that collar placement in the carotid artery also affected plaque growth in aortic root, e.g., by systemic or local immunological or inflammatory changes induced by the collar, changes in blood pressure, or flow disturbances. The latter may be of particular relevance when reviewing the extent of collar-induced plaque growth in the carotid artery, showing an almost complete occlusion of the artery. In summary, the experimental model, mouse model, composition of the diet, antibody, and its administration regime were different, all of which may underlie differences in plaque formation and explain conflicting data. Notably, pDC depletion by the PDCA1 antibody in Apoe−/− mice with already established lesions, starting after 4 weeks of high fat diet, and analysis of lesion formation in the aortic root and aorta after an additional 4 weeks did not reveal any alterations in the progression of lesion formation in our hands (Döring and Zernecke, unpublished observations).
In general it is now established that pDCs not only represent a first line of defense to pathogens by releasing vast amounts of type I IFNs, but that this cell type is also capable of exerting typical “cDC functions”. The contribution of pDCs to various autoimmune diseases, however, my primarily correspond to functions as an IFN-producing cell type (Gilliet et al., 2008). Tolerogenic functions of pDCs, and the presentation of antigen and stimulation of Treg responses, in contrast, may mostly be confined to specific pDC subsets (Ochando et al., 2006; Hadeiba et al., 2008), e.g., related to mature (Ochando et al., 2006) and/or tissue restricted Ccr9-expressing pDC populations (Hadeiba et al., 2008).
In atherosclerosis, pDCs may influence disease development in both ways, and trigger type I IFN release, but also induce T-cell activation, which could ultimately also include triggering of Treg responses. It may be conceivable that pDCs contribute to the initiation of early lesion development by rapidly secreting type I IFNs as an innate cell type, whereas during more advanced stages of lesion formation, pDCs exert atheroprotective functions and decelerate disease development. However, these notions clearly require further investigations. Given the function of Tregs as powerful inhibitors of atherosclerosis, antigen-specific tolerogenic functions of pDCs seem highly desirable, and elucidation of mechanisms triggering these responses would be of primary interest.
Autoimmune Activation of pDCs
During infection, pDCs sense nucleic acids of foreign origin (but not self-nucleic acids of dying host cells). For this it is critical that the expression of TLR7 and TLR9 is restricted to endosomal compartments within the cell (Barton et al., 2006). However pDCs are able to retain DNA in early endosomes for prolonged periods of time if multimeric DNA complexes are being formed that lead to the sustained activation of IRF7, which dominates the regulation of type I IFN responses in pDCs (Honda et al., 2005). While usually not recognizing self-nucleic acids, free self-DNA can be complexed to cationic LL-37 (or its murine homologue Cramp), a protein overexpressed and released by neutrophils as well as damaged epithelial cells in psoriasis (Lande et al., 2007). These complexes can enter endosomal pDC compartments most likely via lipid rafts (Sandgren et al., 2004), and protect self-DNA from nuclease degradation, resulting in the retention of aggregated self-DNA-LL37 complexes in early endosomes to trigger robust type I IFN production.
Notably, extending findings by Edfeldt et al. (2006) describing the detection of LL-37 in human atherosclerotic lesions, we detected increased Cramp expression in murine atherosclerotic plaques of Apoe−/− mice (Döring et al., 2012; Figure 1). While deficiency in Cramp in bone marrow cells in Ldlr−/− mice protected from atherosclerotic lesion formation, the injection of Apoe−/− mice fed a high fat diet with Cramp (requiring the presence of extracellular DNA) and of preformed DNA-Cramp complexes pDC-dependently enhanced atherosclerotic lesion development (Döring et al., 2012).
Neutrophil extracellular traps (NETs) released during a form of pathogen-induced cell death named NETosis that contain large amounts of Cramp protein (Garcia-Romo et al., 2011; Lande et al., 2011), can furthermore be evidenced within murine plaques, representing another mechanism within atherosclerotic lesions that may drive pDC activation (Döring et al., 2012; Figure 1). This concurs with observations that complexes of NETs that contain DNA, LL37, and HMGB1 trigger pDC activation in SLE and psoriasis (Garcia-Romo et al., 2011; Lande et al., 2011). The pathophysiological relevance of increased NET-formation has furthermore been described for infection-associated thrombosis (Fuchs et al., 2010) and small vessel vasculitis, a chronic autoinflammatory condition characterized by the presence of anti-neutrophil cytoplasmatic autoantibodies (Kessenbrock et al., 2009).
