 Kai Friedrichs1
Kai Friedrichs1
- 1 Department of Electrophysiology, Cardiovascular Research Center, University Heart Center Hamburg, Hamburg, Germany
- 2 Department of General and Interventional Cardiology, Cardiovascular Research Center, University Heart Center Hamburg, Hamburg, Germany
Atrial fibrosis with enhanced turnover and deposition of matrix proteins leads to inhomogeneous atrial electrical conduction and gives rise to electrical reentry circuits resulting in atrial fibrillation. The multifactorial pathogenesis of atrial fibrosis involves resident cardiac cells as well as infiltrating leukocytes, both generating and sequestering matrix metalloproteinases (MMPs), a key enzyme family involved in fibrosis. A growing body of evidence points toward an important role of reactive oxygen species (ROS) in the release and activation of pro-MMPs and the stimulation of pro-fibrotic cascades. Myeloperoxidase (MPO), a bactericidal enzyme released from activated polymorphonuclear neutrophils (PMN) is not only associated with a variety of cardiovascular diseases, but has also been shown to be mechanistically linked to atrial fibrosis and fibrillation. MPO catalyzes the generation of reactive species like hypochlorous acid, which affect intracellular signaling cascades in various cells and advance activation of pro-MMPs and deposition of atrial collagen resulting in atrial arrhythmias. Thus, inflammatory mechanisms effectively promote atrial structural remodeling and importantly contribute to the initiation and perpetuation of atrial fibrillation.
Introduction
Atrial fibrillation (AF) remains the most prevalent rhythm disorder affecting 1–1.5% of the population in western industrialized countries and its prevalence is supposed to double within the next 50 years (Go et al., 2001; Savelieva and Camm, 2008). With thromboembolic events as one of the most hazardous complications of this disease, AF emerges as a substantial contributor to morbidity and mortality (Kannel et al., 1982; Tsang et al., 2003; Wang et al., 2003).
AF may be triggered by spontaneous premature depolarizations of myocytes originating, e.g., from the pulmonary veins, alternatively by rapid electrical activity in the form of pulmonary vein tachycardia. Once apparent, AF causes alterations of electrical properties of the atria such as reduction of refractory period, which further propagate the perpetuation of AF (Haïssaguerre et al., 1998).
Consequently, electrical isolation of the pulmonary veins is often an important and effective therapeutic strategy especially in patients with paroxysmal AF or in patients without cardiovascular comorbidities (“lone atrial fibrillation”). But a significant number of patients particularly with persistent or chronic AF remains refractory to this approach, pointing toward involvement of atrial tissue as a substrate in those types of AF (Allessie, 1998; Calkins et al., 2012). In particular cardiovascular diseases such as arterial hypertension, heart failure, coronary artery disease, valvular, or inflammatory disease are independent risk factors to AF. In fact, these disorders are believed to be mechanistically involved, since they participate in electrical, contractile, and structural remodeling of atrial tissue (Kannel et al., 1982).
Still, intercellular and subcellular mechanisms underlying AF remain incompletely understood: besides electrical abnormalities mainly based on ion channel dysfunction and contractile remodeling yielding atrial hypocontractility and atrial dilatation, structural remodeling owing to atrial fibrosis has emerged as one of the key aspects in the pathophysiology of AF (Goette et al., 1996; Iwasaki et al., 2011).
Accumulating evidence now advocates for a critical mechanistic role of inflammatory processes in the pathogenesis of atrial fibrosis. This review intends to summarize the role of reactive species in the development of atrial fibrosis and to elucidate the impact of the leukocyte-derived enzyme myeloperoxidase (MPO) herein.
Fibrosis in Atrial Fibrillation
Fibrosis – A Substrate for Atrial Fibrillation
Apart from electrical remodeling and contractile dysfunction, alterations in atrial tissue structure, namely atrial fibrosis, have been shown to increase the susceptibility to AF and may serve as a critical substrate in the formation of the arrhythmia. Additionally, atrial fibrosis is hard to reverse and has therefore been considered as a major contributor in the progression from paroxysmal to persistent or permanent AF (Xu et al., 2004). However, those structural abnormalities must not solely be related to AF but may also be present in patients with underlying or coexisting cardiovascular disease without necessarily increasing the vulnerability to AF.
Fibrosis has been observed in atrial tissue of patients with AF (Kostin et al., 2002; Boldt et al., 2004; Saito et al., 2007). Additionally, a correlation of the degree of atrial fibrosis with the persistence of AF in patients was demonstrated. Furthermore, in patients undergoing cardiovascular surgery, the degree of atrial fibrosis correlated with the risk for postoperative AF (Xu et al., 2004).
Of note, experimental and clinical investigations revealed, that prevention of atrial fibrosis attenuates AF stability, as anti-fibrotic therapeutic strategies (statins, ACE inhibitors, and AT1-receptor blocker) effectively limit AF substrate formation (Li et al., 2001; Kumagai et al., 2004; Sakabe et al., 2004; Milliez et al., 2005; Lee et al., 2006).
Mechanistically, experimental studies have demonstrated fibrosis to account for decreased velocity and increased heterogeneity of electrical conduction (Li et al., 1999; Ausma et al., 2003; Verheule et al., 2003, 2004; Lee et al., 2006; Rudolph et al., 2010).
Fibrosis-driven AF implies the disturbance of electrical conductance due to disruptions of intercellular communication of atrial myocytes. Collagen fibers represent electrical barriers, which can cause asynchronous propagation of electrical activation (de Jong et al., 2011b). The electrical barriers establish adjacent regions of diverging refractory periods (Spach and Boineau, 1997; Lin and Pan, 2008; Goudis et al., 2012). This morphological atrial compartmentalization facilitates the formation of micro- and macro-reentry circuits resulting in a higher susceptibility to AF.
Furthermore, the uncoupling of cardiomyocytes by collagen facilitates ectopic activity to arise in atrial tissue. Physiologic impulse conduction depends on a balance of energy for current conduction between a depolarized region (“source” of the current) and an adjacent repolarized region (“sink”; Nattel et al., 2008). A mismatch of this so called “source-to-sink” effect, which occurs by virtue of impaired myocyte coupling, enables the propagation of early or delayed after depolarizations (EADs or DADs). This is due to the fact that in well-coupled myocardium surrounding myocytes minimize voltage differences of neighboring cells and thereby suppress EAD or DAD propagation, whereas a decrease of the source-to-sink effect impairs this compensation (Nattel et al., 2008; Xie et al., 2010).
Interestingly, apart from matrix deposition also direct fibroblast-myocyte interactions contribute to electrophysiological alterations. Activated fibroblasts modify conduction velocities in cultured cardiomyocyte monolayers, and it was proven that a direct heterocellular electrotonic communication between cells can induce depolarization of the myocytes (Miragoli et al., 2007; Vasquez et al., 2010).
These electrical and structural characteristics of fibrotic remodeling give rise to a strong arrhythmogenic potential of this process.
Extracellular Matrix, MMPs, and Fibroblasts
The myocardial extracellular matrix (ECM) consists of matrix proteins such as collagens and fibronectin, proteoglycans, and basement membrane proteins like laminins (Hagiwara, 2010). It further comprises various proteases and signaling molecules (Spinale, 2007). The predominant matrix proteins in myocardial ECM are the collagens type I and III, which are deposited in the myocardial interstitium in a fibrillar architecture to ensure myocardial stability and organization (Spinale, 2007). These proteins are arranged in a physiological balance of synthesis and degradation – with a disturbed balance associating with cardiac disease (Swynghedauw, 1999; de Jong et al., 2011a). Enhanced synthesis and deposition of ECM proteins can occur either as replacement fibrosis related to cell death or as reactive fibrosis. In particular interstitial reactive fibrosis has been identified to be highly pro-arrhythmic in the atrial myocardium (de Jong et al., 2011b). Whereas the myocardial production of ECM proteins is mainly carried out by fibroblasts and to a lesser extent by cardiomyocytes and smooth muscle cells, their degradation occurs primarily via matrix metalloproteinases (MMPs). MMPs comprise a family of proteolytic enzymes with a variety of different substrates. They are produced as inactive pro-enzymes mostly by fibroblasts, but also by cardiomyocytes and leukocytes, and can be activated by cleavage of their pro-peptide. In turn, inhibition of MMPs is due to tissue-inhibitors of MMPs (TIMPs). The interrelation of fibrotic remodeling and MMPs is complex and still not yet fully elucidated. Although active MMPs degrade matrix proteins, pro-fibrotic states are mostly accompanied by increased instead of decreased MMP-activity (Li et al., 2000; Nattel et al., 2005). Whether increased MMP expression and activity during states of myocardial remodeling reflects cause or consequence still remains to be fully determined. Pro-fibrotic molecules like TGF-β1 and the plasmin system are known to activate MMPs (Jugdutt, 2003; de Jong et al., 2011a). Thus, MMP-activity might be merely an epiphenomenon of the fibrotic process. However, accumulating evidence supports the notion of a causal role of MMPs in fibrosis and their activity is regarded to result in enhanced instead of reduced ECM deposition, which is particularly valid for the gelatinases MMP-2 and -9 (de Jong et al., 2011a). This assumption is supported by observations, that long-term inhibition of MMPs suppresses fibrosis (Nattel et al., 2005; Moe et al., 2008). This pro-fibrotic effect of MMPs might be due to the concomitant release of matrix-embedded bioactive molecules like cytokines as TNF-α and IL-1β (Flesch et al., 2003; Diwan et al., 2004), growth factors like TGF-β1 and angiotensin II (ATII), and endothelin-1 (Dell’Italia et al., 1997; Wei et al., 1999; Ergul et al., 2000; Multani et al., 2005), which further spur the fibrotic cascade. Furthermore, fragments of matrix proteins themselves form bioactive molecules, so called matrikines, which serve as ligands of leukocyte integrins and other cell activating receptors and effectively stimulate connective tissue formation (Li et al., 2000; Mott and Werb, 2004). Importantly, protein composition, organization, and cross-links rather than just the extent of deposition is considered relevant for the matrix pro-arrhythmic potential (Li et al., 2000; Pellman et al., 2010). It is known that MMP-degraded physiological collagens are replaced by fibrous interstitial deposits of various unorganized ECM proteins.
