 David A. Hicks1
David A. Hicks1 Natalia N. Nalivaeva1,2 and Anthony J. Turner1*
Natalia N. Nalivaeva1,2 and Anthony J. Turner1*- 1 School of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, Leeds, UK
- 2 I.M. Sechenov Institute of Evolutionary Physiology and Biochemistry RAS, St. Petersburg, Russia
Lipid rafts are membrane domains, more ordered than the bulk membrane and enriched in cholesterol and sphingolipids. They represent a platform for protein-lipid and protein–protein interactions and for cellular signaling events. In addition to their normal functions, including membrane trafficking, ligand binding (including viruses), axonal development and maintenance of synaptic integrity, rafts have also been implicated in the pathogenesis of several neurodegenerative diseases including Alzheimer’s disease (AD). Lipid rafts promote interaction of the amyloid precursor protein (APP) with the secretase (BACE-1) responsible for generation of the amyloid β peptide, Aβ. Rafts also regulate cholinergic signaling as well as acetylcholinesterase and Aβ interaction. In addition, such major lipid raft components as cholesterol and GM1 ganglioside have been directly implicated in pathogenesis of the disease. Perturbation of lipid raft integrity can also affect various signaling pathways leading to cellular death and AD. In this review, we discuss modulation of APP cleavage by lipid rafts and their components, while also looking at more recent findings on the role of lipid rafts in signaling events.
Introduction
It has traditionally been difficult to reach any kind of consensus concerning the definition of lipid rafts. However, in 2006 at the Keystone Symposium on Lipid Rafts and Cell Function in Colorado it was agreed that “membrane rafts are small (10–200 nm), heterogeneous, highly dynamic, sterol, and sphingolipid-enriched domains that compartmentalize cellular processes. Small rafts can sometimes be stabilized to form larger platforms through protein–protein and protein-lipid interactions” (Pike, 2006). The long, saturated acyl chains of sphingolipids allow tight packing hence their juxtaposition with the kinked, unsaturated acyl chains of bulk membrane phospholipids leads to phase separation (Brown and London, 2000). The cholesterol molecules can act as “spacers,” filling any gaps in sphingolipid packing (Simons and Ikonen, 1997).
The notion of lipid rafts, while not new, has never been far from controversy (Pike, 2009), their existence frequently questioned (Munro, 2003). It is almost 30 years since Karnovsky et al. (1982) suggested that protein diffusion in membranes is not free, but somehow constrained. They went on to detail the now familiar concept of the organization of lipids in domains, which may have functional significance. The differential detergent (Triton X-100) solubility of glycosylphosphatidylinositol (GPI)-anchored proteins and transmembrane proteins was first shown by Hooper and Turner (1988). Later work showed the importance of lipid rafts in protein sorting and segregation, with GPI anchored proteins being preferentially localized in lipid rafts (Brown and Rose, 1992; Brown and London, 2000; Pike, 2009). Other lipid modifications of proteins have also been described, such as palmitoylation and myristoylation which may influence raft localization (Brown and London, 2000; Smotrys and Linder, 2004; Pike, 2009). In describing membrane lipid clusters as moving platforms, or rafts, which are enriched in both sphingolipids and cholesterol, perhaps the most important finding was that proteins could be segregated being selectively included or excluded from the rafts (Simons and Ikonen, 1997). In this way, raft localization can serve to facilitate or obstruct protein interactions (Brown and London, 2000; Lingwood and Simons, 2009) or act as a protein scaffold while allowing diffusion (Maxfield and Tabas, 2005).
Despite significant efforts in developing methodology and techniques for lipid raft research, there are still some shortcomings in precise determination of their size, structure, and composition which were recently critically discussed by Simons et al. (2010). The most important issue which still induces debate is related to application of detergents for isolation of lipid rafts and methyl-β-cyclodextrin for extraction of cholesterol from cell membranes which could lead to formation of artificial complexes not existing in the natural environment. It has now been established that detergent-resistant membranes (DRMs) are not identical to lipid rafts and the data on proteins detected within DRMs from various types of cells should be treated with caution and not always considered to be functionally raft localized (Lichtenberg et al., 2005; Chen et al., 2009a). Development of a new technique that purifies nano-meso scale DRMs at 37°C in an ionic buffer that preserves the lamellar phase of the metastable inner leaflet lipids could be a significant step toward purifying individual physiologically relevant rafts (Morris et al., 2011). Although Triton X-100 is still the standard detergent in preparation of lipid rafts some groups claim that use of Brij 96 produces more reliable results although comparing properties of both detergents in the same set of experiments have not produced significantly different results. Moreover it has been suggested that preparation of the rafts in a buffer mimicking the cytoplasmic environment will preserve the structure of the rafts under physiological conditions by retaining the proteins associated with the intracellular part of the membrane (Chen et al., 2009b).
Another source of controversy in lipid raft research which still has to be resolved is determined by the lack of suitable detection techniques in living cells. This resulted in estimation of the putative size of lipid rafts in the range of 5–700 nm. In the last decade a number of new techniques assisting in estimation of raft size and helping in their visualization has been developed and applied (for review see Simons et al., 2010). They include single molecule spectroscopy and super-resolution microscopy such as fluorescence recovery after photobleaching (FRAP), stimulated emission depletion (STED), Förster resonance energy transfer (FRET), total internal reflection fluorescence (TIRF), and fluorescence correlation spectroscopy (FCS) techniques. However, even using these techniques the subwavelength lipid domains have never been directly visualized but their size was predicted to be <20 nm (Eggeling et al., 2009). Despite the limitations these new techniques confirmed the existence of nanoscale cholesterol-based assemblies of lipids and proteins in the membranes of living cells and allowed to characterize further their dynamics and properties.
When comparing the data on the size and precise protein and lipid composition of lipid rafts it is important to bear in mind that they are determined not only by the type of detergent used, the conditions of the experiments and resolution of techniques applied, but also by the type of tissue and cells used for their isolation and visualization. This has to be taken into account when comparing the data and making conclusions about the physiological relevance of proteins detected in lipid rafts. From this point of view a systematic meta-analysis of existing data considering various experimental conditions, tissue specificity, and resolution of the techniques applied might provide a useful tool for understanding raft heterogeneity and for optimizing further research into this intriguing subject.
The best-characterized raft proteins include lipid-modified proteins containing saturated acyl chains, such as GPI-anchored proteins, and doubly acylated proteins, such as Src family kinases and the α subunits of heterotrimeric G proteins (Hooper, 1999; Liang et al., 2001; Oh et al., 2001). Many other physiologically important proteins have been investigated for their possible raft localizations, including key proteins involved in Alzheimer’s disease (AD), such as amyloid precursor protein (APP; Parkin et al., 1999), β-site APP cleaving enzyme (BACE-1; Ehehalt et al., 2003; Kalvodova et al., 2005), the γ-secretase complex (Hur et al., 2008), a disentegrin and metalloprotease ADAM10 (Harris et al., 2009), acetylcholinesterase (AChE; Xie et al., 2010b), angiotensin-converting enzyme (ACE; Parkin et al., 2003), a ligand for the Notch receptor (Jagged1; Parr-Sturgess et al., 2010), and most recently an amyloid β (Aβ)-degrading enzyme, neprilysin (NEP; Sato et al., 2012). However the full list of raft-associated proteins is still far from completion.
The progress in understanding structural and functional diversity of lipid rafts led to the concept of caveolae as a sub-type of lipid rafts, appearing as 50–80 nm pits in the plasma membrane. Although caveolae are sometimes considered to be synonymous with lipid rafts, it is now clear that they represent only a subset of rafts whose properties are defined by their major protein components belonging to the caveolin family and denoted Cav-1, Cav-2, and Cav-3 (Parton and Simons, 1995, 2007). The characterization of PTRF (polymerase I and transcript release factor, originally identified as an RNA Pol I transcription factor, also called Cav-P60 and now termed cavin-1) and subsequently of other cavin family members as important constituents in caveolae formation has revealed new levels of complexity in the biogenesis of these plasma membrane invaginations (Briand et al., 2011). Like the rafts themselves, the caveolae are enriched in cholesterol, glycosphingolipids, and SM. They are the site of several important protein–protein interactions, for example, the neurotrophin receptors, TrkA and p75(NTR), whose respective interactions with caveolin regulates neurotrophin signaling in the brain (Bilderback et al., 1999). Caveolins also regulate G-proteins, MAPK, PI3K, and Src tyrosine kinases (Marin, 2011).
Another group of proteins which was suggested as markers of caveolae are flotillins (also known as reggies) which were discovered as proteins involved in nerve regeneration (Schulte et al., 1997). The flotillins are palmitoylated and myristoylated proteins, anchored to the plasma membrane and are considered to be exclusively raft localized (Schneider et al., 2008). Flotillins belong to a larger class of integral membrane proteins that have an evolutionarily conserved domain called the prohibitin homology domain (PHB) which determines the affinity of proteins carrying it to the lipid rafts (Morrow and Parton, 2005).
In the context of this review it is important to mention another cholesterol and ganglioside enriched membrane domain, which share some similarity but are physically and functionally distinct from the lipid rafts, the tetraspanin-enriched microdomains (TEMs). Tetraspanins are a large family of small membrane-spanning proteins (Yanez-Mo et al., 2011), numbering at least 32 family members in mammals. They are involved in a whole range of cellular processes, from cell morphology and motility to signaling pathways (Hemler, 2005).
In general, lipid rafts can be considered as signaling platforms that bring together various ingredients of the biological membranes determining specificity of the cells and their functioning. They include receptors, channels, recognition molecules, coupling factors and enzymes, facilitating their interaction and supporting signaling. Lipid rafts have been implicated in a plethora of both physiological and pathological processes. Among beneficial processes are axonal growth and branching (Kamiguchi, 2006; Grider et al., 2009; Munderloh et al., 2009) and hence raft disruption impedes axonogenesis (Petro and Schengrund, 2009). Rafts are also involved in the stabilization of synapses (Willmann et al., 2006). Raft components are also involved in cholera toxin entry via GM1 ganglioside (Holmgren et al., 1973), HIV-1 entry (gp120), and conversion of prion protein (PrPc) to its infectious form (PrPSc; Fantini et al., 2002; Vieira et al., 2010). Lipid rafts play a critical role in entry, replication, assembly, and budding of various types of viruses (Suzuki and Suzuki, 2006). In a more general sense, lipid rafts have been suggested to be involved in cardiovascular disease, carcinogenesis, and immune system diseases (Michel and Bakovic, 2007). However, this review will focus on the role of lipid rafts in the pathogenesis of AD, a condition with which lipid rafts have been demonstrated to have multifarious links (Cordy et al., 2006; Cheng et al., 2007; Vetrivel and Thinakaran, 2010).
Amyloid Cascade Hypothesis of AD
In the last two decades the amyloid cascade hypothesis of AD (Hardy and Higgins, 1992) has prevailed leading to accumulation of a significant amount of data on the molecular basis of the disease which have recently been reviewed by those who originally proposed the concept (Selkoe, 2001; Hardy, 2009). From the clinical point of view, the disease is characterized by global cognitive decline, associated with brain pathology involving accumulation of extracellular amyloid aggregates (also known as senile plaques) of amyloid β peptide (Aβ) and intracellular neurofibrillary tangles of hyper-phosphorylated tau protein (Bothwell and Giniger, 2000). According to the amyloid cascade hypothesis, it is the Aβ which is principally responsible for many of the pathological features of the disease (Hardy and Higgins, 1992; Sakono and Zako, 2010) with Aβ oligomers representing the most toxic species (Haass and Selkoe, 2007; Walsh and Selkoe, 2007; Sakono and Zako, 2010). Accumulation of amyloid plaques is accompanied by astrogliosis and microgliosis (Grilli et al., 2003) and the most affected brain areas are the neocortex and hippocampus (Sisodia and Gallagher, 1998). Although there are strong genetic links, including APP and presenilin mutations (Bothwell and Giniger, 2000), as well as the apolipoprotein ε4 allele (Saunders et al., 1993; Deane et al., 2008; Huang, 2010), sporadic AD is the dominant form. From this point of view predominance of AD research based on the mechanisms of early onset disease versus the broader spectrum of the factors leading to the sporadic form might be one of the reasons for the failure of the majority of therapeutic trials and lack of any preventive measures 20 years since the amyloid hypothesis has been proposed.
The Amyloid Precursor Protein (APP)
APP is a type I integral membrane protein. It exists in three isoforms (APP695, APP751, and APP770), generated from differential splicing of exons 7 and 8 (Sandbrink et al., 1996). Exon 7 is homologous to protease inhibitors of the Kunitz type (KPI domain), while exon 8 is related to the MRC OX-2 antigen in thymocytes (Kitaguchi et al., 1988; Sandbrink et al., 1996). APP695 lacks both KPI and OX-2 domains, while APP751 only lacks the OX-2 domain (Henriques et al., 2007). In terms of distribution, APP mRNA is expressed in almost every tissue, where only the isoform ratio differs (Araki et al., 1991). It is APP695 that predominates in neurons (Gralle and Ferreira, 2007). There are two proteolytic pathways of APP processing (Figure 1). Amyloidogenic processing involves sequential cleavage of APP by β- and γ-secretases (for review see Zhang et al., 2012). This process ultimately releases Aβ peptide, responsible in large part for the pathogenesis of AD and a soluble ectodomain, sAPPβ. The second, non-amyloidogenic, pathway involves α-secretase cleavage of APP. This cleavage occurs between Lys16 and Leu17, within the Aβ region (Allinson et al., 2003) and precludes formation of Aβ. There is also the release of the large, soluble ectodomain, the neuroprotective sAPPα, which is shed from the cell surface (Allinson et al., 2004) and has been found in the CSF (Palmert et al., 1989).