Circulating immune complexes that are comprised of self-DNA and auto-antibodies to DNA, nucleoproteins or NET structures, can furthermore trigger the activation of pDCs, as seen in patients with SLE (Ronnblom et al., 2003; Barrat et al., 2005; Lande et al., 2011). In line, isolated immune complexes from SLE patients containing DNA fragments stimulate IFNα secretion by pDCs in vitro (Lovgren et al., 2004). Mechanistically, complexes of self-DNA and DNA-specific antibodies (produced by autoreactive B cells) are bound and internalized by low-affinity Fc receptors for IgG (FcγRIIA), and translocate to TLR9-containing endosomal compartments (Means et al., 2005). pDCs continuously activated by these immune complexes sustain the production of type I IFNs, a mechanism likely contributing to pathophysiologically elevated type I IFN levels in SLE (Guiducci et al., 2010). Production of type I IFNs can in turn furthermore promote autoreactive B- and T-cell stimulation (Blanco et al., 2001; Jego et al., 2003; Eloranta et al., 2009). Increased pDC numbers and elevated IFNα levels in SLE patients may thus serve as one explanation for an increased risk to develop atherosclerosis (Frostegard, 2008). Importantly, a hallmark of SLE diagnostics is the detection of anti-nuclear antibodies (ANAs), including anti-dsDNA antibodies (Banchereau and Pascual, 2006). In 115 tested patients, these autoantibodies were already present 3.4 years before the diagnosis of SLE. A progressive accumulation of specific autoantibodies may thus occur before the onset of disease at a time when patients are still asymptomatic (Arbuckle et al., 2003; Tew et al., 2012).
One of the triggers causing autoantibody production in SLE patients may in addition arise from a molecular mimicry between a peptide from latent viral protein Epstein-Barr virus nuclear antigen-1 (EBNA-1) and a specific lupus auto-antigen, in accordance with an etiologic role for Epstein-Barr virus in SLE (McClain et al., 2005). Given the association of Epstein-Barr virus infection with atherosclerosis (Rupprecht et al., 2001), virus, but then also anti-dsDNA antibody-mediated pDC activation and IFNα production, may likewise contribute to accelerated atherosclerotic lesion formation.
Importantly, Pertovaara et al. (2009) observed that elevated anti-nuclear antibody titers were associated with decreased carotid elasticity in young Finns, which may indicate their contribution to the development of early atherosclerosis. Similarly, we recently detected enhanced anti-dsDNA antibody titers in patients with symptomatic compared with asymptomatic carotid artery stenosis and in atherosclerotic Apoe−/− mice compared with healthy controls but not in mice depleted of pDCs (Döring et al., 2012). In addition, defects in apoptotic cell clearance associated with an accumulation of self-DNA was proposed as a pathogenic trigger in both atherogenesis and SLE in a study using a combined mouse model of lupus and atherosclerosis (Aprahamian et al., 2004).
Concluding Remarks
The complex pathophysiology of atherosclerosis as a chronic inflammatory disease is influenced by innate and adaptive immune mechanisms. Although antigens discussed as being recognized in atherosclerosis are of self-origin, e.g., oxLDL or HSP60, the recognition of atherosclerosis as being partly driven by autoimmune mechanisms is still a matter of debate (Blasi, 2008). The aberrant conversion of self-nucleic acids into TLR7/9 ligands and triggers of pDC activation, however, may represent a common nominator of several pathogenic cascades in autoimmune diseases, such as in psoriasis and SLE (Reizis et al., 2011a). Importantly, both SLE and psoriasis are associated with an increased risk of atherosclerosis (Ghazizadeh et al., 2010; Lopez-Pedrera et al., 2010; Sherer et al., 2010). Thus, this pathogenic mechanism may also be relevant in atherosclerosis, and precipitate vascular disease. pDCs represent a diverse cell population mediating a variety of immune functions in atherosclerosis. This plasticity includes inflammatory responses leading to DC and T-cell stimulation (Niessner et al., 2006, 2007) as well as breakdown of self-tolerance (Döring et al., 2012), both of which increase lesion burden. However, pDCs also seem to be capable of facilitating protective effects by controlling T effector cell responses (Daissormont et al., 2011).
More advanced animal models (e.g., cell specific knock-out mouse models, as recently described by the group of Marco Colonna; Swiecki et al., 2010) that allow the stage-specific depletion of pDCs, and the employment of pDC-specific knock-out mouse models of, e.g., IFNα and IDO, will help to unequivocally scrutinize the role of pDCs in lesion formation and their predominant mediators in promoting or curtailing inflammation. Moreover, a better characterization of pDCs and their potential subsets in steady state and during lesion evolution, as well as mapping their fate during lesion development to determine a possible plasticity and differentiation into cDC-like cells with different functions are to be addressed in future studies.
A detailed understanding of the mechanisms triggering inflammatory versus protective immune responses by pDCs in the context of atherosclerosis should be an important objective in future research (Niessner and Weyand, 2010), and may promote the discovery of novel therapeutic approaches targeting autoimmune disease and atherosclerosis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (FOR809, ZE 827/1-2; SFB 688 TPA12; ZE 827/4-1).
References
Ait-Oufella, H., Salomon, B. L., Potteaux, S., Robertson, A. K., Gourdy, P., Zoll, J., Merval, R., Esposito, B., Cohen, J. L., Fisson, S., Flavell, R. A., Hansson, G. K., Klatzmann, D., Tedgui, A., and Mallat, Z. (2006). Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 12, 178–180.
