


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 13 June 2012
Sec. Cardiac Electrophysiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00204
This article is part of the Research Topic Autonomic regulation of the heart - emerging regulators and arrhythmogenic mechanisms View all 8 articles
Neurotransmitters released from sympathetic and parasympathetic nerve terminals in the sinoatrial node (SAN) exert their effects via G-protein-coupled receptors. Integration of these different G-protein signals within pacemaker cells of the SAN is critical for proper regulation of heart rate and function. For example, excessive parasympathetic signaling can be associated with sinus node dysfunction (SND) and supraventricular arrhythmias. Our previous work has shown that one member of the regulator of G-protein signaling (RGS) protein family, RGS4, is highly and selectively expressed in pacemaker cells of the SAN. Consistent with its role as an inhibitor of parasympathetic signaling, RGS4-knockout mice have reduced basal heart rates and enhanced negative chronotropic responses to parasympathetic agonists. Moreover, RGS4 appears to be an important part of SA nodal myocyte signaling pathways that mediate G-protein-coupled inwardly rectifying potassium channel (GIRK) channel activation/deactivation and desensitization. Since RGS4 acts immediately downstream of M2 muscarinic receptors, it is tempting to speculate that RGS4 functions as a master regulator of parasympathetic signaling upstream of GIRKs, HCNs, and L-type Ca2+ channels in the SAN. Thus, loss of RGS4 function may lead to increased susceptibility to conditions associated with increased parasympathetic signaling, including bradyarrhythmia, SND, and atrial fibrillation.
Sinus node dysfunction (SND) can be associated with bradycardia/bradyarrhythmias, sinus node exit block, and increased susceptibility to atrial fibrillation (Benditt et al., 1995; Mangrum and DiMarco, 2000; Dobrzynski et al., 2007). This condition, also known as sick sinus syndrome, occurs commonly within the aging adult population, but is particularly prevalent in patients with heart disease, on anti-arrhythmic therapy, with high vagal tone, or after surgical correction of congenital heart defects. Genetic predisposition for SND has also been proposed in patients and that have this condition in conjunction with abnormal ion channel and GTPase activating protein (GAP) junction function (Bezzina et al., 1999; Benson et al., 2003; Groenewegen et al., 2003; Schulze-Bahr et al., 2003; Veldkamp et al., 2003; Ueda et al., 2004; Mohler and Bennett, 2005; Smits et al., 2005; Milanesi et al., 2006). Thus in order to treat and prevent SND, it may be important to understand the cast of molecules that coordinate the balance between autonomic signaling pathways within pacemaker cells of the sinoatrial node (SAN).
Heart rate regulation by the autonomic nervous system occurs at the level of specialized autorhythmic (pacemaker) cells in the SAN. Sympathetic neurotransmitters work via Gαs-coupled β-adrenergic receptors (β-ARs) to increase adenylyl cyclase activity, intracellular cAMP concentration, and protein kinase A (PKA) activity. As a result cAMP-regulated effectors such as hyperpolarization-activated cyclic nucleotide-gated cation (HCN), delayed rectifier, and voltage-gated Ca2+ channels are enlisted by sympathetic activity to increase pacemaker cell firing rat (Irisawa et al., 1993; DiFrancesco, 2006). By contrast, vagal parasympathetic activity decreases HR via Gαi/o-coupled cholinergic M2 muscarinic receptors (M2 muscarinic receptor). Several effects, mediated by both Gαi/o and Gβγ subunits contribute to this reduction in HR. Gβγ heterodimers directly activate G-protein-coupled inward rectifying potassium (GIRK) channels, resulting in membrane hyperpolarization. Gαi/o can also inhibit adenylyl cyclase activity to reduce intracellular cAMP levels and PKA activity, thus leading to decreased depolarizing currents carried by HCN and L-type Ca2+ channels (DiFrancesco, 1993; Kubo et al., 1993; Fischmeister et al., 2006). Since dysregulation of parasympathetic activity occurs in SND and selected cardiac arrhythmias (Dobrzynski et al., 2007), it is of clinical interest to identify key molecular regulators of parasympathetic signaling. In particular, our laboratory studies molecules that regulate the activity of G-protein signaling downstream of parasympathetic activation inside SAN cells.