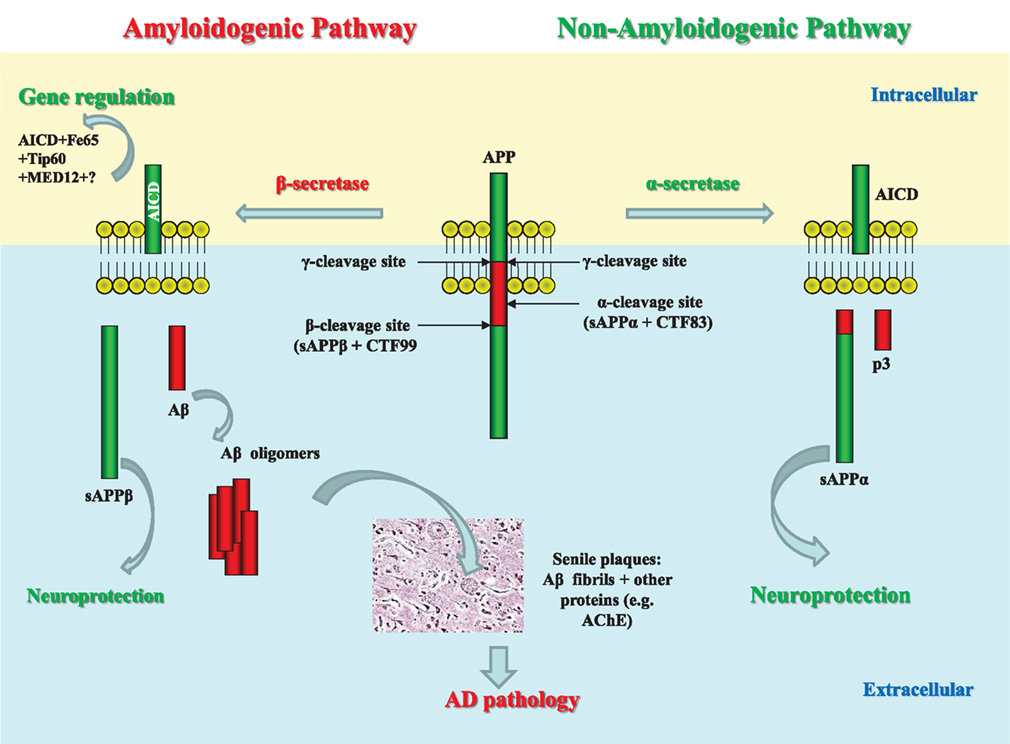
Figure 1. Schematic representation of APP processing and role of its products in AD pathology. The proteolytic processing of the large, transmembrane, amyloid precursor protein (APP) occurs in two distinct amyloidogenic and non-amyloidogenic pathways. The amyloidogenic pathway involves the sequential cleavage of APP by an aspartic proteinase, β-secretase, which releases a soluble ectodomain (sAPPβ) and the C-terminal fragment CTF99. This, in turn, is cleaved by another aspartic proteinase, γ-secretase, generating the transcriptional regulator APP intracellular domain (AICD), and releasing the 39–42 amino acid amyloid-β peptide (Aβ). Due to its very high ability to aggregation, Aβ forms dimers, trimers, and higher level oligomers which are toxic to cells and cause neuronal death. Formation of amyloid plaques from Aβ aggregates in complex with other proteins is a hallmark of AD but is considered as a scavenging process. In the non-amyloidogenic pathway APP molecules are cleaved at the α-secretase site within the Aβ-domain releasing a soluble ectodomain sAPPα and the C-terminal fragment CTF83. Proteolytic cleavage of CTF83 by γ-secretase releases AICD and p3 fragment whose functions are still unknown. The AICD fragment produced in the amyloidogenic pathway binds to a stabilizing factor Fe65 and in a complex with other factors (the histone acetyl transferase, Tip60, and a Mediator complex subunit Med12) can act as transcription factor regulating expression of a variety of genes, including an Aβ-degrading enzyme neprilysin. This process was found to be specific to the neuronal APP695 isoform. AICD produced in the non-amyloidogenic pathway and from other APP isoforms (APP751 and APP770) is most likely to be degraded (e.g., by some intracellular proteases, e.g., insulin-degrading enzyme). Soluble APP ectodomains, sAPPα, and sAPPβ, have been shown to have neuroprotective properties.
At the time when the terms were introduced, α-, β-, and γ-secretases were activities known to cleave APP proteolytically, but not defined as specific enzymes performing the cleavage. It was later found that BACE-1 was responsible for β-secretase activity (Vassar et al., 1999) and that a complex of presenilin, nicastrin, Aph-1, and Pen-2 had γ-secretase activity (Yu et al., 2000; Hooper, 2005). The α-secretase cleavage was found to be mediated by zinc metalloproteases of the disintegrin and metalloprotease (ADAM) family, specifically ADAM10 and ADAM17 (also known as TNFα converting enzyme, TACE). ADAM10 was found to be the dominant enzyme of APP processing in SH-SY5Y cells, given a lesser effect of ADAM17 knockdown (Allinson et al., 2004). Recent investigations have suggested that ADAM10 is the principal APP secretase in primary neurons, with ADAM17 playing more of an auxiliary role (Kuhn et al., 2010; Lichtenthaler, 2010). Allinson et al. (2004) suggested a model whereby the group of metalloproteases would each contribute to greater or lesser extents to APP cleavage in different cells and under different conditions. The ADAMs have a wide number of substrates, including angiotensin-converting enzyme (ACE; Allinson et al., 2004), ACE2 (Lambert et al., 2005), the prion protein (Vincent et al., 2001) as well as each other (Parkin and Harris, 2009). The ADAMs have been reviewed in greater detail elsewhere (Allinson et al., 2003; Edwards et al., 2008; van Goor et al., 2009).
Raft Localization of APP
The idea that lipid rafts may somehow modulate APP cleavage and hence affect the progression of AD has been around for over 10 years and previously reviewed (Cordy et al., 2006; Cheng et al., 2007; Vetrivel and Thinakaran, 2010). APP itself is not generally a raft protein, although a small proportion of APP is localized in lipid rafts (Parkin et al., 1999). Regulation of APP raft localization has been suggested to involve an interaction between the C-terminus of APP and flotillin-1 (Chen et al., 2006). A more recent study suggested that flotillin-2 may also act as a scaffolding protein, clustering APP in lipid rafts. Schneider et al. (2008) postulated that a transient interaction between APP and flotillin-2 may regulate endocytosis of APP, which is important for its processing. It has also been suggested that another raft component, cholesterol, has a role in binding APP promoting its raft localization (Beel et al., 2010) although it was earlier proposed that one of the physiological functions of APP and Aβ is to control cholesterol transport (Yao and Papadopoulos, 2002). There is also evidence that the adaptor protein Disabled1 (Dab1) interacts with APP regulating its processing and that the glycoprotein reelin promotes interaction of APP and Dab1 and their localization to lipid rafts involving phosphorylation by Fyn kinase (Hoe et al., 2009; Minami et al., 2011). APP trafficking to the lipid rafts was shown to be dependent on the low-density lipoprotein receptor-related protein (LRP) which also promotes BACE1-APP interaction (Yoon et al., 2007). On the other hand it was also demonstrated that ApoER2, a member of the low density lipoprotein receptor (LDL-R), negatively affects APP internalization and its expression stimulates Aβ production by shifting the proportion of APP from the non-raft region to the raft membrane domains (Fuentealba et al., 2007).
Dependence of APP Secretases on Raft Localization
In search of an explanation as to how APP could be cleaved in two distinct (amyloidogenic and non-amyloidogenic) pathways, it was suggested that APP was present in two cellular pools (Ehehalt et al., 2003) as schematically presented in Figure 2. Amyloidogenic processing was suggested to be linked to lipid rafts as their integrity was critical for Aβ formation (Simons et al., 1998; Cordy et al., 2003; Ehehalt et al., 2003; Rushworth and Hooper, 2011) and Aβ production was also indicated to be raft-localized (Lee et al., 1998). Furthermore, the precise protein/lipid composition of the rafts was shown to influence Aβ release (Lemkul and Bevan, 2011) and aggregation (Ikeda et al., 2011). Tetraspanins were also shown to regulate APP processing (Yanez-Mo et al., 2011) and perturbation of tetraspanin enriched domains (TEMs) can affect cell signaling pathways (Hemler, 2005). For example, the α-secretase ADAM10 is regulated by tetraspanins and in this way, tetraspanins can affect the non-amyloidogenic processing of APP (Arduise et al., 2008). In addition to this, tetraspanins also regulate the amyloidogenic pathway of APP processing since γ-secretase was shown to be associated with TEMs (Wakabayashi et al., 2009).
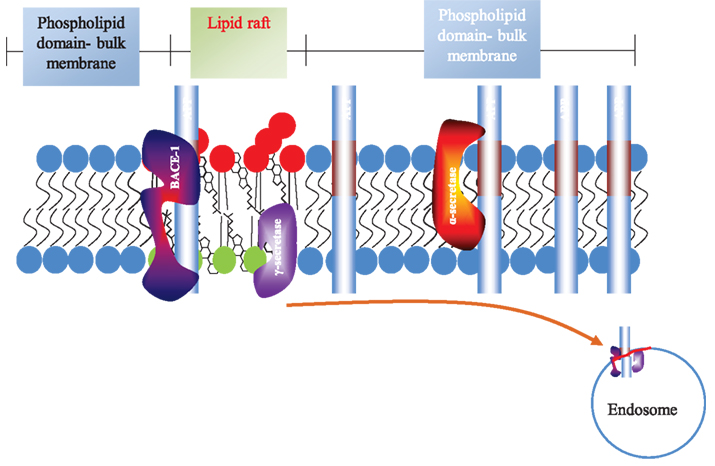
Figure 2. Topography of APP processing in cell membrane and lipid rafts. The lipid raft is shown as part of the plasma membrane. The phospholipid domain (light blue) is separate from the lipid raft. The latter is enriched in glycosphingolipids and sphingomyelin (red) on the exofacial leaflet and glycerolipids (e.g., phosphatidylserine and phosphatidylethanolamine; green) on the cytofacial leaflet. Cholesterol (black) is enriched in both leaflets. The acyl chains in lipid rafts are more able to pack together. APP (Aβ region in maroon) is localized in raft and non-raft fractions, but predominates outside rafts. The α-secretase is not raft-associated, while the β- and γ-secretases predominate in rafts. The cell surface is shown for clarity, although β-cleavage predominantly occurs in endosomes.
BACE-1 was found to be palmitoylated at three residues, which indicated possible raft localization and interaction with raft-resident lipids (Kalvodova et al., 2005; Hattori et al., 2006). When BACE-1 was targeted to lipid rafts via GPI-anchoring, production of Aβ was increased, indicating upregulation of amyloidogenic APP processing (Cordy et al., 2003). A more recent study suggests that GPI-anchorage increases preferential cleavage at the β-site of APP, producing the full length Aβ, whereas wtBACE-1 cleaves APP at two sites (β and β’ sites) producing full length and N-terminally truncated Aβ (Vetrivel et al., 2011). Another study has reported the effects of GPI-anchorage of ADAM10, the principal α-secretase, and showed that no wtADAM10 was raft localized while all GPI-ADAM10 was in lipid rafts. It was associated with a reduction in Aβ as GPI-ADAM10 competed with BACE-1 for the APP substrate (Harris et al., 2009).
In addition to BACE1, it was shown that the subunits of the γ-secretase complex are also enriched in the lipid rafts (Lee et al., 1998; Hur et al., 2008) and that S-palmitoylation plays a role in localization and stability of nicastrin and Aph-1 within the rafts although not affecting the γ-secretase processing of APP (Cheng et al., 2009). While caveolin-1 was shown to be an important regulator of γ-secretase spatial distribution and activity (Kapoor et al., 2010), the proteins of the γ-secretase complex, e.g., PS1, in turn, can induce lipid raft formation and decrease the membrane fluidity (Eckert and Müller, 2009). A subset of proteins, in particular voltage-dependent anion channel 1 and contact in associated protein 1, are also associated with γ-secretase in lipid rafts and affect APP processing (Hur et al., 2012).
Another important lipid-raft associated protein which was shown to play an important role in APP processing is the GPI-anchored prion protein (PrP; Naslavsky et al., 1997). It was demonstrated that PrPc regulates APP processing by inhibiting BACE1 activity and that the effect of PrPc on the β-secretase cleavage of APP requires the localization of PrPc to cholesterol-rich lipid rafts and is mediated by the N-terminal polybasic region of PrPC via interaction with glycosaminoglycans (Parkin et al., 2007). This interaction decreased BACE1 at the cell surface and in endosomes where it preferentially cleaves wild type APP but increased it in the Golgi where it preferentially cleaves APP with the Swedish mutation (APPSwe). Although deletion of PrPc in transgenic mice expressing human mutated APPSwe,Ind had no effect on APP processing and Aβ levels, deletion of PrPc in HEK293 cells expression wild type reduced APP cleavage by BACE1. Because there was no effect of PrPc deletion on processing of APPSwe in HEK cells the authors suggested that PrPc may be a key protective player against sporadic Alzheimer disease versus its less common familiar form (Griffiths et al., 2011). Because there is no detected decrease of PrPc content in the AD brain (Saijo et al., 2011) it is possible to suggest that age- and disease-dependent disruption of lipid rafts might be a cause of decreased ability of PrPc to control BACE1 activity resulting in accumulation of Aβ peptide in the case of sporadic AD.
The amyloid-degrading enzyme, NEP, has been shown to be partially associated with lipid rafts in human synoviocytes in “ectopeptidase-rich membrane microdomains” (Riemann et al., 2001), and also in pre-B and B cell lines (Nalm-6 and Raji; Angelisová et al., 1999). By targeting NEP to different intracellular compartments of neurons, including the lipid rafts, Hama and colleagues demonstrated that the endogenous targeting signal in wild-type NEP is well optimized for the overall neuronal clearance of Aβ (Hama et al., 2004). Only the mature, fully glycosylated form of NEP, preferentially in its dimerized form, can be found in lipid rafts in direct association with phosphatidylserine (Hama et al., 2004). These authors also suggested that the localization of NEP in different intracellular compartments may be involved in the metabolism of distinct pools of Aβ and that the endogenous targeting signal in wild-type NEP is well optimized for the overall neuronal clearance of Aβ. The partitioning of NEP into lipid rafts has also recently been confirmed by Sato and colleagues (Sato et al., 2012). Although another Aβ-degrading enzyme, insulysin (insulin-degrading enzyme, IDE), is primarily a cytosolic protein it is also, in part, associated with lipid rafts where it may facilitate Aβ clearance (Bulloj et al., 2008).
Sub-Cellular Localization of APP Processing
Apart from compartmentalization within the lipid rafts, the amyloidogenic processing pathway was shown to be dependent on endocytosis (Ehehalt et al., 2003). In addition to flotillin, caveolin-1 is responsible for partitioning of γ-secretase between the plasma membrane and endosomes and cells depleted of caveolin-1 had more γ-secretase localized within the clathrin-coated non-caveolar endocytic vesicles, although different caveolins have been shown to have different effects on APP processing (Nishiyama et al., 1999; Kapoor et al., 2010).
Lipid rafts affect APP processing not only through favoring interactions between APP and BACE-1 but also by promoting endocytosis of APP. This process was shown to be APP isoform-dependent with the neuronal APP695 isoform to be mostly processed via the β-secretase pathway whereas APP751 and APP770 mainly undergo α-secretase cleavage (Belyaev et al., 2010). Some of the earliest work about subcellular localization of APP processing indicated that sAPPβ could be detected in neuronal NT2N cell lysates with the absence of sAPPα or p3 fragments. This suggested an intracellular β-secretase pathway (Chyung et al., 1997; Hartmann et al., 1997). It is worth noting that the β-secretase pathway operates differently with wild-type APP (wtAPP) and APP carrying the Swedish mutation (APPSw; Haass et al., 1995; Griffiths et al., 2011). An alternative trafficking pathway has been recently reported, whereby APP can bypass endosomes and be trafficked directly to the lysosomes. This, though, does not occur with either APPSw or APPLondon variants suggesting that these mutations affect APP transit (Lorenzen et al., 2010).
Lipid Rafts and the APP Intracellular Domain (AICD)
Although there is quite a substantial amount of data on the physiological role of N-terminal soluble ectodomains of APP – sAPPα and sAPPβ, for review see Chasseigneaux and Allinquant (2012), the C-terminal products of APP proteolytic cleavage have only recently started to attract special attention. It is now rather well documented (although not without some controversy) that the C-terminal fragment of APP, AICD, can act as a transcription factor (Cao and Sudhof, 2001; Leissring et al., 2002; Schettini et al., 2010). The controversy of the AICD research has been underpinned by utilization of different cell types expressing different APP isoforms and by the difficulties in detecting AICD due to its very short half life (Cupers et al., 2001). However, it was repeatedly demonstrated that functional AICD, which translocates to the cell nucleus and up-regulates expression of a reporter NEP gene, is produced only via the amyloidogenic pathway and only from the APP695 isoform (Goodger et al., 2009; Belyaev et al., 2010). This process was also shown to be neuronal cell specific and lipid raft dependent (Belyaev et al., 2010). In an earlier work by Cao and Sudhof (2001), a yeast 2-hybrid (Y2H) screen was used to identify binding partners of the C-terminal domain of APP which revealed the role of Fe65 and the histone acetyltransferase (HAT) Tip60 in formation of functionally active AICD.
