

 Qing Lu1
Qing Lu1
- 1 Vascular Biology Center, Georgia Health Sciences University, Augusta, GA, USA
- 2 Department of Pharmacology and Toxicology, Georgia Health Sciences University, Augusta, GA, USA
Acute lung injury (ALI) is a life threatening condition associated with hypoxemia, diffuse alveolar damage, inflammation, and loss of lung function. Lipopolysaccharide (LPS; endotoxin) from the outer membrane of Gram-negative bacteria is a major virulence factor involved in the development of ALI. The depletion of glutathione (GSH), an essential intra- and extra-cellular protective antioxidant, by LPS is an important event that contributes to the elevation in reactive oxygen species. Whether restoring GSH homeostasis can effectively ameliorate mitochondrial dysfunction and cellular apoptosis in ALI is unknown and therefore, was the focus of this study. In peripheral lung tissue of LPS-treated mice, hydrogen peroxide and protein nitration levels were significantly increased. Pre-treatment with GSH-ethyl ester (GSH-EE) prevented this increase in oxidative stress. LPS also increased the lactate/pyruvate ratio, attenuated SOD2 protein levels, and decreased ATP levels in the mouse lung indicative of mitochondrial dysfunction. Again, GSH-EE treatment preserved the mitochondrial function. Finally, our studies showed that LPS induced an increase in the mitochondrial translocation of Bax, caspase 3 activation, and nuclear DNA fragmentation and these parameters were all prevented with GSH-EE. Thus, this study suggests that GSH-EE supplementation may reduce the mitochondrial dysfunction associated with ALI.
Introduction
Acute lung injury (ALI) is an acute inflammatory disorder associated with both high morbidity and mortality in afflicted patients even under intensive care. ALI is characterized by non-cardiogenic dyspnea, hypoxemia, pulmonary neutrophil sequestration, and low lung compliance (Martinez et al., 2009). ALI can occur in response to direct insults, such as viral or bacterial infections of the lung, hyperoxia, and acid aspiration, or indirect insults like sepsis, multiple transfusions, and pancreatitis. Lipopolysaccharide (LPS) from the outer cell wall of Gram-negative bacteria that cause sepsis is the most common indirect pulmonary insult leading to ALI (Erickson et al., 2009). LPS provokes damage to the alveolar-capillary membrane and the adhesion, activation, and sequestration of polymorphonuclear neutrophils (PMN), which result in the deterioration of gas exchange (Nagase et al., 2003). Although several therapies, such as corticosteroids, prostacyclins, exogenous surfactants, ketoconazole, and nitric oxide, have shown promising outcomes, the estimated mortality rate in ALI is 38.5% (Rubenfeld et al., 2005). Therefore, there is a further need to study the pathophysiology of ALI and identify therapeutic targets to improve patient outcome.
Glutathione (GSH), a tripeptide thiol, is the most potent antioxidant found in the cell and plays a protective role against reactive oxygen and nitrogen species (ROS and RNS) mediated injury and lung inflammation (Li et al., 1994; van Klaveren et al., 1997). This antioxidant is also involved in the regulation of apoptosis, cell proliferation, and gene transcription (Rahman and MacNee, 2000b; Luppi et al., 2005). GSH also modifies protein sulfdryl groups by a number of reactions: reduction of protein sulfenic acids, formation of protein mixed disulfides, and their subsequent reduction. The conjugation of GSH with electrophilic compounds mediated by glutathione S-transferases (GSTs) and the subsequent excretion of these conjugates from the cell also serve to protect against toxins, such as LPS. GSH and GSH-associated enzymes present in the lower respiratory tract act as the first line of defense against external agents (DeLeve and Kaplowitz, 1990; Pacht et al., 1991). Alterations in alveolar and lung GSH metabolism have been shown to be a central feature of many inflammatory lung diseases, such as acute respiratory distress syndrome (ARDS), cystic fibrosis, and asthma (Rahman and MacNee, 2000a). GSH levels are significantly reduced in bronchoalveolar lavage fluid (BALF) from patients with ALI (Pacht et al., 1991). Further, it has been reported that disruptions in thiol status are associated with ALI (Quinlan et al., 1994, 1997).
Apoptosis, or programmed cell death, is an essential physiological phenomenon responsible for the selective elimination of cells. However, the dysregulation of apoptotic signaling pathways is thought to play an important role in the development of ALI (Z’Graggen et al., 2010). Several studies have shown compelling evidence that increased epithelial/endothelial cell apoptosis significantly contributes to the damage to the pulmonary alveolar-capillary interface in ALI (Kitamura et al., 2001). The inhibition of apoptosis increases survival in animal models of LPS-induced ALI (Kawasaki et al., 2000; Ma et al., 2010). Previously, we have shown that key mechanisms of zinc-mediated apoptotic induction in pulmonary endothelial cells include the disruption of cellular glutathione homeostasis and decreased mitochondrial ATP synthesis (Wiseman et al., 2010). We found that the acute increases in both oxidative and nitrosative stress (similar to that occurring in ALI) in endothelial cells led to a significant increase in cellular apoptosis, which was attenuated by pre-treatment with GSH-ethyl ester (GSH-EE) (Wiseman et al., 2010). Therefore, the focus of this study was to determine whether GSH supplementation can prevent LPS-mediated ROS/RNS generation, mitochondrial dysfunction, and cellular apoptosis in mouse model of LPS-induced ALI.
Materials and Methods
Animal Treatments
Adult male C57BL/6NHsd mice (7–8 weeks; Harlan, Indianapolis, IN, USA) were used in all experiments and housed in a temperature-controlled animal facility with 12-h light–dark cycles. All animal care and experimental procedures were approved by the Committee on Animal Use in Research and Education of the Georgia Health Sciences University (Augusta, GA, USA). Mice were divided into four experimental groups (n = 6/group); Vehicle, LPS, GSH-EE + LPS, and GSH-EE alone, respectively. Mice were injected intraperitoneally either with saline (vehicle) or LPS (6.75 × 104 EU/g body wt), as previously published (Chatterjee et al., 2008). The group of mice receiving both GSH-EE and LPS were injected with two separate doses of 2 mmol GSH-EE/kg body wt (Sigma, St. Louis, MO, USA; Singhal and Jain, 2000). Dose 1 was given 2 h prior to LPS and dose 2 was given along with LPS injection. The pH of the GSH-EE solution was adjusted to 6.8 by the addition of 2 M NaOH immediately before injection. Mice belonging to GSH-EE alone group received similar GSH-EE doses. LPS only and GSH-EE + LPS groups of mice were euthanized 12 h after LPS injection, and the lungs were flushed with ice-cold EDTA-PBS, excised, snap-frozen in liquid nitrogen, and stored at −80°C until used.