Collectively, enhanced matrix turnover affects signaling cascades of cardiomyocytes, fibroblasts, endothelial cells, and leukocytes creating an inflammatory milieu, which results in increased fibrotic remodeling (Figure 1).
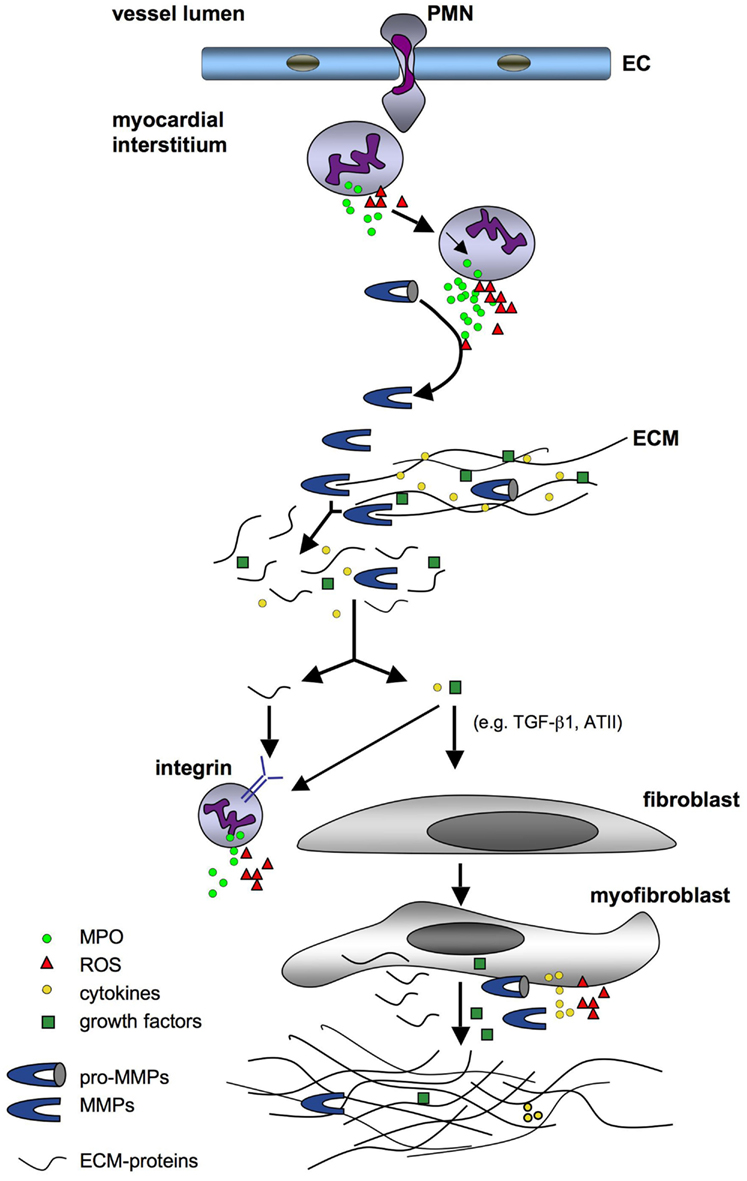
Figure 1. Schematic overview of fibrotic remodeling triggered by reactive species. Neutrophils infiltrating the myocardial interstitium release MPO and ROS, which induce the activation of further neutrophils with sequestration of MPO, ROS, and pro-MMPs. Resident and leukocyte-derived pro-MMPs are converted by MPO and ROS to active MMPs, which degrade the ECM resulting in a release of ECM-protein fragments and embedded cytokines, growth factors, and MMPs. These molecules stimulate leukocytes to secrete further ROS and MPO as well as fibroblasts to differentiate into myofibroblasts. The latter produce cytokines, growth factors, pro-MMPs, and ECM proteins building up a fibrotic matrix. Abbreviations: EC, endothelial cell; ECM, extracellular matrix; MMP, matrix metalloproteinase; MPO, myeloperoxidase; PMN, polymorphonuclear neutrophil; ROS, reactive oxygen species.
The significance of ECM-turnover and MMPs for AF has clearly been pointed out in a number of experimental and clinical studies illustrating that MMP levels are associated with AF in patients with cardiovascular disease (Xu et al., 2004; Gai et al., 2009), demonstrating that MMP inhibition attenuates AF inducibility and maintenance (Moe et al., 2008) and showing that AF recurrence after ablation therapy is associated with MMP serum levels (Kato et al., 2009; Okumura et al., 2011).
Interestingly, the atrial myocardium is especially prone to fibrotic remodeling. Verheule et al. (2003) for example demonstrated that mice with cardiac overexpression of TGF-β1 developed solely atrial but not ventricular fibrosis. Healthy atrial myocardium not only contains more fibroblasts than ventricular myocardium (Hinescu et al., 2006; Burstein et al., 2008), but also displays a more pronounced response to pro-fibrotic stimuli. Atrial fibroblasts exhibit a markedly different gene expression profile and a profoundly increased disposition to proliferate and differentiate in culture and in vivo as compared to ventricular fibroblasts (Burstein et al., 2008). Upon differentiation the active fibroblast phenotype is referred to as myofibroblast. Myofibroblasts differ significantly from fibroblasts and are the principal cell type in the myocardium to produce growth factors, cytokines, MMPs, and ECM proteins (Yue et al., 2011). Moreover, they have a more pronounced disposition to migrate and to proliferate (Vasquez et al., 2011). Thus, the differentiation of fibroblasts to myofibroblasts is a central and important pro-fibrotic event. Besides fibroblasts, cardiac epithelial, and endothelial cells as well as circulating precursors can undergo the differentiation to myofibroblasts (Kis et al., 2011). The differentiation is provoked by mechanical stress, reactive oxygen species (ROS), cytokines, and growth factors. In particular TGF-β1, but also PDGF, ATII, and catecholamines are known to stimulate this process (Rohr, 2009). Apart from promoting the fibrotic cascade myofibroblasts per se may exert highly arrhythmogenic effects. It has been observed under cell-culture conditions and in vivo, that myofibroblasts have distinct electrophysiological properties. On the one hand, they can act as passive electrical conduits, but promote rather slow and discontinuous conduction, since electrical propagation is slower as compared to myocytes (Rohr, 2009). On the other hand, myofibroblasts can lead to ectopic activity by inducing myocyte depolarization. Electrotonic interaction via heterocellular gap-junctional coupling of myofibroblasts with cardiomyocytes has been described to differ significantly from that derived from non-activated fibroblasts (Rohr, 2009; Vasquez et al., 2011; Yue et al., 2011). This further underscores the potentially pro-arrhythmic significance of myofibroblasts.
Oxidative Stress Promotes Atrial Fibrosis
Oxidative stress denotes an unbalanced relation of reactive oxidants and antioxidants and is a hallmark of inflammation. The major sources of reactive species in cardiac cells are mitochondria, xanthine oxidase, uncoupled NO-synthase and in particular NADPH-oxidases (Murdoch et al., 2006). Apart from resident cardiac cells, infiltrated leukocytes account for a large portion of ROS and reactive nitrogen species (RNS) in myocardial tissue via production of superoxide and release of pro-oxidant enzyme systems like MPO.