The activation of the Gαi/o-coupled M2 muscarinic receptor by cholinergic (parasympathetic) activity produces a cascade of physiologic changes within the cell. The timing and duration of these changes are mainly dependent on the lifetime of the activated (GTP-bound) Gαi/o subunit. In the basal state, the quiescent (GDP-bound) Gαi/o subunit is complexed with the Gβγ and coupled to the intracellular surface of the receptor. Specifically, M2 muscarinic receptor activation results in the exchange of GTP for GDP on the Gα subunit and the dissociation of GTP-bound Gα from the Gβγ heterodimer. This condition marks the activated (“ON”) state (Clapham and Neer, 1997; Hamm, 1998) of parasympathetic signaling during which time the Gα and Gβγ subunits are free to engage downstream parasympathetic effector molecules such as GIRK channels and adenylyl cyclase. Effector signaling is terminated following Gα-catalyzed hydrolysis of GTP and reformation of the quiescent (“OFF”) receptor-coupled complex. Importantly, the intrinsic rate of GTP hydrolysis by Gα subunits (the rate limiting step for signal termination) is very slow. Therefore, to produce the rapid ON-OFF kinetic changes needed to modulate autonomic activity in vivo cells require additional factors that increase the rate of GTP hydrolysis of the Gα subunit. This class of molecules is called GAPs. Regulator of G-protein signaling (RGS) proteins (Hepler, 1999; Ross and Wilkie, 2000) are a mammalian family of >35 GAPs for Gα subunits (Berman et al., 1996; Watson et al., 1996). By increasing the rates of GTP hydrolysis by up to 2000-fold RGS proteins are among the most potent inhibitors of G-protein signaling identified to date.
Endogenous RGS proteins have recently been shown to play an important role in the regulation of muscarinic receptor-mediated signaling. In embryonic stem cell-derived cardiomyocytes, expression of RGS-insensitive Gαi2 and Gαo subunits enhanced sensitivity of these cells to the M2 muscarinic and adenosine receptor agonists (Fu et al., 2006). Similar observations were also shown in the intact animal and isolated perfused hearts (Fu et al., 2006, 2007). Despite M2R and adenosine receptor (A1R) both mediating negative chronotropic effects, it appears that these receptors may do so through distinct intracellular mechanisms. Specifically, the M2R appears to signal via Gαi2 and Gαo, whereas the adenosine receptor appears to signal predominantly via Gαo. It may not be surprising, therefore, that the contribution of GIRK currents to M2R- versus A1R-mediated heart rate slowing effects appears to be different. Tertiapin-Q abolished the increased responsiveness of RGS-resistant Gαi2 tissues to carbachol suggesting the importance of GIRK activity as a primary pathway mediating the M2R chronotropic response and establishing a role for endogenous RGS proteins as inhibitors of this pathway. By contrast, the enhanced sensitivity of RGS-resistant Gαo cells to A1R agonist is unaffected by tertiapin-Q, suggesting minimal contribution of GIRK activation to the heart rate slowing effects of A1R agonists. Since, as outlined above, A1R preferentially couple to Gαo it has been suggested that A1R are likely to use a GIRK-independent pathway (e.g., ICa,L or If) to mediate its bradycardic effects. These exciting observations prompted the search for RGS proteins that may selectively regulate the activity of Gαi2 and Gαo or their associated receptors in pacemaker cells of the SAN. A number of different RGS proteins have been identified in the atrial myocardium (Kardestuncer et al., 1998; Doupnik et al., 2001); however, the specific RGS protein responsible for this effect was not characterized in these studies.
Regulator of G-protein signaling 4 was one of the first proteins of this superfamily to be cloned and characterized (Berman et al., 1996; Druey et al., 1996; Watson et al., 1996). Of the main G-protein signaling pathways controlling heart rate, RGS4 shows a remarkable degree of biochemical selectivity for inhibiting Gαi/o relative to Gαs function. RGS4 has been shown to inhibit neural (Pacey et al., 2009), pancreatic (Ruiz de Azua et al., 2010), tumor (Xie et al., 2009), and cardiac cell signaling (Zhang et al., 1998; Mittmann et al., 2002). Overexpression of RGS4 can also prevent hypertrophic growth in cardiomyocytes (Rogers et al., 1999, 2001; Tamirisa et al., 1999; Tokudome et al., 2008). Several papers from Doupnik and colleagues have demonstrated the potential importance of RGS4 as an inhibitor of hyperpolarizing GIRK channels (Doupnik et al., 1997, 2004; Zhang et al., 2002, 2004). Importantly, RGS4 has been shown to selectively associate with the M2 muscarinic receptor and GIRK channels in pulldown experiments (Jaen and Doupnik, 2006). Moreover, because of its biochemical selectivity for inhibiting Gαi/o- but not Gαs-signaling (Berman et al., 1996), RGS4 is well-positioned to regulate parasympathetic signaling without directly affecting sympathetically derived signals at the level of the receptor. Accordingly, through its ability to modulate Gαi/o-mediated regulation of GIRK channel activation, RGS4 was predicted to be an important inhibitor of the negative chronotropic actions of parasympathetic activity.
