
- 1 Experimental Cardiology Group, Heart Failure Research Center, Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 2 Department of Cardiology, St. Antonius Hospital, Nieuwegein, Netherlands
Arrhythmogenic right ventricular cardiomyopathy (ARVC) and Brugada syndrome are distinct clinical entities which diagnostic criteria exclude their coexistence in individual patients. ARVC is a myocardial disorder characterized by fibro-fatty replacement of the myocardium and ventricular arrhythmias. In contrast, the Brugada syndrome has long been considered a functional cardiac disorder: no gross structural abnormalities can be identified in the majority of patients and its electrocardiographic hallmark of coved-type ST-segment elevation in right precordial leads is dynamic. Nonetheless, a remarkable overlap in clinical features has been demonstrated between these conditions. This review focuses on this overlap and discusses its potential causes and consequences.
Introduction
The potential overlap between Brugada syndrome and arrhythmogenic right ventricular cardiomyopathy (ARVC) has been subject of heated debate (Martini et al., 2001; Pérez Riera et al., 2005). Contested in this debate are the myocardial condition and thereby the arrhythmogenic mechanism underlying the Brugada syndrome. This review will described the clinical features and explore the overlap of these features between these syndromes.
Arrhythmogenic Right Ventricular Cardiomyopathy
Marcus et al. (1982) published a case series that for the first time captured the clinical profile of ARVC. In 24 patients, they described the fibro-fatty replacement, the ventricular tachyarrhythmias, and premature ventricular complexes with left bundle branch block morphology, the repolarization abnormalities in the form of inversed T-waves in the right precordial leads, delayed activation on standard or signal averaged ECGs, the morphological features of increased dimensions and wall motion abnormalities of the right ventricle and the familial occurrence of this disorder. The causative mechanism was unknown and was speculated to lie in the development of the right ventricle hence it was termed “arrhythmogenic right ventricular dysplasia” (Marcus et al., 1982).
Since, our understanding of the causative mechanism of ARVC has progressed substantially. An important step was made by the association of ARVC with mutations in the genes encoding the desmosomal proteins plakoglobin (McKoy et al., 2000), desmoplakin (Rampazzo et al., 2002), plakophilin-2 (Gerull et al., 2004), desmoglein-2 (Pilichou et al., 2006), and desmocollin-2 (Heuser et al., 2006). These proteins are located at the intercalated disk in adhering junctions, which in the heart contain both desmosomal and fascia adherens components, and provide mechanical strength to the cell–cell junction (Borrmann et al., 2006; Franke et al., 2006). Mechanical overload of the cell–cell junction is considered to trigger the pathophysiological myocardial changes in ARVC (Delmar and McKenna, 2010). Mutations in these genes can be identified in ∼40% of ARVC patients (Sen-Chowdhry et al., 2007a).
The concept of mechanical overload of the cell–cell junction has been substantiated further by mouse models of ARVC (Garcia-Gras et al., 2006; Kirchhof et al., 2006; Pilichou et al., 2009). Consistent with the concept of mechanical overload of the cell–cell junctions, the development of cardiomyopathy in mouse models can be influenced by altering the mechanical load on the heart: endurance training accelerates (Kirchhof et al., 2006) and pharmacological therapy that lowers the hemodynamic load slows the development of the ARVC phenotype in heterozygous plakoglobin deficient mice (Fabritz et al., 2011). Indeed, ARVC patients engaged in endurance training appear to have a more severe ARVC phenotype than those that are not (Sen-Chowdhry et al., 2007b).
The mutations associated with ARVC have been incorporated in the revised diagnostic Task Force Criteria as published in 2010 (Marcus et al., 2010). These diagnostic criteria are divided in six categories: depolarization/conduction abnormalities, repolarization abnormalities, ventricular arrhythmias, right ventricular structural or functional characteristics as demonstrated by imaging techniques, histological criteria, and familial occurrence of ARVC or presence of mutations associated or probably associated with ARVC (Table 1). In each category, a major or minor criteria can be scored. A definite diagnosis of ARVC is made in the presence of two major, one major and two minor or four minor criteria. The presence of one major and one minor or three minor criteria is classified as “borderline” and one major or two minor criteria as “possible” diagnosis of ARVC.
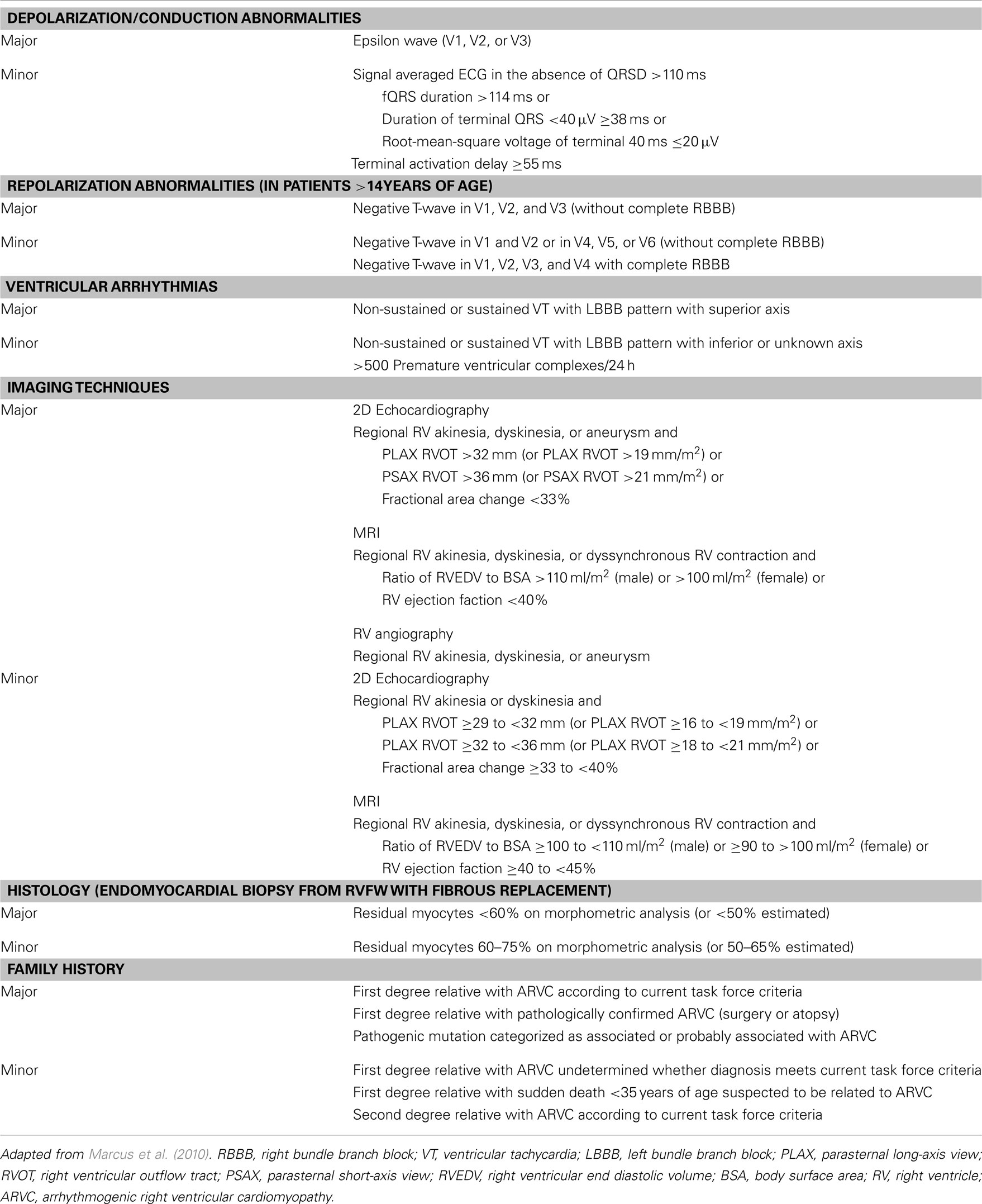
Table 1. Revised task force criteria for arrhythmogenic right ventricular cardiomyopathy.