AICD regulates the transcription of several target genes, some better characterized than others (Beckett et al., 2012; Pardossi-Piquard and Checler, 2012). The most well documented gene up-regulated by AICD is of the amyloid-degrading enzyme neprilysin (Pardossi-Piquard et al., 2005; Belyaev et al., 2009). However, there is also evidence that APP itself (von Rotz et al., 2004), BACE1 (von Rotz et al., 2004), GSK-3β (Kim et al., 2003) and aquaporin-1 (Huysseune et al., 2009) can be regulated by AICD. In addition to APP, regulation of the GSK-3β can be considered as a link between AICD and AD pathology especially taking into account the data on elevated levels of AICD in the brain of AD patients (Ghosal et al., 2009). Moreover, the ability of AICD to regulate expression of APP and BACE1 suggests a feedback mechanism of its own regulation by proteolytic processing of its precursor (Grimm et al., 2012a).
AICD also has a direct link to lipid metabolism as it has been found to suppress the expression of the major lipoprotein receptor LRP1 and as such affect apoE/cholesterol metabolism (Liu et al., 2007). On the other hand AICD controls expression of the alkyldihydroxyacetonephosphate-synthase which regulates plasmalogen synthesis in the cells (Grimm et al., 2011b) and reduced levels of these brain-specific lipids are characteristic of the AD brain (Han et al., 2001; Rothhaar et al., 2012). Reduced plasmalogen levels in the AD brain might have direct effect on production of Aβ since they were shown to inhibit activity of γ-secretase (Rothhaar et al., 2012). There is also evidence that AICD regulates sphingolipid synthesis via serine-palmitoyl transferase (Grimm et al., 2011a), and as such may control composition of lipid rafts and APP processing. The wide range of putative AICD target genes highlights the role of APP signaling in normal brain functioning and in AD pathology.
Lipid Raft Components and Their Changes in AD
Sphingomyelin
The major component of lipid rafts, sphingomyelin (SM), is characteristic only for eukaryotic cells where it comprises about 10–15% of total phospholipids and even more in the brain and peripheral nervous tissue. SM and its metabolites play an important role as second messengers in signal transduction events during development, differentiation and immune response of the organisms (Nalivaeva et al., 2000; Hannun et al., 2001). SM is essential for the activity of some types of receptors, including the α7 nicotinic receptor (Colón-Sáez and Yakel, 2011), NMDA receptors (Wheeler et al., 2009), neurotrophic tyrosine kinase receptor type 2 (Trovò et al., 2011), serotonin1A receptor (Jafurulla et al., 2008) and the urokinase receptor (uPAR; Sahores et al., 2008). It was also found that some disease-related membrane proteins (APP, gp120, and PrP) have a common SM-recognition site which underscores the role of lipid rafts in AD, HIV, and prion diseases (Mahfoud et al., 2002).
Investigation of lipid raft biology was enhanced by the discovery of SM-specific probes, e.g., lysenin, which serve as powerful tools to study the organization and biological function of this lipid in biological membranes (Hullin-Matsuda and Kobayashi, 2007; Shogomori and Kobayashi, 2008). These studies have demonstrated functional and structural diversity of lipid rafts and characterized in the plasma membrane of Jurkat T cells the SM-rich domains which had spatial and functional specificity compared to the GM1-rich domains (Kiyokawa et al., 2005). Formation of SM clusters in the membranes of neuronal cells was shown to depend on localization of SM synthase (SMS) isoforms in various cell compartments and that the activity of SMS is the rate-limiting step in SM cluster formation. In accordance with this it was also demonstrated that SM clusters were formed only in the vicinity of SM synthase proteins. In particular, it was also found that the SMS2 isoform is specific for the dendrites of hippocampal neurons (Kidani et al., 2012). The pattern of individual species of sphingomeylin was also found to be different in various types of cells and even in cell compartments (e.g., in lipid rafts versus detergent-soluble fractions) which testifies to the role of these lipids in determining cell-specific membrane properties (Valsecchi et al., 2007).
In the aging brain and especially in AD pathology, lipid metabolism, and in particular that of SM, undergoes significant changes. By using a shotgun lipidomics approach Han and colleagues have compared levels of over 800 lipid species in control and AD brains and demonstrated a significant decrease in SM and increases in ceramide levels in the affected brain. They suggested a model in which an AD-related increase in SMase activity results in faster SM hydrolysis (and increased ceramide production) which would lead to alterations in lipid raft formation (Han et al., 2011). These authors also hypothesized that this would impair functions of GLUT4, which was previously shown to be functionally linked to lipid rafts (Michel and Bakovic, 2007) and lead to the dysfunctions in energy homeostasis characteristic of the disease. Moreover it was also shown that accumulation of the amyloid peptide in neuronal cells leads to SMase activation and SM depletion which, in turn, affects cellular trafficking and abnormal APP processing (Soreghan et al., 2003). Further work demonstrated that the most toxic form of amyloid peptide Aβ42 can also activate SMase (Grimm et al., 2005; Grosgen et al., 2010). Since excessive SMase-mediated cleavage of SM occurs early in AD it is likely to disrupt a range of protein-lipid interactions and hence downstream signaling pathways (Haughey et al., 2010). In addition to AD-linked SMase over-activity, there have been reports of deficiencies of the enzymes responsible for sphingolipid synthesis (Piccinini et al., 2010).
Gangliosides
Gangliosides are glycosphingolipids with one or more sialic acid residues. They are essential components of all animal cell membranes where they are anchored in the external leaflet by the hydrophobic ceramide part of their molecule while the oligosaccharide chain protrudes into the extracellular medium. Gangliosides are particularly abundant in the plasma membranes of neuronal cells (Sonnino et al., 2007) have been implicated in various cellular functions since the high heterogeneity of their oligosaccharide structures allows specific interactions with various proteins and oligosaccharide chains of other molecules at the cell membrane surface. Gangliosides to a great extent determine the fine structure of membranes, e.g., lateral diffusion of its components, as well as the organization of lipid rafts (Cantù et al., 2011). Disruption of ganglioside metabolism leads to various neurological diseases (for review see Walkley, 2003). Studies of ganglioside-knockout mice have demonstrated that depletion of various classes of gangliosides results in development of neurodegeneration and pathology similar to Alzheimer’s or Parkinson’s diseases (Furukawa et al., 2011; Wu et al., 2011). In the aging brain, and especially in AD patients, ganglioside content significantly decreases in various brain structures, especially in the areas of the brain related to the pathogenesis of the disease (Kracun et al., 1991).
The link of gangliosides with AD pathology has been mostly related to their localization in lipid rafts which have been suggested to act as a platform for GM1-induced aggregation of Aβ peptide (Parton, 1994; Zha et al., 2004; Ariga et al., 2008). Labeled Aβ shows high affinity for GM1 containing membranes, suggesting that GM1 acts as an Aβ binding molecule (Kakio et al., 2004). It has been suggested that the N-terminal region of Aβ interacts with GM1 clusters through hydrogen bonding and electrostatic interactions (Lemkul and Bevan, 2011). It was also suggested that cholesterol may facilitate GM1 clustering (Kakio et al., 2001). Extraction of cortical lipid rafts from human AD brains showed an increase in both GM1 and GM2 (Molander-Melin et al., 2005) despite an overall reduction of gangliosides (Ariga et al., 2008). There are data indicating that gangliosides accumulate in senile plaques and that they may be involved in the conversion of Aβ to a neurotoxic oligomeric form (Molander-Melin et al., 2005; Okada et al., 2008).
Various synaptosomal and liposomal studies have suggested that GM1 accelerates formation of amyloid fibrils (Yamamoto et al., 2004; Zha et al., 2004). Further analysis of the role of lipid rafts in Aβ aggregation have confirmed that raft proteins do not play a significant role in this process as proteinase K or SDS treatment did not affect the aggregation-promoting capacity of the rafts. Moreover, the cholesterol-depleting agent methyl-β-cyclodextrin (MβCD) also did not have an effect on Aβ oligomerization (Kim et al., 2006). Further cell studies reinforce the role of gangliosides in Aβ aggregation. Rafts taken from ganglioside rich cells (C2C12) were able to induce Aβ aggregation more potently than ganglioside-poor cells (HeLa and SK-N-MC). Similarly, rafts isolated from brain tissue rich in gangliosides were able to increase Aβ aggregation compared to rafts isolated from the liver with lower ganglioside content. Also, CHO-K1 cells genetically deficient in the synthesis of complex gangliosides had lower levels of Aβ aggregation than the wild type cells (Kim et al., 2006).
An examination of the effects of GM1 on APP processing in SH-SY5Y and COS7 cells showed an inhibition of α-cleavage (Zha et al., 2004). On the other hand, Aβ1–40 oligomers were found to stimulate the amyloidogenic processing of APP by reducing membrane fluidity and complexing with GM1 ganglioside (Peters et al., 2009). Although precise mechanisms of GM1 changes with age are still unknown it was demonstrated that GM1 content in neuronal membranes, particularly in DRM microdomains, increases with age and this increase was more pronounced in the brain of apoE4 knock-in mice compared to apoE3 knock-in animals (Yamamoto et al., 2004).
Recently two novel mechanisms linking gangliosides with AD have been described by Grimm et al. (2012b). They discovered that Aβ binds to GD3-synthase (GD3S, the key-enzyme in converting a-series gangliosides to major brain specific b-series) and inhibits its activity. On the other hand, the APP intracellular domain AICD, together with Fe65, was found to down-regulate expression of the GD3S gene. This provides an explanation for age-dependent and AD-related changes in brain ganglioside patterns and supports an essential role of APP in ganglioside homeostasis. Another link between gangliosides and AD lies in their ability to alter APP processing. As observed by the authors, GM3 was able to decrease production of Aβ while GD3 was shown to increase its levels in COS7 cells. This might be related to the changes in the properties of lipid rafts containing different amounts of GM3 or GD3 ganglioside species and their ability to regulate activity of β- and γ-secretases.
Taking into account the important role of gangliosides in normal brain function, their involvement in the pathogenesis of AD should be treated with caution since different species of gangliosides will have a different impact on the integrity and properties of neuronal membranes. Recently, by using G3 synthase KO mice (lacking only b-series gangliosides) and GM2/GD2 synthase KO mice (which lack almost all gangliosides except GM3 and GD3) it was found that these ganglioside species are important for neuroprotection and anti-inflammatory response via maintenance of lipid rafts (Ohmi et al., 2011). Intraventricular treatment of AD patients with GM1 was shown to stop the progression of cognitive deterioration and improve motor performance and neuropsychological assessments (Svennerholm et al., 2002). Peripheral utilization of GM1 for prevention of Aβ aggregation in the brain via establishing peripheral/brain dynamics of Aβ was suggested as a possible therapeutic approach for AD since in the PS/APP mice this was shown to reduce Aβ accumulation in the brain (Matsuoka et al., 2003). However, due to the antigenic properties of gangliosides this might provoke some side-effects (López-Requena et al., 2007).
Cholesterol
Another important component of cellular membranes and of lipid rafts, cholesterol, has been recently extensively reviewed in relation to its role in AD (Marzolo and Bu, 2009; Burns and Rebeck, 2010; Mathew et al., 2011). The picture is by no means a simple one with cholesterol being ascribed both positive and negative roles. Indeed, other diseases are also linked to cholesterol, while it remains unclear which are specifically raft-associated. Such pathologies include Smith–Lemli–Opitz syndrome, Huntington’s disease and Niemann–Pick Type C disease (Korade and Kenworthy, 2008). In terms of normal aging, the data seem to show that the changes in cholesterol are highly dependent on the brain region and cell types used in the studies (Martin et al., 2010).
Cholesterol levels have been shown to be elevated in AD patients as well as the levels of cholesterol precursors in the mevalonate pathway, farnesylpyrophosphate, and geranylgeranylpyrophosphate (Hooff et al., 2010; Kolsch et al., 2010). However, this view is by no means unanimous and other groups have found lower levels of cholesterol in AD brains (Kolsch et al., 2010; Leduc et al., 2010), in addition to its precursors lanosterol and lathosterol (Kolsch et al., 2010), with lower levels of its synthesizing enzyme, HMG CoA reductase (Leduc et al., 2010). However, quantification of global cholesterol levels are not necessarily reflective of the number or distribution of lipid rafts (Leduc et al., 2010).
As cholesterol is integral to ordered lipid rafts, the consequences of cholesterol depletion are widely regarded as effects of raft disruption (Hao et al., 2001; Mondal et al., 2009). In this context the cholesterol content in the membranes has an inversely proportional relationship with the membrane-perturbing effects of Aβ oligomers (Cecchi et al., 2009). As such, if cholesterol increases do elevate lipid raft abundance, then it would increase Aβ formation (Simons et al., 1998; Ehehalt et al., 2003) and, on the contrary, low cholesterol levels will lead to up-regulation of the activity of the α-secretase, ADAM10 (Kojro et al., 2001). Cholesterol depletion in cells by MβCD was shown to affect APP-processing and formation of functionally active AICD resulting in reduced levels of expression of the amyloid-degrading enzyme, neprilysin (Belyaev et al., 2010). On the other hand, cholesterol has been shown to bind C99, which promotes amyloidogenic processing (Beel et al., 2010) and the increase of Aβ levels, in turn, can cause changes in cholesterol homeostasis in the Golgi and plasma membrane (Igbavboa et al., 2009). Formation of the Aβ “seed” and initiation of Aβ aggregation was also shown to be cholesterol dependent (Mizuno et al., 1999; Kakio et al., 2001; Simons et al., 2001). In a more biophysical sense, raised cholesterol has been implicated in facilitating the insertion of Aβ into the plasma membrane. In so doing, Aβ then destroys the cells’ membrane integrity (Ji et al., 2002).
Just as cholesterol and lipid rafts have been shown to affect APP processing, APP-derived species have been shown to impact on cholesterol and lipid homeostasis. Aβ peptides modulate the metabolism of cholesterol, in particular its esterification rate, and of phospholipids in hepatocytes, neuronal cells, and in the entire brain (Koudinov et al., 1996; Koudinova et al., 1996, 2000). It was also found that Aβ peptides alter vesicle trafficking and cholesterol homeostasis (Liu et al., 1998). On the other hand, it was shown that cholesterol binds to APP at the α-secretase cleavage sites and Aβ itself can bind cholesterol and prevent its interaction with low-density lipoprotein (Yao and Papadopoulos, 2002). This confirmed that Aβ might act as a component of lipoprotein complexes and affect reverse cholesterol transport from neuronal tissue to the periphery in addition to its role in cholesterol synthesis and intracellular dynamics (Koudinov et al., 2001; Michikawa et al., 2001). In a later study Aβ40 has been shown to inhibit a key enzyme in the biosynthesis of cholesterol, HMG CoA reductase (Grimm et al., 2005). Further to this, APP intracellular domain AICD was found to regulate cholesterol levels via LRP1 (Grosgen et al., 2010). However, there also are data reporting that cholesterol in physiological concentrations can protect neuronal cells against Aβ-induced toxicity and slow down the process of formation of toxic aggregates of Aβ with metal ions, in particular with aluminum (Granzotto et al., 2011). This correlates with the data suggesting that cholesterol may have a protective effect against membrane disruption by amyloid species (in this case, Aβ-derived diffusible ligands: ADDLs). Cholesterol supplemented SH-SY5Y cells were shown to display reduced binding of ADDLs to the plasma membrane, while oligomers increased in membrane presence after treatment with the cholesterol-depleting agent MβCD (Cecchi et al., 2009).