Using a RGS4-knockout mouse strain with a LacZ reporter cassette, our laboratory showed that RGS4 is highly and selectively expressed in the SAN compared to other regions of the heart (Cifelli et al., 2008). Its high relative expression in the SAN was evident from whole-mount LacZ staining of intact hearts and right atrial preparations as well as from correlation of LacZ patterns with HCN4 immunolabeling. Consistent with an important role for RGS4 as an inhibitor of parasympathetic signaling in the SAN, RGS4-null mice show increased bradycardic responses to carbachol (parasympathetic agonist mimetic) in waking animals and lower baseline heart rates in anesthetized models. Retrograde-perfused hearts from RGS4-null mice show enhanced negative chronotropic responses to carbachol, while isolated pacemaker cells of the SAN show greater sensitivity to carbachol-mediated reduction in the action potential firing rate. Moreover, RGS4-null SAN cells show decreased levels of G-protein-coupled inward rectifying potassium (GIRK) channel desensitization (i.e., greater channel activity with prolonged stimulus), and altered modulation of GIRK channel current (IKACh) kinetics following both carbachol addition and removal. In particular, the IKACh persists much longer after carbachol washout in the RGS4-null compared to wild type cells, consistent with the loss of a key kinetic inhibitor of Gαi/o signaling. Lastly, RGS4-null pacemaker cells of the SAN treated with carbachol showed marked membrane hyperpolarization and loss of spontaneous action potential activity, reminiscent of the loss in SA node automaticity observed in patients with SND. Figure 1 summarizes the potential role of RGS4 as a master regulator of parasympathetic signaling downstream of the M2 muscarinic receptor. As summarized by the comparison of M2 muscarinic receptor-dependent signaling events in Figures 1A,B, the loss of RGS4 function in pacemaker cells of the SAN may be expected to result in increased GIRK channel currents, decreased adenylyl cyclase activity/cAMP production, leading to decreased activation of HCN4, protein kinase A, and L-type calcium channel currents. As discussed above, loss of RGS4 is known to increase M2 muscarinic receptor-dependent GIRK currents, however the question of whether HCN4 and L-type calcium channel currents are affected in any meaningful way remains to be determined.
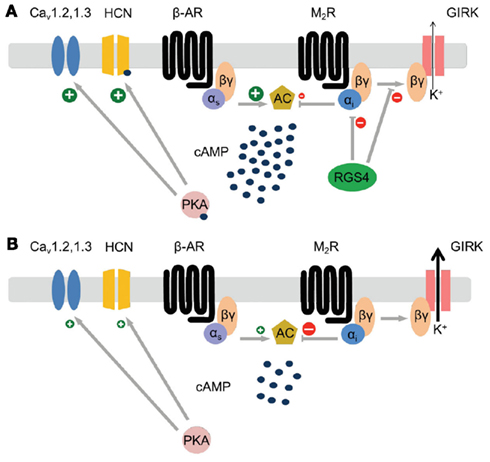
Figure 1. Modeling parasympathetic signaling pathways in pacemaker cells of the SAN in the presence (A) and absence (B) of RGS4 function. Note the potential for RGS4 to act as a master upstream regulator of the primary effectors of the M2 muscarinic receptor (GIRK channels, and adenylyl cyclase). (A) In the model where RGS4 is functional, Gαi signaling is attenuated resulting in decreased GIRK channel activity and increased cAMP concentrations that can directly stimulate increased HCN4 channel activity and indirectly (via PKA activation) increases CaV channel activity. (B) In the model where RGS4 function is absent Gαi signaling is increased resulting in increased GIRK channel activity and decreased cAMP concentrations. This condition is expected to result in lower HCN4 and CaV channel activity.
More recently, RGS6 was also shown to be an important regulator of M2R-dependent signaling in the SAN. Specifically, two groups working independently showed that RGS6-deficient mice, displayed enhanced bradycardic responses to carbachol in intact animals, isolated hearts, and cultured SAN cells (Posokhova et al., 2010; Yang et al., 2010). As was the case for RGS4-deficiency, the phenotypes associated with loss of RGS6 appeared consistent with its ability to regulate GIRK channel activity. It remains to be determined whether RGS6 will also be capable of regulating GIRK channel independent effectors of negative chronotropy.