Lung Tissue Homogenates
Lung protein extracts were prepared by homogenizing mouse lung tissues in Triton lysis buffer (50 mM Tris–HCL, pH 7.6, 0.5% Triton X-100, 20% glycerol) containing a protease inhibitor cocktail (Sigma). Extracts were then clarified by centrifugation (14,000 rpm for 10 min at 4°C). Supernatant fractions were then assayed for protein concentration using the Bradford reagent (Bio-Rad, Richmond, CA, USA). Mitochondria were isolated from all four groups of mice lungs using the Mitochondria Isolation Kit (Thermo Fisher Scientific Inc., Rockford, IL, USA) and analyzed by western blot to detect the Bax protein levels.
Measurement of GSH Levels
Tissue samples were homogenized in 5% sulfosalicylic acid (SSA) and centrifuged at 12,000 × g for 15 min. Hundred microliters of supernatant was brought to neutral pH by using triethanolamine and split into two tubes, one for the total GSH/GSSG and another to measure the GSSG levels alone (Griffith, 1980). Two microliters of 2-vinylpyridine (2-VP) was added to the sample tube measuring GSSG levels alone and placed on a rocker for an hour. 2-VP masks GSH and prevents its reaction with 5,5′-dithio-bis(2-nitrobenzoic acid; DTNB), thereby measuring only oxidized GSSG levels. Five microliters of 50 U/mL glutathione reductase was added to both the tubes to convert GSSG to GSH. The reaction was then initiated in both tubes by adding, 50 μL of 6 mM DTNB and 350 μL of 0.3 mM NADPH. All solutions were made with 125 mM NaHPO4 buffer (pH 7.5) containing 6.3 mM EDTA. The increase in the absorbance was measured at 412 nm. Reduced GSH levels were extrapolated from the GSH/GSSG ratio and the oxidized GSSG levels and then normalized to tissue wet weight.
In situ Detection of H2O2 Levels
The Amplex Red Reagent (Molecular Probes) was used to detect H2O2 levels in fresh lung tissue samples obtained from mice treated with LPS in the presence or absence of GSH-EE supplementation. Briefly, an equal amount (∼10 mg/sample) of lung tissue was incubated at 37°C for 30 min in master mix solution containing Amplex Red reagent, horseradish peroxidase, and a buffer solution. Supernatant was then collected, fluorescence was read at excitation/emission 530/590 nm, and concentrations of H2O2 were determined through extrapolation from a standard curve. Protein in the tissue samples was estimated by BCA assay and used to normalize the detected H2O2 levels.
Measurement of Protein Nitration
The total nitrated protein levels were measured in the lung homogenates of mice treated with LPS in the presence or absence of GSH-EE via a dot blot procedure. Briefly, 30 μg protein was applied to a nitrocellulose membrane pre-soaked with Tris-buffered saline (TBS). After the protein samples were completely transferred, the membrane was blocked in 5% fat-free milk for 1 h, washed with TBS, and incubated with mouse anti-3-nitrotyrosine (1:100, Calbiochem) antibody overnight. Finally, the membrane was incubated with goat anti-mouse IgG for 2 h. The reactive dots were visualized using chemiluminescence (Pierce Laboratories) on a Kodak 440CF image station (New Haven, CT, USA). The band intensity was quantified using Kodak 1D image processing software. The protein expression was normalized by re-probing with mouse anti β-actin antibody.
Western Blot Analyses
The lung tissue sample homogenates containing 25–50 μg proteins were separated on 4–20% denaturing polyacrylamide gels and transferred to Immunoblot-PVDF membranes (Bio-Rad Lab, Hercules, CA, USA). The membranes were blocked with 5% non-fat dry milk in TBS containing 0.1% Tween (TBST). After blocking, each membrane was incubated overnight at 4°C with either SOD1 (1:500, custom-made), SOD2 (1:1000, LS Biochemicals), Bax (1:1000, Cell Signaling, Danvers, MA, USA), cleaved caspase 3 (1:1000, Cell Signaling, Danvers, MA, USA), cleaved caspase 7 (1:1000, Cell Signaling, Danvers, MA, USA), Voltage-dependent anion channels (VDAC; 1:500, Cell Signaling, Danvers, MA, USA), or mouse β-actin (1:5000, Sigma), washed with TBST, and then incubated with a IgG-horseradish peroxidase. The reactive bands were visualized using chemiluminescence (SuperSignal West Femto substrate kit; Pierce) on a Kodak 440CF image station. The band intensity was quantified using Kodak 1D image processing software. To normalize for equal protein loading, the blots were re-probed with either β-actin or VDAC.
Determination of Lactate and Pyruvate Levels
The lung tissues were homogenized in ice-cold 0.5 M perchloric acid and centrifuged at 14,000 rpm for 20 min. The collected supernatants were then neutralized with 3 M KHCO3 and used for the lactate and pyruvate assays. The relative changes in lactate levels were measured using a lactate assay kit (BioVision). The pyruvate levels were determined using the spectrophotometric enzymatic measurement assay at 340 nm, as previously published (Sharma et al., 2008). NADH was used as a cofactor and lactate dehydrogenase as the coenzyme.
ATP Determination
The ATP levels were quantified with a commercially available kit (Invitrogen) based on the firefly luciferin-luciferase reaction. In this reaction, ATP is consumed and light is emitted when firefly luciferase catalyzes the oxidation of luciferin. The amount of light emitted during the reaction is proportional to the availability of ATP. The luminescence was determined using a Fluoroscan Ascent plate luminometer (Thermo Electron, Corp.), and the ATP levels reported in nanomoles per milligram protein.
Measurement of Caspase 3/7 Activity
Caspase 3/7 activity was detected with the caspase-Glo 3/7 assay kit (Promega Corporation, Australia). Briefly, 100 μL caspase 3/7 reagents were added to each well with 50 μL peripheral lung homogenates containing equal amount of protein and incubated for 1 h on rotary shaker at room temperature. The luminescence intensity was measured using a Multiskan Microplate reader according to the manufacturer’s instructions.
Determination of Lung Apoptosis
The lung tissues were washed in PBS, fixed in 4% paraformaldehyde (RT, 1 h), then placed in 30% sucrose (RT, 1 h), embedded in O.C.T embedding medium (Tissue-Tek, Sakura Finetechnical, Tokyo, Japan), and stored at −80°C overnight. The embedded tissue blocks were sectioned to 5 μm slices and were analyzed for the presence of apoptotic nuclei using the DeadEnd Fluorometric TUNEL System (Promega, Madison, WI, USA). The DNA breaks were labeled with fluorescein-12-dUTP (green), and the nuclei were stained with propidium iodide (red). The vascular endothelial cell nuclei were labeled with anti-Von Willebrand Factor (1:500; Abcam, Cambridge, MA, USA), and Alexa Fluor 350 (blue) was used as the secondary antibody (Invitrogen, Carlsbad, CA, USA). The sections were examined under a fluorescence microscope (Olympus, Japan). All images were captured by Image-Pro Plus ver 5.0 (Media Cybernetics, Silver Spring, MD, USA). The quantification of the TUNEL positive endothelia nuclei and total endothelia nuclei was processed by Image-Pro software and presented as a percentage.