Statins
Statins are a class of drugs which inhibit HMG CoA reductase, a key enzyme in the biosynthesis of cholesterol. Given the links between cholesterol and AD, it is not surprising that there are multiple investigations on the effects of statins in AD pathology. The current state of the field and problems have been recently discussed and the role of statins has been reviewed (Wang et al., 2010; Wood et al., 2010). The authors point out that the effects of statins are not only related to APP processing nor specific for the AD pathology. Furthermore, the physiological effects of statins are not solely due to inhibition of cholesterol biosynthesis but include perturbation of other mevalonate-dependent pathways such as protein prenylation.
A number of studies have shown positive effects of statins in AD (Jick et al., 2000; Buxbaum et al., 2001, 2002). This is in agreement with the previous studies demonstrating that depletion of cholesterol reduces Aβ in cultured neurons (Simons et al., 1998). One recent study has suggested that fluvastatin is able to modify the trafficking of APP. Use of this drug was stated to increase lysosomal degradation of APP C-terminal fragments (CTFs) and hence facilitate Aβ clearance (Shinohara et al., 2010). However, the reduction in Aβ levels is not necessarily linked with the cognitive benefits. A recent randomized, double-blinded, placebo-controlled trial of simvastatin showed reduced Aβ in treated patients, but no corresponding improvement in cognitive performance (ADAS-Cog score; Sano et al., 2011). The biochemical studies are fairly uniform in that cholesterol depletion results in reductions in key AD-related markers. However, the results of epidemiological research are unequivocal. The Cochrane Dementia and Cognitive Improvement Group study reported that statin treatment had no effect on the prevention or treatment of dementia (McGuinness et al., 2009a,b). However, taking into account the highly variable relationship between the initiation of statin therapy and the time and severity of the AD, it is very difficult to get a conclusive assessment of the accumulated data on the beneficial effect of statins and any such study should use a defined set of criteria during epidemiological meta-analysis (Shepardson et al., 2011a,b).
Lipid Rafts and Cell Signaling
Lipid rafts have been implicated as the sites for a great number of signaling pathways (Allen et al., 2007). Perturbation of, or changes, in lipid rafts could therefore affect neuronal signaling, including cholinergic transmission. As cholinergic hypofunction is key to the pathogenesis of AD (Schliebs, 2005; Schliebs and Arendt, 2006), there are strong links between lipid rafts, neuronal signaling pathways, and AD.
Lipid Mediators
Various lipids, such as eicosanoids, docosanoids, and cannabinoids, can act as signaling mediators and their abnormal metabolism has implications in AD (Farooqui, 2011). These lipid mediators can modulate the metabolism of sphingolipids through activation of SMases. Furthermore, sphingolipid-derived molecules such as ceramide and ceramide 1-phosphate can act as lipid mediators and accumulate in the AD brain. This accumulation of sphingolipid derivatives can activate cytosolic phospholipase A2 (cPLA2), which leads to changes in membrane fluidity and permeability with concomitant alterations in ion homeostasis. Furthermore, the degradation products of cPLA2 metabolism are often pro-inflammatory (Frisardi et al., 2011). This work shows how the lipid raft constituents, in this case sphingolipids, can affect AD processes in an APP-independent manner. Here, accumulation and action of these lipid mediators promotes inflammation, a process characteristic of AD (Akiyama et al., 2000).
Neurotransmitter Signaling
Numerous neurotransmitter signaling systems, especially receptor function, are influenced by lipid rafts. The range of receptors involved is all-encompassing, including ionotropic receptors such as α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA receptors), γ-aminobutyric acid (GABA) receptors, N-methyl-D-aspartate (NMDA), and nicotinic acetylcholine (nACh) receptors. The influence of lipid rafts also extends to metabotropic G-protein coupled receptors, such as the muscarinic acetylcholine receptor (mAChR; Allen et al., 2007; Rushworth and Hooper, 2011). Although all these signaling systems can be linked to AD, it is the cholinergic system which is of central importance, as its hypofunction is considered one of the hallmarks of the disease (Coyle et al., 1983; Auld et al., 2002; Schliebs and Arendt, 2006) and its involvement is therefore principally considered here.
The Cholinergic System and the α7 Nicotinic Acetylcholine Receptor
Lipid rafts have been implicated in regulating the clustering of nAChRs via the myristoylated peripheral membrane protein, rapsyn, which is constitutively lipid raft localized. Perturbation of the rafts impedes the interaction between rapsyn and nAChRs, which reduces clustering of the receptors and hence their function (Zhu et al., 2006). Protein reconstitution studies using lipid vesicles imply that the partitioning of the nAChR into raft domains is not solely due to the intrinsic biophysical properties of the receptor but requires a signaling event to translocate the protein into specific membrane domains (Bermúdez et al., 2010). Furthermore, cholesterol affects the properties, both structural and functional, of some acetylcholine receptors (Barrantes et al., 2010). Agrin signaling represents one pathway which can enhance the translocation of the nAChR into lipid rafts (Campagna and Fallon, 2006; Zhu et al., 2006).
In particular, one of the most prominent examples of a lipid raft-linked signaling protein is the α7 nicotinic acetylcholine receptor (nAChR), a Ca2+ channel (Albuquerque et al., 2009), which has a close involvement with Aβ and cognitive decline (Albuquerque et al., 2009; Jurgensen and Ferreira, 2009; Hernandez et al., 2010). On balance, the α7 nAChR seems to have a neuroprotective role in model systems. Significant amounts of the α7 nAChR are associated with lipid rafts and these rafts are essential for maintenance of function (Bruses et al., 2001). Lipid rafts appear to assist the interaction between soluble Aβ and the receptor (Khan et al., 2010). Although α7 KO mice show no apparent cognitive defects, Aβ42 is enriched and Aβ oligomers are enhanced in hippocampi from these animals (Hernandez et al., 2010).
The α7 nAChR regulates, in part, the pleiotropic intracellular signaling effects of Ca2+. In addition to this, the receptor is closely linked to the signaling of cAMP (via adenylyl cyclase) as well as the kinases Fyn and PI3K (Oshikawa et al., 2003). Disruption of lipid rafts with MβCD or SMase has significant effects on receptor desensitization kinetics (Colón-Sáez and Yakel, 2011) and hence the many important downstream signaling events. Additional interactions of the α7 nAChR have been assessed using a proteomic screen, which identified several proteins involved in neurite outgrowth and maintenance, namely α-catenin 2, BASP1/NAP-22, gelsolin, homer 1, and neuromodulin (Paulo et al., 2009). Further to this, the AD therapeutic donepezil protects against glutamate toxicity in part through stimulation of the α7 nAChR (Shen et al., 2010). Given the central role of the α7 nAChR, it has been suggested promoting its activation via novel agonists may represent a new therapeutic approach in AD (Wang, 2010).
Acetylcholinesterase (AChE)
Lipid raft localization has recently been linked to another protein of the cholinergic system, namely acetylcholinesterase (AChE), although the functional implications of this are as yet unclear (Xie et al., 2010b; Hicks et al., 2011). AChE inhibition by compounds such as rivastigmine or galantamine represents the major therapeutic option for treating the cognitive impairment seen in the early stages of AD yet the relationship between AChE and AD represents something of a paradox. AChE exists in a number of different molecular forms (G1, G2, G4) of which the tetrameric G4 form is predominant in brain. In AD, brain G4 AChE levels are seen to fall as the disease progresses, while G1 and G2 levels rise somewhat, as compared to normal brains (Atack et al., 1983; Garcia-Ayllon et al., 2010). AChE is found associated with amyloid plaques, leading to the suggestion that AChE may promote Aβ aggregation (Moran et al., 1993; Inestrosa et al., 1996). In some brain regions with AD pathology, virtually all of the AChE is localized in these complexes (Mesulam et al., 1987). A direct interaction between Aβ and AChE has been proposed, with binding occurring at the peripheral anionic site (PAS) of the enzyme. Those AChE inhibitors which occupy the PAS (e.g., propidium) show the most significant reductions in fibril formation (Bartolini et al., 2003) since the catalytic site is not required for interaction with Aβ (Inestrosa et al., 1996). Furthermore, monoclonal antibodies directed against the PAS inhibit fibril formation (Reyes et al., 1997), which has led to the development of PAS blockers, such as the DUO compounds, that also occupy the active site. They show inhibitory activity on AChE as well as inhibition of Aβ40 fibril formation (Alptuzun et al., 2010) and have been suggested as potential novel AD therapeutics targeting two facets of the disease. The related enzyme butyrylcholinesterase (BuChE), as well as a synthetic peptide derived from the BuChE C terminus (BSP41), have also been shown to reduce amyloid fibril formation (Diamant et al., 2006). The corresponding AChE synthetic peptide, however, did not significantly affect Aβ fibril formation.
AChE is not a transmembrane protein, rather it is anchored to the plasma membrane by the proline rich membrane anchor (PRiMA) which is a 20-kDa type I transmembrane protein which can be acylated (Henderson et al., 2010; Xie et al., 2010b). PRiMA contains a CRAC (cholesterol recognition amino acid consensus) motif which sequesters PRiMA into lipid rafts and hence AChE is also partly associated with rafts although the functional significance of this interaction is unclear (Xie et al., 2010a). It has been suggested that the lipid raft localization and shedding of AChE may have a certain role in the pathology of AD (Hicks et al., 2011).
PrP Signaling in AD
PrP is another component of lipid rafts which was shown to act as a cellular receptor and change Ca2+ signaling upon activation by some ligands, e.g., by antibody cross-linking in T-lymphocytes (Stuermer et al., 2005). PrPc was also shown to be a receptor for Aβ oligomers at nanomolar concentrations and binding of Aβ oligomers to PrPc results in the blockage of hippocampal LTP. Incubation of hippocampal slices with PrP antibody abolishes the effect of Aβ oligomers and rescues synaptic plasticity (Lauren et al., 2009). Most recently it was demonstrated PrPc also interacts with the NMDA receptor complex in a copper-dependent manner to allosterically reduce glycine affinity for the receptor and that Aβ(1–42), copper chelators or PrPc inactivation all enhance the activity of glycine at the receptor resulting in steady-state NMDAR currents and neurotoxicity. It was suggested that the physiological role of PrPc might be related to limitation of excessive NMDAR activity which might cause neuronal damage (You et al., 2012). Together with the data on BACE1 regulation by PrPc this provides a unifying molecular mechanism explaining the interplay between toxic Aβ species, NMDA receptor-mediated toxicity and copper homeostasis in pathogenesis of AD (Rushworth and Hooper, 2011).
The “Signalosome”
It is possible for lipid rafts to modulate signaling in a general way by affecting the activities of signaling molecules involved in multiple pathways. These include, for example, the pleiotropic src kinases (Arcaro et al., 2007) with effects on the PI3K-Akt signaling pathway. Other raft-localized signaling proteins include the epidermal growth factor receptor (EGFR), which associates with caveolins (Couet et al., 1997) and is involved in diverse processes including cell cycle regulation, endocytosis, and the MAPK cascade (Oda et al., 2005). This has led to the concept of the signalosome, containing interacting components of signaling pathways (e.g., EGFR) within lipid rafts along with other scaffolding proteins, such as caveolins, and sequestering the complex from other interacting proteins that may disrupt the signaling process. A prime example is the CD40 signalosome associated with cell growth in B cell lymphomas (Pham et al., 2002).
Similar signaling platforms operate in neuronal systems, such as that involving estrogen receptor (ER) interactions (reviewed in Marin, 2011). This signaling mechanism is linked to neurogenesis, neuronal differentiation, synaptic plasticity, and neuroprotection (including against Aβ). Recent research indicates that lipid rafts are the site of formation of a complex between the ER, insulin growth factor 1 receptor (IGF-1R), Cav-1, and a voltage gated anion channel, VDAC. The formation of this signaling complex is neuroprotective but also lipid raft dependent (Marin, 2011). The “signalosome” paradigm was developed further by Chadwick et al. (2011) who isolated lipid rafts in control and 3xTg AD mice and compared their respective proteomes by mass spectrometry. Proteins so identified were then clustered into specific signaling pathways, which allowed an appraisal of which lipid raft signaling pathways may be altered in AD, rather than changes in individual proteins. This systems biology approach indicated that, in lipid rafts, wild-type mice had higher activation of pro-survival pathways such as PTEN and Wnt/β-catenin, whereas 3xTg mice showed activation of p53 and JNK signaling pathways. In addition, 3xTg mice had a deficit in growth factor signaling, neurodevelopmental signaling and signaling through the sonic hedgehog pathway (Chadwick et al., 2011). Another proteomic analysis extracted post-synaptic lipid rafts and used LC-MS/MS to analyze their protein content. They found an enrichment of cell adhesion molecules, channels/transporters and G-protein related species. Their data linked lipid rafts to cell adhesion with cell-cell contact regions and cell adhesion points being enriched in rafts. Further to this, H+-ATPase and Na+-K+ ATPase are enriched in lipid rafts, being responsible for maintaining ionic gradients and modulating neuronal excitability. This study also found the post-synaptic density to be associated with lipid rafts (Suzuki et al., 2011). Among channels localized in lipid rafts and involved in maintenance of neuronal cell homeostasis, in particular of astrocytes, are K+-buffering inwardly rectifying Kir4.1 channels and the water channel AQP4 (Hibino and Kurachi, 2007). Residence in the lipid rafts was also shown to be important for the activity of a member of the chloride channel family, ClC-2, which is widely expressed in the brain and other organs (Cornejo et al., 2009).
Concluding Remarks
Ever since the initial descriptions and characterizations of lipid rafts, the field has been beset by controversy (Pike, 2009) from their very existence down to the very specific aspects of how to isolate these structures in the laboratory. However, current consensus is that lipid rafts do represent dynamic structural components of cellular membranes integrating signaling events and regulating cell functioning and that their dysregulation can lead to disease.
This review has largely concentrated on the links between lipid rafts and AD pathology since processing of AD-related APP and production of Aβ peptide are clearly affected by lipid rafts. Also Aβ signaling involves interactions of proteins resident to lipid rafts. The mechanism underlying these effects has been examined in some detail although numerous gaps in knowledge still remain. Although the attempts to modulate lipid rafts and hence amyloidogenic processing have failed to translate into successful drugs, some epidemiological studies still indicate that inhibition of cholesterol synthesis through statin treatment might be beneficial if applied early (Solomon and Kivipelto, 2009; Shepardson et al., 2011a,b). The complexity of AD pathology and etiology dictates to consider involvement of lipid rafts in its pathogenesis in a more generic way, not only as a simplistic link between cholesterol levels, amyloid burden and cognition. More important for normal brain functioning is to maintain lipid metabolism in the aging brain at its normal rate and integrity.