Aprahamian, T., Rifkin, I., Bonegio, R., Hugel, B., Freyssinet, J. M., Sato, K., Castellot, J. J. Jr., and Walsh, K. (2004). Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J. Exp. Med. 199, 1121–1131.
Arbuckle, M. R., Mcclain, M. T., Rubertone, M. V., Scofield, R. H., Dennis, G. J., James, J. A., and Harley, J. B. (2003). Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 349, 1526–1533.
Asselin-Paturel, C., Brizard, G., Pin, J. J., Briere, F., and Trinchieri, G. (2003). Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J. Immunol. 171, 6466–6477.
Banchereau, J., and Pascual, V. (2006). Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25, 383–392.
Barrat, F. J., Meeker, T., Gregorio, J., Chan, J. H., Uematsu, S., Akira, S., Chang, B., Duramad, O., and Coffman, R. L. (2005). Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 202, 1131–1139.
Barton, G. M., Kagan, J. C., and Medzhitov, R. (2006). Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat. Immunol. 7, 49–56.
Binder, C. J., Hartvigsen, K., Chang, M. K., Miller, M., Broide, D., Palinski, W., Curtiss, L. K., Corr, M., and Witztum, J. L. (2004). IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J. Clin. Invest. 114, 427–437.
Blanco, P., Palucka, A. K., Gill, M., Pascual, V., and Banchereau, J. (2001). Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 294, 1540–1543.
Blasius, A. L., Cella, M., Maldonado, J., Takai, T., and Colonna, M. (2006a). Siglec-H is an IPC-specific receptor that modulates type I IFN secretion through DAP12. Blood 107, 2474–2476.
Blasius, A. L., Giurisato, E., Cella, M., Schreiber, R. D., Shaw, A. S., and Colonna, M. (2006b). Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 177, 3260–3265.
Blomberg, S., Eloranta, M. L., Cederblad, B., Nordlin, K., Alm, G. V., and Ronnblom, L. (2001). Presence of cutaneous interferon-alpha producing cells in patients with systemic lupus erythematosus. Lupus 10, 484–490.
Bobryshev, Y. V., and Lord, R. S. (1995). Ultrastructural recognition of cells with dendritic cell morphology in human aortic intima. Contacting interactions of vascular dendritic cells in athero-resistant and athero-prone areas of the normal aorta. Arch. Histol. Cytol. 58, 307–322.
Braunersreuther, V., Zernecke, A., Arnaud, C., Liehn, E. A., Steffens, S., Shagdarsuren, E., Bidzhekov, K., Burger, F., Pelli, G., Luckow, B., Mach, F., and Weber, C. (2007). Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 27, 373–379.
Choi, J. H., Cheong, C., Dandamudi, D. B., Park, C. G., Rodriguez, A., Mehandru, S., Velinzon, K., Jung, I. H., Yoo, J. Y., Oh, G. T., and Steinman, R. M. (2011). Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 35, 819–831.
Choi, J. H., Do, Y., Cheong, C., Koh, H., Boscardin, S. B., Oh, Y. S., Bozzacco, L., Trumpfheller, C., Park, C. G., and Steinman, R. M. (2009). Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J. Exp. Med. 206, 497–505.
Corrales, J. J., Almeida, M., Burgo, R. M., Hernandez, P., Miralles, J. M., and Orfao, A. (2007). Decreased production of inflammatory cytokines by circulating monocytes and dendritic cells in type 2 diabetic men with atherosclerotic complications. J. Diabetes Complicat. 21, 41–49.
Cybulsky, M. I., and Jongstra-Bilen, J. (2010). Resident intimal dendritic cells and the initiation of atherosclerosis. Curr. Opin. Lipidol. 21, 397–403.
Daissormont, I. T., Christ, A., Temmerman, L., Sampedro Millares, S., Seijkens, T., Manca, M., Rousch, M., Poggi, M., Boon, L., van Der Loos, C., Daemen, M., Lutgens, E., Halvorsen, B., Aukrust, P., Janssen, E., and Biessen, E. A. (2011). Plasmacytoid dendritic cells protect against atherosclerosis by tuning T-cell proliferation and activity. Circ. Res. 109, 1387–1395.
Denny, M. F., Thacker, S., Mehta, H., Somers, E. C., Dodick, T., Barrat, F. J., Mccune, W. J., and Kaplan, M. J. (2007). Interferon-alpha promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood 110, 2907–2915.
Döring, Y., Manthey, H., Drechsler, M., Lievens, D., Megens, R., Soehnlein, O., Busch, M., Manca, M., Koenen, R. R., Pelisek, J., Daemen, M. J., Zenke, M., Lutgens, E., Binder, C. J., Weber, C., and Zernecke, A. (2012). Autoantigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 125, 1673–1683.
Drechsler, M., Megens, R. T., van Zandvoort, M., Weber, C., and Soehnlein, O. (2010). Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 122, 1837–1845.