The Brugada Syndrome
Brugada and Brugada (1992) introduced a new clinical entity characterized by right precordial ST-segment elevation followed by a negative T-wave and sudden cardiac arrest by ventricular fibrillation in otherwise healthy individuals. No underlying cause of the arrhythmias in the form of electrolyte disturbances, coronary, or structural heart disease could be identified in these now called Brugada syndrome patients.
The characteristic electrocardiographic feature (Brugada ECG pattern) is dynamic and can even disappear temporarily (Brugada and Brugada, 1997). Prior to the initiation of ventricular fibrillation however, the Brugada ECG pattern reappears in these patients (Kasanuki et al., 1997). The Brugada ECG pattern is most pronounced at night or at rest (Mizumaki et al., 2004), during febrile illness (Amin et al., 2008), and after consumption of a copious meal (Ikeda et al., 2006) which also are known triggers of ventricular tachyarrhythmias and sudden death in Brugada syndrome patients (Matsuo et al., 1999; Ikeda et al., 2006; Amin et al., 2008; Takigawa et al., 2008). Furthermore, pharmacological agents have been demonstrated to modulate the Brugada ECG pattern. Sodium channel blockers are known to augment or provoke the Brugada ECG pattern (Miyazaki et al., 1996). Other drugs can ameliorate the Brugada ECG pattern such as isoproterenol (Miyazaki et al., 1996) and quinidine (Alings et al., 2001) and have been used to prevent arrhythmias in observational studies and case reports (Tanaka et al., 2001; Belhassen et al., 2004). These findings suggested the Brugada syndrome to be a functional electrical cardiac disorder (Ackerman et al., 2011).
Thus far, mutations in 11 genes have been associated with the Brugada syndrome. Most of these mutations reduce the cardiac sodium current (INa) and are located in SCN5A (Chen et al., 1998), the gene encoding the cardiac sodium channel, its β-subunits SCN1B (Watanabe et al., 2008) and SCN3B (Hu et al., 2009) or in GPD1L (London et al., 2007) and MOG1 (Kattygnarath et al., 2011) which are thought to impair trafficking of the cardiac sodium channel to the cell membrane. Other mutations associated with the Brugada syndrome reduce the L-type calcium current (ICaL) and are located in CACNA1C, CACNB2b (Antzelevitch et al., 2007) and CACNA2D1 (Burashnikov et al., 2010) which encode the α1-, β2b-, and α2δ-subunit of the L-type calcium channel. Lastly, mutations in KCNE3 which encodes MiRP2, a β-subunit of several potassium channels (Delpon et al., 2009), and in KCNJ8 encoding the ATP-sensitive potassium channel (Barajas-Martínez et al., 2012) have been associated with the Brugada syndrome. These mutations have been suggested to increase repolarizing currents activated early during the action potential (Delpon et al., 2009; Barajas-Martínez et al., 2012).
The role these mutations play in the Brugada syndrome is subject of debate. The most common of these mutations appear to be located in SCN5A which can be identified in ∼25% of Brugada syndrome patients (Probst et al., 2010). Even in these patients, the role of loss-of-function mutations in SCN5A appears to be more complex than that of a dominant mutation with incomplete penetrance. In families affected by the Brugada syndrome in which the index patient carried a mutation in SCN5A, a significant number of Brugada syndrome patients (∼1 in 8) does not carry the mutation in SCN5A (Probst et al., 2009; Santos et al., 2012).
The mutations associated with Brugada syndrome have not been incorporated in the diagnostic criteria for Brugada syndrome as put forward in the 2005 consensus report (Table 2; Antzelevitch et al., 2005).

Table 2. Diagnostic criteria of Brugada syndrome.
Overlapping Features between Brugada Syndrome and ARVC
As apparent from the diagnostic criteria, Brugada syndrome and ARVC are distinct clinical entities with a different scope of clinical features. Nonetheless, a remarkable number of publications have shown that features of both syndromes occur in conjunction in some patients.
Brugada Syndrome Features in ARVC Patients
Prior to the introduction of the Brugada syndrome, Martini et al. (1989) presented a patient with right precordial ST-segment elevation and idiopathic ventricular fibrillation. One patient demonstrated what was later to be called the Brugada ECG pattern. Careful clinical re-evaluation demonstrated enlargement of the right ventricular outflow tract and right ventricular wall motion abnormalities. This caused the authors to conclude that a concealed form of ARVC was present in this patient. Since, more histological findings consistent with ARVC have been demonstrated at autopsy series of patients with the Brugada ECG (Corrado et al., 1996, 2001; Tada et al., 1998). The largest series was presented by Corrado et al. (2001) and consisted of 13 young sudden cardiac death victims with a Brugada ECG pattern on their last recorded ECG. Of these patients, 12 demonstrated structural heart disease consistent with ARVC at autopsy. Similar to Brugada syndrome patients (Matsuo et al., 1999), most of these patients died at night or at rest (Tada et al., 1998; Corrado et al., 2001). Furthermore, polymorphic VTs as observed in the Brugada syndrome were recorded in some these ARVC patients (Corrado et al., 2001).
Secondly, sodium channel blockers can induce the Brugada ECG pattern ∼1 in 6 patients previously diagnosed with ARVC (Peters et al., 2004; Peters, 2008). These data indicate the Brugada ECG pattern, the associated ventricular arrhythmias are present and can be modulated by INa in some ARVC patients.