Impact of Reactive Species on MMPs and Cardiac Cells
Reactive oxygen and nitrogen species exert direct effects on MMPs both on a transcriptional and posttranslational level. The former implies primarily activation of mitogen-activated protein kinases (MAPK) and the Jak/Stat kinase pathway in cardiac cells, which leads to activation of transcription factors inducing MMP release (Siwik and Colucci, 2004; Spinale, 2007). Several in vitro studies demonstrate that hydrogen peroxide (H2O2) induces an increase in MMP release from endothelial cells or cardiac fibroblasts (Belkhiri et al., 1997; Siwik et al., 2001). Moreover, cytokine-induced superoxide production in cardiomyocytes or macrophages increased MMP transcription via MAPK activation (Gurjar et al., 2001; Yoo et al., 2002; Spallarossa et al., 2006). Apart from that, the impact of ROS and RNS on MMP transcription, in particular on MMP-2 and -9, has been proven in vivo and ex vivo in ischemia and reperfusion models or isolated perfused hearts (Wang et al., 2002, 2005; Scholz et al., 2004; Wainwright, 2004).
Many studies have documented posttranslational effects of ROS and RNS on MMPs (Rajagopalan et al., 1996; Fu et al., 2001; Gu et al., 2002; Wang et al., 2002; Yoon et al., 2002; Zhang et al., 2002; Spinale, 2007). Not only in vitro and in cell-culture, but also in vivo and ex vivo the cleavage of the pro-peptide is known to be directly mediated by reactive species. Activation of pro-MMP-2 and -9 has been observed in the presence of H2O2 (Rajagopalan et al., 1996; Siwik et al., 2001; Yoon et al., 2002), peroxynitrite (Rajagopalan et al., 1996; Wang et al., 2002), and hypochlorous acid (HOCl; Fu et al., 2001, 2003).
As mentioned above, ROS and RNS modulate the activation state of cardiac cells, in particular by activating MAPKs and other redox-sensitive signaling pathways. Apart from the release of MMPs, further pro-fibrotic pathways are induced thereby: H2O2 and the superoxide-generating system xanthine/xanthine oxidase led to modulation of collagen synthesis in cardiac fibroblasts (Siwik et al., 2001). Interestingly, a reduced production of collagen was observed herein. However, in vivo studies proved increased deposition of collagen in dependence on ROS, as it was observed in rat hearts following ATII-dependent superoxide production (Zhao et al., 2008). Here, treatment with antioxidants simultaneously to ATII-application reduced the myocardial collagen volume and prevented in part the expression of TGF-β1 and the differentiation of fibroblasts to myofibroblasts. The pro-fibrotic differentiation of fibroblasts to myofibroblasts – as described above – is amongst others also provoked by ROS (Hecker et al., 2009; Barnes and Gorin, 2011). Peroxynitrite for example induced nuclear translocation of NFκ-B in fibroblasts, which led to the differentiation to myofibroblasts with enhanced production of TGF-β, fibronectin, and collagen I (Ichikawa et al., 2008). Furthermore, it has been disclosed, that TGF-β1-induced expression of the NADPH-oxidase isoform Nox4 is mainly responsible for the TGF-β1-dependent differentiation of fibroblasts. In fact, the released superoxide led to activation of the well-known downstream target of TGF-β1, i.e., Smad 2/3 (Cucoranu et al., 2005). Likewise, Nox2 was identified in different animal models to mediate pro-fibrotic effects (Murdoch et al., 2006). Interstitial fibrosis induced by ATII- or aldosterone-administration or transaortic banding has been described to be markedly reduced in Nox2-deficient mice, which was related to changes in expression of collagen and growth factors and activation of NF-κB (Bendall et al., 2002; Sun et al., 2002; Grieve et al., 2006; Johar et al., 2006). Moreover, the involvement of NADPH-oxidases in the activation of pro-fibrotic cascades in cardiac cells has been observed in mice with cardiac deficiency or overexpression of the NADPH-oxidase subunit Rac-1 (Li et al., 2010; Reil et al., 2010; Adam et al., 2011). Interestingly, ROS are described to provoke the formation of an inflammasome – a cytosolic multiprotein complex, which leads to the expression of IL-1β in cardiac fibroblasts with subsequent enhancement of fibrosis (Kawaguchi et al., 2011).
Leukocytes in Atrial Fibrillation
Leukocytes account for the major source of ROS at inflammatory sites with polymorphonuclear neutrophils (PMN) being the first cell type to infiltrate affected tissue.
Whereas the impact of signaling cascades and ROS-formation within cardiac cells was solidified, the role of leukocytes for the development of AF remained undefined. Recent observations however now provide evidence for a causal role of these cells in AF. Elevated atrial leukocyte infiltration is not only found in patients with AF with an underlying structural heart disease (Yamashita, 2007; Chen et al., 2008) as well as in patients with lone AF (Frustaci et al., 1997), but also independently predicts post-surgery AF (Lamm et al., 2006; Ramlawi et al., 2007). Moreover, leukocyte adhesion molecules like VCAM-1 are elevated in animal models and in patients with AF (Goette et al., 2008). So far, it remains unclear whether this is cause or consequence of the atrial arrhythmia. However, the leukocytes’ armament of cytokines like IL-6 and TNF-α, proteases like MMP-8 and -9 and peroxidases like MPO, all of which have been shown to be involved in atrial remodeling (Han et al., 2001; Saba et al., 2005; Spinale, 2007; Rudolph et al., 2010), advocates for an important role of leukocytes in AF.
Role of Myeloperoxidase
MPO and Reactive Species
As stated above, cardiovascular disorders like hypertension, coronary artery disease, and heart failure are important risk factors for AF. Myeloperoxidase (MPO) is not only secreted in a variety of these diseases and in AF itself, but has also been identified to be mechanistically linked to these disorders (Nicholls and Hazen, 2005; Lau and Baldus, 2006), which calls for a deeper understanding of MPO’s contribution.
MPO is a heme peroxidase expressed in myeloid cells – in particular in neutrophils MPO makes up to 5% of the protein dry weight. It is stored in azurophilic granules, which secrete the enzyme into the phagosome or extracellular environment upon activation of the PMN. H2O2 is the essential substrate necessary to convert MPO to an oxidized form (compound I). This in turn reacts with various oxidizable molecules resulting in the generation of ROS and RNS. Primarily (pseudo)halides serve as substrates, with chloride being the most abundant one in physiological environments leading to the formation of HOCl, which is the strongest bactericidal oxidant produced by PMN (Figure 2). However, deficiency of MPO in mice and humans is unexpectedly hardly related to increased susceptibility for infections (Klebanoff, 1970, 2005). Importantly, besides bacterial proteins, HOCl affects various biological targets within the host, including intracellular signaling proteins (Midwinter et al., 2001; Lane et al., 2010), heme proteins, glycoproteins, and lipids (Rees et al., 2010; Szuchman-Sapir et al., 2010; Maitra et al., 2011). In particular the chlorination of tyrosine residues leading to the formation of 3- or 3-,5-(di)chlorotyrosine is considered a marker of MPO’s activity. In addition, these posttranslational modifications also influence structure and function of the affected molecules. For example, MPO-derived oxidants were proven to alter MAPK signaling pathways in macrophages by oxidation of protein thiol residues (Lane et al., 2010). Furthermore, thiol group oxidation by MPO inhibited sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) activity and perturbed Ca2+ homeostasis in endothelial cells (Cook et al., 2012).
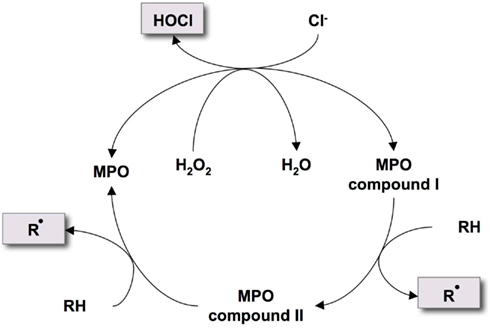
Figure 2. Halogenation and peroxidase cycle of MPO. MPO reacts with H2O2 to form compound I, which oxidizes chloride to build the reactive oxidant hypochlorous acid (HOCl). Alternatively, compound I reacts with oxidizable molecules (RH) like ascorbate or tyrosine to form radical intermediates (Rbullet) via compound II.