Statistical Analysis
The experimental results are expressed as mean ± standard error of the mean (SEM) for n = 5–6 for each group. The statistical analysis was done using GraphPad Prism version 4.01 for Windows (GraphPad Software, San Diego, CA, USA) using one-way analysis of variance (ANOVA) and a Newman–Keuls multiple comparison post hoc test. A value of P < 0.05 was considered significant.
Results
GSH-EE Supplementation Attenuates LPS-induced Oxidative and Nitrosative Stress in the Mouse Lungs
To determine the effect of LPS exposure on oxidant and antioxidant homeostasis, we first measured the glutathione levels in the peripheral lung tissue of mice treated with or without LPS. There was a significant (approximately threefold) reduction in GSH levels in LPS-treated mouse lung when compared to controls (Figure 1A) that was preserved with GSH-EE supplementation. Earlier studies have provided evidence indicating that ROS and RNS, particularly H2O2 and ONOO−, contributes to the pathogenesis of ALI (Lang et al., 2002; Tasaka et al., 2008). Therefore, we determined whether GSH-EE pre-treatment attenuated LPS-induced oxidative and nitrosative stress in the mouse lung mice. We found elevated H2O2 levels (Figure 1B) and increased protein nitration (Figure 1C). GSH-EE pre-treatment preserved the LPS-induced H2O2, and nitrated protein levels in the mouse lung (Figure 1).
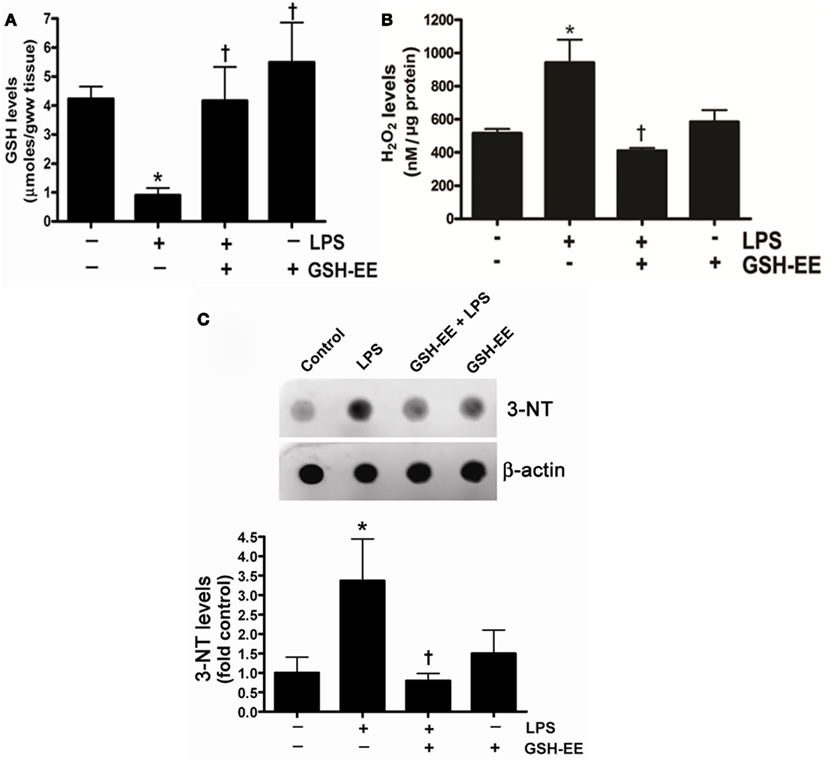
Figure 1. GSH-EE supplementation improves LPS-mediated decrease in GSH levels and attenuates LPS-induced oxidative and nitrosative stress. Lungs from vehicle, LPS, GSH-EE + LPS, and GSH-EE treated mice were used for the analysis of GSH, H2O2, and nitrated proteins levels. LPS significantly decreased GSH levels but GSH-EE pre-treatment prevented the loss of GSH in these animals (A). The Amplex Red assay measurement indicated a twofold increase in H2O2 levels in LPS-treated mice lungs as compared to controls (B). Total nitrated protein levels were measured by dot blot analysis (C). LPS caused significant increases in the formation of H2O2 and nitrated proteins, whereas GSH-EE pre-treatment prevented the LPS-induced increase in these oxidative and nitrosative stress parameters. Values are mean ± SEM; n = 6/group. *P < 0.05 vs. vehicle; †P < 0.05 vs. LPS alone.
GSH-EE Supplementation Preserves Mitochondrial Function
The loss of mitochondrial function upon exposure to LPS has been implicated in the development of ALI (Kuwabara and Imajoh-Ohmi, 2004). Superoxide dismutase-1 and 2 (SOD1 and SOD2) are the two important antioxidant enzymes actively involved in quenching harmful superoxide radicals. The lungs of LPS-treated mice had a approximately twofold decrease in SOD1 and SOD2 protein levels (Figures 2A,B), while pre-treatment with GSH-EE preserved both SOD1 and SOD2 protein levels. Under physiological conditions where mitochondrial function is optimal, pyruvate levels are higher than lactate, and thus any increase in lactate levels will decrease the pyruvate-to-lactate ratio and can be extrapolated to suggest a switch in mitochondrial ATP generation from gluconeogenesis to glycolysis (Shinde et al., 2005). The LPS-exposed mice had a significantly higher lactate/pyruvate ratio, thus indicating a disruption in lung mitochondrial ATP generation from gluconeogenesis (Figure 2C) and this was confirmed by an overall decrease in ATP levels in the LPS-treated mouse lung (Figure 2D). Pre-treatment with GSH-EE blocked the increase in the lactate/pyruvate ratio (Figure 2C) and preserved ATP generation (Figure 2D).