ARVC Features in Brugada Syndrome Patients
Diagnostic ARVC criteria (Marcus et al., 2010) have vice-versa been demonstrated in Brugada syndrome patients. Thus far, the diagnostic criteria of ARVC have been validated to distinguish ARVC from idiopathic right ventricular outflow tract tachycardias (Nasir et al., 2004; Cox et al., 2008) but not from Brugada syndrome.
Ventricular activation abnormalities in the Brugada syndrome are well known (Postema et al., 2010). Thus far, Letsas et al. (2011) published the only paper that described the presence of electrocardiographic ARVC criteria in Brugada syndrome patients in the absence of sodium channel blockers. Epsilon waves appear to be rare in Brugada syndrome patients and were found in 2 of 47 patients. Other conduction abnormalities appear to be more common with 40% demonstrating terminal activation delay >55 ms (Letsas et al., 2011). Late potential on signal averaged ECG also appear to be common in Brugada syndrome patients and could be identified by Ikeda et al. (2001) in the majority of patients despite using more restrictive cut-off values than the revised Task Force Criteria of ARVC (≥2 LP criteria vs ≥1).
The monomorphic VTs with left bundle branch block are rare in Brugada syndrome patients but have been described in case reports (Viskin and Belhassen, 1995). Premature ventricular complexes are rare in Brugada syndrome patients (Krittayaphong et al., 2003). Ventricular arrhythmias therefore, are unlikely to cause suspicion of ARVC in Brugada syndrome patients.
Imaging studies have revealed moderate structural abnormalities in Brugada syndrome patients despite exclusion of structural heart disease in the process of diagnosing the Brugada syndrome. Magnetic resonance imaging (MRI) in 20 Brugada syndrome patients demonstrated a larger right ventricular outflow tract area compared with control subjects and a high intramyocardial T1 signal suggestive for fatty infiltration in 4 patients (Papavassiliu et al., 2004). A more elaborate MRI study also identified right ventricular wall motion abnormalities and increased right ventricular end-systolic and inflow tract diameters in Brugada syndrome patients (Catalano et al., 2009). The structural and functional abnormalities on MRI appear to be more outspoken in patients with spontaneous than a drug-induced Brugada ECG pattern (Papavassiliu et al., 2010). Right ventricular angiography is rarely used in the work-up of Brugada syndrome patients, however microaneurysms during angiography were identified by Frustaci et al. (2005) in 7 out of 18 patients undergoing RV endocardial biopsy.
Thus far, three studies systematically analyzed endocardial biopsies of Brugada syndrome patients (Frustaci et al., 2005; Zumhagen et al., 2009; Ohkubo et al., 2010). These studies produced varied findings. In a series of septal right and left ventricular biopsies 1 out of 18 patient was consistent with ARVC in the series by Frustaci et al. (2005) The biopsies in other patients demonstrated lymphocytic infiltration in 14 and hypertrophy and diffuse vacuolization in 3 patients. No microaneurysms during right ventricular angiography or signs of myocarditis in endocardial biopsies from multiple RV locations were detected by Zumhagen et al. (2009) However, mild myocardial abnormalities were detected in endocardial biopsies of most patients. These changes consisted of hypertrophy, fatty replacement, or fibrosis. The combination of fatty replacement and fibrosis as required for the diagnosis of ARVC, was present in 2 patients (Zumhagen et al., 2009). A biopsy series from Japan, 5 out of 25 patients moderate to severe fatty replacement was present in RV septal biopsies which were accompanied by fibrosis in 4 patients (Ohkubo et al., 2010).
Mutations in genes associated with ARVC appear to be rare in Brugada syndrome patients. No mutations in the gene encoding plakophilin-2, the gene most commonly involved in ARVC (Kapplinger et al., 2011), could be identified in screening of 38 Brugada syndrome patients in whom SCN5A were previously excluded (Koopmann et al., 2007).
Potential Causes of the Overlapping Features between Brugada Syndrome and ARVC Patients
Arrhythmogenic right ventricular cardiomyopathy and Brugada syndrome show an overlap: (1) the Brugada ECG pattern and the associated polymorphic VTs are present or can be provoked by sodium channel blockers in a subgroup of ARVC patients and (2) three of the diagnostic categories of the Task Force Criteria of ARVC namely conduction abnormalities, wall motion abnormalities and histology of the right ventricle are present in Brugada syndrome patients. The question arises at what level these conditions interact.
Firstly, these syndromes could interact at a genetic level. Mutations in genes associated with ARVC patients could cause electrophysiological changes that facilitate the pathophysiological mechanism of the Brugada syndrome. Support for this hypothesis can be found in cellular models of reduced mechanical coupling of cardiomyocytes. The adhering junctions, cardiac sodium channels and gap-junctions, responsible for the electrical coupling between cardiomyocytes, colocalize at the intercalated disk. Mechanical stability of the intercalated disk is thought to be important for the function of cardiac sodium channels and gap-junctions (Delmar and McKenna, 2010). In cultured neonatal rat ventricular myocytes a reduction in the expression of plakophilin-2 is associated with a reduction in the INa (Sato et al., 2009) and electrical coupling (Oxford et al., 2007). However, no indications of altered INa characteristics could be found recently in a murine model of heterozygous desmoplakin knockout (Gomes et al., 2012). Reversely, loss-of-function in SCN5A associated with Brugada syndrome may predispose patients to develop structural myocardial derangements as observed in ARVC. Support for such an hypothesis can be found in heterozygous SCN5A-knockout mice which develop cardiac fibrosis upon aging (Royer et al., 2005). The mechanism by which the reduced expression of the cardiac sodium channel causes fibrosis is currently unknown. Toxicity studies with the sodium channel blocker flecainide question whether the observed cardiac fibrosis is the direct result of reduced INa. Long-term treatment with flecainide did not result in cardiac fibrosis in mice and was even associated with a reduced prevalence of cardiac fibrosis in rats (Case et al., 1984). It is therefore uncertain whether these observations in cellular or mouse models can be extrapolated to man. In case these mechanisms are operational in man, interaction at a genetic level is unlikely to fully account for the overlap between ARVC and Brugada syndrome. As indicated above, no mutations in the gene encoding plakophilin-2 could be identified in screening of Brugada syndrome patients (Koopmann et al., 2007) and a mutation in SCN5A could be identified in only 1 out of 17 ARVC patients with a drug-induced Brugada ECG pattern (Peters, 2008). Furthermore, structural abnormalities in Brugada syndrome patients could also be identified in the absence of SCN5A mutations (Frustaci et al., 2005; Zumhagen et al., 2009).