Apart from the oxidation of halides MPO compound I also reacts via compound II with various organic and inorganic substrates like aromatic amino acids or ascorbate producing radical intermediates (Eiserich et al., 2002; Figure 2). An important finding in the field of cardiovascular disease is the prevalent reaction of these radical intermediates with nitric oxide (NO; Eiserich et al., 2002). This not only leads to the reduction of NO-bioavailability, but also forms the reactive nitrating agent nitric dioxide. In cardiomyocytes, MPO-dependent protein nitration was shown to effectively promote apoptosis (Yan et al., 2010). As a further consequence of NO-oxidation, impaired endothelial function became obvious in experimental ex vivo (Eiserich et al., 2002) and in vivo studies and in human subjects (Rudolph et al., 2007, 2011).
Thus, chlorination, nitration, and other MPO-induced oxidative modifications of host tissue disturb the physiological function of endothelial and myocardial components. Apart from MPO’s potency to generate reactive species, also its high affinity to biological structures, which is due to its cationic charge, reinforces the pathophysiological significance of the enzyme.
MPO Promotes Fibrosis
As described above, ROS and RNS mediate fibrotic remodeling via activation of MMPs and various intracellular signaling cascades in cardiac cells. Of note, both of these mechanisms also apply to MPO. Fu et al. (2001) described the activation of MMP-7 by HOCl via oxidation of a cysteine residue in the active site leading to autolytic cleavage of the pro-peptide. MPO’s regulatory effect on MMP expression and activation might furthermore be related to the consumption of NO, as a decrease in myocardial NO-bioavailability has been shown to induce MMP-2 release (Wang et al., 2005). In addition, oxidation of a critical cysteine residue induced the inactivation of TIMP-1 (Wang et al., 2007; Figure 3). In vivo, wild-type (WT) mice treated with ATII displayed enhanced activity of MMP-2 and MMP-9 and elevated MMP-9 protein amount in atrial tissue as compared to MPO-deficient (Mpo−/−) mice (Rudolph et al., 2010). This was accompanied by a significant increase in atrial fibrosis in ATII-treated WT mice, which was blunted in Mpo−/−.
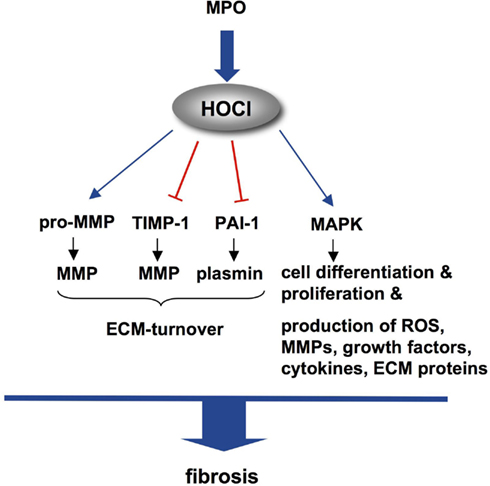
Figure 3. Molecular targets of HOCl mediating fibrosis. MPO-derived HOCl leads to conversion of inactive pro-MMP to active MMP, inactivates TIMP-1 and PAI-1, which results in enhanced ECM-turnover. Activation of MAPKs in various cells initiates pro-fibrotic cascades. Abbreviations: ECM, extracellular matrix; HOCl, hypochlorous acid; MAPK, mitogen-activated protein kinase; MMP, matrix metalloproteinase; PAI-1, plasmin activator-inhibitor-1; ROS, reactive oxygen species; TIMP-1, tissue-inhibitor of MMPs.
In contrast, in a model of acute myocardial infarction the level of pro-MMP-9 and MMP-2 in myocardial tissue was decreased in WT as compared to Mpo−/− mice on day 3 after ligation of the left descending coronary artery (Askari et al., 2003). The authors interpreted these unexpected findings by MPO-dependent consumption of H2O2: increased bioavailability of H2O2 in Mpo−/− mice was assumed to lead to MMP expression and activation. However, collagen deposition was markedly delayed in Mpo−/− as compared to WT mice upon AMI in this study. This observation underlines the complexity of the interrelation of ECM deposition and MMP-activity – in particular in the setting of myocardial infarction (Jugdutt, 2003). The differences between WT and Mpo−/− mice in myocardial remodeling in this model were related to MPO-dependent inhibition of the plasmin activator-inhibitor-1 (PAI-1), which resulted in elevated plasmin activity in WT myocardium. As the fibrinolytic enzyme plasmin has been shown to activate TGF-β1 as well as MMPs (Loskutoff and Quigley, 2000; Zheng and Harris, 2004), and as PAI-1−/− mice display enhanced fibrotic remodeling as compared to WT (Xu et al., 2010), MPO was noted for affecting myocardial remodeling via PAI-1-inhibition (Figure 3). Whether this mechanism is also of significance in atrial myocardium remains to be investigated.
Apart from MPO’s impact on proteolytic enzyme systems, additional mechanisms are initiated by MPO, which may account for its pro-fibrotic properties: given that leukocytes with their high potency to generate reactive species and to secrete cytokines and MMPs are strongly suggested to contribute to fibrosis formation, MPO’s ability to enhance the recruitment of leukocytes (Klinke et al., 2011) might be a pro-fibrotic event. Moreover, MPO not only leads to infiltration of leukocyte but also directly activates MAPK and NADPH-oxidase of PMN via interaction with their integrins CD11b/CD18 (Lau et al., 2005). In addition, activation of macrophages with enhanced production of cytokines is also mediated by MPO (Lefkowitz et al., 1992). Recent data now suggest that MPO-derived HOCl also activates MAPK in cardiomyocytes and endothelial cells (unpublished; Figure 4).
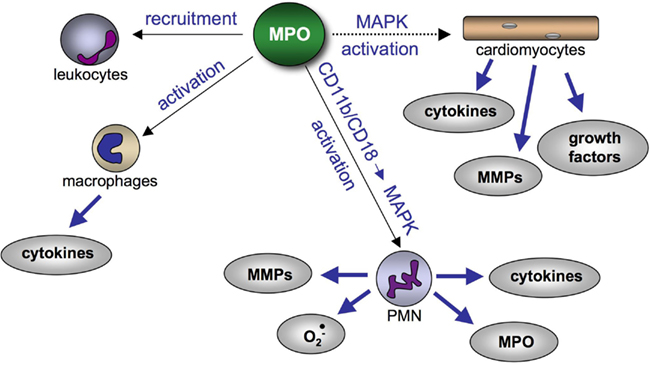
Figure 4. Effect of MPO on cardiac cells and leukocytes contributing to pro-fibrotic remodeling. MPO promotes the recruitment of leukocytes and initiates cytokine secretion from macrophages. Furthermore, MPO activates MAPKs in PMN via CD11b/CD18 integrins, which induces secretion of MMPs, superoxide, cytokines, and MPO. Potentially (dashed line), MPO activates pro-fibrotic pathways in cardiomyocytes via MAPKs. Abbreviations: MAPK, mitogen-activated protein kinase; MMP, matrix metalloproteinase; MPO, myeloperoxidase.
Whether MPO-derived reactive species affect fibroblast differentiation, collagen expression, and other processes as described for ROS remains elusive so far.
MPO in Atrial Fibrillation
MPO’s potency to generate ROS and RNS, its impact on fibrotic remodeling, and its high abundance in leukocytes, which are found to accumulate in fibrillating atria, suggest the enzyme to critically contribute to the formation and maintenance of AF.
In a canine surgical incision model, MPO deposition in the atria was found to correlate with an inhomogeneity of electrical conduction (Ishii et al., 2005). Likewise, surgical induction of pericarditis in dogs lead to marked epicardial injury with reduced expression of connexin-40 and -43 and pronounced accumulation of MPO-positive leukocytes (Ryu et al., 2007; Goldstein et al., 2008). Prednisone treatment in this model prevented the occurrence of atrial flutter, which was accompanied by profoundly reduced MPO immunostaining in the atrial tissue. Furthermore, in patients following AF-ablation an early recurrence of AF was associated with increased MPO plasma levels (Richter et al., 2012). Likewise, increased plasma levels and enhanced atrial deposition of MPO were detected in patients with paroxysmal AF (Rudolph et al., 2010). Whereas these findings remain descriptive without proving a causal role, a clear mechanistic link between MPO and AF development was disclosed within the latter study (Rudolph et al., 2010). As described above, ATII-treated Mpo−/− mice were found to be protected of fibrotic atrial remodeling as compared to WT mice. These animals showed markedly attenuated vulnerability to AF upon programmed and burst atrial electrophysiological stimulation as compared to MPO-competent animals. This observation was accompanied by inhomogeneous atrial electrical conduction in ATII-treated WT but not in Mpo−/− mice assessed by an electrophysiological mapping technique. MPO-activity and resultant modifications in atrial tissue following ATII-treatment in WT mice was evidenced by mass-spectromic analysis of chlorotyrosine. Of note, chronic intravenous infusion of MPO led to a profound and dose-dependent increase in AF vulnerability in WT and Mpo−/− mice. These data underscore the importance of MPO for atrial arrhythmias, but also shed light on the function of ATII herein: whereas ATII exerts direct effects on cardiac fibroblasts and myocytes and stimulates the release of MMPs, its ability to activate leukocytes appears to be of great significance. ATII was proven to induce PMN degranulation in the current study (Rudolph et al., 2010) with elevation of MPO concentrations in plasma and atrial tissue of WT mice. The attenuated atrial fibrosis and fibrillation in MPO-naïve animals suggests, that MPO is in part mandatory for ATII-dependent effects.