It is interesting to consider whether there may exist heart-intrinsic mechanisms that would allow for rapid changes in RGS protein activity, and thus rapid changes in heart rate and cardiac output in response to certain physiologic stimuli. One stimulus that is believed to regulate heart rate at the level of the SAN is atrial stretch. This pathway has been proposed to be one of the intrinsic mechanisms whereby increased venous return may result in rapid upregulation of cardiac output via alterations of the autonomic signaling balance. Specifically, small increases in atrial pressure have been shown to reduce the heart’s response to vagal stimulation resulting in rapid heart rate increase (Bolter and Wilson, 1999). Of potential relevance to this article is the observation that the increased heart rate effect is apparently mediated by rapid inhibition of GIRK channel activity in response to atrial stretch. Indeed, it was shown that Tertiapin-Q could eliminate the mechanosensitive component of muscarinic control in rat atria (Han et al., 2010). Although the cellular mechanisms by which stretch is sensed and exerts its effects on the SAN are not known it is tempting to speculate that the rapid regulation of GIRK channels in response to changing atrial pressures may be partly explained by changes in RGS protein activity. While, there are wide number of pathways that have been proposed to regulate the function of RGS4, an intriguing possibility for linking mechanosensation to increased RGS4 function is the Ca2+/Calmodulin pathway. Mechanical stretch has been associated with increased intracellular calcium levels in atrial tissues, and rapid activation of RGS4 activity was shown to be associated with Ca2+/calmodulin binding to an allosteric site on the RGS4 protein (Popov et al., 2000; Ishii et al., 2001, 2002). Thus, atrial stretch might be expected to increase RGS4 activity, resulting in decreased parasympathetic signals and heart rate increase. It remains to be determined whether other RGS proteins important for regulation of GIRKs in the SAN (i.e., RGS6) may also be regulated by mechanosensitive pathways.
In animal models of AF, stimulation with the muscarinic agonist carbachol can facilitate the induction of AF (Wakimoto et al., 2001), whereas inhibition of Gi/o signaling can reduce susceptibility to vagal-mediated AF (Aistrup et al., 2009, 2011). In the clinic, Coumel (1996) have described cases of AF where vagal activity preceded the onset of atrial arrhythmias (Herweg et al., 1998). Several observations suggest that activation of IKAch may be a key component of parasympathetic pathway-mediated initiation and maintenance of AF. Firstly, studies show that reducing IKAch using both genetic disruption (Kovoor et al., 2001) and pharmacological inhibition (Hashimoto et al., 2006) of GIRK currents confers resistance to AF, while increasing IKAch facilitates induction of AF (Kovoor et al., 2001). Furthermore, Voigt et al. (2008) demonstrated the presence of agonist-independent IKAch activity in a canine model of chronic AF. Importantly, evidence from human studies confirms the presence of constitutively active (agonist-independent) IKAch in patients with persistent AF (Dobrev et al., 2005). Human studies identified a genetic polymorphism in the β3 subunit of heterotrimeric G-proteins which was associated with reduced IKAch in human atrial myocytes (Dobrev et al., 2000) and protection from AF in humans (Schreieck et al., 2004). These findings suggest that a reduction of IKAch could be beneficial in AF prevention and treatment and makes endogenous inhibitors of IKAch, such as RGS4, potentially interesting anti-arrhythmic targets. Indeed, much work in the area of pharmacologic targeting of RGS proteins in cardiovascular and other tissues has already been carried out. Several of the key discoveries in this field are discussed elsewhere in a comprehensive series of recent reviews on this topic (Sjogren and Neubig, 2010; Sjogren et al., 2010; Kimple et al., 2011; Zhang et al., 2012).
Thus, two important aspects of the phenotype observed in RGS4-deficient hearts suggest that they may be expected to show increased susceptibility to AF. First, the characteristic bradyarrhythmia that was found to be associated with vagal stimulation may significantly increase the chance that an ectopic pacemaker could act to trigger an arrhythmia (Schuessler et al., 1992; Chou et al., 2008). Second, to the extent that RGS4 is required for inhibition of parasympathetic signaling in the atrial syncytium, loss of RGS4 may predispose hearts to regional changes in repolarization kinetics that might support re-entrant circuits in the presence of M2 muscarinic receptor activation. Specifically, the increased GIRK channel activity that is characteristic of RGS4-deficient hearts may be expected to contribute to a decrease in the action potential duration of atrial myocytes and thus lead to increased susceptibility to rotor formation. Future studies will be required to determine whether this is indeed the case. It is currently not known whether RGS4 expression is decreased in the SAN or atria of patients with SND or AF, but it would be tempting to examine these possibilities. Likewise, certain SNPs in the non-coding regions of the RGS4 gene have been associated with schizophrenia in the general population (Chowdari et al., 2002; Morris et al., 2004) so it would be of interest to determine whether individuals with those SNPs may also show increased susceptibility to SAN dysfunction and supraventricular arrhythmias.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to two anonymous reviewers for their valuable contributions to this manuscript. This work was supported by funding from the Canada Research Chairs Program and the Heart and Stroke Foundation of Ontario (T6799, Scott P. Heximer).