Dzionek, A., Sohma, Y., Nagafune, J., Cella, M., Colonna, M., Facchetti, F., Gunther, G., Johnston, I., Lanzavecchia, A., Nagasaka, T., Okada, T., Vermi, W., Winkels, G., Yamamoto, T., Zysk, M., Yamaguchi, Y., and Schmitz, J. (2001). BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J. Exp. Med. 194, 1823–1834.
Edfeldt, K., Agerberth, B., Rottenberg, M. E., Gudmundsson, G. H., Wang, X. B., Mandal, K., Xu, Q., and Yan, Z. Q. (2006). Involvement of the antimicrobial peptide LL-37 in human atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 26, 1551–1557.
Eloranta, M. L., Lovgren, T., Finke, D., Mathsson, L., Ronnelid, J., Kastner, B., Alm, G. V., and Ronnblom, L. (2009). Regulation of the interferon-alpha production induced by RNA-containing immune complexes in plasmacytoid dendritic cells. Arthritis Rheum. 60, 2418–2427.
Facchetti, F., and Vergoni, F. (2000). The plasmacytoid monocyte: from morphology to function. Adv. Clin. Path. 4, 187–190.
Fitzgerald-Bocarsly, P., Dai, J., and Singh, S. (2008). Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 19, 3–19.
Fuchs, T. A., Brill, A., Duerschmied, D., Schatzberg, D., Monestier, M., Myers, D. D. Jr., Wrobleski, S. K., Wakefield, T. W., Hartwig, J. H., and Wagner, D. D. (2010). Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. U.S.A. 107, 15880–15885.
Galkina, E., Kadl, A., Sanders, J., Varughese, D., Sarembock, I. J., and Ley, K. (2006). Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J. Exp. Med. 203, 1273–1282.
Galkina, E., and Ley, K. (2009). Immune and inflammatory mechanisms of atherosclerosis (*). Annu. Rev. Immunol. 27, 165–197.
Garcia-Romo, G. S., Caielli, S., Vega, B., Connolly, J., Allantaz, F., Xu, Z., Punaro, M., Baisch, J., Guiducci, C., Coffman, R. L., Barrat, F. J., Banchereau, J., and Pascual, V. (2011). Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra20.
Gautier, E. L., Huby, T., Saint-Charles, F., Ouzilleau, B., Pirault, J., Deswaerte, V., Ginhoux, F., Miller, E. R., Witztum, J. L., Chapman, M. J., and Lesnik, P. (2009). Conventional dendritic cells at the crossroads between immunity and cholesterol homeostasis in atherosclerosis. Circulation 119, 2367–2375.
Geissmann, F., Manz, M. G., Jung, S., Sieweke, M. H., Merad, M., and Ley, K. (2010). Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661.
Gerlini, G., Mariotti, G., Bianchi, B., and Pimpinelli, N. (2006). Massive recruitment of type I interferon producing plasmacytoid dendritic cells in varicella skin lesions. J. Invest. Dermatol. 126, 507–509.
Ghazizadeh, R., Shimizu, H., Tosa, M., and Ghazizadeh, M. (2010). Pathogenic mechanisms shared between psoriasis and cardiovascular disease. Int. J. Med. Sci. 7, 284–289.
Gilliet, M., Cao, W., and Liu, Y. J. (2008). Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8, 594–606.
Goossens, P., Gijbels, M. J., Zernecke, A., Eijgelaar, W., Vergouwe, M. N., van Der Made, I., Vanderlocht, J., Beckers, L., Buurman, W. A., Daemen, M. J., Kalinke, U., Weber, C., Lutgens, E., and De Winther, M. P. (2010). Myeloid type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell Metab. 12, 142–153.
Gotsman, I., Grabie, N., Gupta, R., Dacosta, R., Macconmara, M., Lederer, J., Sukhova, G., Witztum, J. L., Sharpe, A. H., and Lichtman, A. H. (2006). Impaired regulatory T-cell response and enhanced atherosclerosis in the absence of inducible costimulatory molecule. Circulation 114, 2047–2055.
Grouard, G., Rissoan, M. C., Filgueira, L., Durand, I., Banchereau, J., and Liu, Y. J. (1997). The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J. Exp. Med. 185, 1101–1111.
Guiducci, C., Gong, M., Xu, Z., Gill, M., Chaussabel, D., Meeker, T., Chan, J. H., Wright, T., Punaro, M., Bolland, S., Soumelis, V., Banchereau, J., Coffman, R. L., Pascual, V., and Barrat, F. J. (2010). TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 465, 937–941.
Hadeiba, H., Sato, T., Habtezion, A., Oderup, C., Pan, J., and Butcher, E. C. (2008). CCR9 expression defines tolerogenic plasmacytoid dendritic cells able to suppress acute graft-versus-host disease. Nat. Immunol. 9, 1253–1260.
Hansson, G. K., and Hermansson, A. (2011). The immune system in atherosclerosis. Nat. Immunol. 12, 204–212.