Taken together, these findings point out that inflammatory processes and in particular MPO-derived reactive species are critically involved in the pathogenesis of AF.
Outlook
Atrial fibrosis is a critical contributor in AF initiation and perpetuation. The pathogenesis of atrial fibrosis is multifactorial and oftentimes related to hypertension, heart failure, or coronary artery disease. So far, the detailed mechanisms, which trigger fibrotic remodeling and account for enhanced deposition of ECM proteins in the atria remain incompletely understood. MPO, with its high potency to generate reactive species and its impact on cellular signaling cascades has evolved as an important mediator in atrial fibrosis and fibrillation. Inhibition of MPO’s catalytic activity under conditions of established risk factors for AF might emerge as a potential therapeutic target. However, MPO secretion occurs under a large variety of inflammatory conditions and MPO-derived reactive species exhibit a broad range of biological targets. Thus, further studies are necessary to elucidate MPO’s distinct mechanistic implications and therapeutic potentials in AF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adam, O., Theobald, K., Lavall, D., Grube, M., Kroemer, H. K., Ameling, S., Schäfers, H. J., Böhm, M., and Laufs, U. (2011). Increased lysyl oxidase expression and collagen cross-linking during atrial fibrillation. J. Mol. Cell. Cardiol. 50, 678–685.
Allessie, M. A. (1998). Atrial electrophysiologic remodeling: another vicious circle? J. Cardiovasc. Electrophysiol. 9, 1378–1393.
Askari, A. T., Brennan, M. L., Zhou, X., Drinko, J., Morehead, A., Thomas, J. D., Topol, E. J., Hazen, S. L., and Penn, M. S. (2003). Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction. J. Exp. Med. 197, 615–624.
Ausma, J., van der Velden, H. M., Lenders, M. H., van Ankeren, E. P., Jongsma, H. J., Ramaekers, F. C., Borgers, M., and Allessie, M. A. (2003). Reverse structural and gap-junctional remodeling after prolonged atrial fibrillation in the goat. Circulation 107, 2051–2058.
Barnes, J. L., and Gorin, Y. (2011). Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. 79, 944–956.
Belkhiri, A., Richards, C., Whaley, M., McQueen, S. A., and Orr, F. W. (1997). Increased expression of activated matrix metalloproteinase-2 by human endothelial cells after sublethal H2O2 exposure. Lab. Invest. 77, 533–539.
Bendall, J. K., Cave, A. C., Heymes, C., Gall, N., and Shah, A. M. (2002). Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 105, 293–296.
Boldt, A., Wetzel, U., Lauschke, J., Weigl, J., Gummert, J., Hindricks, G., Kottkamp, H., and Dhein, S. (2004). Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart 90, 400–405.
Burstein, B., Libby, E., Calderone, A., and Nattel, S. (2008). Differential behaviors of atrial versus ventricular fibroblasts: a potential role for platelet-derived growth factor in atrial-ventricular remodeling differences. Circulation 117, 1630–1641.
Calkins, H., Kuck, K. H., Cappato, R., Brugada, J., Camm, A. J., Chen, S. A., Crijns, H. J., Damiano, R. J., Davies, D. W., DiMarco, J., Edgerton, J., Ellenbogen, K., Ezekowitz, M. D., Haines, D. E., Haissaguerre, M., Hindricks, G., Iesaka, Y., Jackman, W., Jalife, J., Jais, P., Kalman, J., Keane, D., Kim, Y. H., Kirchhof, P., Klein, G., Kottkamp, H., Kumagai, K., Lindsay, B. D., Mansour, M., Marchlinski, F. E., McCarthy, P. M., Mont, J. L., Morady, F., Nademanee, K., Nakagawa, H., Natale, A., Nattel, S., Packer, D. L., Pappone, C., Prystowsky, E., Raviele, A., Reddy, V., Ruskin, J. N., Shemin, R. J., Tsao, H. M., and Wilber, D. Heart Rhythm Society Task Force on Catheter and Surgical Ablation of Atrial Fibrillation. (2012). 2012 HRS/EHRA/ECAS Expert Consensus Statement on Catheter and Surgical Ablation of Atrial Fibrillation: Recommendations for Patient Selection, Procedural Techniques, Patient Management and Follow-up, Definitions, Endpoints, and Research Trial Design: A report of the Heart Rhythm Society (HRS) Task Force on Catheter and Surgical Ablation of Atrial Fibrillation. Developed in partnership with the European Heart Rhythm Association (EHRA), a registered branch of the European Society of Cardiology (ESC) and the European Cardiac Arrhythmia Society (ECAS); and in collaboration with the American College of Cardiology (ACC), American Heart Association (AHA), the Asia Pacific Heart Rhythm Society (APHRS), and the Society of Thoracic Surgeons (STS). Endorsed by the governing bodies of the American College of Cardiology Foundation, the American Heart Association, the European Cardiac Arrhythmia Society, the European Heart Rhythm Association, the Society of Thoracic Surgeons, the Asia Pacific Heart Rhythm Society, and the Heart Rhythm Society. Heart Rhythm. [Epub ahead of print].
Chen, M. C., Chang, J. P., Liu, W. H., Yang, C. H., Chen, Y. L., Tsai, T. H., Wang, Y. H., and Pan, K. L. (2008). Increased inflammatory cell infiltration in the atrial myocardium of patients with atrial fibrillation. Am. J. Cardiol. 102, 861–865.
Cook, N. L., Viola, H. M., Sharov, V. S., Hool, L. C., Schöneich, C., and Davies, M. J. (2012). Myeloperoxidase-derived oxidants inhibit sarco/endoplasmic reticulum Ca2+-ATPase activity and perturb Ca2+ homeostasis in human coronary artery endothelial cells. Free Radic. Biol. Med. 52, 951–961.
Cucoranu, I., Clempus, R., Dikalova, A., Phelan, P. J., Ariyan, S., Dikalov, S., and Sorescu, D. (2005). NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 97, 900–907.
de Jong, S., van Veen, T. A., de Bakker, J. M., Vos, M. A., and van Rijen, H. V. (2011a). Biomarkers of myocardial fibrosis. J. Cardiovasc. Pharmacol. 57, 522–535.
de Jong, S., van Veen, T. A., van Rijen, H. V., and de Bakker, J. M. (2011b). Fibrosis and cardiac arrhythmias. J. Cardiovasc. Pharmacol. 57, 630–638.
Dell’Italia, L. J., Meng, Q. C., Balcells, E., Wei, C. C., Palmer, R., Hageman, G. R., Durand, J., Hankes, G. H., and Oparil, S. (1997). Compartmentalization of angiotensin II generation in the dog heart. Evidence for independent mechanisms in intravascular and interstitial spaces. J. Clin. Invest. 100, 253–258.
Diwan, A., Dibbs, Z., Nemoto, S., DeFreitas, G., Carabello, B. A., Sivasubramanian, N., Wilson, E. M., Spinale, F. G., and Mann, D. L. (2004). Targeted overexpression of noncleavable and secreted forms of tumor necrosis factor provokes disparate cardiac phenotypes. Circulation 109, 262–268.
Eiserich, J. P., Baldus, S., Brennan, M. L., Ma, W., Zhang, C., Tousson, A., Castro, L., Lusis, A. J., Nauseef, W. M., White, C. R., and Freeman, B. A. (2002). Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 296, 2391–2394.
Ergul, A., Walker, C. A., Goldberg, A., Baicu, S. C., Hendrick, J. W., King, M. K., and Spinale, F. G. (2000). ET-1 in the myocardial interstitium: relation to myocyte ECE activity and expression. Am. J. Physiol. Heart Circ. Physiol. 278, H2050–H2056.