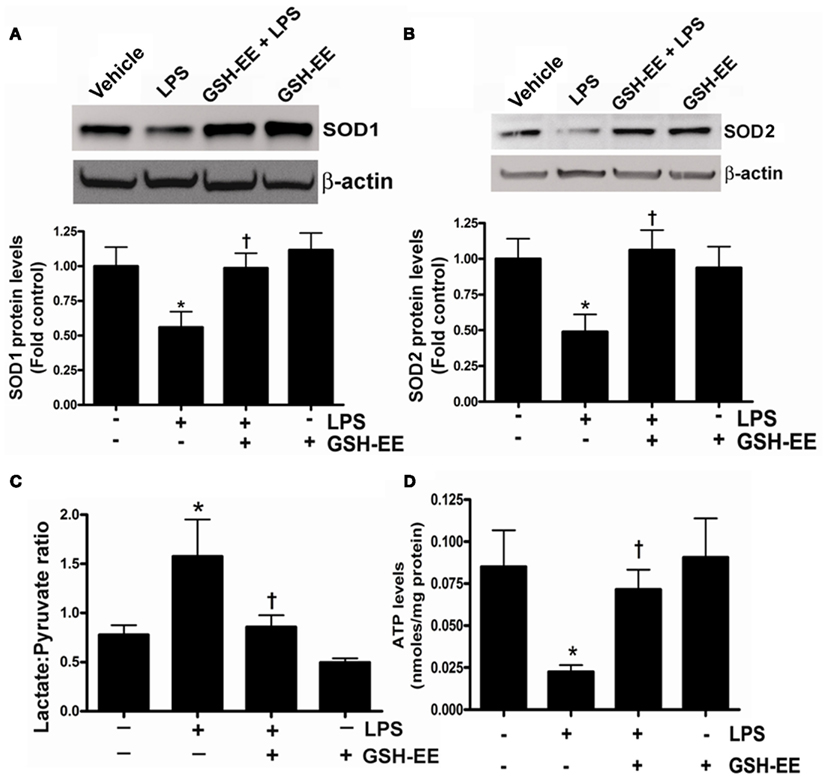
Figure 2. GSH-EE supplementation prevents LPS-mediated mitochondrial dysfunction. Protein extracts prepared from peripheral lungs of vehicle, LPS, GSH-EE + LPS, and GSH-EE treated mice were analyzed by Western blot analysis using a specific antiserum raised against SOD1 (A) or SOD2 (B). Protein levels were normalized for loading using β-actin. A representative blot and normalized densitometric values are shown. There was a significant decrease in SOD1 and SOD2 protein levels in LPS-treated mice and this was prevented by GHS-EE pre-treatment. The lactate/pyruvate ratio was also determined in all four groups (C). LPS-exposed mice had a significantly higher lactate/pyruvate ratio (C). Pre-treatment with GSH-EE preserved the lactate/pyruvate ratio. There was a significant reduction in lung ATP levels after LPS exposure (D). However, the LPS-mediated decrease in ATP levels was not observed in mice pre-treated with GSH-EE (D). Values are mean ± SEM; n = 6/group. *P < 0.05 vs. vehicle; †P < 0.05 vs. LPS.
GSH-EE Supplementation Prevents the LPS-induced Mitochondrial Translocation of Bax and Caspase 3/7 Activation
Bax, a member of the Bcl-2 family of proteins, promotes apoptosis. The translocation of Bax from the cytoplasm to the outer mitochondrial membrane stimulates apoptosis (Sarkar et al., 2003). We found higher Bax protein levels in the mitochondria isolated from the LPS-treated mice compared to the control mice (Figure 3A). GSH-EE prevented the LPS-induced mitochondrial translocation of Bax (Figure 3A). Activated caspases 3 and 7 are the executors of apoptotic cell death. Western blot analysis demonstrated a twofold increase in activated caspase 3 (Figure 3B), and a threefold increase in activated caspase 7 (Figure 3C), protein levels after LPS exposure. However, pre-treatment with GSH-EE significantly reduced only activated caspase 3, and not activated caspase 7, levels (Figures 3B,C). Furthermore, using a luminescent assay, we found a fivefold increase in caspase 3/7 activity in LPS-treated mice, as compared to the controls (Figure 3D). GSH-EE pre-treatment partially decreased caspase 3/7 activity (Figure 3D). These results suggest that GSH-EE pre-treatment attenuated the LPS-mediated increase in caspase 3/7 activity primarily by decreasing active caspase 3 levels.
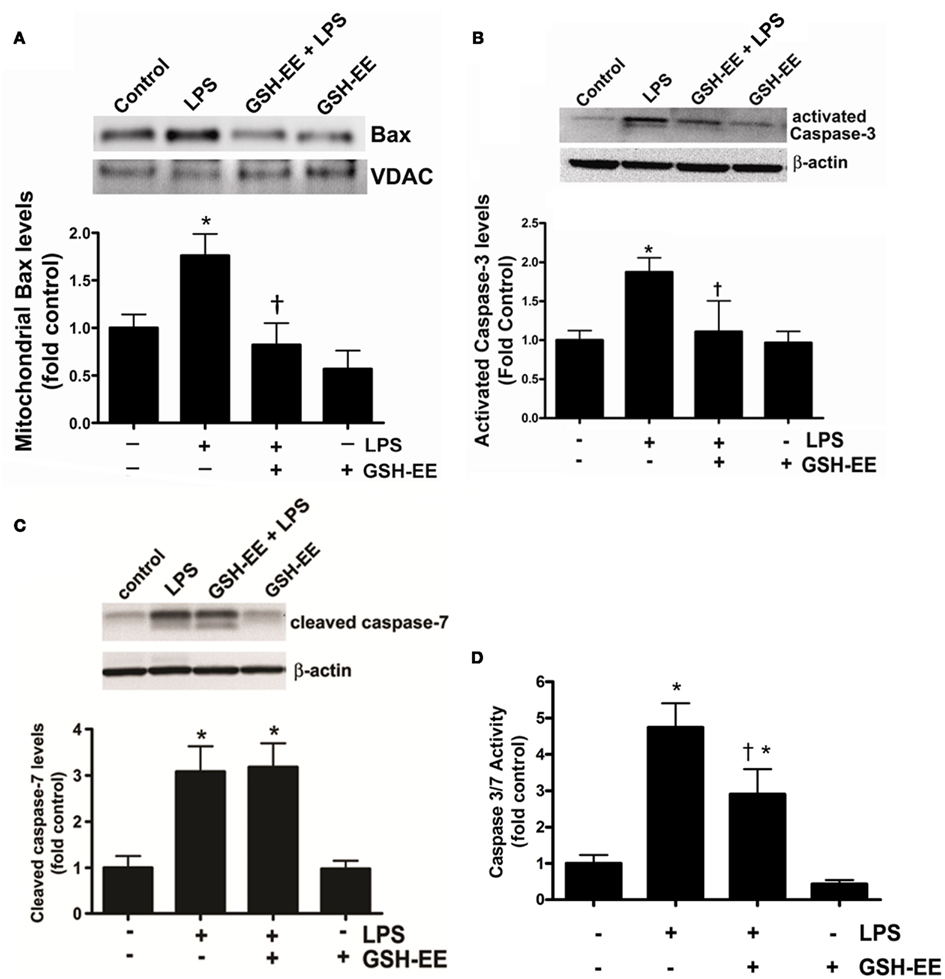
Figure 3. GSH supplementation prevents the LPS-mediated translocation of Bax and caspase 3 activation. Bax protein levels were measured in the mitochondria isolated from lung homogenates of vehicle, LPS, LPS + GSH-EE, and GSH-EE treated mice using Western blot analysis (A). Mitochondrial Bax protein levels from LPS-treated mouse lungs were significantly higher and GSH-EE prevented this translocation (A). Activated caspase 3 and 7 protein levels and activity were also determined in lung homogenates from all four groups. LPS-treated mice had significantly increased activated caspase 3 protein levels that was reduced by GSH-EE pre-treatment (B). The activated caspase 7 protein levels were also upregulated in LPS-treated mice, but did not change after GSH-EE pre-treatment (C). The increase in activated caspase 3 protein levels correlated with an increase in caspase 3/7 activity in lungs of mice exposed to LPS, which was significantly decreased in presence of GSH-EE (D). Values are mean ± SEM; n = 3–6/group. *P < 0.05 vs. vehicle; †P < 0.05 vs. LPS.