Aistrup, G. L., Cokic, I., Ng, J., Gordon, D., Koduri, H., Browne, S., Arapi, D., Segon, Y., Goldstein, J., Angulo, A., Wasserstrom, J. A., Goldberger, J. J., Kadish, A. H., and Arora, R. (2011). Targeted nonviral gene-based inhibition of Galpha(i/o)-mediated vagal signaling in the posterior left atrium decreases vagal-induced atrial fibrillation. Heart Rhythm 8, 1722–1729.
Aistrup, G. L., Villuendas, R., Ng, J., Gilchrist, A., Lynch, T. W., Gordon, D., Cokic, I., Mottl, S., Zhou, R., Dean, D. A., Wasserstrom, J. A., Goldberger, J. J., Kadish, A. H., and Arora, R. (2009). Targeted G-protein inhibition as a novel approach to decrease vagal atrial fibrillation by selective parasympathetic attenuation. Cardiovasc. Res. 83, 481–492.
Benditt, D. G., Sakaguchi, S., Goldstein, M. A., Lurie, K. G., Gornick, C. C., and Adler, S. W. (1995). “Sinus node dysfunction: pathophysiology, clinical features, evaluation, and treatment,” in Cardiac Electrophysiology: From Cell to Bedside, 2nd Edn, eds D. P. Zipes and J. Jalife (Philadelphia, PA: WB Saunders Company), 1215–1247.
Benson, D. W., Wang, D. W., Dyment, M., Knilans, T. K., Fish, F. A., Strieper, M. J., Rhodes, T. H., and George, A. L. Jr. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028.
Berman, D. M., Wilkie, T. M., and Gilman, A. G. (1996). GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell 86, 445–452.
Bezzina, C., Veldkamp, M. W., van Den Berg, M. P., Postma, A. V., Rook, M. B., Viersma, J. W., van Langen, I. M., Tan-Sindhunata, G., Bink-Boelkens, M. T., van Der Hout, A. H., Mannens, M. M., and Wilde, A. A. (1999). A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 85, 1206–1213.
Bolter, C. P., and Wilson, S. J. (1999). Influence of right atrial pressure on the cardiac pacemaker response to vagal stimulation. Am. J. Physiol. 276, R1112–R1117.
Chou, C. C., Nguyen, B. L., Tan, A. Y., Chang, P. C., Lee, H. L., Lin, F. C., Yeh, S. J., Fishbein, M. C., Lin, S. F., Wu, D., Wen, M. S., and Chen, P. S. (2008). Intracellular calcium dynamics and acetylcholine-induced triggered activity in the pulmonary veins of dogs with pacing-induced heart failure. Heart Rhythm 5, 1170–1177.
Chowdari, K. V., Mirnics, K., Semwal, P., Wood, J., Lawrence, E., Bhatia, T., Deshpande, S. N., Thelma, B. K., Ferrell, R. E., Middleton, F. A., Devlin, B., Levitt, P., Lewis, D. A., and Nimgaonkar, V. L. (2002). Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum. Mol. Genet. 11, 1373–1380.
Cifelli, C., Rose, R. A., Zhang, H., Voigtlaender-Bolz, J., Bolz, S. S., Backx, P. H., and Heximer, S. P. (2008). RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ. Res. 103, 527–535.
Clapham, D. E., and Neer, E. J. (1997). G protein beta gamma subunits. Annu. Rev. Pharmacol. Toxicol. 37, 167–203.
Coumel, P. (1996). Autonomic influences in atrial tachyarrhythmias. J. Cardiovasc. Electrophysiol. 7, 999–1007.
DiFrancesco, D. (2006). Funny channels in the control of cardiac rhythm and mode of action of selective blockers. Pharmacol. Res. 53, 399–406.
Dobrev, D., Friedrich, A., Voigt, N., Jost, N., Wettwer, E., Christ, T., Knaut, M., and Ravens, U. (2005). The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706.
Dobrev, D., Wettwer, E., Himmel, H. M., Kortner, A., Kuhlisch, E., Schuler, S., Siffert, W., and Ravens, U. (2000). G-Protein beta(3)-subunit 825T allele is associated with enhanced human atrial inward rectifier potassium currents. Circulation 102, 692–697.
Dobrzynski, H., Boyett, M. R., and Anderson, R. H. (2007). New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115, 1921–1932.
Doupnik, C. A., Davidson, N., Lester, H. A., and Kofuji, P. (1997). RGS proteins reconstitute the rapid gating kinetics of gbetagamma- activated inwardly rectifying K+ channels. Proc. Natl. Acad. Sci. U.S.A. 94, 10461–10466.