Flesch, M., Höper, A., Dell’Italia, L., Evans, K., Bond, R., Peshock, R., Diwan, A., Brinsa, T. A., Wei, C. C., Sivasubramanian, N., Spinale, F. G., and Mann, D. L. (2003). Activation and functional significance of the renin-angiotensin system in mice with cardiac restricted overexpression of tumor necrosis factor. Circulation 108, 598–604.
Frustaci, A., Chimenti, C., Bellocci, F., Morgante, E., Russo, M. A., and Maseri, A. (1997). Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation 96, 1180–1184.
Fu, X., Kassim, S. Y., Parks, W. C., and Heinecke, J. W. (2001). Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J. Biol. Chem. 276, 41279–41287.
Fu, X., Kassim, S. Y., Parks, W. C., and Heinecke, J. W. (2003). Hypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammation. J. Biol. Chem. 278, 28403–28409.
Gai, X., Lan, X., Luo, Z., Wang, F., Liang, Y., Zhang, H., Zhang, W., Hou, J., and Huang, M. (2009). Association of MMP-9 gene polymorphisms with atrial fibrillation in hypertensive heart disease patients. Clin. Chim. Acta 408, 105–109.
Go, A. S., Hylek, E. M., Phillips, K. A., Chang, Y., Henault, L. E., Selby, J. V., and Singer, D. E. (2001). Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 285, 2370–2375.
Goette, A., Bukowska, A., Lendeckel, U., Erxleben, M., Hammwöhner, M., Strugala, D., Pfeiffenberger, J., Röhl, F. W., Huth, C., Ebert, M. P., Klein, H. U., and Röcken, C. (2008). Angiotensin II receptor blockade reduces tachycardia-induced atrial adhesion molecule expression. Circulation 117, 732–742.
Goette, A., Honeycutt, C., and Langberg, J. J. (1996). Electrical remodeling in atrial fibrillation. Time course and mechanisms. Circulation 94, 2968–2974.
Goldstein, R. N., Ryu, K., Khrestian, C., van Wagoner, D. R., and Waldo, A. L. (2008). Prednisone prevents inducible atrial flutter in the canine sterile pericarditis model. J. Cardiovasc. Electrophysiol. 19, 74–81.
Goudis, C. A., Kallergis, E. M., and Vardas, P. E. (2012). Extracellular matrix alterations in the atria: insights into the mechanisms and perpetuation of atrial fibrillation. Europace 14, 623–630.
Grieve, D. J., Byrne, J. A., Siva, A., Layland, J., Johar, S., Cave, A. C., and Shah, A. M. (2006). Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J. Am. Coll. Cardiol. 47, 817–826.
Gu, Z., Kaul, M., Yan, B., Kridel, S. J., Cui, J., Strongin, A., Smith, J. W., Liddington, R. C., and Lipton, S. A. (2002). S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297, 1186–1190.
Gurjar, M. V., Deleon, J., Sharma, R. V., and Bhalla, R. C. (2001). Role of reactive oxygen species in IL-1 beta-stimulated sustained ERK activation and MMP-9 induction. Am. J. Physiol. Heart Circ. Physiol. 281, H2568–H2574.
Haïssaguerre, M., Jaïs, P., Shah, D. C., Takahashi, A., Hocini, M., Quiniou, G., Garrigue, S., Le Mouroux, A., Le Métayer, P., and Clémenty, J. (1998). Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659–666.
Han, Y. P., Tuan, T. L., Wu, H., Hughes, M., and Garner, W. L. (2001). TNF-alpha stimulates activation of pro-MMP-2 in human skin through NF-(kappa)B mediated induction of MT1-MMP. J. Cell. Sci. 114, 131–139.
Hecker, L., Vittal, R., Jones, T., Jagirdar, R., Luckhardt, T. R., Horowitz, J. C., Pennathur, S., Martinez, F. J., and Thannickal, V. J. (2009). NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 15, 1077–1081.
Hinescu, M. E., Gherghiceanu, M., Mandache, E., Ciontea, S. M., and Popescu, L. M. (2006). Interstitial Cajal-like cells (ICLC) in atrial myocardium: ultrastructural and immunohistochemical characterization. J. Cell. Mol. Med. 10, 243–257.
Ichikawa, T., Sugiura, H., Koarai, A., Yanagisawa, S., Kanda, M., Hayata, A., Furukawa, K., Akamatsu, K., Hirano, T., Nakanishi, M., Matsunaga, K., Minakata, Y., and Ichinose, M. (2008). Peroxynitrite augments fibroblast-mediated tissue remodeling via myofibroblast differentiation. Am. J. Physiol. Lung Cell Mol. Physiol. 295, L800–L808.
Ishii, Y., Schuessler, R. B., Gaynor, S. L., Yamada, K., Fu, A. S., Boineau, J. P., and Damiano, R. J. (2005). Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation. Circulation 111, 2881–2888.
Iwasaki, Y. K., Nishida, K., Kato, T., and Nattel, S. (2011). Atrial fibrillation pathophysiology: implications for management. Circulation 124, 2264–2274.
Johar, S., Cave, A. C., Narayanapanicker, A., Grieve, D. J., and Shah, A. M. (2006). Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 20, 1546–1548.
Jugdutt, B. I. (2003). Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation 108, 1395–1403.
Kannel, W. B., Abbott, R. D., Savage, D. D., and McNamara, P. M. (1982). Epidemiologic features of chronic atrial fibrillation: the Framingham study. N. Engl. J. Med. 306, 1018–1022.
Kato, K., Fujimaki, T., Yoshida, T., Oguri, M., Yajima, K., Hibino, T., and Murohara, T. (2009). Impact of matrix metalloproteinase-2 levels on long-term outcome following pharmacological or electrical cardioversion in patients with atrial fibrillation. Europace 11, 332–337.
Kawaguchi, M., Takahashi, M., Hata, T., Kashima, Y., Usui, F., Morimoto, H., Izawa, A., Takahashi, Y., Masumoto, J., Koyama, J., Hongo, M., Noda, T., Nakayama, J., Sagara, J., Taniguchi, S., and Ikeda, U. (2011). Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123, 594–604.
Kis, K., Liu, X., and Hagood, J. S. (2011). Myofibroblast differentiation and survival in fibrotic disease. Expert Rev. Mol. Med. 13, e27.
Klebanoff, S. J. (1970). Myeloperoxidase: contribution to the microbicidal activity of intact leukocytes. Science 169, 1095–1097.
Klinke, A., Nussbaum, C., Kubala, L., Friedrichs, K., Rudolph, T. K., Rudolph, V., Paust, H. J., Schröder, C., Benten, D., Lau, D., Szocs, K., Furtmüller, P. G., Heeringa, P., Sydow, K., Duchstein, H.-J., Ehmke, H., Schumacher, U., Meinertz, T., Sperandio, M., and Baldus, S. (2011). Myeloperoxidase attracts neutrophils by physical forces. Blood 117, 1350–1358.
Kostin, S., Klein, G., Szalay, Z., Hein, S., Bauer, E. P., and Schaper, J. (2002). Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 54, 361–379.
Kumagai, K., Ogawa, M., Noguchi, H., Yasuda, T., Nakashima, H., and Saku, K. (2004). Electrophysiologic properties of pulmonary veins assessed using a multielectrode basket catheter. J. Am. Coll. Cardiol. 43, 2281–2289.
Lamm, G., Auer, J., Weber, T., Berent, R., Ng, C., and Eber, B. (2006). Postoperative white blood cell count predicts atrial fibrillation after cardiac surgery. J. Cardiothorac. Vasc. Anesth. 20, 51–56.
Lane, A. E., Tan, J. T., Hawkins, C. L., Heather, A. K., and Davies, M. J. (2010). The myeloperoxidase-derived oxidant HOSCN inhibits protein tyrosine phosphatases and modulates cell signalling via the mitogen-activated protein kinase (MAPK) pathway in macrophages. Biochem. J. 430, 161–169.
Lau, D., and Baldus, S. (2006). Myeloperoxidase and its contributory role in inflammatory vascular disease. Pharmacol. Ther. 111, 16–26.
Lau, D., Mollnau, H., Eiserich, J. P., Freeman, B. A., Daiber, A., Gehling, U. M., Brummer, J., Rudolph, V., Munzel, T., Heitzer, T., Meinertz, T., and Baldus, S. (2005). Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc. Natl. Acad. Sci. U.S.A. 102, 431–436.
Lee, K. W., Everett, T. H., Rahmutula, D., Guerra, J. M., Wilson, E., Ding, C., and Olgin, J. E. (2006). Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation 114, 1703–1712.
Lefkowitz, D. L., Mills, K., Morgan, D., and Lefkowitz, S. S. (1992). Macrophage activation and immunomodulation by myeloperoxidase. Proc. Soc. Exp. Biol. Med. 199, 204–210.