Secondly, the electrophysiological mechanism of the Brugada syndrome could cause structural derangements. Support for this hypothesis can be found in MRI studies. Right ventricular derangements are more outspoken in patients with a spontaneous than those with a drug-induced Brugada ECG pattern (Papavassiliu et al., 2010). Patients with a spontaneous Brugada ECG pattern are known to be at increased risk of arrhythmic events (Probst et al., 2010). One could therefore also argue that patients with more structural derangements are more severely affected and are more likely to demonstrate a spontaneous Brugada ECG pattern. The fact that sodium channel blockers can provoke the Brugada ECG pattern in some ARVC patients indicate that the electrophysiological mechanism depends on structural derangements and not the other way around (Peters et al., 2004).
Thirdly, structural abnormalities could facilitate the development of the Brugada syndrome and that the structural abnormalities inherent to ARVC underlie its overlap with the Brugada syndrome. Thus far, two hypotheses of the pathophysiological mechanism of the Brugada syndrome have been coined. One suggests that that a functional change causes subepicardial abbreviation of action potential (“repolarization hypothesis”; Yan and Antzelevitch, 1999), while the other suggest that a structural change in the subepicardium causes conduction abnormalities that lead to activation delay (Coronel et al., 2005) or excitation failure (Hoogendijk et al., 2010a; “depolarization hypothesis”) underlies the Brugada syndrome. Both hypotheses account for the modulation of the Brugada syndrome by pharmacological or genetically determined changes in the INa (Miyazaki et al., 1996; Chen et al., 1998; London et al., 2007; Watanabe et al., 2008; Hu et al., 2009; Kattygnarath et al., 2011), ICaL (Miyazaki et al., 1996; Antzelevitch et al., 2007; Burashnikov et al., 2010), and transient outward current (Ito; Alings et al., 2001; Delpon et al., 2009). In the repolarization hypothesis, the balance during phase-1 of the action potential between depolarizing (INa and ICaL) and repolarizing (Ito) influence the action potential shortening and ST-segment similarly as in Brugada syndrome patients (Yan and Antzelevitch, 1999). In the depolarization hypothesis, regional activation delay in structural discontinuous myocardium make conduction sensitive to currents activated after the upstroke of the action potential. Both depolarizing (INa and ICaL) and repolarizing (Ito) influence the current available for conduction during this phase of the action potential and can modulate the Brugada ECG pattern (Hoogendijk et al., 2011).
Testing of these hypothesis has been troublesome because of the epicardial localization of the arrhythmogenic substrate in the Brugada syndrome. Until recently measurements were limited to electrograms recorded through the conus artery which runs over the right ventricular outflow tract (Shimizu et al., 2001; Nagase et al., 2002, 2008), monophasic action potentials during open chest surgery (Kurita et al., 2002), and a rare explanted heart in the setting of cardiac transplantation (Coronel et al., 2005). None of these studies was able to demonstrate ST-segment elevation in local electrograms underlying the Brugada ECG pattern. Recently, Nademanee et al. (2011) did demonstrate ST-segment elevation in unipolar electrograms in an epicardial mapping and ablation study in nine highly symptomatic Brugada syndrome patients. The ST-segment elevation in unipolar electrograms strongly resembled the Brugada EGG pattern. These electrograms were located at low-voltage areas with fractionated activation that markedly exceeding the QRS complex. Ablation at these sites resulted in a marked reduction in arrhythmic events and disappearance of the Brugada ECG pattern in eight out of nine patients. Morphologically identical unipolar electrograms were previously provoked by sodium channel blockade in the explanted heart of a SCN5A mutation carrier at sites were the subepicardium was interspersed by fibrous and adipose tissue (Hoogendijk et al., 2010a). However, no data are available on asymptomatic Brugada syndrome patients and the mechanism underlying the Brugada syndrome is still at debate.
Consequences of the Overlapping Features of ARVC and Brugada Syndrome
In conclusion, the clinical features of Brugada syndrome and ARVC coincide frequently in patients. The recent publication of the first epicardial electrograms underlying the Brugada ECG pattern support the notion that conduction disturbances in structural discontinuous myocardium underlie the Brugada syndrome (Hoogendijk et al., 2010a; Nademanee et al., 2011). The structural right ventricular abnormalities in ARVC therefore most likely predispose patients to develop the Brugada ECG pattern and the associated ventricular arrhythmias (Hoogendijk et al., 2010b). The strict exclusion of structural heart disease in the Brugada syndrome as advocated in the 2005 consensus report (Antzelevitch et al., 2005) appear arbitrary. These conclusions plead for a broadening of the diagnostic criteria by making a distinction in patients with the Brugada features in the presence and absence (Brugada syndrome) of identifiable underlying structural heart disease.
The treatment of these patients with the Brugada features in the setting of structural heart disease remains challenging. The arrhythmogenic risk in these patients has not been clearly established and may prove to be difficult to determine in the future. First of all, this patients group is heterogeneous and incorporates various underlying cardiac conditions besides ARVC (Corrado et al., 1996, 2001; Tada et al., 1998; Peters et al., 2004): the Brugada ECG pattern can also occur in the setting of Chagas’ disease (Chiale et al., 1982; Brito et al., 2010). Thus far, prospective data are available of only 17 ARVC patients with drug-induced Brugada ECG (Peters, 2008) which is associated with a low arrhythmogenic risk in the setting of Brugada syndrome (Probst et al., 2010). Only one monomorphic ventricular was recorded during follow-up (Peters, 2008). Secondly, the myocardial condition and arrhythmogenic substrate that facilitates the Brugada features may change over time in patients with and underlying cardiomyopathy. In the ARVC follow-up study by Peters, the reproducibility of drug-induced Brugada ECG pattern was four out of eight (Peters, 2008). Until the arrhythmogenic risk in patients with the Brugada ECG pattern in the setting of structural heart disease has been assessed, it appears reasonable to avoid or treat known triggers arrhythmias in Brugada syndrome patients such as certain pharmacological agents (Postema et al., 2009) and fever (Amin et al., 2008).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ackerman, M. J., Priori, S. G., Willems, S., Berul, C., Brugada, R., Calkins, H., Camm, A. J., Ellinor, P. T., Gollob, M., Hamilton, R., Hershberger, R. E., Judge, D. P., Le Marec, H., McKenna, W. J., Schulze-Bahr, E., Semsarian, C., Towbin, J. A., Watkins, H., Wilde, A., Wolpert, C., and Zipes, D. P. (2011). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 8, 1308–1339.
Alings, M., Dekker, L. R. C., Sadee, A., and Wilde, A. A. M. (2001). Quinidine induced electrocardiographic normalization in two patients with Brugada syndrome. Pacing Clin. Electrophysiol. 24, 1420–1422.