GSH-EE Supplementation Protects Against LPS-Induced Apoptosis
TUNEL staining was carried out to determine the effect of GSH-EE pre-treatment on apoptosis in the LPS-exposed mouse lungs. Sections were co-stained with TUNEL, a nuclei marker (propidium iodide), and endothelial cell marker (VWF; Figure 4A). The quantitation of TUNEL positive cells indicated increased endothelial cell apoptosis in LPS-treated mice lungs, which was attenuated with GSH-EE administration (Figure 4B). In animals receiving vehicle or GSH-EE alone, no apoptotic nuclei were observed (Figure 4).
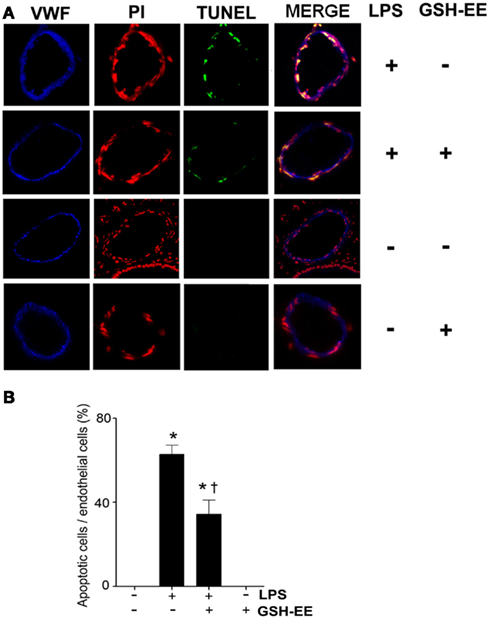
Figure 4. GSH supplementation inhibits the LPS-induced endothelial cell apoptosis in the mouse lung. Lung tissue sections from vehicle, LPS, LPS + GSH-EE, and GSH-EE treated mice were analyzed for the presence of apoptotic nuclei using the DeadEnd Fluorometric TUNEL System. The DNA breaks were labeled with fluorescein-12-dUTP (TUNEL; green), and the nuclei were stained using propidium iodide (PI; red). Vascular endothelial cells were labeled with anti-Von Willebrand Factor (VWF; blue) (A). The quantification of the TUNEL positive endothelial nuclei and total endothelial nuclei was processed by Image-Pro software and presented as a percentage. LPS treatment caused a significant increase in endothelial apoptosis and this was attenuated by GSH-EE pre-treatment (B). Values are mean ± SEM; n = 4/group. *P < 0.05 vs. vehicle; †P < 0.05 vs. LPS.
Discussion
The data presented in this study demonstrate that GSH-EE supplementation can protect the mouse lung against LPS-induced mitochondrial dysfunction and subsequent endothelial cell apoptosis. The prior administration of GSH-EE: preserved LPS-mediated attenuation of GSH levels; reduced the LPS-induced oxidative and nitrosative stress; mitigated the LPS-mediated disruption of mitochondrial ATP generation; prevented the LPS-induced loss of the mitochondrial antioxidant enzyme SOD2; decreased the LPS-mediated mitochondrial translocation of Bax, caspase 3 activation; and prevented endothelial apoptosis.
GSH has been shown to protect cells against toxins, ROS, and RNS (Messana et al., 1988). The depletion of GSH is an important pathological event in the development of many neurological, hepatic, and pulmonary diseases (Lee et al., 2004; Vargas et al., 2011). Although the cause of GSH depletion is not clear, traditionally, it is thought that the decrease in GSH levels is a direct consequence of increased production of ROS and RNS. For example, a common pathological hallmark in various neurodegenerative disorders, such as amyotrophic lateral sclerosis, Alzheimer’s, and Parkinson’s diseases, is the increase in oxidative stress and a subsequent decrease in the GSH content. The intraperitoneal administration of the GSH precursor, N-acetylcysteine (NAC), to rats has been shown to replenish GSH levels and provide protection against oxidative damage to brain proteins in these neurodegenerative disorders (Pocernich et al., 2001). In addition, drugs, infections, and inflammation in the liver increase the generation of ROS and RNS with concomitant decreases in GSH levels. Liver injury induced by acetaminophen also involves the severe depletion of both cytosolic and mitochondrial GSH pools, increased ROS generation, and cell death. NAC has been proven to be effective in treating acetaminophen and non-acetaminophen induced liver injury that, conversely, is potentiated by GSH depletors (Jaeschke and Bajt, 2006; Lee et al., 2009). In human studies, high-dose of NAC significantly improved the antioxidant profile of the lungs by elevating GSH levels and elicited an antioxidant effect at the alveolar surface in patients with fibrosing alveolitis. These biochemical changes were accompanied by an improvement in the pulmonary function tests of these patients (Behr et al., 1997). In placebo-controlled trials, orally administered NAC (≥600 mg/day) has been shown to significantly improve the response to steroids, increase the general well-being, and decrease exacerbation rates, emergency room visits, days of illness, costs of hospitalization, and work time loss in patients with chronic lung diseases (Grandjean et al., 2000; Kasielski and Nowak, 2001; Poole and Black, 2001).
