
 Aki Sugano2
Aki Sugano2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 07 May 2012
Sec. Gastrointestinal Sciences
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00126
This article is part of the Research Topic Recent advances in Pancreatology View all 10 articles
Studies on hereditary pancreatitis have provided evidence in favor of central role for trypsin activity in the disease. Identification of genetic variants of trypsinogen linked the protease to the onset of pancreatitis, and biochemical characterization proposed an enzymatic gain of function as the initiating mechanism. Mutations of serine protease inhibitor Kazal type 1 gene (SPINK1) are shown to be associated with hereditary pancreatitis. We previously reported that Spink3 (a mouse homolog gene of human SPINK1) deficient mice showed excessive autophagy, followed by inappropriate trypsinogen activation in the exocrine pancreas. These data indicate that the role of SPINK1/Spink3 is not only trypsin inhibitor, but also negative regulator of autophagy. On the other hand, recent studies showed that high levels of SPINK1 protein detected in a serum or urine were associated with adverse outcome in various cancer types. It has been suggested that expression of SPINK1 and trypsin is balanced in normal tissue, but this balance could be disrupted during tumor progression. Based on the structural similarity between SPINK1 and epidermal growth factor (EGF), we showed that SPINK1 protein binds and activates EGF receptor, thus acting as a growth factor on tumor cell lines. In this review, we summarize the old and new roles of SPINK1/Spink3 in trypsin inhibition, autophagy, and cancer cell growth. These new functions of SPINK1/Spink3 may be related to the development of chronic pancreatitis.
Chronic pancreatitis (CP) is a common disease characterized by progressive, destructive, and inflammatory process of multifactorial etiology that leads to irreversible obliteration of the exocrine and endocrine pancreatic tissues and to its replacement by fibrous tissue, which ultimately results in the clinical manifestations typical of an “end-stage” disorder of pancreatic function (Steer et al., 1995; Mergener and Baillie, 1997; Braganza et al., 2011). Furthermore, CP is a well-described risk factor for pancreatic adenocarcinoma (Whitcomb, 2004; Lowenfels and Maisonneuve, 2005), especially in cases of hereditary pancreatitis (HP; Lowenfels et al., 1997). In the Western countries, alcohol is generally considered as an important risk factor for the development of CP (Gullo et al., 1988). In addition, other metabolic, anatomical, obstructive, and autoimmune etiological factors have also been recognized (Steer et al., 1995; Etemad and Whitcomb, 2001). Furthermore, in recent years, several genetic risk factors for CP have been identified. HP is a very rare form of early onset CP. With the exception of the young age at diagnosis and a slower progression, the clinical course, morphological features, and laboratory findings of HP do not differ from those of patients with alcoholic CP. Gene mutations of cationic trypsinogen (protease serine 1; PRSS1), anionic trypsinogen (protease serine 2; PRSS2), pancreatic secretory trypsin inhibitor (PSTI; serine protease inhibitor Kazal type 1; SPINK1), cystic fibrosis transmembrane conductance regulator (CFTR), chymotrypsinogen C (CTRC), and calcium-sensing receptor (CASR) have been shown to be associated with HP (Whitcomb, 2010). Although the pathogenesis of CP, including HP, is not completely understood, the necrosis–fibrosis concept is supported by both clinical and experimental data. Necrosis–fibrosis concept is that repeated attacks of acute pancreatitis (AP) induce CP. Animal models of CP have been developed by inducing repeated episodes of AP in the pancreas using an administration of cerulein, an analog of cholecystokinin (Neuschwander-Tetri et al., 2000), or choline-deficient ethionine-supplemented diet (Ida et al., 2010).
The main mechanism in the onset of AP is believed to be the autodigestion of pancreatic structural cells by various proteases that are activated in response to the ectopic (intrapancreatic) activation of trypsinogen (trypsin production). A relationship between the trypsinogen gene mutations and the onset of pancreatitis was initially reported in 1996 (Whitcomb et al., 1996). The effect of mutations in SPINK1 gene on the onset of pancreatitis was reported in 2000 (Witt et al., 2000). Mutations in PRSS1 gene, encoding cationic trypsinogen, play a causative role in HP (Whitcomb et al., 1996). It has been shown that PRSS1 mutations increase autocatalytic conversion of trypsinogen to active trypsin, and thus probably cause premature, intrapancreatic trypsinogen activation disturbing the intrapancreatic balance of proteases and their inhibitors (Whitcomb et al., 1996).