Hansson, G. K., Robertson, A. K., and Soderberg-Naucler, C. (2006). Inflammation and atherosclerosis. Annu. Rev. Pathol. 1, 297–329.
Hermansson, A., Johansson, D. K., Ketelhuth, D. F., Andersson, J., Zhou, X., and Hansson, G. K. (2011). Immunotherapy with tolerogenic apolipoprotein B-100-loaded dendritic cells attenuates atherosclerosis in hypercholesterolemic mice. Circulation 123, 1083–1091.
Hermansson, A., Ketelhuth, D. F., Strodthoff, D., Wurm, M., Hansson, E. M., Nicoletti, A., Paulsson-Berne, G., and Hansson, G. K. (2010). Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J. Exp. Med. 207, 1081–1093.
Honda, K., Ohba, Y., Yanai, H., Negishi, H., Mizutani, T., Takaoka, A., Taya, C., and Taniguchi, T. (2005). Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434, 1035–1040.
Ito, T., Amakawa, R., Inaba, M., Ikehara, S., Inaba, K., and Fukuhara, S. (2001). Differential regulation of human blood dendritic cell subsets by IFNs. J. Immunol. 166, 2961–2969.
Ito, T., Kanzler, H., Duramad, O., Cao, W., and Liu, Y. J. (2006). Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 107, 2423–2431.
Jego, G., Palucka, A. K., Blanck, J. P., Chalouni, C., Pascual, V., and Banchereau, J. (2003). Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19, 225–234.
Johnson, B. A. 3rd, Baban, B., and Mellor, A. L. (2009). Targeting the immunoregulatory indoleamine 2,3 dioxygenase pathway in immunotherapy. Immunotherapy 1, 645–661.
Jongstra-Bilen, J., Haidari, M., Zhu, S. N., Chen, M., Guha, D., and Cybulsky, M. I. (2006). Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J. Exp. Med. 203, 2073–2083.
Ju, X., Clark, G., and Hart, D. N. (2010). Review of human DC subtypes. Methods Mol. Biol. 595, 3–20.
Kadowaki, N., Antonenko, S., Lau, J. Y., and Liu, Y. J. (2000). Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J. Exp. Med. 192, 219–226.
Kadowaki, N., Ho, S., Antonenko, S., Malefyt, R. W., Kastelein, R. A., Bazan, F., and Liu, Y. J. (2001). Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 194, 863–869.
Kessenbrock, K., Krumbholz, M., Schonermarck, U., Back, W., Gross, W. L., Werb, Z., Grone, H. J., Brinkmann, V., and Jenne, D. E. (2009). Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15, 623–625.
Kleemann, R., Zadelaar, S., and Kooistra, T. (2008). Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc. Res. 79, 360–376.
Koltsova, E. K., and Ley, K. (2011). How dendritic cells shape atherosclerosis. Trends Immunol. 32, 540–547.
Kunjathoor, V. V., Febbraio, M., Podrez, E. A., Moore, K. J., Andersson, L., Koehn, S., Rhee, J. S., Silverstein, R., Hoff, H. F., and Freeman, M. W. (2002). Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 277, 49982–49988.
Lande, R., Gafa, V., Serafini, B., Giacomini, E., Visconti, A., Remoli, M. E., Severa, M., Parmentier, M., Ristori, G., Salvetti, M., Aloisi, F., and Coccia, E. M. (2008). Plasmacytoid dendritic cells in multiple sclerosis: intracerebral recruitment and impaired maturation in response to interferon-beta. J. Neuropathol. Exp. Neurol. 67, 388–401.
Lande, R., Ganguly, D., Facchinetti, V., Frasca, L., Conrad, C., Gregorio, J., Meller, S., Chamilos, G., Sebasigari, R., Riccieri, V., Bassett, R., Amuro, H., Fukuhara, S., Ito, T., Liu, Y. J., and Gilliet, M. (2011). Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 3, 73ra19.
Lande, R., and Gilliet, M. (2010). Plasmacytoid dendritic cells: key players in the initiation and regulation of immune responses. Ann. N. Y. Acad. Sci. 1183, 89–103.
Lande, R., Gregorio, J., Facchinetti, V., Chatterjee, B., Wang, Y. H., Homey, B., Cao, W., Su, B., Nestle, F. O., Zal, T., Mellman, I., Schroder, J. M., Liu, Y. J., and Gilliet, M. (2007). Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449, 564–569.
Le Bon, A., Schiavoni, G., D’Agostino, G., Gresser, I., Belardelli, F., and Tough, D. F. (2001). Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14, 461–470.
Lee, P. Y., Li, Y., Richards, H. B., Chan, F. S., Zhuang, H., Narain, S., Butfiloski, E. J., Sobel, E. S., Reeves, W. H., and Segal, M. S. (2007). Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum. 56, 3759–3769.
Lennert, K., and Remmele, W. (1958). [Karyometric research on lymph node cells in man. I. Germinoblasts, lymphoblasts & lymphocytes. Acta Haematol. 19, 99–113.