In recent years, several studies have delineated the role of GSH depletion and subsequent increase in oxidative stress and cytokine production in the development of ALI. Although, the role of GSH in the LPS-induced pulmonary inflammation may be cell specific. For example, in murine macrophage-like cells (RAW 264.7), LPS elevated GSH levels but when these LPS-treated cells were supplemented with GSH precursor, NAC, there was attenuation in the LPS-dependent inflammation. However, the co-administration of NAC with the anti-inflammatory agent, ethyl pyruvate, partially reversed the anti-inflammatory effects of ethyl pyruvate suggesting a dual role of GSH in these murine macrophage-like cells (Song et al., 2004). In alveolar type II epithelial cells of rat lungs, LPS decreased GSH, which resulted in an increase in TNF-α and nuclear factor (NF)-kappa B levels (Zhang et al., 2008). Furthermore, the inhibition of GSH biosynthesis by buthionine sulfoximine (BSO) increased intracellular oxidative stress and enhanced the production of IL-1β, IL-6, and TNF-α in the alveolar epithelium (Haddad et al., 2001). In polymicrobial sepsis induced by cecal ligation and puncture, exogenous GSH protected mice from the lethal quantities of ROS and RNS produced by neutrophils. In these mice, GSH stimulated the migration of neutrophils to the site of infection but prevented their infiltration to distant sites, thereby promoting the bactericidal, while limiting the cytolytic, properties of neutrophils (Villa et al., 2002). Interestingly, tempol, a membrane permeable free radical scavenger, ameliorated LPS-induced GSH depletion in a mouse model of ALI (El-Sayed et al., 2011), suggesting that oxidants potentiate the cellular reduction of GSH levels. In support of these studies, our present data demonstrated that LPS exposure significantly diminished GSH levels in the mouse lung, which correlated with an increase in pulmonary oxidative and nitrosative stress. Exogenous supplementation with GSH-EE in the presence of LPS restored the GSH levels and subsequently attenuated the LPS-induced elevation of both H2O2 and total protein nitration in the mouse lung. We also found that LPS-challenge led to the attenuation of SOD1 and SOD2 protein levels, whereas this decrease was not observed in GSH-EE pre-treated mice. Although it is unclear how LPS treatment led to the down-regulation of these enzymes in the mouse lung, based upon previous studies, we can speculate that the activation of protein kinase B (PKB)/Akt by LPS and also by H2O2 leads to the down-regulation of SOD levels (Wang et al., 2000; Monick et al., 2001). Akt has been shown to phosphorylate and repress the DNA binding activity of the transcription factor, FOXO3a (Forkhead box class O), and consequently decrease the gene expression of SOD2 (Li et al., 2006).
Intracellular redox status regulates various aspects of cellular function. Mitochondrial function is particularly susceptible to oxidative damage. For example, unopposed oxidative stress selectively inhibits mitochondrial respiratory-chain enzymes, thereby decreasing ATP synthesis (Drahota et al., 2005). Studies in Parkinson’s and Alzheimer’s diseases have shown that if ONOO− is not scavenged, e.g., by antioxidants, such as reduced GSH, it leads to irreversible damage to critical cellular enzymes of the mitochondrial electron transport chain, alpha ketoglutarate dehydrogenase, and pyruvate dehydrogenase (Foxton et al., 2007). Biochemically, LPS increases the NADH:NAD ratio, thereby shifting the equilibrium of the lactate dehydrogenase reaction toward lactate and causing profound lactic acidosis. The resultant increase in the lactate/pyruvate ratio is characteristic of attenuated oxidative phosphorylation in the mitochondria. Interestingly, our data demonstrate that mice supplemented with GSH-EE prior to LPS exposure are protected against the LPS-dependent increase in the lactate/pyruvate ratio and maintain normal ATP levels. These results compliment other studies that have shown that GSH protects the mitochondria against oxidative insult under various pathologic conditions such as reperfusion injury in the liver, ethanol-induced toxicity, and diabetes (Sommer et al., 2012).
Mitochondrial regulation of apoptosis is central to cell survival. A recent study demonstrated that LPS-mediated cellular apoptosis is dependent upon mitochondrial dysfunction in human monoblastic U937 cells (Kuwabara and Imajoh-Ohmi, 2004). Mitochondria regulate apoptosis by maintaining a delicate balance between pro- and anti-apoptotic Bcl-2 proteins. The pro-apoptotic member of the Bcl-2 protein family, Bax, is normally found in the cytosol where it acts as a sensor for cellular stress. LPS-induced oxidative and nitrosative stress activates Bax, which then translocates to the outer mitochondrial membrane where it interacts with anti-apoptotic proteins (Mishra and Dhali, 2007). This interaction between Bax and anti-apoptotic proteins disrupts their normal function, which leads to the formation of pores in the mitochondria, allowing the exit of cytochrome c. In addition, ROS independently oxidize mitochondrial pores further contributing to cytochrome c release upon GSH depletion (Costantini et al., 1996). Cytochrome c released from the mitochondria activates the caspase cascade, which then induces apoptosis. Recent studies have shown that LPS triggers the release of cytochrome c from the mitochondria to the cytoplasm, thereby activating the caspase cascade and DNA fragmentation (Chuang et al., 2011). The pre-treatment of human alveolar epithelial A549 cells with NAC significantly decreased LPS-mediated caspase activation and DNA damage, suggesting an important role of GSH in ameliorating LPS-induced cell death (Chuang et al., 2011). The loss of GSH and subsequent decrease in ATP and ensuing cell death by dopamine metabolites is prevented by GSH-EE and NAC supplementation, suggesting a critical role of GSH in maintaining functional mitochondria in Parkinson’s disease (Nunes et al., 2011). GSH also exhibited anti-apoptotic effects in selenium induced apoptosis in HSC-3 human oral squamous cell carcinoma cells and arsenic trioxide-induced apoptosis in lymphoma cells (Takahashi et al., 2005). Thus, our findings in the mouse model of ALI concur with these studies and demonstrate that GSH-EE pre-treatment prevents LPS-induced pulmonary endothelial cell apoptosis.
In conclusion, the results presented in this study suggest that the modulation of the cellular redox equilibrium by GSH preserves oxidative phosphorylation in the mitochondria in response to LPS-induced oxidative damage and subsequent cellular apoptosis. Although clinical trials involving the use of antioxidants have not shown promising results in chronically ill patients, the acute burst of oxidative, and nitrosative stress seen in ALI may be more susceptible to antioxidant therapy. Based upon past studies and our present data, we conclude that GSH depletion plays a vital role in the pathogenesis of ALI, and therefore, GSH supplementation may act as an important adjuvant therapy to reverse the disease process.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Imran Rehmani, Suphin Kallarackal, Johnny Wright, and Sumant Ponnala for their excellent technical assistance. This research was supported in part by a Beginning Grant in Aid (09BGIA2310050) from the Southeast Affiliates, and a Scientist Development Grant (11SDG7460024; all to Shruti Sharma) from the American Heart Association; 1P01HL101902-01A, HL60190, HL67841, and HL101902 (all to Stephen M. Black) from the National Institutes of Health.