Doupnik, C. A., Jaen, C., and Zhang, Q. (2004). Measuring the modulatory effects of RGS proteins on GIRK channels. Meth. Enzymol. 389, 131–154.
Doupnik, C. A., Xu, T., and Shinaman, J. M. (2001). Profile of RGS expression in single rat atrial myocytes. Biochim. Biophys. Acta 1522, 97–107.
Druey, K. M., Blumer, K. J., Kang, V. H., and Kehrl, J. H. (1996). Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature 379, 742–746.
Fischmeister, R., Castro, L. R., Bi-Gerges, A., Rochais, F., Jurevicius, J., Leroy, J., and Vandecasteele, G. (2006). Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ. Res. 99, 816–828.
Fu, Y., Huang, X., Piao, L., Lopatin, A. N., and Neubig, R. R. (2007). Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: implications for sick sinus syndrome and AV block. Am. J. Physiol. Heart Circ. Physiol. 292, H2532–H2539.
Fu, Y., Huang, X., Zhong, H., Mortensen, R. M., D’Alecy, L. G., and Neubig, R. R. (2006). Endogenous RGS proteins and Galpha subtypes differentially control muscarinic and adenosine-mediated chronotropic effects. Circ. Res. 98, 659–666.
Groenewegen, W. A., Firouzi, M., Bezzina, C. R., Vliex, S., Van Langen, I. M., Sandkuijl, L., Smits, J. P., Hulsbeek, M., Rook, M. B., Jongsma, H. J., and Wilde, A. A. (2003). A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ. Res. 92, 14–22.
Han, S., Wilson, S. J., and Bolter, C. P. (2010). Tertiapin-Q removes a mechanosensitive component of muscarinic control of the sinoatrial pacemaker in the rat. Clin. Exp. Pharmacol. Physiol. 37, 900–904.
Hashimoto, N., Yamashita, T., and Tsuruzoe, N. (2006). Tertiapin, a selective IKACh blocker, terminates atrial fibrillation with selective atrial effective refractory period prolongation. Pharmacol. Res. 54, 136–141.
Hepler, J. R. (1999). Emerging roles for RGS proteins in cell signalling. Trends Pharmacol. Sci. 20, 376–382.
Herweg, B., Dalal, P., Nagy, B., and Schweitzer, P. (1998). Power spectral analysis of heart period variability of preceding sinus rhythm before initiation of paroxysmal atrial fibrillation. Am. J. Cardiol. 82, 869–874.
Irisawa, H., Brown, H. F., and Giles, W. (1993). Cardiac pacemaking in the sinoatrial node. Physiol. Rev. 73, 197–227.
Ishii, M., Inanobe, A., Fujita, S., Makino, Y., Hosoya, Y., and Kurachi, Y. (2001). Ca(2+) elevation evoked by membrane depolarization regulates G protein cycle via RGS proteins in the heart. Circ. Res. 89, 1045–1050.
Ishii, M., Inanobe, A., and Kurachi, Y. (2002). PIP3 inhibition of RGS protein and its reversal by Ca2+/calmodulin mediate voltage-dependent control of the G protein cycle in a cardiac K+ channel. Proc. Natl. Acad. Sci. U.S.A. 99, 4325–4330.
Jaen, C., and Doupnik, C. A. (2006). RGS3 and RGS4 differentially associate with G protein-coupled receptor-Kir3 channel signaling complexes revealing two modes of RGS modulation. Precoupling and collision coupling. J. Biol. Chem. 281, 34549–34560.
Kardestuncer, T., Wu, H., Lim, A. L., and Neer, E. J. (1998). Cardiac myocytes express mRNA for ten RGS proteins: changes in RGS mRNA expression in ventricular myocytes and cultured atria. FEBS Lett. 438, 285–288.
Kimple, A. J., Bosch, D. E., Giguere, P. M., and Siderovski, D. P. (2011). Regulators of G-protein signaling and their Galpha substrates: promises and challenges in their use as drug discovery targets. Pharmacol. Rev. 63, 728–749.
Kovoor, P., Wickman, K., Maguire, C. T., Pu, W., Gehrmann, J., Berul, C. I., and Clapham, D. E. (2001). Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 37, 2136–2143.
Kubo, Y., Reuveny, E., Slesinger, P. A., Jan, Y. N., and Jan, L. Y. (1993). Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature 364, 802–806.
Mangrum, J. M., and DiMarco, J. P. (2000). The evaluation and management of bradycardia. N. Engl. J. Med. 342, 703–709.
Milanesi, R., Baruscotti, M., Gnecchi-Ruscone, T., and Difrancesco, D. (2006). Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N. Engl. J. Med. 354, 151–157.