In terms of future progress, lipid raft research might open new avenues in regulation of the proteolytic and signaling processes involved in AD pathology. The most recent discovery of the role of lipid raft disruption in decreased production of the functionally active transcriptional regulator AICD which might lead to an aberrant expression of its target genes including amyloid-degrading enzymes neprilysin (Howell et al., 1995; Carson and Turner, 2002; Belyaev et al., 2009; Liu et al., 2011), suggests that any therapeutics aimed at manipulation of lipid raft composition should be treated with caution. The role of the lipid components in cell membrane functioning and their structural variability and adaptive potential is extremely important for normal functioning of cells and organisms and much of the recent work in this area is both novel and revealing. However, it is important to note that these discoveries should be recognized as important advances in cell science and not seen as stepping stones to a therapeutic panacea.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the U.K. Medical Research Council and the Russian Academy of Science Programme “Fundamental Sciences to Medicine” and Russian Foundation for Basic Research (10-04-01156) for their financial assistance, and the Biotechnology and Biological Sciences Research Council for their Ph.D. studentship support for David A. Hicks.
References
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G. M., Cooper, N. R., Eikelenboom, P., Emmerling, M., Fiebich, B. L., Finch, C. E., Frautschy, S., Griffin, W. S., Hampel, H., Hull, M., Landreth, G., Lue, L., Mrak, R., Mackenzie, I. R., McGeer, P. L., O’Banion, M. K., Pachter, J., Pasinetti, G., Plata-Salaman, C., Rogers, J., Rydel, R., Shen, Y., Streit, W., Strohmeyer, R., Tooyoma, I., Van Muiswinkel, F. L., Veerhuis, R., Walker, D., Webster, S., Wegrzyniak, B., Wenk, G., and Wyss-Coray, T. (2000). Inflammation and Alzheimer’s disease. Neurobiol. Aging 21, 383–421.
Albuquerque, E. X., Pereira, E. F., Alkondon, M., and Rogers, S. W. (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89, 73–120.
Allen, J. A., Halverson-Tamboli, R. A., and Rasenick, M. M. (2007). Lipid raft microdomains and neurotransmitter signalling. Nat. Rev. Neurosci. 8, 128–140.
Allinson, T. M., Parkin, E. T., Condon, T. P., Schwager, S. L., Sturrock, E. D., Turner, A. J., and Hooper, N. M. (2004). The role of ADAM10 and ADAM17 in the ectodomain shedding of angiotensin converting enzyme and the amyloid precursor protein. Eur. J. Biochem. 271, 2539–2547.
Allinson, T. M., Parkin, E. T., Turner, A. J., and Hooper, N. M. (2003). ADAMs family members as amyloid precursor protein α-secretases. J. Neurosci. Res. 74, 342–352.
Alptuzun, V., Prinz, M., Horr, V., Scheiber, J., Radacki, K., Fallarero, A., Vuorela, P., Engels, B., Braunschweig, H., Erciyas, E., and Holzgrabe, U. (2010). Interaction of (benzylidene-hydrazono)-1,4-dihydropyridines with β-amyloid, acetylcholine, and butyrylcholine esterases. Bioorg. Med. Chem. 18, 2049–2059.
Angelisová, P., Drbal, K., Horejsí, V., and Cerný, J. (1999). Association of CD10/neutral endopeptidase 24.11 with membrane microdomains rich in glycosylphosphatidylinositol-anchored proteins and Lyn kinase. Blood 93, 1437–1439.
Araki, W., Kitaguchi, N., Tokushima, Y., Ishii, K., Aratake, H., Shimohama, S., Nakamura, S., and Kimura, J. (1991). Trophic effect of β-amyloid precursor protein on cerebral cortical neurons in culture. Biochem. Biophys. Res. Commun. 181, 265–271.
Arcaro, A., Aubert, M., Espinosa del Hierro, M. E., Khanzada, U. K., Angelidou, S., Tetley, T. D., Bittermann, A. G., Frame, M. C., and Seckl, M. J. (2007). Critical role for lipid raft-associated Src kinases in activation of PI3K-Akt signalling. Cell. Signal. 19, 1081–1092.
Arduise, C., Abache, T., Li, L., Billard, M., Chabanon, A., Ludwig, A., Mauduit, P., Boucheix, C., Rubinstein, E., and Le Naour, F. (2008). Tetraspanins regulate ADAM10-mediated cleavage of TNF-α and epidermal growth factor. J. Immunol. 181, 7002–7013.
Ariga, T., McDonald, M. P., and Yu, R. K. (2008). Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease – a review. J. Lipid Res. 49, 1157–1175.
Atack, J. R., Perry, E. K., Bonham, J. R., Perry, R. H., Tomlinson, B. E., Blessed, G., and Fairbairn, A. (1983). Molecular forms of acetylcholinesterase in senile dementia of Alzheimer type: selective loss of the intermediate (10S) form. Neurosci. Lett. 40, 199–204.
Auld, D. S., Kornecook, T. J., Bastianetto, S., and Quirion, R. (2002). Alzheimer’s disease and the basal forebrain cholinergic system: relations to β-amyloid peptides, cognition, and treatment strategies. Prog. Neurobiol. 68, 209–245.
Barrantes, F. J., Borroni, V., and Valles, S. (2010). Neuronal nicotinic acetylcholine receptor-cholesterol crosstalk in Alzheimer’s disease. FEBS Lett. 584, 1856–1863.
Bartolini, M., Bertucci, C., Cavrini, V., and Andrisano, V. (2003). β-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem. Pharmacol. 65, 407–416.
Beckett, C., Nalivaeva, N. N., Belyaev, N. D., and Turner, A. J. (2012). Nuclear signalling by membrane protein intracellular domains: the AICD enigma. Cell Signal. 24, 402–409.
Beel, A. J., Sakakura, M., Barrett, P. J., and Sanders, C. R. (2010). Direct binding of cholesterol to the amyloid precursor protein: an important interaction in lipid-Alzheimer’s disease relationships? Biochim. Biophys. Acta 1801, 975–982.
Belyaev, N. D., Kellett, K. A., Beckett, C., Makova, N. Z., Revett, T. J., Nalivaeva, N. N., Hooper, N. M., and Turner, A. J. (2010). The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a β-secretase dependent pathway. J. Biol. Chem. 285, 41443–41454.
Belyaev, N. D., Nalivaeva, N. N., Makova, N. Z., and Turner, A. J. (2009). Neprilysin gene expression requires binding of the amyloid precursor protein intracellular domain to its promoter: implications for Alzheimer disease. EMBO Rep. 10, 94–100.
Bermúdez, V., Antollini, S. S., Fernández Nievas, G. A., Aveldaño, M. I., and Barrantes, F. J. (2010). Partition profile of the nicotinic acetylcholine receptor in lipid domains upon reconstitution. J. Lipid Res. 51, 2629–2641.
Bilderback, T. R., Gazula, V. R., Lisanti, M. P., and Dobrowsky, R. T. (1999). Caveolin interacts with Trk A and p75(NTR) and regulates neurotrophin signaling pathways. J. Biol. Chem. 274, 257–263.
Bothwell, M., and Giniger, E. (2000). Alzheimer’s disease: neurodevelopment converges with neurodegeneration. Cell 102, 271–273.
Briand, N., Dugail, I., and Le Lay, S. (2011). Cavin proteins: new players in the caveolae field. Biochimie 93, 71–77.
Brown, D. A., and London, E. (2000). Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275, 17221–17224.
Brown, D. A., and Rose, J. K. (1992). Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68, 533–544.
Bruses, J. L., Chauvet, N., and Rutishauser, U. (2001). Membrane lipid rafts are necessary for the maintenance of the α7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J. Neurosci. 21, 504–512.
Bulloj, A., Leal, M. C., Surace, E. I., Zhang, X., Xu, H., Ledesma, M. D., Castaño, E. M., and Morelli, L. (2008). Detergent resistant membrane-associated IDE in brain tissue and cultured cells: relevance to Aβ and insulin degradation. Mol. Neurodegener. 3, 22.
Burns, M. P., and Rebeck, G. W. (2010). Intracellular cholesterol homeostasis and amyloid precursor protein processing. Biochim. Biophys. Acta 1801, 853–859.
Buxbaum, J. D., Cullen, E. I., and Friedhoff, L. T. (2002). Pharmacological concentrations of the HMG-CoA reductase inhibitor lovastatin decrease the formation of the Alzheimer β-amyloid peptide in vitro and in patients. Front. Biosci. 7, a50–a59.
Buxbaum, J. D., Geoghagen, N. S., and Friedhoff, L. T. (2001). Cholesterol depletion with physiological concentrations of a statin decreases the formation of the Alzheimer amyloid Aβ peptide. J. Alzheimers Dis. 3, 221–229.
Campagna, J. A., and Fallon, J. (2006). Lipid rafts are involved in C95 (4,8) agrin fragment-induced acetylcholine receptor clustering. Neuroscience 138, 123–132.
Cantù, L., Del Favero, E., Sonnino, S., and Prinetti, A. (2011). Gangliosides and the multiscale modulation of membrane structure. Chem. Phys. Lipids 164, 796–810.
Cao, X., and Sudhof, T. C. (2001). A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120.
Carson, J. A., and Turner, A. J. (2002). β-amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? J. Neurochem. 81, 1–8.
Cecchi, C., Nichino, D., Zampagni, M., Bernacchioni, C., Evangelisti, E., Pensalfini, A., Liguri, G., Gliozzi, A., Stefani, M., and Relini, A. (2009). A protective role for lipid raft cholesterol against amyloid-induced membrane damage in human neuroblastoma cells. Biochim. Biophys. Acta 1788, 2204–2216.
Chadwick, W., Brenneman, R., Martin, B., and Maudsley, S. (2011). Complex and multidimensional lipid raft alterations in a murine model of Alzheimer’s disease. Int. J. Alzheimers Dis. 2010, 604792.
Chasseigneaux, S., and Allinquant, B. (2012). Functions of Aβ, sAPPα and sAPPβ: similarities and differences. J. Neurochem. 120(Suppl. 1), 99–108.
Chen, T. Y., Liu, P. H., Ruan, C. T., Chiu, L., and Kung, F. L. (2006). The intracellular domain of amyloid precursor protein interacts with flotillin-1, a lipid raft protein. Biochem. Biophys. Res. Commun. 342, 266–272.
Chen, X., Lawrence, M. J., Barlow, D. J., Morris, R. J., Heenan, R. K., and Quinn, P.J. (2009a). The structure of detergent-resistant membrane vesicles from rat brain cells. Biochim. Biophys. Acta 1788, 477–483.
Chen, X., Jen, A., Warley, A., Lawrence, M. J., Quinn, P. J., and Morris, R. J. (2009b). Isolation at physiological temperature of detergent-resistant membranes with properties expected of lipid rafts: the influence of buffer composition. Biochem. J. 417, 525–533.
Cheng, H., Vetrivel, K. S., Drisdel, R. C., Meckler, X., Gong, P., Leem, J. Y., Li, T., Carter, M., Chen, Y., Nguyen, P., Iwatsubo, T., Tomita, T., Wong, P. C., Green, W. N., Kounnas, M. Z., and Thinakaran, G. (2009). S-palmitoylation of γ-secretase subunits nicastrin and APH-1. J. Biol. Chem. 284, 1373–1384.
Cheng, H., Vetrivel, K. S., Gong, P., Meckler, X., Parent, A., and Thinakaran, G. (2007). Mechanisms of disease: new therapeutic strategies for Alzheimer’s disease–targeting APP processing in lipid rafts. Nat. Clin. Pract. Neurol. 3, 374–382.
Chyung, A. S., Greenberg, B. D., Cook, D. G., Doms, R. W., and Lee, V. M. (1997). Novel β-secretase cleavage of β-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment of NT2N cells. J. Cell Biol. 138, 671–680.
Colón-Sáez, J. O., and Yakel, J. L. (2011). The 7 nicotinic acetylcholine receptor function in hippocampal neurons is regulated by the lipid composition of the plasma membrane. J. Physiol. 589, 3163–3174.
Cordy, J. M., Hooper, N. M., and Turner, A. J. (2006). The involvement of lipid rafts in Alzheimer’s disease. Mol. Membr. Biol. 23, 111–122.
Cordy, J. M., Hussain, I., Dingwall, C., Hooper, N. M., and Turner, A. J. (2003). Exclusively targeting β-secretase to lipid rafts by GPI-anchor addition up-regulates β-site processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 100, 11735–11740.
Cornejo, I., Niemeyer, M. I., Zúñiga, L., Yusef, Y. R., Sepúlveda, F. V., and Cid, L. P. (2009). Rapid recycling of ClC-2 chloride channels between plasma membrane and endosomes: role of a tyrosine endocytosis motif in surface retrieval. J. Cell Physiol. 221, 650–657.
Couet, J., Sargiacomo, M., and Lisanti, M. P. (1997). Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J. Biol. Chem. 272, 30429–30438.
Coyle, J. T., Price, D. L., and DeLong, M. R. (1983). Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 219, 1184–1190.
Cupers, P., Orlans, I., Craessaerts, K., Annaert, W., and De Strooper, B. (2001). The amyloid precursor protein (APP)-cytoplasmic fragment generated by γ-secretase is rapidly degraded but distributes partially in a nuclear fraction of neurones in culture. J. Neurochem. 78, 1168–1178.
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., Holtzman, D. M., and Zlokovic, B. V. (2008). apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013.
Diamant, S., Podoly, E., Friedler, A., Ligumsky, H., Livnah, O., and Soreq, H. (2006). Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. U.S.A. 103, 8628–8633.
Eckert, G. P., and Müller, W. E. (2009). Presenilin 1 modifies lipid raft composition of neuronal membranes. Biochem. Biophys. Res. Commun. 382, 673–677.
Edwards, D. R., Handsley, M. M., and Pennington, C. J. (2008). The ADAM metalloproteinases. Mol. Aspects Med. 29, 258–289.
Eggeling, C., Ringemann, C., Medda, R., Schwarzmann, G., Sandhoff, K., Polyakova, S., Belov, V. N., Hein, B., von Middendorff, C., Schönle, A., and Hell, S. W. (2009). Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 457, 1159–1162.
Ehehalt, R., Keller, P., Haass, C., Thiele, C., and Simons, K. (2003). Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123.
Fantini, J., Garmy, N., Mahfoud, R., and Yahi, N. (2002). Lipid rafts: structure, function and role in HIV, Alzheimer’s and prion diseases. Expert Rev. Mol. Med. 4, 1–22.
Farooqui, A. A. (2011). Lipid mediators and their metabolism in the nucleous: implications for Alzheimer’s Disease. J. Alzheimers Dis. [Epub ahead of print]. PMID:21955817.
Frisardi, V., Panza, F., Seripa, D., Farooqui, T., and Farooqui, A. A. (2011). Glycerophospholipids and glycerophospholipid-derived lipid mediators: a complex meshwork in Alzheimer’s disease pathology. Prog. Lipid Res. 50, 313–330.
Fuentealba, R. A., Barría, M. I., Lee, J., Cam, J., Araya, C., Escudero, C. A., Inestrosa, N. C., Bronfman, F. C., Bu, G., and Marzolo, M. P. (2007). ApoER2 expression increases Aβ production while decreasing Amyloid Precursor Protein (APP) endocytosis: possible role in the partitioning of APP into lipid rafts and in the regulation of γ-secretase activity. Mol. Neurodegener. 2, 14.
Furukawa, K., Ohmi, Y., Ohkawa, Y., Tokuda, N., Kondo, Y., Tajima, O., and Furukawa, K. (2011). Regulatory mechanisms of nervous systems with glycosphingolipids. Neurochem. Res. 36, 1578–1586.