Amin, A. S., Meregalli, P. G., Bardai, A., Wilde, A. A. M., and Tan, H. L. (2008). Fever increases the risk for cardiac arrest in the Brugada syndrome. Ann. Intern. Med. 149, 216–218.
Antzelevitch, C., Brugada, P., Borggrefe, M., Brugada, J., Brugada, R., Corrado, D., Gussak, I., LeMarec, H., Nademanee, K., Pérez Riera, A. R., Shimizu, W., Schulze-Bahr, E., Tan, H., and Wilde, A. (2005). Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 111, 659–670.
Antzelevitch, C., Pollevick, G. D., Cordeiro, J. M., Casis, O., Sanguinetti, M. C., Aizawa, Y., Guerchicoff, A., Pfeiffer, R., Oliva, A., Wollnik, B., Gelber, P., Bonaros, E. P. Jr., Burashnikov, E., Wu, Y., Sargent, J. D., Schickel, S., Oberheiden, R., Bhatia, A., Hsu, L. F., Haïssaguerre, M., Schimpf, R., Borggrefe, M., and Wolpert, C. (2007). Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442–449.
Barajas-Martínez, H., Hu, D., Ferrer, T., Onetti, C. G., Wu, Y., Burashnikov, E., Boyle, M., Surman, T., Urrutia, J., Veltmann, C., Schimpf, R., Borggrefe, M., Wolpert, C., Ibrahim, B. B., Sánchez-Chapula, J. A., Winters, S., Haïssaguerre, M., and Antzelevitch, C. (2012). Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm 9, 548–555.
Belhassen, B., Glick, A., and Viskin, S. (2004). Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation 110, 1731–1737.
Borrmann, C. M., Grund, C., Kuhn, C., Hofmann, I., Pieperhoff, S., and Franke, W. W. (2006). The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adherens molecules in the intercalated disk. Eur. J. Cell Biol. 85, 469–485.
Brito, M. R., Miranda, C. E., Rabelo, W., and Marino, R. L. (2010). Type 1 electrocardiographic Brugada pattern in a woman with Chagas disease: a case report. Europace 12, 1345–1346.
Brugada, J., and Brugada, P. (1997). Further characterization of the syndrome of right bundle branch block, ST segment elevation, and sudden cardiac death. J. Cardiovasc. Electrophysiol. 8, 325–331.
Brugada, P., and Brugada, J. (1992). Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 20, 1391–1396.
Burashnikov, E., Pfeiffer, R., Barajas-Martinez, H., Delpón, E., Hu, D., Desai, M., Borggrefe, M., Haïssaguerre, M., Kanter, R., Pollevick, G. D., Guerchicoff, A., Laiño, R., Marieb, M., Nademanee, K., Nam, G. B., Robles, R., Schimpf, R., Stapleton, D. D., Viskin, S., Winters, S., Wolpert, C., Zimmern, S., Veltmann, C., and Antzelevitch, C. (2010). Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872–1882.
Case, M. T., Sibinski, L. J., and Steffen, G. R. (1984). Chronic oral toxicity and oncogenicity studies of flecainide, an antiarrhythmic, in rats and mice. Toxicol. Appl. Pharmacol. 73, 232–242.
Catalano, O., Antonaci, S., Moro, G., Mussida, M., Frascaroli, M., Baldi, M., Cobelli, F., Baiardi, P., Nastoli, J., Bloise, R., Monteforte, N., Napolitano, C., and Priori, S. G. (2009). Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur. Heart J. 30, 2241–2248.
Chen, Q., Kirsch, G. E., Zhang, D., Brugada, R., Brugada, J., Brugada, P., Potenza, D., Moya, A., Borggrefe, M., Breithardt, G., Ortiz-Lopez, R., Wang, Z., Antzelevitch, C., O’Brien, R. E., Schulze-Bahr, E., Keating, M. T., Towbin, J. A., and Wang, Q. (1998). Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296.
Chiale, P. A., Przybylski, J., Laiño, R. A., Halpern, M. S., Sánchez, R. A., Gabrieli, A., Elizari, M. V., and Rosenbaum, M. B. (1982). Electrocardiographic changes evoked by ajmaline in chronic Chagas’ disease without manifest myocarditis. Am. J. Cardiol. 49, 14–20.
Coronel, R., Casini, S., Koopmann, T. T., Wilms-Schopman, F. J. G., Verkerk, A. O., de Groot, J. R., Bhuiyan, Z., Bezzina, C. R., Veldkamp, M. W., Linnenbank, A. C., van der Wal, A. C., Tan, H. L., Brugada, P., Wilde, A. A. M., and de Bakker, J. M. T. (2005). Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 112, 2769–2777.
Corrado, D., Basso, C., Buja, G., Nava, A., Rossi, L., and Thiene, G. (2001). Right bundle branch block, right precordial ST-segment elevation, and sudden death in young people. Circulation 103, 710–717.
Corrado, D., Nava, A., Buja, G., Martini, B., Fasoli, G., Oselladore, L., Turrini, P., and Thiene, G. (1996). Familial cardiomyopathy underlies syndrome of right bundle branch block, ST segment elevation and sudden death. J. Am. Coll. Cardiol. 27, 443–448.
Cox, M. G., Nelen, M. R., Wilde, A. A., Wiesfeld, A. C., Smagt, J. J., Loh, P., Cramer, M. J., Doevendans, P. A., Tintelen, J. P., de Bakker, J. M., and Hauer, R. N. (2008). Activation delay and VT parameters in arrhythmogenic right ventricular dysplasia/cardiomyopathy: toward improvement of diagnostic ECG criteria. J. Cardiovasc. Electrophysiol. 19, 775–781.
Delmar, M., and McKenna, W. J. (2010). The cardiac desmosome and arrhythmogenic cardiomyopathies. Circ. Res. 107, 700–714.
Delpon, E., Cordeiro, J., Nunez, L., Thomsen, P., Guerchicoff, A., Pollevick, G., Wu, Y., Kanters, J., Larsen, C., Burashnikov, E., Christiansen, M., and Antzelevitch, C. (2009). Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 1, 209–218.
Fabritz, L., Hoogendijk, M. G., Scicluna, B. P., van Amersfoorth, S. C. M., Fortmueller, L., Wolf, S., Laakmann, S., Kreienkamp, N., Piccini, I., Breithardt, G., Ruiz Noppinger, P., Witt, H., Ebnet, K., Wichter, T., Levkau, B., Franke, W. W., Pieperhoff, S., de Bakker, J. M. T., Coronel, R., and Kirchhof, P. (2011). Load-reducing therapy prevents development of arrhythmogenic right ventricular cardiomyopathy in plakoglobin-deficient mice. J. Am. Coll. Cardiol. 57, 740–750.