Li, D., Fareh, S., Leung, T. K., and Nattel, S. (1999). Promotion of atrial fibrillation by heart failure in dogs: atrial remodeling of a different sort. Circulation 100, 87–95.
Li, D., Shinagawa, K., Pang, L., Leung, T. K., Cardin, S., Wang, Z., and Nattel, S. (2001). Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation 104, 2608–2614.
Li, J., Zhu, H., Shen, E., Wan, L., Arnold, J. M., and Peng, T. (2010). Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 59, 2033–2042.
Li, Y. Y., McTiernan, C. F., and Feldman, A. M. (2000). Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc. Res. 46, 214–224.
Lin, C. S., and Pan, C. H. (2008). Regulatory mechanisms of atrial fibrotic remodeling in atrial fibrillation. Cell. Mol. Life Sci. 65, 1489–1508.
Loskutoff, D. J., and Quigley, J. P. (2000). PAI-1, fibrosis, and the elusive provisional fibrin matrix. J. Clin. Invest. 106, 1441–1443.
Maitra, D., Banerjee, J., Shaeib, F., Souza, C. E., and Abu-Soud, H. M. (2011). Melatonin can mediate its vascular protective effect by modulating free iron level by inhibiting hypochlorous acid-mediated hemoprotein heme destruction. Hypertension 57, e22.
Midwinter, R. G., Vissers, M. C., and Winterbourn, C. C. (2001). Hypochlorous acid stimulation of the mitogen-activated protein kinase pathway enhances cell survival. Arch. Biochem. Biophys. 394, 13–20.
Milliez, P., Deangelis, N., Rucker-Martin, C., Leenhardt, A., Vicaut, E., Robidel, E., Beaufils, P., Delcayre, C., and Hatem, S. N. (2005). Spironolactone reduces fibrosis of dilated atria during heart failure in rats with myocardial infarction. Eur. Heart J. 26, 2193–2199.
Miragoli, M., Salvarani, N., and Rohr, S. (2007). Myofibroblasts induce ectopic activity in cardiac tissue. Circ. Res. 101, 755–758.
Moe, G. W., Laurent, G., Doumanovskaia, L., Konig, A., Hu, X., and Dorian, P. (2008). Matrix metalloproteinase inhibition attenuates atrial remodeling and vulnerability to atrial fibrillation in a canine model of heart failure. J. Card. Fail. 14, 768–776.
Mott, J. D., and Werb, Z. (2004). Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 16, 558–564.
Multani, M. M., Ikonomidis, J. S., Kim, P. Y., Miller, E. A., Payne, K. J., Mukherjee, R., Dorman, B. H., and Spinale, F. G. (2005). Dynamic and differential changes in myocardial and plasma endothelin in patients undergoing cardiopulmonary bypass. J. Thorac. Cardiovasc. Surg. 129, 584–590.
Murdoch, C. E., Zhang, M., Cave, A. C., and Shah, A. M. (2006). NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 71, 208–215.
Nattel, S., Burstein, B., and Dobrev, D. (2008). Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ. Arrhythm. Electrophysiol. 1, 62–73.
Nattel, S., Shiroshita-Takeshita, A., Cardin, S., and Pelletier, P. (2005). Mechanisms of atrial remodeling and clinical relevance. Curr. Opin. Cardiol. 20, 21–25.
Nicholls, S. J., and Hazen, S. L. (2005). Myeloperoxidase and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 25, 1102–1111.
Okumura, Y., Watanabe, I., Nakai, T., Ohkubo, K., Kofune, T., Kofune, M., Nagashima, K., Mano, H., Sonoda, K., Kasamaki, Y., and Hirayama, A. (2011). Impact of biomarkers of inflammation and extracellular matrix turnover on the outcome of atrial fibrillation ablation: importance of matrix matalloproteinase-2 as a predictor of atrial fibrillation recurrence. J. Cardiovasc. Electrophysiol. 22, 987–993.
Pellman, J., Lyon, R. C., and Sheikh, F. (2010). Extracellular matrix remodeling in atrial fibrosis: mechanisms and implications in atrial fibrillation. J. Mol. Cell. Cardiol. 48, 461–467.
Rajagopalan, S., Meng, X. P., Ramasamy, S., Harrison, D. G., and Galis, Z. S. (1996). Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Invest. 98, 2572–2579.
Ramlawi, B., Otu, H., Mieno, S., Boodhwani, M., Sodha, N. R., Clements, R. T., Bianchi, C., and Sellke, F. W. (2007). Oxidative stress and atrial fibrillation after cardiac surgery: a case-control study. Ann. Thorac. Surg. 84, 1166–1172; discussion 1172–1173.
Rees, M. D., Whitelock, J. M., Malle, E., Chuang, C. Y., Iozzo, R. V., Nilasaroya, A., and Davies, M. J. (2010). Myeloperoxidase-derived oxidants selectively disrupt the protein core of the heparan sulfate proteoglycan perlecan. Matrix Biol. 29, 63–73.
Reil, J. C., Hohl, M., Oberhofer, M., Kazakov, A., Kaestner, L., Mueller, P., Adam, O., Maack, C., Lipp, P., Mewis, C., Allessie, M., Laufs, U., Böhm, M., and Neuberger, H. R. (2010). Cardiac Rac1 overexpression in mice creates a substrate for atrial arrhythmias characterized by structural remodelling. Cardiovasc. Res. 87, 485–493.
Richter, B., Gwechenberger, M., Socas, A., Zorn, G., Albinni, S., Marx, M., Bergler-Klein, J., Binder, T., Wojta, J., and Gössinger, H. D. (2012). Markers of oxidative stress after ablation of atrial fibrillation are associated with inflammation, delivered radiofrequency energy and early recurrence of atrial fibrillation. Clin. Res. Cardiol. 101, 217–225.
Rohr, S. (2009). Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm 6, 848–856.
Rudolph, T. K., Wipper, S., Reiter, B., Rudolph, V., Coym, A., Detter, C., Lau, D., Klinke, A., Friedrichs, K., Rau, T., Pekarova, M., Russ, D., Knöll, K., Kolk, M., Schroeder, B., Wegscheider, K., Andresen, H., Schwedhelm, E., Boeger, R., Ehmke, H., and Baldus, S. (2011). Myeloperoxidase deficiency preserves vasomotor function in humans. Eur. Heart J. [Epub ahead of print]. doi: 10.1093/eurheartj/ehr193.
Rudolph, V., Andrié, R. P., Rudolph, T. K., Friedrichs, K., Klinke, A., Hirsch-Hoffmann, B., Schwoerer, A. P., Lau, D., Fu, X., Klingel, K., Sydow, K., Didié, M., Seniuk, A., von Leitner, E. C., Szoecs, K., Schrickel, J. W., Treede, H., Wenzel, U., Lewalter, T., Nickenig, G., Zimmermann, W. H., Meinertz, T., Böger, R. H., Reichenspurner, H., Freeman, B. A., Eschenhagen, T., Ehmke, H., Hazen, S. L., Willems, S., and Baldus, S. (2010). Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat. Med. 16, 470–474.
Rudolph, V., Rudolph, T. K., Hennings, J. C., Blankenberg, S., Schnabel, R., Steven, D., Haddad, M., Knittel, K., Wende, S., Wenzel, J., Munzel, T., Heitzer, T., Meinertz, T., Hübner, C., and Baldus, S. (2007). Activation of polymorphonuclear neutrophils in patients with impaired left ventricular function. Free Radic. Biol. Med. 43, 1189–1196.
Ryu, K., Li, L., Khrestian, C. M., Matsumoto, N., Sahadevan, J., Ruehr, M. L., Van Wagoner, D. R., Efimov, I. R., and Waldo, A. L. (2007). Effects of sterile pericarditis on connexins 40 and 43 in the atria: correlation with abnormal conduction and atrial arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 293, H1231–H1241.
Saba, S., Janczewski, A. M., Baker, L. C., Shusterman, V., Gursoy, E. C., Feldman, A. M., Salama, G., McTiernan, C. F., and London, B. (2005). Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-α. Am. J. Physiol. Heart Circ. Physiol. 289, H1456–H1467.
Saito, T., Tamura, K., Uchida, D., Saito, T., Togashi, M., Nitta, T., and Sugisaki, Y. (2007). Histopathological features of the resected left atrial appendage as predictors of recurrence after surgery for atrial fibrillation in valvular heart disease. Circ. J. 71, 70–78.
Sakabe, M., Fujiki, A., Nishida, K., Sugao, M., Nagasawa, H., Tsuneda, T., Mizumaki, K., and Inoue, H. (2004). Enalapril prevents perpetuation of atrial fibrillation by suppressing atrial fibrosis and over-expression of connexin43 in a canine model of atrial pacing-induced left ventricular dysfunction. J. Cardiovasc. Pharmacol. 43, 851–859.