Garcia-Ayllon, M. S., Riba-Llena, I., Serra-Basante, C., Alom, J., Boopathy, R., and Saez-Valero, J. (2010). Altered levels of acetylcholinesterase in Alzheimer plasma. PLoS ONE 5, e8701. doi:10.1371/journal.pone.0008701
Goodger, Z. V., Rajendran, L., Trutzel, A., Kohli, B. M., Nitsch, R. M., and Konietzko, U. (2009). Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. J. Cell. Sci. 122, 3703–3714.
Ghosal, K., Vogt, D. L., Liang, M., Shen, Y., Lamb, B. T., and Pimplikar, S. W. (2009). Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. U. S. A. 106, 18367–18372.
Gralle, M., and Ferreira, S. T. (2007). Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog. Neurobiol. 82, 11–32.
Granzotto, A., Suwalsky, M., and Zatta, P. (2011). Physiological cholesterol concentration is a neuroprotective factor against β-amyloid and β-amyloid-metal complexes toxicity. J. Inorg. Biochem. 105, 1066–1072.
Grider, M. H., Park, D., Spencer, D. M., and Shine, H. D. (2009). Lipid raft-targeted Akt promotes axonal branching and growth cone expansion via mTOR and Rac1, respectively. J. Neurosci. Res. 87, 3033–3042.
Griffiths, H. H., Whitehouse, I. J., Baybutt, H., Brown, D., Kellett, K. A., Jackson, C. D., Turner, A. J., Piccardo, P., Manson, J. C., and Hooper, N. M. (2011). Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J. Biol. Chem. 286, 33489–33500.
Grilli, M., Ferrari Toninelli, G., Uberti, D., Spano, P., and Memo, M. (2003). Alzheimer’s disease linking neurodegeneration with neurodevelopment. Funct. Neurol. 18, 145–148.
Grimm, M. O., Grimm, H. S., Patzold, A. J., Zinser, E. G., Halonen, R., Duering, M., Tschape, J. A., De Strooper, B., Muller, U., Shen, J., and Hartmann, T. (2005). Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nat. Cell Biol. 7, 1118–1123.
Grimm, M. O., Grösgen, S., Rothhaar, T. L., Burg, V. K., Hundsdörfer, B., Haupenthal, V. J., Friess, P., Müller, U., Fassbender, K., Riemenschneider, M., Grimm, H. S., and Hartmann, T. (2011a). Intracellular APP domain regulates serine-palmitoyl-CoA transferase expression and is affected in Alzheimer’s Disease. Int. J. Alzheimers Dis. 2011, 695413.
Grimm, M. O., Kuchenbecker, J., Rothhaar, T. L., Grosgen, S., Hundsdorfer, B., Burg, V. K., Friess, P., Muller, U., Grimm, H. S., Riemenschneider, M., and Hartmann, T. (2011b). Plasmalogen synthesis is regulated via alkyldihydroxyacetonephosphate-synthase by amyloid precursor protein processing and is affected in Alzheimer’s disease. J. Neurochem. 116, 916–925.
Grimm, M. O., Rothhaar, T. L., and Hartmann, T. (2012a). The role of APP proteolytic processing in lipid metabolism. Exp. Brain Res. 217, 365–375.
Grimm, M. O., Zinser, E. G., Grösgen, S., Hundsdörfer, B., Rothhaar, T. L., Burg, V. K., Kaestner, L., Bayer, T. A., Lipp, P., Müller, U., Grimm, H. S., and Hartmann, T. (2012b). Amyloid precursor protein (APP) mediated regulation of ganglioside homeostasis linking Alzheimer’s disease pathology with ganglioside metabolism. PLoS ONE 7, e34095. doi:10.1371/journal.pone.0034095
Grosgen, S., Grimm, M. O., Friess, P., and Hartmann, T. (2010). Role of amyloid β in lipid homeostasis. Biochim. Biophys. Acta 1801, 966–974.
Haass, C., Lemere, C. A., Capell, A., Citron, M., Seubert, P., Schenk, D., Lannfelt, L., and Selkoe, D. J. (1995). The Swedish mutation causes early-onset Alzheimer’s disease by β-secretase cleavage within the secretory pathway. Nat. Med. 1, 1291–1296.
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112.
Hama, E., Shirotani, K., Iwata, N., and Saido, T. C. (2004). Effects of neprilysin chimeric proteins targeted to subcellular compartments on amyloid β peptide clearance in primary neurons. J. Biol. Chem. 279, 30259–30264.
Han, X., Holtzman, D. M., and McKeel, D. W. Jr. (2001). Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 77, 1168–1180.
Han, X., Rozen, S., Boyle, S. H., Hellegers, C., Cheng, H., Burke, J. R., Welsh-Bohmer, K. A., Doraiswamy, P. M., and Kaddurah-Daouk, R. (2011). Metabolomics in early Alzheimer’s disease: identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS ONE 6, e21643. doi:10.1371/journal.pone.0021643
Hannun, Y. A., Luberto, C., and Argraves, K. M. (2001). Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry 40, 4893–4903.
Hao, M., Mukherjee, S., and Maxfield, F. R. (2001). Cholesterol depletion induces large scale domain segregation in living cell membranes. Proc. Natl. Acad. Sci. U.S.A. 98, 13072–13077.
Hardy, J. (2009). The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J. Neurochem. 110, 1129–1134.
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185.
Harris, B., Pereira, I., and Parkin, E. (2009). Targeting ADAM10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res. 1296, 203–215.
Hartmann, T., Bieger, S. C., Bruhl, B., Tienari, P. J., Ida, N., Allsop, D., Roberts, G. W., Masters, C. L., Dotti, C. G., Unsicker, K., and Beyreuther, K. (1997). Distinct sites of intracellular production for Alzheimer’s disease A β40/42 amyloid peptides. Nat. Med. 3, 1016–1020.
Hattori, C., Asai, M., Onishi, H., Sasagawa, N., Hashimoto, Y., Saido, T. C., Maruyama, K., Mizutani, S., and Ishiura, S. (2006). BACE1 interacts with lipid raft proteins. J. Neurosci. Res. 84, 912–917.
Haughey, N. J., Bandaru, V. V., Bae, M., and Mattson, M. P. (2010). Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim. Biophys. Acta 1801, 878–886.
Hemler, M. E. (2005). Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 6, 801–811.
Henderson, Z., Matto, N., John, D., Nalivaeva, N. N., and Turner, A. J. (2010). Co-localization of PRiMA with acetylcholinesterase in cholinergic neurons of rat brain: an immunocytochemical study. Brain Res.
Henriques, A. G., Vieira, S. I., Rebelo, S., Domingues, S. C., da Cruz e Silva, E. F., and da Cruz e Silva, O. A. (2007). Isoform specific amyloid-β protein precursor metabolism. J. Alzheimers Dis. 11, 85–95.
Hernandez, C. M., Kayed, R., Zheng, H., Sweatt, J. D., and Dineley, K. T. (2010). Loss of α7 nicotinic receptors enhances β-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 30, 2442–2453.
Hibino, H., and Kurachi, Y. (2007). Distinct detergent-resistant membrane microdomains (lipid rafts) respectively harvest K(+) and water transport systems in brain astroglia. Eur. J. Neurosci. 26, 2539–2555.
Hicks, D., John, D., Makova, N. Z., Henderson, Z., Nalivaeva, N. N., and Turner, A. J. (2011). Membrane targeting, shedding and protein interactions of brain acetylcholinesterase. J. Neurochem. 116, 742–746.
Hoe, H. S., Lee, K. J., Carney, R. S., Lee, J., Markova, A., Lee, J. Y., Howell, B. W., Hyman, B. T., Pak, D. T., Bu, G., and Rebeck, G. W. (2009). Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 29, 7459–7473.
Holmgren, J., Lönnroth, I., and Svennerholm, L. (1973). Tissue receptor for cholera exotoxin: postulated structure from studies with GM1 ganglioside and related glycolipids. Infect. Immun. 8, 208–214.
Hooff, G. P., Peters, I., Wood, W. G., Muller, W. E., and Eckert, G. P. (2010). Modulation of cholesterol, farnesylpyrophosphate, and geranylgeranylpyrophosphate in neuroblastoma SH-SY5Y-APP695 cells: impact on amyloid β-protein production. Mol. Neurobiol. 41, 341–350.
Hooper, N. M. (1999). Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae. Mol. Membr. Biol. 16, 145–156.
Hooper, N. M. (2005). Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein. Biochem. Soc. Trans. 33, 335–338.
Hooper, N. M., and Turner, A. J. (1988). Ectoenzymes of the kidney microvillar membrane. Differential solubilization by detergents can predict a glycosyl-phosphatidylinositol membrane anchor. Biochem. J. 250, 865–869.
Howell, S., Nalbantoglu, J., and Crine, P. (1995). Neutral endopeptidase can hydrolyze β-amyloid(1-40) but shows no effect on β-amyloid precursor protein metabolism. Peptides 16, 647–652.
Huang, Y. (2010). Aβ-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer’s disease. Trends. Mol. Med. 16, 287–294.
Hullin-Matsuda, F., and Kobayashi, T. (2007). Monitoring the distribution and dynamics of signaling microdomains in living cells with lipid-specific probes. Cell Mol. Life Sci. 64, 2492–2504.
Hur, J. Y., Teranishi, Y., Kihara, T., Yamamoto, N. G., Inoue, M., Hosia, W., Hashimoto, M., Winblad, B., Frykman, S., and Tjernberg, L. O. (2012). Identification of novel γ-secretase-associated proteins in detergent-resistant membranes from brain. J. Biol. Chem. 287, 11991–112005.
Hur, J. Y., Welander, H., Behbahani, H., Aoki, M., Franberg, J., Winblad, B., Frykman, S., and Tjernberg, L. O. (2008). Active γ-secretase is localized to detergent-resistant membranes in human brain. FEBS J. 275, 1174–1187.
Huysseune, S., Kienlen-Campard, P., Hebert, S., Tasiaux, B., Leroy, K., Devuyst, O., Brion, J. P., De Strooper, B., and Octave, J. N. (2009). Epigenetic control of aquaporin 1 expression by the amyloid precursor protein. FASEB J. 23, 4158–4167.
Igbavboa, U., Sun, G. Y., Weisman, G. A., He, Y., and Wood, W. G. (2009). Amyloid β-protein stimulates trafficking of cholesterol and caveolin-1 from the plasma membrane to the Golgi complex in mouse primary astrocytes. Neuroscience 162, 328–338.
Ikeda, K., Yamaguchi, T., Fukunaga, S., Hoshino, M., and Matsuzaki, K. (2011). Mechanism of amyloid β-protein aggregation mediated by GM1 ganglioside clusters. Biochemistry 50, 6433–6440.
Inestrosa, N. C., Alvarez, A., Perez, C. A., Moreno, R. D., Vicente, M., Linker, C., Casanueva, O. I., Soto, C., and Garrido, J. (1996). Acetylcholinesterase accelerates assembly of amyloid-(-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron 16, 881–891.
Jafurulla, M., Pucadyil, T. J., and Chattopadhyay, A. (2008). Effect of sphingomyelinase treatment on ligand binding activity of human serotonin1A receptors. Biochim. Biophys. Acta 1778, 2022–2025.
Ji, S. R., Wu, Y., and Sui, S. F. (2002). Cholesterol is an important factor affecting the membrane insertion of β-amyloid peptide (Aβ 1-40), which may potentially inhibit the fibril formation. J. Biol. Chem. 277, 6273–6279.
Jick, H., Zornberg, G. L., Jick, S. S., Seshadri, S., and Drachman, D. A. (2000). Statins and the risk of dementia. Lancet 356, 1627–1631.
Jurgensen, S., and Ferreira, S. T. (2009). Nicotinic receptors, amyloid-β, and synaptic failure in Alzheimer’s disease. J. Mol. Neurosci. 40, 421–429.
Kakio, A., Nishimoto, S. I., Yanagisawa, K., Kozutsumi, Y., and Matsuzaki, K. (2001). Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J. Biol. Chem. 276, 24985–24990.
Kakio, A., Yano, Y., Takai, D., Kuroda, Y., Matsumoto, O., Kozutsumi, Y., and Matsuzaki, K. (2004). Interaction between amyloid β-protein aggregates and membranes. J. Pept. Sci. 10, 612–621.
Kalvodova, L., Kahya, N., Schwille, P., Ehehalt, R., Verkade, P., Drechsel, D., and Simons, K. (2005). Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J. Biol. Chem. 280, 36815–36823.
Kamiguchi, H. (2006). The region-specific activities of lipid rafts during axon growth and guidance. J. Neurochem. 98, 330–335.
Kapoor, A., Hsu, W. M., Wang, B. J., Wu, G. H., Lin, T. Y., Lee, S. J., Yen, C. T., Liang, S. M., and Liao, Y. F. (2010). Caveolin-1 regulates γ-secretase-mediated AβPP processing by modulating spatial distribution of γ-secretase in membrane. J. Alzheimers Dis. 22, 423–442.
Karnovsky, M. J., Kleinfeld, A. M., Hoover, R. L., and Klausner, R. D. (1982). The concept of lipid domains in membranes. J. Cell Biol. 94, 1–6.
Khan, G. M., Tong, M., Jhun, M., Arora, K., and Nichols, R. A. (2010). β-Amyloid activates presynaptic α7 nicotinic acetylcholine receptors reconstituted into a model nerve cell system: involvement of lipid rafts. Eur. J. Neurosci. 31, 788–796.
Kidani, Y., Ohshima, K., Sakai, H., Kohno, T., Baba, A., and Hattori, M. (2012). Differential localization of sphingomyelin synthase isoforms in neurons regulates sphingomyelin cluster formation. Biochem. Biophys. Res. Commun. 417, 1014–1017.
Kim, H. S., Kim, E. M., Lee, J. P., Park, C. H., Kim, S., Seo, J. H., Chang, K. A., Yu, E., Jeong, S. J., Chong, Y. H., and Suh, Y.-H. (2003). C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3β expression. FASEB J. 17, 1951–1953.
Kim, S. I., Yi, J. S., and Ko, Y. G. (2006). Amyloid β oligomerization is induced by brain lipid rafts. J. Cell. Biochem. 99, 878–889.
Kitaguchi, N., Takahashi, Y., Tokushima, Y., Shiojiri, S., and Ito, H. (1988). Novel precursor of Alzheimer’s disease amyloid protein shows protease inhibitory activity. Nature 331, 530–532.
Kiyokawa, E., Baba, T., Otsuka, N., Makino, A., Ohno, S., and Kobayashi, T. (2005). Spatial and functional heterogeneity of sphingolipid-rich membrane domains. J. Biol. Chem. 280, 24072–24084.
Kojro, E., Gimpl, G., Lammich, S., Marz, W., and Fahrenholz, F. (2001). Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the α-secretase ADAM 10. Proc. Natl. Acad. Sci. U.S.A. 98, 5815–5820.
Kolsch, H., Heun, R., Jessen, F., Popp, J., Hentschel, F., Maier, W., and Lutjohann, D. (2010). Alterations of cholesterol precursor levels in Alzheimer’s disease. Biochim. Biophys. Acta 1801, 945–950.
Korade, Z., and Kenworthy, A. K. (2008). Lipid rafts, cholesterol, and the brain. Neuropharmacology 55, 1265–1273.