Levy, Z., Rachmani, R., Trestman, S., Dvir, A., Shaish, A., Ravid, M., and Harats, D. (2003). Low-dose interferon-alpha accelerates atherosclerosis in an LDL receptor-deficient mouse model. Eur. J. Intern. Med. 14, 479–483.
Li, J., Fu, Q., Cui, H., Qu, B., Pan, W., Shen, N., and Bao, C. (2011). Interferon-alpha priming promotes lipid uptake and macrophage-derived foam cell formation: a novel link between interferon-alpha and atherosclerosis in lupus. Arthritis Rheum. 63, 492–502.
Lopez-Pedrera, C., Aguirre, M. A., Barbarroja, N., and Cuadrado, M. J. (2010). Accelerated atherosclerosis in systemic lupus erythematosus: role of proinflammatory cytokines and therapeutic approaches. J. Biomed. Biotechnol. 2010.
Lovgren, T., Eloranta, M. L., Bave, U., Alm, G. V., and Ronnblom, L. (2004). Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 50, 1861–1872.
Maganto-Garcia, E., Tarrio, M. L., Grabie, N., Bu, D. X., and Lichtman, A. H. (2011). Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation 124, 185–195.
Mallat, Z., Besnard, S., Duriez, M., Deleuze, V., Emmanuel, F., Bureau, M. F., Soubrier, F., Esposito, B., Duez, H., Fievet, C., Staels, B., Duverger, N., Scherman, D., and Tedgui, A. (1999). Protective role of interleukin-10 in atherosclerosis. Circ. Res. 85, e17–e24.
Manthey, H. D., and Zernecke, A. (2011). Dendritic cells in atherosclerosis: functions in immune regulation and beyond. Thromb. Haemost. 106, 772–778.
McClain, M. T., Heinlen, L. D., Dennis, G. J., Roebuck, J., Harley, J. B., and James, J. A. (2005). Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat. Med. 11, 85–89.
Means, T. K., Latz, E., Hayashi, F., Murali, M. R., Golenbock, D. T., and Luster, A. D. (2005). Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest. 115, 407–417.
Miloud, T., Hammerling, G. J., and Garbi, N. (2010). Review of murine dendritic cells: types, location, and development. Methods Mol. Biol. 595, 21–42.
Moore, K. J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355.
Naik, S. H. (2008). Demystifying the development of dendritic cell subtypes, a little. Immunol. Cell Biol. 86, 439–452.
Nakajima, K., Yamashita, T., Kita, T., Takeda, M., Sasaki, N., Kasahara, K., Shinohara, M., Rikitake, Y., Ishida, T., Yokoyama, M., and Hirata, K. (2011). Orally administered eicosapentaenoic acid induces rapid regression of atherosclerosis via modulating the phenotype of dendritic cells in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 31, 1963–1972.
Nickel, T., Schmauss, D., Hanssen, H., Sicic, Z., Krebs, B., Jankl, S., Summo, C., Fraunberger, P., Walli, A. K., Pfeiler, S., and Weis, M. (2009). oxLDL uptake by dendritic cells induces upregulation of scavenger-receptors, maturation and differentiation. Atherosclerosis 205, 442–450.
Niessner, A., Sato, K., Chaikof, E. L., Colmegna, I., Goronzy, J. J., and Weyand, C. M. (2006). Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation 114, 2482–2489.
Niessner, A., Shin, M. S., Pryshchep, O., Goronzy, J. J., Chaikof, E. L., and Weyand, C. M. (2007). Synergistic proinflammatory effects of the antiviral cytokine interferon-alpha and Toll-like receptor 4 ligands in the atherosclerotic plaque. Circulation 116, 2043–2052.
Niessner, A., and Weyand, C. M. (2010). Dendritic cells in atherosclerotic disease. Clin. Immunol. 134, 25–32.
Noels, H., and Weber, C. (2011). Catching up with important players in atherosclerosis: type I interferons and neutrophils. Curr. Opin. Lipidol. 22, 144–145.
Ochando, J. C., Homma, C., Yang, Y., Hidalgo, A., Garin, A., Tacke, F., Angeli, V., Li, Y., Boros, P., Ding, Y., Jessberger, R., Trinchieri, G., Lira, S. A., Randolph, G. J., and Bromberg, J. S. (2006). Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat. Immunol. 7, 652–662.
Perrins, C. J., and Bobryshev, Y. V. (2011). Current advances in understanding of immunopathology of atherosclerosis. Virchows Arch. 458, 117–123.
Pertovaara, M., Kahonen, M., Juonala, M., Laitinen, T., Taittonen, L., Lehtimaki, T., Viikari, J. S., Raitakari, O. T., and Hurme, M. (2009). Autoimmunity and atherosclerosis: the presence of antinuclear antibodies is associated with decreased carotid elasticity in young women. The Cardiovascular Risk in Young Finns Study. Rheumatology 48, 1553–1556.
Pestka, S., Langer, J. A., Zoon, K. C., and Samuel, C. E. (1987). Interferons and their actions. Annu. Rev. Biochem. 56, 727–777.