References
Behr, J., Maier, K., Degenkolb, B., Krombach, F., and Vogelmeier, C. (1997). Antioxidative and clinical effects of high-dose N-acetylcysteine in fibrosing alveolitis. Adjunctive therapy to maintenance immunosuppression. Am. J. Respir. Crit. Care Med. 156, 1897–1901.
Chatterjee, A., Snead, C., Yetik-Anacak, G., Antonova, G., Zeng, J., and Catravas, J. D. (2008). Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L755–L763.
Chuang, C. Y., Chen, T. L., Cherng, Y. G., Tai, Y. T., Chen, T. G., and Chen, R. M. (2011). Lipopolysaccharide induces apoptotic insults to human alveolar epithelial A549 cells through reactive oxygen species-mediated activation of an intrinsic mitochondrion-dependent pathway. Arch. Toxicol. 85, 209–218.
Costantini, P., Chernyak, B. V., Petronilli, V., and Bernardi, P. (1996). Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. J. Biol. Chem. 271, 6746–6751.
DeLeve, L. D., and Kaplowitz, N. (1990). Importance and regulation of hepatic glutathione. Semin. Liver Dis. 10, 251–266.
Drahota, Z., Krivakova, P., Cervinkova, Z., Kmonickova, E., Lotkova, H., Kucera, O., and Houstek, J. (2005). Tert-butyl hydroperoxide selectively inhibits mitochondrial respiratory-chain enzymes in isolated rat hepatocytes. Physiol. Res. 54, 67–72.
El-Sayed, N. S., Mahran, L. G., and Khattab, M. M. (2011). Tempol, a membrane-permeable radical scavenger, ameliorates lipopolysaccharide-induced acute lung injury in mice: a key role for superoxide anion. Eur. J. Pharmacol. 663, 68–73.
Erickson, S. E., Martin, G. S., Davis, J. L., Matthay, M. A., and Eisner, M. D. (2009). Recent trends in acute lung injury mortality: 1996-2005. Crit. Care Med. 37, 1574–1579.
Foxton, R. H., Land, J. M., and Heales, S. J. (2007). Tetrahydrobiopterin availability in Parkinson’s and Alzheimer’s disease; potential pathogenic mechanisms. Neurochem. Res. 32, 751–756.
Grandjean, E. M., Berthet, P., Ruffmann, R., and Leuenberger, P. (2000). Efficacy of oral long-term N-acetylcysteine in chronic bronchopulmonary disease: a meta-analysis of published double-blind, placebo-controlled clinical trials. Clin. Ther. 22, 209–221.
Griffith, O. W. (1980). Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem. 106, 207–212.
Haddad, J. J., Safieh-Garabedian, B., Saade, N. E., and Land, S. C. (2001). Thiol regulation of pro-inflammatory cytokines reveals a novel immunopharmacological potential of glutathione in the alveolar epithelium. J. Pharmacol. Exp. Ther. 296, 996–1005.
Jaeschke, H., and Bajt, M. L. (2006). Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol. Sci. 89, 31–41.
Kasielski, M., and Nowak, D. (2001). Long-term administration of N-acetylcysteine decreases hydrogen peroxide exhalation in subjects with chronic obstructive pulmonary disease. Respir. Med. 95, 448–456.
Kawasaki, M., Kuwano, K., Hagimoto, N., Matsuba, T., Kunitake, R., Tanaka, T., Maeyama, T., and Hara, N. (2000). Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am. J. Pathol. 157, 597–603.
Kitamura, Y., Hashimoto, S., Mizuta, N., Kobayashi, A., Kooguchi, K., Fujiwara, I., and Nakajima, H. (2001). Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am. J. Respir. Crit. Care Med. 163, 762–769.
Kuwabara, T., and Imajoh-Ohmi, S. (2004). LPS-induced apoptosis is dependent upon mitochondrial dysfunction. Apoptosis 9, 467–474.
Lang, J. D., McArdle, P. J., O’reilly, P. J., and Matalon, S. (2002). Oxidant-antioxidant balance in acute lung injury. Chest 122, 314S–320S.
Lee, T. D., Sadda, M. R., Mendler, M. H., Bottiglieri, T., Kanel, G., Mato, J. M., and Lu, S. C. (2004). Abnormal hepatic methionine and glutathione metabolism in patients with alcoholic hepatitis. Alcohol. Clin. Exp. Res. 28, 173–181.
Lee, W. M., Hynan, L. S., Rossaro, L., Fontana, R. J., Stravitz, R. T., Larson, A. M., Davern, T. J. II, Murray, N. G., McCashland, T., Reisch, J. S., and Robuck, P. R. (2009). Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 137, 856.e1–864.e1.
Li, M., Chiu, J. F., Mossman, B. T., and Fukagawa, N. K. (2006). Down-regulation of manganese-superoxide dismutase through phosphorylation of FOXO3a by Akt in explanted vascular smooth muscle cells from old rats. J. Biol. Chem. 281, 40429–40439.
Li, X. Y., Donaldson, K., Rahman, I., and MacNee, W. (1994). An investigation of the role of glutathione in increased epithelial permeability induced by cigarette smoke in vivo and in vitro. Am. J. Respir. Crit. Care Med. 149, 1518–1525.
Luppi, F., Aarbiou, J., Van Wetering, S., Rahman, I., De Boer, W. I., Rabe, K. F., and Hiemstra, P. S. (2005). Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: role of glutathione. Respir. Res. 6, 140.
Ma, X., Xu, D., Ai, Y., Ming, G., and Zhao, S. (2010). Fas inhibition attenuates lipopolysaccharide-induced apoptosis and cytokine release of rat type II alveolar epithelial cells. Mol. Biol. Rep. 37, 3051–3056.
Martinez, O., Nin, N., and Esteban, A. (2009). Prone position for the treatment of acute respiratory distress syndrome: a review of current literature. Arch. Bronconeumol. 45, 291–296.
Messana, J. M., Cieslinski, D. A., O’connor, R. P., and Humes, H. D. (1988). Glutathione protects against exogenous oxidant injury to rabbit renal proximal tubules. Am. J. Physiol. 255, F874–F884.
Mishra, D. P., and Dhali, A. (2007). Endotoxin induces luteal cell apoptosis through the mitochondrial pathway. Prostaglandins Other Lipid Mediat. 83, 75–88.
Monick, M. M., Carter, A. B., Robeff, P. K., Flaherty, D. M., Peterson, M. W., and Hunninghake, G. W. (2001). Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of beta-catenin. J. Immunol. 166, 4713–4720.
Nagase, T., Uozumi, N., Aoki-Nagase, T., Terawaki, K., Ishii, S., Tomita, T., Yamamoto, H., Hashizume, K., Ouchi, Y., and Shimizu, T. (2003). A potent inhibitor of cytosolic phospholipase A2, arachidonyl trifluoromethyl ketone, attenuates LPS-induced lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 284, L720–L726.