Koudinov, A. R., Berezov, T. T., and Koudinova, N. V. (2001). The levels of soluble amyloid b in different high density lipoprotein subfractions distinguish Alzheimer’s and normal aging cerebrospinal fluid: implication for brain cholesterol pathology? Neurosci. Lett. 314, 115–118.
Koudinov, A. R., Koudinova, N., and Berezov, T. T. (1996). Alzheimer’s peptides Aβ 1-40 and Aβ 1-28 inhibit the plasma cholesterol esterification rate. Biochem. Mol. Biol. Int. 38(4):747–52 38, 747–752.
Koudinova, N. V., Berezov, T. T., and Koudinov, A. R. (1996). Multiple inhibitory effects of Alzheimer’s peptide Ab1-40 on lipid biosynthesis in cultured human HepG2 cells. FEBS Lett. 395, 204–206.
Koudinova, N. V., Koudinov, A. R., and Yavin, E.. (2000). Alzheimer’s Aβ1-40 peptide modulates lipid synthesis in neuronal cultures and intact rat fetal brain under normoxic and oxidative stress conditions. Neurochem. Res. 25, 653–660.
Kracun, I., Rosner, H., Drnovsek, V., Heffer-Lauc, M., Cosović, C., and Lauc, G. (1991). Human brain gangliosides in development, aging and disease. Int. J. Dev. Biol. 35, 289–295.
Kuhn, P. H., Wang, H., Dislich, B., Colombo, A., Zeitschel, U., Ellwart, J. W., Kremmer, E., Rossner, S., and Lichtenthaler, S. F. (2010). ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 29, 3020–3032.
Lambert, D. W., Yarski, M., Warner, F. J., Thornhill, P., Parkin, E. T., Smith, A. I., Hooper, N. M., and Turner, A. J. (2005). Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 280, 30113–30119.
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132.
Leduc, V., Jasmin-Belanger, S., and Poirier, J. (2010). APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol. Med. 16, 469–477.
Lee, S. J., Liyanage, U., Bickel, P. E., Xia, W., Lansbury, P. T. Jr., and Kosik, K. S. (1998). A detergent-insoluble membrane compartment contains Aβ in vivo. Nat. Med. 4, 730–734.
Leissring, M. A., Murphy, M. P., Mead, T. R., Akbari, Y., Sugarman, M. C., Jannatipour, M., Anliker, B., Muller, U., Saftig, P., De Strooper, B., Wolfe, M. S., Golde, T. E., and LaFerla, F. M. (2002). A physiologic signaling role for the γ-secretase-derived intracellular fragment of APP. Proc. Natl. Acad. Sci. U.S.A. 99, 4697–4702.
Lemkul, J. A., and Bevan, D. R. (2011). Lipid composition influences the release of Alzheimer’s amyloid β-peptide from membranes. Protein Sci. 20, 1530–1545.
Liang, X., Nazarian, A., Erdjument-Bromage, H., Bornmann, W., Tempst, P., and Resh, M. D. (2001). Heterogeneous fatty acylation of Src family kinases with polyunsaturated fatty acids regulates raft localization and signal transduction. J. Biol. Chem. 276, 30987–30994.
Lichtenberg, D., Goñi, F. M., and Heerklotz, H. (2005). Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem. Sci. 30, 430–436.
Lichtenthaler, S. F. (2010). α-secretase in Alzheimer’s disease: molecular identity, regulation and therapeutic potential. J. Neurochem. 116, 10–21.
Lingwood, D., and Simons, K. (2009). Lipid rafts as a membrane-organizing principle. Science 327, 46–50.
Liu, Q., Zerbinatti, C. V., Zhang, J., Hoe, H. S., Wang, B., Cole, S. L., Herz, J., Muglia, L., and Bu, G. (2007). Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56, 66–78.
Liu, Y., Peterson, D. A., and Schubert, D. (1998). Amyloid β peptide alters intracellular vesicle trafficking and cholesterol homeostasis. Proc. Natl. Acad. Sci. U.S.A. 95, 13266–13271.
Liu, Y., Studzinski, C., Beckett, T., Murphy, M. P., Klein, R. L., and Hersh, L. B. (2011). Circulating neprilysin clears brain amyloid. Mol. Cell. Neurosci. 45, 101–107.
López-Requena, A., De Acosta, C. M., Moreno, E., González, M., Puchades, Y., Talavera, A., Vispo, N. S., Vázquez, A. M., and Pérez, R. (2007). Gangliosides, Ab1 and Ab2 antibodies I. Towards a molecular dissection of an idiotype-anti-idiotype system. Mol. Immunol. 44, 423–433.
Lorenzen, A., Samosh, J., Vandewark, K., Anborgh, P. H., Seah, C., Magalhaes, A. C., Cregan, S. P., Ferguson, S. S., and Pasternak, S. H. (2010). Rapid and direct transport of cell surface APP to the lysosome defines a novel selective pathway. Mol Brain 3, 11.
Mahfoud, R., Garmy, N., Maresca, M., Yahi, N., Puigserver, A., and Fantini, J. (2002). Identification of a common sphingolipid-binding domain in Alzheimer, prion, and HIV-1 proteins. J. Biol. Chem. 277, 11292–11296.
Marin, R. (2011). Signalosomes in the brain: relevance in the development of certain neuropathologies such as Alzheimer’s disease. Front. Physiol. 2, 23.
Martin, M., Dotti, C. G., and Ledesma, M. D. (2010). Brain cholesterol in normal and pathological aging. Biochim. Biophys. Acta 1801, 934–944.
Marzolo, M. P., and Bu, G. (2009). Lipoprotein receptors and cholesterol in APP trafficking and proteolytic processing, implications for Alzheimer’s disease. Semin. Cell Dev. Biol. 20, 191–200.
Mathew, A., Yoshida, Y., Maekawa, T., and Sakthi Kumar, D. (2011). Alzheimer’s disease: cholesterol a menace? Brain Res. Bull. 86, 1–12.
Matsuoka, Y., Saito, M., LaFrancois, J., Saito, M., Gaynor, K., Olm, V., Wang, L., Casey, E., Lu, Y., Shiratori, C., Lemere, C., and Duff, K. (2003). Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-amyloid. J. Neurosci. 23, 29–33.
Maxfield, F. R., and Tabas, I. (2005). Role of cholesterol and lipid organization in disease. Nature 438, 612–621.
McGuinness, B., Craig, D., Bullock, R., and Passmore, P. (2009a). Statins for the prevention of dementia. Cochrane Database Syst. Rev., CD003160.
McGuinness, B., O’Hare, J., Craig, D., Bullock, R., Malouf, R., and Passmore, P. (2009b). Statins for the treatment of dementia. Cochrane Database Syst Rev, CD007514.
Mesulam, M. M., Geula, C., and Moran, M. A. (1987). Anatomy of cholinesterase inhibition in Alzheimer’s disease: effect of physostigmine and tetrahydroaminoacridine on plaques and tangles. Ann. Neurol. 22, 683–691.
Michikawa, M., Gong, J. S., Fan, Q. W., Sawamura, N., and Yanagisawa, K. (2001). A novel action of Alzheimer’s amyloid b-protein (Ab): oligomeric Ab promotes lipid release. J. Neurosci. 21, 7226–7235.
Minami, S. S., Hoe, H. S., and Rebeck, G. W. (2011). Fyn kinase regulates the association between amyloid precursor protein and Dab1 by promoting their localization to detergent-resistant membranes. J. Neurochem. 118, 879–890.
Mizuno, T., Nakata, M., Naiki, H., Michikawa, M., Wang, R., Haass, C., and Yanagisawa, K. (1999). Cholesterol-dependent generation of a seeding amyloid β-protein in cell culture. J. Biol. Chem. 274, 15110–15114.
Molander-Melin, M., Blennow, K., Bogdanovic, N., Dellheden, B., Mansson, J. E., and Fredman, P. (2005). Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J. Neurochem. 92, 171–182.
Mondal, M., Mesmin, B., Mukherjee, S., and Maxfield, F. R. (2009). Sterols are mainly in the cytoplasmic leaflet of the plasma membrane and the endocytic recycling compartment in CHO cells. Mol. Biol. Cell 20, 581–588.
Moran, M. A., Mufson, E. J., and Gomez-Ramos, P. (1993). Colocalization of cholinesterases with β amyloid protein in aged and Alzheimer’s brains. Acta Neuropathol. 85, 362–369.
Morris, R. J., Jen, A., and Warley, A. (2011). Isolation of nano-meso scale detergent resistant membrane that has properties expected of lipid ‘rafts’. J. Neurochem. 116, 671–677.
Morrow, I. C., and Parton, R. G. (2005). Flotillins and the PHB domain protein family: rafts, worms and anaesthetics. Traffic 6, 725–740.
Munderloh, C., Solis, G. P., Bodrikov, V., Jaeger, F. A., Wiechers, M., Malaga-Trillo, E., and Stuermer, C. A. (2009). Reggies/flotillins regulate retinal axon regeneration in the zebrafish optic nerve and differentiation of hippocampal and N2a neurons. J. Neurosci. 29, 6607–6615.
Nalivaeva, N. N., Rybakina, E. G., Pivanovich, I. Yu., Kozinets, I. A., Shanin, S. N., and Bartfai, T. (2000). Activation of neutral sphingomyelinase by IL-1β requires the type 1 interleukin 1 receptor. Cytokine 12, 229–232.
Naslavsky, N., Stein, R., Yanai, A., Friedlander, G., and Taraboulos, A. (1997). Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J. Biol. Chem. 272, 6324–6331.
Nishiyama, K., Trapp, B. D., Ikezu, T., Ransohoff, R. M., Tomita, T., Iwatsubo, T., Kanazawa, I., Hsiao, K. K., Lisanti, M. P., and Okamoto, T. (1999). Caveolin-3 upregulation activates β-secretase-mediated cleavage of the amyloid precursor protein in Alzheimer’s disease. J. Neurosci. 19, 6538–6548.
Oda, K., Matsuoka, Y., Funahashi, A., and Kitano, H. (2005). A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 1, 2005 0010.
Oh, P., and Schnitzer, J. E. (2001). Segregation of heterotrimeric G proteins in cell surface microdomains. G(q) binds caveolin to concentrate in caveolae, whereas G(i) and G(s) target lipid rafts by default. Mol. Biol. Cell 12, 685–698.
Ohmi, Y., Tajima, O., Ohkawa, Y., Yamauchi, Y., Sugiura, Y., Furukawa, K., and Furukawa, K. (2011). Gangliosides are essential in the protection of inflammation and neurodegeneration via maintenance of lipid rafts: elucidation by a series of ganglioside-deficient mutant mice. J. Neurochem. 116, 926–935.
Okada, T., Ikeda, K., Wakabayashi, M., Ogawa, M., and Matsuzaki, K. (2008). Formation of toxic Aβ(1-40) fibrils on GM1 ganglioside-containing membranes mimicking lipid rafts: polymorphisms in Aβ(1-40) fibrils. J. Mol. Biol. 382, 1066–1074.
Oshikawa, J., Toya, Y., Fujita, T., Egawa, M., Kawabe, J., Umemura, S., and Ishikawa, Y. (2003). Nicotinic acetylcholine receptor α 7 regulates cAMP signal within lipid rafts. Am. J. Physiol., Cell Physiol. 285, C567–C574.
Palmert, M. R., Podlisny, M. B., Witker, D. S., Oltersdorf, T., Younkin, L. H., Selkoe, D. J., and Younkin, S. G. (1989). The β-amyloid protein precursor of Alzheimer disease has soluble derivatives found in human brain and cerebrospinal fluid. Proc. Natl. Acad. Sci. U.S.A. 86, 6338–6342.
Pardossi-Piquard, R., and Checler, F. (2012). The physiology of the β-amyloid precursor protein intracellular domain AICD. J. Neurochem. 120(Suppl. 1), 109–124.
Pardossi-Piquard, R., Petit, A., Kawarai, T., Sunyach, C., Alves da Costa, C., Vincent, B., Ring, S., D’Adamio, L., Shen, J., Muller, U., St George Hyslop, P., and Checler, F. (2005). Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of (APP and APLP. Neuron 46, 541–554.
Parkin, E., and Harris, B. (2009). A disintegrin and metalloproteinase (ADAM)-mediated ectodomain shedding of ADAM10. J. Neurochem. 108, 1464–1479.
Parkin, E. T., Tan, F., Skidgel, R. A., Turner, A. J., and Hooper, N. M. (2003). The ectodomain shedding of angiotensin-converting enzyme is independent of its localisation in lipid rafts. J. Cell. Sci. 116, 3079–3087.
Parkin, E. T., Turner, A. J., and Hooper, N. M. (1999). Amyloid precursor protein, although partially detergent-insoluble in mouse cerebral cortex, behaves as an atypical lipid raft protein. Biochem. J. 344(Pt 1), 23–30.
Parkin, E. T., Watt, N. T., Hussain, I., Eckman, E. A., Eckman, C. B., Manson, J. C., Baybutt, H. N., Turner, A. J., and Hooper, N. M. (2007). Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A. 104, 11062–11067.
Parr-Sturgess, C. A., Rushton, D. J., and Parkin, E. T. (2010). Ectodomain shedding of the Notch ligand Jagged1 is mediated by ADAM17 but is not a lipid raft-associated event. Biochem. J. 432, 283–294.
Parton, R. G. (1994). Ultrastructural localization of gangliosides; GM1 is concentrated in caveolae. J. Histochem. Cytochem. 42, 155–166.
Parton, R. G., and Simons, K. (2007). The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8, 185–194.
Paulo, J. A., Brucker, W. J., and Hawrot, E. (2009). Proteomic analysis of an α7 nicotinic acetylcholine receptor interactome. J. Proteome Res. 8, 1849–1858.
Peters, I., Igbavboa, U., Schütt, T., Haidari, S., Hartig, U., Rosello, X., Böttner, S., Copanaki, E., Deller, T., Kögel, D., Wood, W. G., Müller, W. E., and Eckert, G. P. (2009). The interaction of β-amyloid protein with cellular membranes stimulates its own production. Biochim. Biophys. Acta 1788, 964–972.
Petro, K. A., and Schengrund, C. L. (2009). Membrane raft disruption promotes axonogenesis in n2a neuroblastoma cells. Neurochem. Res. 34, 29–37.
Pham, L.V., Tamayo, A. T., Yoshimura, L. C., Lo, P., Terry, N., Reid, P. S., and Ford, R. J. (2002). A CD40 Signalosome anchored in lipid rafts leads to constitutive activation of NF-kappaB and autonomous cell growth in B cell lymphomas. Immunity 16, 37–50.
Piccinini, M., Scandroglio, F., Prioni, S., Buccinna, B., Loberto, N., Aureli, M., Chigorno, V., Lupino, E., Demarco, G., Lomartire, A., Rinaudo, M. T., Sonnino, S., and Prinetti, A. (2010). Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol. Neurobiol. 41, 314–340.
Pike, L. J. (2006). Rafts defined: a report on the keystone symposium on lipid rafts and cell function. J. Lipid Res. 47, 1597–1598.
Reyes, A. E., Perez, D. R., Alvarez, A., Garrido, J., Gentry, M. K., Doctor, B. P., and Inestrosa, N. C. (1997). A monoclonal antibody against acetylcholinesterase inhibits the formation of amyloid fibrils induced by the enzyme. Biochem. Biophys. Res. Commun. 232, 652–655.