Savelieva, I., and Camm, J. (2008). Update on atrial fibrillation: part I. Clin. Cardiol. 31, 55–62.
Scholz, H., Aukrust, P., Damås, J. K., Tonstad, S., Sagen, E. L., Kolset, S. O., Hall, C., Yndestad, A., and Halvorsen, B. (2004). 8-Isoprostane increases scavenger receptor A and matrix metalloproteinase activity in THP-1 macrophages, resulting in long-lived foam cells. Eur. J. Clin. Invest. 34, 451–458.
Siwik, D. A., and Colucci, W. S. (2004). Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail. Rev. 9, 43–51.
Siwik, D. A., Pagano, P. J., and Colucci, W. S. (2001). Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 280, C53–C60.
Spach, M. S., and Boineau, J. P. (1997). Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: a major mechanism of structural heart disease arrhythmias. Pacing Clin. Electrophysiol. 20, 397–413.
Spallarossa, P., Altieri, P., Garibaldi, S., Ghigliotti, G., Barisione, C., Manca, V., Fabbi, P., Ballestrero, A., Brunelli, C., and Barsotti, A. (2006). Matrix metalloproteinase-2 and -9 are induced differently by doxorubicin in H9c2 cells: the role of MAP kinases and NAD(P)H oxidase. Cardiovasc. Res. 69, 736–745.
Spinale, F. G. (2007). Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol. Rev. 87, 1285–1342.
Sun, Y., Zhang, J., Lu, L., Chen, S. S., Quinn, M. T., and Weber, K. T. (2002). Aldosterone-induced inflammation in the rat heart: role of oxidative stress. Am. J. Pathol. 161, 1773–1781.
Szuchman-Sapir, A. J., Pattison, D. I., Davies, M. J., and Witting, P. K. (2010). Site-specific hypochlorous acid-induced oxidation of recombinant human myoglobin affects specific amino acid residues and the rate of cytochrome b5-mediated heme reduction. Free Radic. Biol. Med. 48, 35–46.
Tsang, T. S., Petty, G. W., Barnes, M. E., O’Fallon, W. M., Bailey, K. R., Wiebers, D. O., Sicks, J. D., Christianson, T. J., Seward, J. B., and Gersh, B. J. (2003). The prevalence of atrial fibrillation in incident stroke cases and matched population controls in Rochester, Minnesota: changes over three decades. J. Am. Coll. Cardiol. 42, 93–100.
Vasquez, C., Benamer, N., and Morley, G. E. (2011). The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts. J. Cardiovasc. Pharmacol. 57, 380–388.
Vasquez, C., Mohandas, P., Louie, K. L., Benamer, N., Bapat, A. C., and Morley, G. E. (2010). Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ. Res. 107, 1011–1020.
Verheule, S., Sato, T., Everett, T., Engle, S. K., Otten, D., Rubart-von der Lohe, M., Nakajima, H. O., Nakajima, H., Field, L. J., and Olgin, J. E. (2004). Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ. Res. 94, 1458–1465.
Verheule, S., Wilson, E., Everett, T., Shanbhag, S., Golden, C., and Olgin, J. (2003). Alterations in atrial electrophysiology and tissue structure in a canine model of chronic atrial dilatation due to mitral regurgitation. Circulation 107, 2615–2622.
Wainwright, C. L. (2004). Matrix metalloproteinases, oxidative stress and the acute response to acute myocardial ischaemia and reperfusion. Curr. Opin. Pharmacol. 4, 132–138.
Wang, T. J., Massaro, J. M., Levy, D., Vasan, R. S., Wolf, P. A., D’Agostino, R. B., Larson, M. G., Kannel, W. B., and Benjamin, E. J. (2003). A risk score for predicting stroke or death in individuals with new-onset atrial fibrillation in the community: the Framingham Heart Study. JAMA 290, 1049–1056.
Wang, W., Sawicki, G., and Schulz, R. (2002). Peroxynitrite-induced myocardial injury is mediated through matrix metalloproteinase-2. Cardiovasc. Res. 53, 165–174.
Wang, W., Viappiani, S., Sawicka, J., and Schulz, R. (2005). Inhibition of endogenous nitric oxide in the heart enhances matrix metalloproteinase-2 release. Br. J. Pharmacol. 145, 43–49.
Wang, Y., Rosen, H., Madtes, D. K., Shao, B., Martin, T. R., Heinecke, J. W., and Fu, X. (2007). Myeloperoxidase inactivates TIMP-1 by oxidizing its N-terminal cysteine residue: an oxidative mechanism for regulating proteolysis during inflammation. J. Biol. Chem. 282, 31826–31834.
Wei, C. C., Meng, Q. C., Palmer, R., Hageman, G. R., Durand, J., Bradley, W. E., Farrell, D. M., Hankes, G. H., Oparil, S., and Dell’Italia, L. J. (1999). Evidence for angiotensin-converting enzyme- and chymase-mediated angiotensin II formation in the interstitial fluid space of the dog heart in vivo. Circulation 99, 2583–2589.
Xie, Y., Sato, D., Garfinkel, A., Qu, Z., and Weiss, J. N. (2010). So little source, so much sink: requirements for after depolarizations to propagate in tissue. Biophys. J. 99, 1408–1415.
Xu, J., Cui, G., Esmailian, F., Plunkett, M., Marelli, D., Ardehali, A., Odim, J., Laks, H., and Sen, L. (2004). Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation 109, 363–368.
Xu, Z., Castellino, F. J., and Ploplis, V. A. (2010). Plasminogen activator inhibitor-1 (PAI-1) is cardioprotective in mice by maintaining microvascular integrity and cardiac architecture. Blood 115, 2038–2047.
Yamashita, T. (2007). Molecular basis of thromboembolism in association with atrial fibrillation. Circ. J. 71(Suppl. A), A40–A44.
Yan, Z., Liang, F., Guo, L., Wang, J., Wang, X. L., Cheng, X. L., Ma, X. L., and Liu, H. R. (2010). Myeloperoxidase increased cardiomyocyte protein nitration in mice subjected to nonlethal mechanical trauma. Biochem. Biophys. Res. Commun. 393, 531–535.
Yoo, H. G., Shin, B. A., Park, J. S., Lee, K. H., Chay, K. O., Yang, S. Y., Ahn, B. W., and Jung, Y. D. (2002). IL-1beta induces MMP-9 via reactive oxygen species and NF-kappaB in murine macrophage RAW 264.7 cells. Biochem. Biophys. Res. Commun. 298, 251–256.
Yoon, S. O., Park, S. J., Yoon, S. Y., Yun, C. H., and Chung, A. S. (2002). Sustained production of H(2)O(2) activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-kappa B pathway. J. Biol. Chem. 277, 30271–30282.
Yue, L., Xie, J., and Nattel, S. (2011). Molecular determinants of cardiac fibroblast electrical function and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 89, 744–753.
Zhang, H. J., Zhao, W., Venkataraman, S., Robbins, M. E., Buettner, G. R., Kregel, K. C., and Oberley, L. W. (2002). Activation of matrix metalloproteinase-2 by overexpression of manganese superoxide dismutase in human breast cancer MCF-7 cells involves reactive oxygen species. J. Biol. Chem. 277, 20919–20926.
Zhao, W., Zhao, T., Chen, Y., Ahokas, R. A., and Sun, Y. (2008). Oxidative stress mediates cardiac fibrosis by enhancing transforming growth factor-beta1 in hypertensive rats. Mol. Cell. Biochem. 317, 43–50.
Keywords: atrial fibrillation, fibrosis, reactive oxygen species, myeloperoxidase
Citation: Friedrichs K, Baldus S and Klinke A (2012) Fibrosis in atrial fibrillation – role of reactive species and MPO. Front. Physio. 3:214. doi: 10.3389/fphys.2012.00214
Received: 21 March 2012; Accepted: 30 May 2012;
Published online: 20 June 2012.
Edited by:
David R. Van Wagoner, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USAReviewed by:
David R. Van Wagoner, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USAHeather S. Duffy, Harvard Medical School, USA; Ali A. Sovari, University of Illinois at Chicago, USA
Copyright: © 2012 Friedrichs, Baldus and Klinke. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Anna Klinke, Department of Cardiology and Cardiovascular Research Center, University Medical Center Hamburg Eppendorf, University Heart Center Hamburg, Martinistrasse 52, 20246 Hamburg, Germany. e-mail:YWtsaW5rZUB1a2UudW5pLWhhbWJ1cmcuZGU=