Mittmann, C., Chung, C. H., Hoppner, G., Michalek, C., Nose, M., Schuler, C., Schuh, A., Eschenhagen, T., Weil, J., Pieske, B., Hirt, S., and Wieland, T. (2002). Expression of ten RGS proteins in human myocardium: functional characterization of an upregulation of RGS4 in heart failure. Cardiovasc. Res. 55, 778–786.
Mohler, P. J., and Bennett, V. (2005). Ankyrin-based cardiac arrhythmias: a new class of channelopathies due to loss of cellular targeting. Curr. Opin. Cardiol. 20, 189–193.
Morris, D. W., Rodgers, A., McGhee, K. A., Schwaiger, S., Scully, P., Quinn, J., Meagher, D., Waddington, J. L., Gill, M., and Corvin, A. P. (2004). Confirming RGS4 as a susceptibility gene for schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 125B, 50–53.
Pacey, L. K., Heximer, S. P., and Hampson, D. R. (2009). Increased GABA(B) receptor-mediated signaling reduces the susceptibility of fragile X knockout mice to audiogenic seizures. Mol. Pharmacol. 76, 18–24.
Popov, S. G., Krishna, U. M., Falck, J. R., and Wilkie, T. M. (2000). Ca2+/Calmodulin reverses phosphatidylinositol 3,4, 5-trisphosphate-dependent inhibition of regulators of G protein-signaling GTPase-activating protein activity. J. Biol. Chem. 275, 18962–18968.
Posokhova, E., Wydeven, N., Allen, K. L., Wickman, K., and Martemyanov, K. A. (2010). RGS6/Gss5 complex accelerates IKACh gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ. Res. 107, 1350–1354.
Rogers, J. H., Tamirisa, P., Kovacs, A., Weinheimer, C., Courtois, M., Blumer, K. J., Kelly, D. P., and Muslin, A. J. (1999). RGS4 causes increased mortality and reduced cardiac hypertrophy in response to pressure overload. J. Clin. Invest. 104, 567–576.
Rogers, J. H., Tsirka, A., Kovacs, A., Blumer, K. J., Dorn, G. W., and Muslin, A. J. (2001). RGS4 reduces contractile dysfunction and hypertrophic gene induction in Galpha q overexpressing mice. J. Mol. Cell. Cardiol. 33, 209–218.
Ross, E. M., and Wilkie, T. M. (2000). GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 69, 795–827.
Ruiz de Azua, I., Scarselli, M., Rosemond, E., Gautam, D., Jou, W., Gavrilova, O., Ebert, P. J., Levitt, P., and Wess, J. (2010). RGS4 is a negative regulator of insulin release from pancreatic beta-cells in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 7999–8004.
Schreieck, J., Dostal, S., Von Beckerath, N., Wacker, A., Flory, M., Weyerbrock, S., Koch, W., Schomig, A., and Schmitt, C. (2004). C825T polymorphism of the G-protein beta3 subunit gene and atrial fibrillation: association of the TT genotype with a reduced risk for atrial fibrillation. Am. Heart J. 148, 545–550.
Schuessler, R. B., Grayson, T. M., Bromberg, B. I., Cox, J. L., and Boineau, J. P. (1992). Cholinergically mediated tachyarrhythmias induced by a single extrastimulus in the isolated canine right atrium. Circ. Res. 71, 1254–1267.
Schulze-Bahr, E., Neu, A., Friederich, P., Kaupp, U. B., Breithardt, G., Pongs, O., and Isbrandt, D. (2003). Pacemaker channel dysfunction in a patient with sinus node disease. J. Clin. Invest. 111, 1537–1545.
Sjogren, B., Blazer, L. L., and Neubig, R. R. (2010). Regulators of G protein signaling proteins as targets for drug discovery. Prog. Mol. Biol. Transl. Sci. 91, 81–119.
Sjogren, B., and Neubig, R. R. (2010). Thinking outside of the “RGS box”: new approaches to therapeutic targeting of regulators of G protein signaling. Mol. Pharmacol. 78, 550–557.
Smits, J. P., Koopmann, T. T., Wilders, R., Veldkamp, M. W., Opthof, T., Bhuiyan, Z. A., Mannens, M. M., Balser, J. R., Tan, H. L., Bezzina, C. R., and Wilde, A. A. (2005). A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J. Mol. Cell. Cardiol. 38, 969–981.
Tamirisa, P., Blumer, K. J., and Muslin, A. J. (1999). RGS4 inhibits G-protein signaling in cardiomyocytes. Circulation 99, 441–447.