Riemann, D., Hansen, G. H., Niels-Christiansen, L., Thorsen, E., Immerdal, L., Santos, A. N., Kehlen, A., Langner, J., and Danielsen, E. M. (2001). Caveolae/lipid rafts in fibroblast-like synoviocytes: ectopeptidase-rich membrane microdomains. Biochem. J. 354, 47–55.
Rothhaar, T. L., Grösgen, S., Haupenthal, V. J., Burg, V. K., Hundsdörfer, B., Mett, J., Riemenschneider, M., Grimm, H. S., Hartmann, T., and Grimm, M. O. (2012). Plasmalogens inhibit APP processing by directly affecting γ-secretase activity in Alzheimer’s disease. Sci. World J. 2012, 141240.
Rushworth, J. V., and Hooper, N. M. (2011). Lipid rafts: linking Alzheimer’s amyloid-β production, aggregation, and toxicity at neuronal membranes. Int. J. Alzheimers Dis. 2011, 603052.
Sahores, M., Prinetti, A., Chiabrando, G., Blasi, F., and Sonnino, S. (2008). uPA binding increases UPAR localization to lipid rafts and modifies the receptor microdomain composition. Biochim. Biophys. Acta 1778, 250–259.
Sakono, M., and Zako, T. (2010). Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 277, 1348–1358.
Saijo, E., Scheff, S. W., and Telling, G. C. (2011). Unaltered prion protein expression in Alzheimer disease patients. Prion 5, 109–116.
Sandbrink, R., Masters, C. L., and Beyreuther, K. (1996). APP gene family. Alternative splicing generates functionally related isoforms. Ann. NY Acad. Sci. 777, 281–287.
Sano, M., Bell, K. L., Galasko, D., Galvin, J. E., Thomas, R. G., van Dyck, C. H., and Aisen, P. S. (2011). A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 77, 556–563.
Sato, K., Tanabe, C., Yonemura, Y., Watahiki, H., Zhao, Y., Yagishita, S., Ebina, M., Suo, S., Futai, E., Murata, M., and Ishiura, S. (2012). Localization of mature neprilysin in lipid rafts. J. Neurosci. Res. 90, 870–877.
Saunders, A. M., Strittmatter, W. J., Schmechel, D., George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., Rosi, B. L., Gusella, J. F., Crapper-MacLachlan, D. R., Alberts, M. J., et al. (1993). Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472.
Schettini, G., Govoni, S., Racchi, M., and Rodriguez, G. (2010). Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins: signal transduction and/or transcriptional role–relevance for Alzheimer pathology. J. Neurochem. 115, 1299–1308.
Schliebs, R. (2005). Basal forebrain cholinergic dysfunction in Alzheimer’s disease–interrelationship with β-amyloid, inflammation and neurotrophin signaling. Neurochem. Res. 30, 895–908.
Schliebs, R., and Arendt, T. (2006). The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural Transm. 113, 1625–1644.
Schneider, A., Rajendran, L., Honsho, M., Gralle, M., Donnert, G., Wouters, F., Hell, S. W., and Simons, M. (2008). Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J. Neurosci. 28, 2874–2882.
Shen, H., Kihara, T., Hongo, H., Wu, X., Kem, W. R., Shimohama, S., Akaike, A., Niidome, T., and Sugimoto, H. (2010). Neuroprotection by donepezil against glutamate excitotoxicity involves stimulation of α7 nicotinic receptors and internalization of NMDA receptors. Br. J. Pharmacol. 161, 127–139.
Shinohara, M., Sato, N., Kurinami, H., Takeuchi, D., Takeda, S., Shimamura, M., Yamashita, T., Uchiyama, Y., Rakugi, H., and Morishita, R. (2010). Reduction of brain Aβ by fluvastatin, an HMG-CoA reductase inhibitor, through increase in degradation of APP-CTFs and Aβ clearance. J. Biol. Chem. 285, 22091–22102.
Shogomori, H., and Kobayashi, T. (2008). Lysenin: a sphingomyelin specific pore-forming toxin. Biochim. Biophys. Acta 1780, 612–618.
Schulte, T., Paschke, K. A., Laessing, U., Lottspeich, F., and Stuermer, C. A. (1997). Reggie-1 and reggie-2, two cell surface proteins expressed by retinal ganglion cells during axon regeneration. Development 124, 577–587.
Shepardson, N. E., Shankar, G. M., and Selkoe, D. J. (2011a). Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch. Neurol. 68, 1239–1244.
Shepardson, N. E., Shankar, G. M., and Selkoe, D. J. (2011b). Cholesterol level and statin use in Alzheimer disease: II. Review of human trials and recommendations. Arch. Neurol. 68, 1385–1392.
Simons, K., and Gerl, M. J. (2010). Revitalizing membrane rafts: new tools and insights. Nat. Rev. Mol. Cell Biol. 11, 688–699.
Simons, M., Keller, P., De Strooper, B., Beyreuther, K., Dotti, C. G., and Simons, K. (1998). Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 95, 6460–6464.
Simons, M., Keller, P., Dichgans, J., and Schulz, J. B. (2001). Cholesterol and Alzheimer’s disease: is there a link? Neurology 57, 1089–1093.
Sisodia, S. S., and Gallagher, M. (1998). A role for the amyloid precursor protein in memory? Proc. Natl. Acad. Sci. U.S.A. 95, 12074–12076.
Smotrys, J. E., and Linder, M. E. (2004). Palmitoylation of intracellular signaling proteins: regulation and function. Annu. Rev. Biochem. 73, 559–587.
Solomon, A., and Kivipelto, M. (2009). Cholesterol-modifying strategies for Alzheimer’s disease. Expert Rev. Neurother. 9, 695–709.
Sonnino, S., Mauri, L., Chigorno, V., and Prinetti, A. (2007). Gangliosides as components of lipid membrane domains. Glycobiology 17, 1R–13R.
Soreghan, B., Thomas, S. N., and Yang, A. J. (2003). Aberrant sphingomyelin/ceramide metabolic-induced neuronal endosomal/lysosomal dysfunction: potential pathological consequences in age-related neurodegeneration. Adv. Drug Deliv. Rev. 55, 1515–1524.
Stuermer, C. A., and Plattner, H. (2005). The ’lipid raft’ microdomain proteins reggie-1 and reggie-2 (flotillins) are scaffolds for protein interaction and signalling. Biochem. Soc. Symp. 72, 109–118.
Svennerholm, L., Bråne, G., Karlsson, I., Lekman, A., Ramström, I., and Wikkelsö, C. (2002). Alzheimer disease - effect of continuous intracerebroventricular treatment with GM1 ganglioside and a systematic activation programme. Dement. Geriatr. Cogn. Disord. 14, 128–136.
Suzuki, T., and Suzuki, Y. (2006). Virus infection and lipid rafts. Biol. Pharm. Bull. 29, 1538–1541.
Suzuki, T., Zhang, J., Miyazawa, S., Liu, Q., Farzan, M. R., and Yao, W. D. (2011). Association of membrane rafts and postsynaptic density: proteomics, biochemical, and ultrastructural analyses. J. Neurochem. 119, 64–77.
Trovò, L., Van Veldhoven, P. P., Martín, M. G., and Dotti, C. G. (2011). Sphingomyelin upregulation in mature neurons contributes to TrkB activity by Rac1 endocytosis. J. Cell Sci. 124, 1308–1315.
Valsecchi, M., Mauri, L., Casellato, R., Prioni, S., Loberto, N., Prinetti, A., Chigorno, V., and Sonnino, S. (2007). Ceramide and sphingomyelin species of fibroblasts and neurons in culture. J. Lipid Res. 48, 417–424.
van Goor, H., Melenhorst, W. B., Turner, A. J., and Holgate, S. T. (2009). Adamalysins in biology and disease. J. Pathol. 219, 277–286.
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., Teplow, D. B., Ross, S., Amarante, P., Loeloff, R., Luo, Y., Fisher, S., Fuller, J., Edenson, S., Lile, J., Jarosinski, M. A., Biere, A. L., Curran, E., Burgess, T., Louis, J. C., Collins, F., Treanor, J., Rogers, G., and Citron, M. (1999). β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741.
Vetrivel, K. S., Barman, A., Chen, Y., Nguyen, P. D., Wagner, S. L., Prabhakar, R., and Thinakaran, G. (2011). Loss of cleavage at β’-site contributes to apparent increase in β-amyloid peptide (Aβ) secretion by β-secretase (BACE1)-glycosylphosphatidylinositol (GPI) processing of amyloid precursor protein. J. Biol. Chem. 286, 26166–26177.
Vetrivel, K. S., and Thinakaran, G. (2010). Membrane rafts in Alzheimer’s disease β-amyloid production. Biochim. Biophys. Acta 1801, 860–867.
Vieira, F. S., Correa, G., Einicker-Lamas, M., and Coutinho-Silva, R. (2010). Host-cell lipid rafts: a safe door for micro-organisms? Biol. Cell 102, 391–407.
Vincent, B., Paitel, E., Saftig, P., Frobert, Y., Hartmann, D., De Strooper, B., Grassi, J., Lopez-Perez, E., and Checler, F. (2001). The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J. Biol. Chem. 276, 37743–37746.
von Rotz, R. C., Kohli, B. M., Bosset, J., Meier, M., Suzuki, T., Nitsch, R. M., and Konietzko, U. (2004). The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell. Sci. 117, 4435–4448.
Wakabayashi, T., Craessaerts, K., Bammens, L., Bentahir, M., Borgions, F., Herdewijn, P., Staes, A., Timmerman, E., Vandekerckhove, J., Rubinstein, E., Boucheix, C., Gevaert, K., and De Strooper, B. (2009). Analysis of the γ-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat. Cell Biol. 11, 1340–1346.
Walkley, S. U. (2003). Neurobiology and cellular pathogenesis of glycolipid storage diseases. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 358, 893–904.
Walsh, D. M., and Selkoe, D. J. (2007). Aβ oligomers – a decade of discovery. J. Neurochem. 101, 1172–1184.
Wang, H. Y. (2010). S 24795 Limits β-amyloid–α7 nicotinic receptor interaction and reduces Alzheimer’s disease-like pathologies. Biol. Psychiatry 67, 522–530.
Wang, Q., Yan, J., Chen, X., Li, J., Yang, Y., Weng, J., Deng, C., and Yenari, M. A. (2010). Statins: multiple neuroprotective mechanisms in neurodegenerative diseases. Exp. Neurol. 230, 27–34.
Wheeler, D., Knapp, E., Bandaru, V. V., Wang, Y., Knorr, D., Poirier, C., Mattson, M. P., Geiger, J. D., and Haughey, N. J. (2009). Tumor necrosis factor-α-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J. Neurochem. 109, 1237–1249.
Willmann, R., Pun, S., Stallmach, L., Sadasivam, G., Santos, A. F., Caroni, P., and Fuhrer, C. (2006). Cholesterol and lipid microdomains stabilize the postsynapse at the neuromuscular junction. EMBO J. 25, 4050–4060.
Wood, W. G., Eckert, G. P., Igbavboa, U., and Muller, W. E. (2010). Statins and neuroprotection: a prescription to move the field forward. Ann. N. Y. Acad. Sci. 1199, 69–76.
Wu, G., Lu, Z. H., Kulkarni, N., Amin, R., and Ledeen, R. W. (2011) Mice lacking major brain gangliosides develop parkinsonism. Neurochem. Res. 36, 1706–1714.
Xie, H. Q., Leung, K. W., Chen, V. P., Chan, G. K., Xu, S. L., Guo, A. J., Zhu, K. Y., Zheng, K. Y., Bi, C. W., Zhan, J. Y., Chan, W. K., Choi, R. C., and Tsim, K. W. (2010a). PRiMA directs a restricted localization of tetrameric AChE at synapses. Chem. Biol. Interact. 187, 78–83.
Xie, H. Q., Liang, D., Leung, K. W., Chen, V. P., Zhu, K. Y., Chan, W. K., Choi, R. C., Massoulie, J., and Tsim, K. W. (2010b). Targeting acetylcholinesterase (AChE) to membrane rafts: a function mediated by the proline rich membrane anchor (PRiMA) in neurons. J. Biol. Chem. 285, 11537–11546.
Yamamoto, N., Igbabvoa, U., Shimada, Y., Ohno-Iwashita, Y., Kobayashi, M., Wood, W. G., Fujita, S. C., and Yanagisawa, K. (2004). Accelerated A (aggregation in the presence of GM1-ganglioside-accumulated synaptosomes of aged apoE4-knock-in mouse brain. FEBS Lett. 569, 135–139.
Yanez-Mo, M., Gutierrez-Lopez, M. D., and Cabanas, C. (2011). Functional interplay between tetraspanins and proteases. Cell. Mol. Life Sci. 68, 3323–3335.
Yao, Z. X., and Papadopoulos, V. (2002). Function of β-amyloid in cholesterol transport: a lead to neurotoxicity. FASEB J. 16, 1677–1679.
Yoon, I. S., Chen, E., Busse, T., Repetto, E., Lakshmana, M. K., Koo, E. H., and Kang, D. E. (2007). Low-density lipoprotein receptor-related protein promotes amyloid precursor protein trafficking to lipid rafts in the endocytic pathway. FASEB J. 21, 2742–2752.
Yu, G., Nishimura, M., Arawaka, S., Levitan, D., Zhang, L., Tandon, A., Song, Y. Q., Rogaeva, E., Chen, F., Kawarai, T., Supala, A., Levesque, L., Yu, H., Yang, D. S., Holmes, E., Milman, P., Liang, Y., Zhang, D. M., Xu, D. H., Sato, C., Rogaev, E., Smith, M., Janus, C., Zhang, Y., Aebersold, R., Farrer, L. S., Sorbi, S., Bruni, A., Fraser, P., and St George-Hyslop, P. (2000). Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 407, 48–54.
You, H., Tsutsui, S., Hameed, S., Kannanayakal, T. J., Chen, L., Xia, P., Engbers, J. D., Lipton, S. A., Stys, P. K., and Zamponi, G. W. (2012). Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. U. S. A. 109, 1737–1742.
Zha, Q., Ruan, Y., Hartmann, T., Beyreuther, K., and Zhang, D. (2004). GM1 ganglioside regulates the proteolysis of amyloid precursor protein. Mol. Psychiatry 9, 946–952.
Zhang, H., Ma, Q., Zhang, Y. W., and Xu, H. (2012). Proteolytic processing of Alzheimer’s β-amyloid precursor protein. J. Neurochem. 120(Suppl. 1), 9–21.
Keywords: AChE, Alzheimer’s disease, amyloid β peptide, APP, AICD, BACE-1, cholesterol, neprilysin
Citation: Hicks DA, Nalivaeva NN and Turner AJ (2012) Lipid rafts and Alzheimer’s disease: protein-lipid interactions and perturbation of signaling. Front. Physio. 3:189. doi: 10.3389/fphys.2012.00189
Received: 29 March 2012; Paper pending published: 13 April 2012;
Accepted: 21 May 2012; Published online: 22 June 2012.
Edited by:
Alessandro Prinetti, University of Milano, ItalyReviewed by:
Marco Parenti, University of Milano-Bicocca, ItalyAmitabha Chattopadhyay, Centre for Cellular and Molecular Biology, India
Copyright: © 2012 Hicks, Nalivaeva and Turner. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Anthony J. Turner, School of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, Leeds LS2 9JT, UK e-mail:YS5qLnR1cm5lckBsZWVkcy5hYy51aw==