Franke, W. W., Borrmann, C. M., Grund, C., and Pieperhoff, S. (2006). The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur. J. Cell Biol. 85, 69–82.
Frustaci, A., Priori, S. G., Pieroni, M., Chimenti, C., Napolitano, C., Rivolta, I., Sanna, T., Bellocci, F., and Russo, M. A. (2005). Cardiac histological substrate in patients with clinical phenotype of Brugada syndrome. Circulation 112, 3680–3687.
Garcia-Gras, E., Lombardi, R., Giocondo, M. J., Willerson, J. T., Schneider, M. D., Khoury, D. S., and Marian, A. J. (2006). Suppression of canonical Wnt/β-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Invest. 116, 2012–2021.
Gerull, B., Heuser, A., Wichter, T., Paul, M., Basson, C. T., McDermott, D. A., Lerman, B. B., Markowitz, S. M., Ellinor, P. T., MacRae, C. A., Peters, S., Grossmann, K. S., Michely, B., Sasse-Klaassen, S., Birchmeier, W., Dietz, R., Breithardt, G., Schulze-Bahr, E., and Thierfelder, L. (2004). Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 36, 1162–1164.
Gomes, J., Finlay, M., Ahmed, A. K., Ciaccio, E. J., Asimaki, A., Saffitz, J. E., Quarta, G., Nobles, M., Syrris, P., Chaubey, S., McKenna, W. J., Tinker, A., and Lambiase, P. D. (2012). Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur. Heart J. doi:10.1093/eurheartj/ehr472
Heuser, A., Plovie, E. R., Ellinor, P. T., Grossmann, K. S., Shin, J. T., Wichter, T., Basson, C. T., Lerman, B. B., Sasse-Klaassen, S., Thierfelder, L., MacRae, C. A., and Gerull, B. (2006). Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 79, 1081–1088.
Hoogendijk, M. G., Potse, M., Linnenbank, A. C., Verkerk, A. O., den Ruijter, H. M., van Amersfoorth, S. C. M., Klaver, E. C., Beekman, L., Bezzina, C. R., Postema, P. G., Tan, H. L., Reimer, A. G., van der Wal, A. C., ten Harkel, A. D. J., Dalinghaus, M., Vinet, A., Wilde, A. A. M., de Bakker, J. M. T., and Coronel, R. (2010a). Mechanism of right precordial ST-segment elevation in structural heart disease: excitation failure by current-to-load mismatch. Heart Rhythm 7, 238–248.
Hoogendijk, M. G., Opthof, T., Postema, P. G., Wilde, A. A. M., de Bakker, J. M. T., and Coronel, R. (2010b). The Brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ. Arrhythm. Electrophysiol. 3, 283–290.
Hoogendijk, M. G., Potse, M., Vinet, A., de Bakker, J. M. T., and Coronel, R. (2011). ST segment elevation by current-to-load mismatch: an experimental and computational study. Heart Rhythm 8, 111–118.
Hu, D., Barajas-Martinez, H., Burashnikov, E., Springer, M., Wu, Y., Varro, A., Pfeiffer, R., Koopmann, T., Cordeiro, J., Guerchicoff, A., Pollevick, G., and Antzelevitch, C. A. (2009). Mutation in the β3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ. Arrhythm. Electrophysiol. 2, 270–278.
Ikeda, T., Abe, A., Yusu, S., Nakamura, K., Ishiguro, H., Mera, H., Yotsukura, M., and Yoshino, H. (2006). The full stomach test as a novel diagnostic technique for identifying patients at risk of Brugada syndrome. J. Cardiovasc. Electrophysiol. 17, 602–607.
Ikeda, T., Sakurada, H., Sakabe, K., Sakata, T., Takami, M., Tezuka, N., Nakae, T., Noro, M., Enjoji, Y., Tejima, T., Sugi, K., and Yamaguchi, T. (2001). Assessment of noninvasive markers in identifying patients at risk in the Brugada syndrome: insight into risk stratification. J. Am. Coll. Cardiol. 37, 1628–1634.
Kapplinger, J. D., Landstrom, A. P., Salisbury, B. A., Callis, T. E., Pollevick, G. D., Tester, D. J., Cox, M. G., Bhuiyan, Z., Bikker, H., Wiesfeld, A. C. P., Hauer, R. N., van Tintelen, J. P., Jongbloed, J. D., Calkins, H., Judge, D. P., Wilde, A. A., and Ackerman, M. J. (2011). Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia–associated mutations from background genetic noise. J. Am. Coll. Cardiol. 57, 2317–2327.
Kasanuki, H., Ohnishi, S., Ohtuka, M., Matsuda, N., Nirei, T., Isogai, R., Shoda, M., Toyoshima, Y., and Hosoda, S. (1997). Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease. Circulation 95, 2277–2285.
Kattygnarath, D., Maugenre, S., Neyroud, N., Balse, E., Ichai, C., Denjoy, I., Dilanian, G., Martins, R. l. P., Fressart, V., Berthet, M., Schott, J. J., Leenhardt, A., Probst, V., Le Marec, H., Hainque, B., Coulombe, A., Hatem, S. P. N., and Guicheney, P. (2011). MOG1: a new susceptibility gene for Brugada syndrome. Circ. Cardiovasc. Genet. 4, 261–268.
Kirchhof, P., Fabritz, L., Zwiener, M., Witt, H., Schafers, M., Zellerhoff, S., Paul, M., Athai, T., Hiller, K. H., Baba, H. A., Breithardt, G., Ruiz, P., Wichter, T., and Levkau, B. (2006). Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation 114, 1799–1806.
Koopmann, T. T., Beekman, L., Alders, M., Meregalli, P. G., Mannens, M. M. A. M., Moorman, A. F. M., Wilde, A. A. M., and Bezzina, C. R. (2007). Exclusion of multiple candidate genes and large genomic rearrangements in SCN5A in a Dutch Brugada syndrome cohort. Heart Rhythm 4, 752–755.
Krittayaphong, R., Veerakul, G., Nademanee, K., and Kangkagate, C. (2003). Heart rate variability in patients with Brugada syndrome in Thailand. Eur. Heart J. 24, 1771–1778.
Kurita, T., Shimizu, W., Inagaki, M., Suyama, K., Taguchi, A., Satomi, K., Aihara, N., Kamakura, S., Kobayashi, J., and Kosakai, Y. (2002). The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J. Am. Coll. Cardiol. 40, 330–334.
Letsas, K. P., Efremidis, M., Weber, R., Korantzopoulos, P., Protonotarios, N., Prappa, E., Kounas, S. P., Evagelidou, E. N., Xydonas, S., Kalusche, D., Sideris, A., and Arentz, T. (2011). Epsilon-like waves and ventricular conduction abnormalities in subjects with type 1 ECG pattern of Brugada syndrome. Heart Rhythm 8, 874–878.