Nunes, C., Barbosa, R. M., Almeida, L., and Laranjinha, J. (2011). Nitric oxide and DOPAC-induced cell death: from GSH depletion to mitochondrial energy crisis. Mol. Cell. Neurosci. 48, 94–103.
Pacht, E. R., Timerman, A. P., Lykens, M. G., and Merola, A. J. (1991). Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest 100, 1397–1403.
Pocernich, C. B., Cardin, A. L., Racine, C. L., Lauderback, C. M., and Butterfield, D. A. (2001). Glutathione elevation and its protective role in acrolein-induced protein damage in synaptosomal membranes: relevance to brain lipid peroxidation in neurodegenerative disease. Neurochem. Int. 39, 141–149.
Poole, P. J., and Black, P. N. (2001). Oral mucolytic drugs for exacerbations of chronic obstructive pulmonary disease: systematic review. BMJ 322, 1271–1274.
Quinlan, G. J., Evans, T. W., and Gutteridge, J. M. (1994). Oxidative damage to plasma proteins in adult respiratory distress syndrome. Free Radic. Res. 20, 289–298.
Quinlan, G. J., Lamb, N. J., Tilley, R., Evans, T. W., and Gutteridge, J. M. (1997). Plasma hypoxanthine levels in ARDS: implications for oxidative stress, morbidity, and mortality. Am. J. Respir. Crit. Care Med. 155, 479–484.
Rahman, I., and MacNee, W. (2000a). Oxidative stress and regulation of glutathione in lung inflammation. Eur. Respir. J. 16, 534–554.
Rahman, I., and MacNee, W. (2000b). Regulation of redox glutathione levels and gene transcription in lung inflammation: therapeutic approaches. Free Radic. Biol. Med. 28, 1405–1420.
Rubenfeld, G. D., Caldwell, E., Peabody, E., Weaver, J., Martin, D. P., Neff, M., Stern, E. J., and Hudson, L. D. (2005). Incidence and outcomes of acute lung injury. N. Engl. J. Med. 353, 1685–1693.
Sarkar, F. H., Rahman, K. M., and Li, Y. (2003). Bax translocation to mitochondria is an important event in inducing apoptotic cell death by indole-3-carbinol (I3C) treatment of breast cancer cells. J. Nutr. 133, 2434S–2439S.
Sharma, S., Sud, N., Wiseman, D. A., Carter, A. L., Kumar, S., Hou, Y., Rau, T., Wilham, J., Harmon, C., Oishi, P., Fineman, J. R., and Black, S. M. (2008). Altered carnitine homeostasis is associated with decreased mitochondrial function and altered nitric oxide signaling in lambs with pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L46–L56.
Shinde, S., Golam, K., Kumar, P., Patil, N., and Sadacharan, K. (2005). Perioperative blood lactate levels, pyruvate levels, and lactate-pyruvate ratio in children undergoing cardiopulmonary bypass for congenital heart disease. Indian J. Crit. Care Med. 9, 145–150.
Singhal, R. K., and Jain, A. (2000). Glutathione ethyl ester supplementation prevents mortality in newborn rats exposed to hyperoxia. Biol. Neonate 77, 261–266.
Sommer, S. P., Sommer, S., Sinha, B., Walter, D., Aleksic, I., Gohrbandt, B., Otto, C., and Leyh, R. G. (2012). Glutathione preconditioning ameliorates mitochondria dysfunction during warm pulmonary ischemia-reperfusion injury. Eur. J. Cardiothorac. Surg. 41, 140–148.
Song, M., Kellum, J. A., Kaldas, H., and Fink, M. P. (2004). Evidence that glutathione depletion is a mechanism responsible for the anti-inflammatory effects of ethyl pyruvate in cultured lipopolysaccharide-stimulated RAW 264.7 cells. J. Pharmacol. Exp. Ther. 308, 307–316.
Takahashi, M., Sato, T., Shinohara, F., Echigo, S., and Rikiishi, H. (2005). Possible role of glutathione in mitochondrial apoptosis of human oral squamous cell carcinoma caused by inorganic selenium compounds. Int. J. Oncol. 27, 489–495.
Tasaka, S., Amaya, F., Hashimoto, S., and Ishizaka, A. (2008). Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid. Redox Signal. 10, 739–753.
van Klaveren, R. J., Demedts, M., and Nemery, B. (1997). Cellular glutathione turnover in vitro, with emphasis on type II pneumocytes. Eur. Respir. J. 10, 1392–1400.
Vargas, M. R., Johnson, D. A., and Johnson, J. A. (2011). Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1(G93A) mice model. Neurobiol. Dis. 43, 543–551.
Villa, P., Saccani, A., Sica, A., and Ghezzi, P. (2002). Glutathione protects mice from lethal sepsis by limiting inflammation and potentiating host defense. J. Infect. Dis. 185, 1115–1120.
Wang, X., McCullough, K. D., Franke, T. F., and Holbrook, N. J. (2000). Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J. Biol. Chem. 275, 14624–14631.
Wiseman, D. A., Sharma, S., and Black, S. M. (2010). Elevated zinc induces endothelial apoptosis via disruption of glutathione metabolism: role of the ADP translocator. Biometals 23, 19–30.
Z’Graggen, B. R., Tornic, J., Muller-Edenborn, B., Reyes, L., Booy, C., and Beck-Schimmer, B. (2010). Acute lung injury: apoptosis in effector and target cells of the upper and lower airway compartment. Clin. Exp. Immunol. 161, 324–331.
Keywords: acute lung injury, mitochondrial dysfunction, apoptosis, lipopolysaccharide, glutathione ethyl ester
Citation: Aggarwal S, Dimitropoulou C, Lu Q, Black SM and Sharma S (2012) Glutathione supplementation attenuates lipopolysaccharide-induced mitochondrial dysfunction and apoptosis in a mouse model of acute lung injury. Front. Physio. 3:161. doi: 10.3389/fphys.2012.00161
Received: 06 February 2012; Accepted: 07 May 2012;
Published online: 28 May 2012.
Edited by:
Gregg Rokosh, University of Louisville, USAReviewed by:
Scarlet Y. Shi, Medical College of Wisconsin, USAWenbo Zhang, The University of Texas Medical Branch, USA
Copyright: © 2012 Aggarwal, Dimitropoulou, Lu, Black and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Shruti Sharma, Vascular Biology Center, Georgia Health Sciences University, 1459 Laney Walker Blvd, Augusta, GA 30912, USA. e-mail:c2hzaGFybWFAZ2VvcmdpYWhlYWx0aC5lZHU=