Tokudome, T., Kishimoto, I., Horio, T., Arai, Y., Schwenke, D. O., Hino, J., Okano, I., Kawano, Y., Kohno, M., Miyazato, M., Nakao, K., and Kangawa, K. (2008). Regulator of G-protein signaling subtype 4 mediates antihypertrophic effect of locally secreted natriuretic peptides in the heart. Circulation 117, 2329–2339.
Ueda, K., Nakamura, K., Hayashi, T., Inagaki, N., Takahashi, M., Arimura, T., Morita, H., Higashiuesato, Y., Hirano, Y., Yasunami, M., Takishita, S., Yamashina, A., Ohe, T., Sunamori, M., Hiraoka, M., and Kimura, A. (2004). Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J. Biol. Chem. 279, 27194–27198.
Veldkamp, M. W., Wilders, R., Baartscheer, A., Zegers, J. G., Bezzina, C. R., and Wilde, A. A. (2003). Contribution of sodium channel mutations to bradycardia and sinus node dysfunction in LQT3 families. Circ. Res. 92, 976–983.
Voigt, N., Maguy, A., Yeh, Y. H., Qi, X., Ravens, U., Dobrev, D., and Nattel, S. (2008). Changes in I K, ACh single-channel activity with atrial tachycardia remodelling in canine atrial cardiomyocytes. Cardiovasc. Res. 77, 35–43.
Wakimoto, H., Maguire, C. T., Kovoor, P., Hammer, P. E., Gehrmann, J., Triedman, J. K., and Berul, C. I. (2001). Induction of atrial tachycardia and fibrillation in the mouse heart. Cardiovasc. Res. 50, 463–473.
Watson, N., Linder, M. E., Druey, K. M., Kehrl, J. H., and Blumer, K. J. (1996). RGS family members: GTPase-activating proteins for heterotrimeric G-protein alpha-subunits. Nature 383, 172–175.
Xie, Y., Wolff, D. W., Wei, T., Wang, B., Deng, C., Kirui, J. K., Jiang, H., Qin, J., Abel, P. W., and Tu, Y. (2009). Breast cancer migration and invasion depend on proteasome degradation of regulator of G-protein signaling 4. Cancer Res. 69, 5743–5751.
Yang, J., Huang, J., Maity, B., Gao, Z., Lorca, R. A., Gudmundsson, H., Li, J., Stewart, A., Swaminathan, P. D., Ibeawuchi, S. R., Shepherd, A., Chen, C. K., Kutschke, W., Mohler, P. J., Mohapatra, D. P., Anderson, M. E., and Fisher, R. A. (2010). RGS6, a modulator of parasympathetic activation in heart. Circ. Res. 107, 1345–1349.
Zhang, H., Gu, S., Al-Sabeq, B., Wang, S., He, J., Tam, A., Cifelli, C., Mathalone, N., Tirgari, S., Boyd, S., and Heximer, S. P. (2012). Origin-specific epigenetic program correlates with vascular bed-specific differences in Rgs5 expression. FASEB J. 26, 181–191.
Zhang, Q., Dickson, A., and Doupnik, C. A. (2004). Gbetagamma-activated inwardly rectifying K(+) (GIRK) channel activation kinetics via Galphai and Galphao-coupled receptors are determined by Galpha-specific interdomain interactions that affect GDP release rates. J. Biol. Chem. 279, 29787–29796.
Zhang, Q., Pacheco, M. A., and Doupnik, C. A. (2002). Gating properties of GIRK channels activated by Galpha(o)- and Galpha(i)-coupled muscarinic m2 receptors in Xenopus oocytes: the role of receptor precoupling in RGS modulation. J. Physiol. (Lond.) 545, 355–373.
Keywords: RGS protein, sinoatrial node, parasympathetic signaling, GIRK channels, bradyarrhythmia
Citation: Mighiu AS and Heximer SP (2012) Controlling parasympathetic regulation of heart rate: a gatekeeper role for RGS proteins in the sinoatrial node. Front. Physio. 3:204. doi: 10.3389/fphys.2012.00204
Received: 07 February 2012; Paper pending published: 04 March 2012;
Accepted: 23 May 2012; Published online: 13 June 2012.
Edited by:
Craig Doupnik, University of South Florida College of Medicine, USAReviewed by:
T. Alexander Quinn, University of Oxford, UKCopyright: © 2012 Mighiu and Heximer. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Scott P. Heximer, Department of Physiology, Heart and Stroke/Richard Lewar Centre of Excellence in Cardiovascular Research, University of Toronto, 1 King’s College Circle, Toronto, ON, Canada M5S 1A8. e-mail:c2NvdHQuaGV4aW1lckB1dG9yb250by5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.