London, B., Michalec, M., Mehdi, H., Zhu, X., Kerchner, L., Sanyal, S., Viswanathan, P. C., Pfahnl, A. E., Shang, L. L., Madhusudanan, M., Baty, C. J., Lagana, S., Aleong, R., Gutmann, R., Ackerman, M. J., McNamara, D. M., Weiss, R., and Dudley, S. C. Jr. (2007). Mutation in glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116, 2260–2268.
Marcus, F. I., Fontaine, G. H., Guiraudon, G., Frank, R., Laurenceau, J. L., Malergue, C., and Grosgogeat, Y. (1982). Right ventricular dysplasia: a report of 24 adult cases. Circulation 65, 384–398.
Marcus, F. I., McKenna, W. J., Sherrill, D., Basso, C., Bauce, B., Bluemke, D. A., Calkins, H., Corrado, D., Cox, M. G. P. J., Daubert, J. P., Fontaine, G., Gear, K., Hauer, R., Nava, A., Picard, M. H., Protonotarios, N., Saffitz, J. E., Sanborn, D. M. Y., Steinberg, J. S., Tandri, H., Thiene, G., Towbin, J. A., Tsatsopoulou, A., Wichter, T., and Zareba, W. (2010). Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Circulation 121, 1533–1541.
Martini, B., Cannas, S., and Nava, A. (2001). Brugada by any other name? Eur. Heart J. 22, 1835–1836.
Martini, B., Nava, A., Thiene, G., Buja, G. F., Canciani, B., Scognamiglio, R., Daliento, L., and La Volta, S. (1989). Ventricular fibrillation without apparent heart disease: description of six cases. Am. Heart J. 118, 1203–1209.
Matsuo, K., Kurita, T., Inagaki, M., Kakishita, M., Aihara, N., Shimizu, W., Taguchi, A., Suyama, K., Kamakura, S., and Shimomura, K. (1999). The circadian pattern of the development of ventricular fibrillation in patients with Brugada syndrome. Eur. Heart J. 20, 465–470.
McKoy, G., Protonotarios, N., Crosby, A., Tsatsopoulou, A., Anastasakis, A., Coonar, A., Norman, M., Baboonian, C., Jeffery, S., and McKenna, W. J. (2000). Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355, 2119–2124.
Miyazaki, T., Mitamura, H., Miyoshi, S., Soejima, K., Ogawa, S., and Aizawa, Y. (1996). Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J. Am. Coll. Cardiol. 27, 1061–1070.
Mizumaki, K., Fujiki, A., Tsuneda, T., Sakabe, M., Nishida, K., Sugao, M., and Inoue, H. (2004). Vagal activity modulates spontaneous augmentation of ST elevation in the daily life of patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 15, 667–673.
Nademanee, K., Veerakul, G., Chandanamattha, P., Chaothawee, L., Ariyachaipanich, A., Jirasirirojanakorn, K., Likittanasombat, K., Bhuripanyo, K., and Ngarmukos, T. (2011). Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 123, 1270–1279.
Nagase, S., Kusano, K. F., Morita, H., Fujimoto, Y., Kakishita, M., Nakamura, K., Emori, T., Matsubara, H., and Ohe, T. (2002). Epicardial electrogram of the right ventricular outflow tract in patients with the Brugada syndrome: using the epicardial lead. J. Am. Coll. Cardiol. 39, 1992–1995.
Nagase, S., Kusano, K. F., Morita, H., Nishii, N., Banba, K., Watanabe, A., Hiramatsu, S., Nakamura, K., Sakuragi, S., and Ohe, T. (2008). Longer repolarization in the epicardium at the right ventricular outflow tract causes type 1 electrocardiogram in patients with Brugada syndrome. J. Am. Coll. Cardiol. 51, 1154–1161.
Nasir, K., Bomma, C., Tandri, H., Roguin, A., Dalal, D., Prakasa, K., Tichnell, C., James, C., Jspevak, P., Marcus, F., and Calkins, H. (2004). Electrocardiographic features of arrhythmogenic right ventricular dysplasia/cardiomyopathy according to disease severity. Circulation 110, 1527–1534.
Ohkubo, K., Watanabe, I., Okumura, Y., Takagi, Y., Ashino, S., Kofune, M., Sugimura, H., Nakai, T., Kasamaki, Y., Hirayama, A., and Morimoto, S. I. (2010). Right ventricular histological substrate and conduction delay in patients with Brugada syndrome. Int. Heart J. 51, 17–23.
Oxford, E. M., Musa, H., Maass, K., Coombs, W., Taffet, S. M., and Delmar, M. (2007). Connexin43 remodeling caused by inhibition of plakophilin-2 expression in cardiac cells. Circ. Res. 101, 703–711.
Papavassiliu, T., Veltmann, C., Doesch, C., Haghi, D., Germans, T., Schoenberg, S. O., van Rossum, A. C., Schimpf, R., Brade, J., Wolpert, C., and Borggrefe, M. (2010). Spontaneous type 1 electrocardiographic pattern is associated with cardiovascular magnetic resonance imaging changes in Brugada syndrome. Heart Rhythm 7, 1790–1796.
Papavassiliu, T., Wolpert, C., Fluchter, S., Schimpf, R., Neff, W., Haase, K. K., Duber, C., and Borggrefe, M. (2004). Magnetic resonance imaging findings in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 15, 1133–1138.
Pérez Riera, A. R., Antzelevitch, C., Schapacknik, E., Dubner, S., and Ferreira, C. (2005). Is there an overlap between Brugada syndrome and arrhythmogenic right ventricular cardiomyopathy/dysplasia? J. Electrocardiol. 38, 260–263.
Peters, S. (2008). Arrhythmogenic right ventricular dysplasia-cardiomyopathy and provocable coved-type ST-segment elevation in right precordial leads: clues from long-term follow-up. Europace 10, 816–820.
Peters, S., Trummel, M., Denecke, S., and Koehler, B. (2004). Results of ajmaline testing in patients with arrhythmogenic right ventricular dysplasia-cardiomyopathy. Int. J. Cardiol. 95, 207–210.
Pilichou, K., Nava, A., Basso, C., Beffagna, G., Bauce, B., Lorenzon, A., Frigo, G., Vettori, A., Valente, M., Towbin, J., Thiene, G., Danieli, G. A., and Rampazzo, A. (2006). Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113, 1171–1179.
Pilichou, K., Remme, C. A., Basso, C., Campian, M. E., Rizzo, S., Barnett, P., Scicluna, B. P., Bauce, B., van den Hoff, M. J. B., de Bakker, J. M. T., Tan, H. L., Valente, M., Nava, A., Wilde, A. A. M., Moorman, A. F. M., Thiene, G., and Bezzina, C. R. (2009). Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J. Exp. Med. 206, 1787–1802.