Puccetti, P., and Grohmann, U. (2007). IDO and regulatory T cells: a role for reverse signalling and non-canonical NF-kappaB activation. Nat. Rev. Immunol. 7, 817–823.
Puddu, P., Puddu, G. M., Cravero, E., Muscari, S., and Muscari, A. (2010). The functional role of dendritic cells in atherogenesis (review). Mol. Med. Reports 3, 551–554.
Reizis, B., Bunin, A., Ghosh, H. S., Lewis, K. L., and Sisirak, V. (2011a). Plasmacytoid dendritic cells: recent progress and open questions. Annu. Rev. Immunol. 29, 163–183.
Reizis, B., Colonna, M., Trinchieri, G., Barrat, F., and Gilliet, M. (2011b). Plasmacytoid dendritic cells: one-trick ponies or workhorses of the immune system? Nat. Rev. Immunol. 11, 558–565.
Ronnblom, L. (2011). The type I interferon system in the etiopathogenesis of autoimmune diseases. Ups. J. Med. Sci. 116, 227–237.
Ronnblom, L., Eloranta, M. L., and Alm, G. V. (2003). Role of natural interferon-alpha producing cells (plasmacytoid dendritic cells) in autoimmunity. Autoimmunity 36, 463–472.
Ross, R. (1993). The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362, 801–809.
Rupprecht, H. J., Blankenberg, S., Bickel, C., Rippin, G., Hafner, G., Prellwitz, W., Schlumberger, W., and Meyer, J. (2001). Impact of viral and bacterial infectious burden on long-term prognosis in patients with coronary artery disease. Circulation 104, 25–31.
Sandgren, S., Wittrup, A., Cheng, F., Jonsson, M., Eklund, E., Busch, S., and Belting, M. (2004). The human antimicrobial peptide LL-37 transfers extracellular DNA plasmid to the nuclear compartment of mammalian cells via lipid rafts and proteoglycan-dependent endocytosis. J. Biol. Chem. 279, 17951–17956.
Sherer, Y., Zinger, H., and Shoenfeld, Y. (2010). Atherosclerosis in systemic lupus erythematosus. Autoimmunity 43, 98–102.
Shi, H., Ge, J., Fang, W., Yao, K., Sun, A., Huang, R., Jia, Q., Wang, K., Zou, Y., and Cao, X. (2007). Peripheral-blood dendritic cells in men with coronary heart disease. Am. J. Cardiol. 100, 593–597.
Shortman, K., and Naik, S. H. (2007). Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7, 19–30.
Siegal, F. P., Kadowaki, N., Shodell, M., Fitzgerald-Bocarsly, P. A., Shah, K., Ho, S., Antonenko, S., and Liu, Y. J. (1999). The nature of the principal type 1 interferon-producing cells in human blood. Science 284, 1835–1837.
Sorrentino, R., Morello, S., and Pinto, A. (2010). Plasmacytoid dendritic cells: from heart to vessels. Int. J. Vasc. Med. 2010, 430318.
Stemme, S., Faber, B., Holm, J., Wiklund, O., Witztum, J. L., and Hansson, G. K. (1995). T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl. Acad. Sci. U.S.A. 92, 3893–3897.
Swiecki, M., Gilfillan, S., Vermi, W., Wang, Y., and Colonna, M. (2010). Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity 33, 955–966.
Swirski, F. K., Libby, P., Aikawa, E., Alcaide, P., Luscinskas, F. W., Weissleder, R., and Pittet, M. J. (2007). Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Invest. 117, 195–205.
Tacke, F., Alvarez, D., Kaplan, T. J., Jakubzick, C., Spanbroek, R., Llodra, J., Garin, A., Liu, J., Mack, M., van Rooijen, N., Lira, S. A., Habenicht, A. J., and Randolph, G. J. (2007). Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 117, 185–194.
Taleb, S., Romain, M., Ramkhelawon, B., Uyttenhove, C., Pasterkamp, G., Herbin, O., Esposito, B., Perez, N., Yasukawa, H., van Snick, J., Yoshimura, A., Tedgui, A., and Mallat, Z. (2009). Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J. Exp. Med. 206, 2067–2077.
Tew, J. G., El Shikh, M. E., El Sayed, R. M., and Schenkein, H. A. (2012). Dendritic cells, antibodies reactive with oxLDL, and inflammation. J. Dent. Res. 91, 8–16.