Postema, P. G., van Dessel, P. F. H. M., Kors, J. A., Linnenbank, A. C., van Herpen, G., Ritsema van Eck, H. J., van Geloven, N., de Bakker, J. M. T., Wilde, A. A. M., and Tan, H. L. (2010). Local depolarization abnormalities are the dominant pathophysiologic mechanism for type 1 electrocardiogram in Brugada syndrome: a study of electrocardiograms, vectorcardiograms, and body surface potential maps during ajmaline provocation. J. Am. Coll. Cardiol. 55, 789–797.
Postema, P. G., Wolpert, C., Amin, A. S., Probst, V., Borggrefe, M., Roden, D. M., Priori, S. G., Tan, H. L., Hiraoka, M., Brugada, J., and Wilde, A. A. M. (2009). Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website (www.brugadadrugs.org). Heart Rhythm 6, 1335–1341.
Probst, V., Veltmann, C., Eckardt, L., Meregalli, P. G., Gaita, F., Tan, H. L., Babuty, D., Sacher, F., Giustetto, C., Schulze-Bahr, E., Borggrefe, M., Haïssaguerre, M., Mabo, P., Le Marec, H., Wolpert, C., and Wilde, A. A. M. (2010). Long-term prognosis of patients diagnosed with Brugada syndrome. Circulation 121, 635–643.
Probst, V., Wilde, A. A. M., Barc, J., Sacher, F., Babuty, D., Mabo, P., Mansourati, J., Le Scouarnec, S., Kyndt, F., Le Caignec, C., Guicheney, P., Gouas, L., Albuisson, J., Meregalli, P. G., Le Marec, H., Tan, H. L., and Schott, J. J. (2009). SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ. Cardiovasc. Genet. 2, 552–557.
Rampazzo, A., Nava, A., Malacrida, S., Beffagna, G., Bauce, B., Rossi, V., Zimbello, R., Simionati, B., Basso, C., Thiene, G., Towbin, J. A., and Danieli, G. A. (2002). Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 71, 1200–1206.
Royer, A., van Veen, T. A. B., Le Bouter, S., Marionneau, C., Griol-Charhbili, V., Leoni, A. L., Steenman, M., van Rijen, H. V. M., Demolombe, S., Goddard, C. A., Richer, C., Escoubet, B., Jarry-Guichard, T., Colledge, W. H., Gros, D., de Bakker, J. M. T., Grace, A. A., Escande, D., and Charpentier, F. (2005). Mouse model of SCN5A-linked hereditary Lenegre’s disease: age-related conduction slowing and myocardial fibrosis. Circulation 111, 1738–1746.
Santos, L. F., Rodrigues, B., Moreira, D., Correia, E., Nunes, L., Costa, A., Elvas, L., Pereira, T., Machado, J. C., Castedo, S., Henriques, C., Matos, A., and Santos, J. O. (2012). Criteria to predict carriers of a novel SCN5A mutation in a large Portuguese family affected by the Brugada syndrome. Europace. doi:10.1093/europace/eur421
Sato, P. Y., Musa, H., Coombs, W., Guerrero-Serna, G., Patino, G. A., Taffet, S. M., Isom, L. L., and Delmar, M. (2009). Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ. Res. 105, 523–526.
Sen-Chowdhry, S., Syrris, P., and McKenna, W. J. (2007a). Role of genetic analysis in the management of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 50, 1813–1821.
Sen-Chowdhry, S., Syrris, P., Ward, D., Asimaki, A., Sevdalis, E., and McKenna, W. J. (2007b). Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 115, 1710–1720.
Shimizu, W., Aiba, T., Kurita, T., and Kamakura, S. (2001). Paradoxic abbreviation of repolarization in epicardium of the right ventricular outflow tract during augmentation of Brugada-type ST segment elevation. J. Cardiovasc. Electrophysiol. 12, 1418–1421.
Tada, H., Aihara, N., Ohe, T., Yutani, C., Hamada, S., Miyanuma, H., Takamiya, M., and Kamakura, S. (1998). Arrhythmogenic right ventricular cardiomyopathy underlies syndrome of right bundle branch block, ST-segment elevation, and sudden death. Am. J. Cardiol. 81, 519–522.
Takigawa, M., Noda, T., Shimizu, W., Miyamoto, K., Okamura, H., Satomi, K., Suyama, K., Aihara, N., Kamakura, S., and Kurita, T. (2008). Seasonal and circadian distributions of ventricular fibrillation in patients with Brugada syndrome. Heart Rhythm 5, 1523–1527.
Tanaka, H., Kinoshita, O., Uchikawa, S., Kasai, H., Nakamura, M., Izawa, A., Yokoseki, O., Kitabayashi, H., Takahashi, W., Yazaki, Y., Watanabe, N., Imamura, H., and Kubo, K. (2001). Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. Pacing Clin. Electrophysiol. 24, 1293–1294.
Watanabe, H., Koopmann, T. T., Le Scouarnec, S., Yang, T., Ingram, C. R., Schott, J., Demolombe, S., Probst, V., Anselme, F., Escande, D., Wiesfeld, A., Pfeurer, A., Kaab, S., Wichmann, H., Hasdemir, C., Aizawa, Y., Wilde, A. A. M., Roden, D., and Bezzina, C. (2008). Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268.
Yan, G. X., and Antzelevitch, C. (1999). Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation 100, 1660–1666.
Keywords: Brugada syndrome, arrhythmogenic right ventricular cardiomyopathy, sudden cardiac death, desmosome, sodium channel, structural heart disease
Citation: Hoogendijk MG (2012) Diagnostic dilemmas: overlapping features of Brugada syndrome and arrhythmogenic right ventricular cardiomyopathy. Front. Physio. 3:144. doi: 10.3389/fphys.2012.00144
Received: 16 February 2012; Paper pending published: 06 March 2012;
Accepted: 29 April 2012; Published online: 23 May 2012.
Edited by:
Ruben Coronel, Academic Medical Center, NetherlandsReviewed by:
Sebastian Pieperhoff, University of Edinburgh, ScotlandLuigi Venetucci, University of Manchester, UK
Pier Lambiase, University College London, UK
Guillaume Duthoit, Groupe Hospitalier Pitié-Salpêtrière, France
Copyright: © 2012 Hoogendijk. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mark G. Hoogendijk, Experimental Cardiology Group, Heart Failure Research Center, Academic Medical Center, University of Amsterdam, Meibergdreef 9, 1105 AZ Amsterdam, Netherlands. e-mail:bWFya2hvb2dlbmRpamtAaG90bWFpbC5jb20=