Thacker, S. G., Berthier, C. C., Mattinzoli, D., Rastaldi, M. P., Kretzler, M., and Kaplan, M. J. (2010). The detrimental effects of IFN-alpha on vasculogenesis in lupus are mediated by repression of IL-1 pathways: potential role in atherogenesis and renal vascular rarefaction. J. Immunol. 185, 4457–4469.
van Brussel, I., van Vre, E. A., De Meyer, G. R., Vrints, C. J., Bosmans, J. M., and Bult, H. (2010). Expression of dendritic cell markers CD11c/BDCA-1 and CD123/BDCA-2 in coronary artery disease upon activation in whole blood. J. Immunol. Methods 362, 168–175.
van Brussel, I., van Vre, E. A., De Meyer, G. R., Vrints, C. J., Bosmans, J. M., and Bult, H. (2011). Decreased numbers of peripheral blood dendritic cells in patients with coronary artery disease are associated with diminished plasma Flt3 ligand levels and impaired plasmacytoid dendritic cell function. Clin. Sci. 120, 415–426.
van Es, T., van Puijvelde, G. H., Ramos, O. H., Segers, F. M., Joosten, L. A., van Den Berg, W. B., Michon, I. M., De Vos, P., van Berkel, T. J., and Kuiper, J. (2009). Attenuated atherosclerosis upon IL-17R signaling disruption in LDLr deficient mice. Biochem. Biophys. Res. Commun. 388, 261–265.
van Vre, E. A., Hoymans, V. Y., Bult, H., Lenjou, M., van Bockstaele, D. R., Vrints, C. J., and Bosmans, J. M. (2006). Decreased number of circulating plasmacytoid dendritic cells in patients with atherosclerotic coronary artery disease. Coron. Artery Dis. 17, 243–248.
Weber, C., Meiler, S., Doring, Y., Koch, M., Drechsler, M., Megens, R. T., Rowinska, Z., Bidzhekov, K., Fecher, C., Ribechini, E., van Zandvoort, M. A., Binder, C. J., Jelinek, I., Hristov, M., Boon, L., Jung, S., Korn, T., Lutz, M. B., Forster, I., Zenke, M., Hieronymus, T., Junt, T., and Zernecke, A. (2011). CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J. Clin. Invest. 121, 2898–2910.
Weber, C., and Noels, H. (2011). Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 17, 1410–1422.
Weber, C., Zernecke, A., and Libby, P. (2008). The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat. Rev. Immunol. 8, 802–815.
Wen, J., Wen, Y., Zhiliang, L., Lingling, C., Longxing, C., Ming, W., and Qiang, F. (2012). A decrease in the percentage of circulating mDC precursors in patients with coronary heart disease: a relation to the severity and extent of coronary artery lesions? Heart Vessels. PMID:22227998 [Epub ahead of print].
Wigren, M., Kolbus, D., Duner, P., Ljungcrantz, I., Soderberg, I., Bjorkbacka, H., Fredrikson, G. N., and Nilsson, J. (2011). Evidence for a role of regulatory T cells in mediating the atheroprotective effect of apolipoprotein B peptide vaccine. J. Intern. Med. 269, 546–556.
Yilmaz, A., Lochno, M., Traeg, F., Cicha, I., Reiss, C., Stumpf, C., Raaz, D., Anger, T., Amann, K., Probst, T., Ludwig, J., Daniel, W. G., and Garlichs, C. D. (2004). Emergence of dendritic cells in rupture-prone regions of vulnerable carotid plaques. Atherosclerosis 176, 101–110.
Yilmaz, A., Rowley, A., Schulte, D. J., Doherty, T. M., Schroder, N. W., Fishbein, M. C., Kalelkar, M., Cicha, I., Schubert, K., Daniel, W. G., Garlichs, C. D., and Arditi, M. (2007). Activated myeloid dendritic cells accumulate and co-localize with CD3+ T cells in coronary artery lesions in patients with Kawasaki disease. Exp. Mol. Pathol. 83, 93–103.
Yilmaz, A., Schaller, T., Cicha, I., Altendorf, R., Stumpf, C., Klinghammer, L., Ludwig, J., Daniel, W. G., and Garlichs, C. D. (2009). Predictive value of the decrease in circulating dendritic cell precursors in stable coronary artery disease. Clin. Sci. 116, 353–363.
Zernecke, A., Bot, I., Djalali-Talab, Y., Shagdarsuren, E., Bidzhekov, K., Meiler, S., Krohn, R., Schober, A., Sperandio, M., Soehnlein, O., Bornemann, J., Tacke, F., Biessen, E. A., and Weber, C. (2008). Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ. Res. 102, 209–217.
Keywords: atherosclerosis, type I IFN, plasmacytoid dendritic cells
Citation: Döring Y and Zernecke A (2012) Plasmacytoid Dendritic Cells in Atherosclerosis. Front. Physio. 3:230. doi: 10.3389/fphys.2012.00230
Received: 26 February 2012; Paper pending published: 04 April 2012;
Accepted: 07 June 2012; Published online: 28 June 2012.
Edited by:
Klaus Ley, La Jolla Institute for Allergy and Immunology, USACopyright: © 2012 Döring and Zernecke. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Alma Zernecke, Rudolf Virchow Center, DFG-Research Center for Experimental Medicine, University of Würzburg, Josef-Schneider Street 2, 97080 Würzburg, Germany. e-mail:YWxtYS56ZXJuZWNrZUB2aXJjaG93LnVuaS13dWVyemJ1cmcuZGU=