Whitcomb et al. (1996) determined the sequence of five exons of the PRSS1 and PRSS2 genes using genomic DNA from patients with HP. To explain why the R122H mutation might cause pancreatitis, Whitcomb proposed that the Arg122-Val123 autolytic peptide bond in trypsin plays an important role in the degradation of prematurely activated trypsin in the pancreas (Whitcomb et al., 1996). Destruction of this “failsafe mechanism” by the R122H mutation would increase intrapancreatic trypsin activity and disturb the protease–antiprotease equilibrium and eventually precipitate pancreatitis. Although, in the following years, further mutations of PRSS1 gene were discovered in patients with hereditary or idiopathic CP (Teich et al., 2006), the R122H, and the N29I mutations are the most common PRSS1 mutations worldwide. They have been frequently reported from Europe, North America, and Asia (Nishimori et al., 1999).
Several mechanisms have been proposed to explain how mutations in the cationic trypsinogen gene can lead to increase of trypsin activity. At first, R122H mutation prevents inactivation (autolysis) of activated trypsin (Whitcomb et al., 1996) and also leads to an increase in the autoactivation of trypsinogen (Sahin-Toth and Toth, 2000). N29I mutation was found to have no effect on trypsin activity and trypsinogen stability by biochemical analysis using recombinant trypsinogen. N29I mutation is hypothesized to change the higher-order structure of trypsin, resulting in decreased SPINK1 binding and increased autoactivation. As it has been shown that changes in the sequence of the cationic trypsinogen N-terminal peptide increased its rate of degradation, mutations A16V, D22G, and K23R (all of which change the signal peptide cleavage site of trypsin) may lead to increased autoactivation of trypsinogen to trypsin. 228delTCC has been hypothesized to enhance cationic trypsinogen transcription, thereby increasing activity. On the basis of these mechanisms, any of these gene mutations can increase trypsin activity in the pancreas.
To examine the link between PRSS1 mutations and the initiation and progression of HP, Archer et al. (2006) generated a transgenic mouse that carries a missense mutation (R122H mutation) in the PRSS1 gene. The pancreas from these transgenic mice displayed early onset acinar cell injury and inflammatory cell infiltration. With progressing age, the transgenic mice developed pancreatic fibrosis and display acinar cell dedifferentiation. Moreover, the expression of mutated PRSS1 transgene is associated with enhanced response to cerulein-induced pancreatitis.
Although increased proteolytic activity caused by the PRSS1 gene mutations enhances the risk for CP, it was thought that mutations in the PRSS2 gene may predispose to disease (Witt et al., 2006). In vitro studies showed that recombinant mutated PRSS2 protein (G191R) showed a loss of trypsin activity due to a novel tryptic cleavage site that renders the enzyme hypersensitive to autocatalytic proteolysis. Thus, it appears that the PRSS2 variant (G191R) mitigates intrapancreatic trypsin activity, thereby playing a protective role against CP. Although the overall contribution of G191R to disease pathogenesis is low, the functional characterization of G191R provides the first example in pancreatitis for a disease-protective genetic variant.
Biological consequences of intracellular trypsinogen activation have not been directly examined. Gaiser et al. (2011) generated mice, which were engineered to conditionally express an endogenously activated trypsinogen within pancreatic acinar cells. The mice can express trypsin activity conditionally in the pancreatic acinar cells. Although initiation of AP was observed with high levels of active trypsin expression, chronic inflammation, or fibrosis did not develop in this mice model, suggesting that intra-acinar activation of trypsinogen is sufficient to initiate AP, but not to CP. Because CP is sometimes observed without evidence of associated necrosis, mutated trypsinogen may have other functions, and cause HP.
SPINK1 molecule was found by two independent groups. At first, Kazal et al. extracted SPINK1 from bovine pancreas as a pancreatic secretory trypsin inhibitor (PSTI) in 1948 (Kazal et al., 1948). The animal pancreas usually has two types of trypsin inhibitors, basic pancreatic trypsin inhibitor (BPTI) and PSTI. Docking site of SPINK1 and PRSS1 which is predicted by using biological information, ZDOCK, was very similar to that of BPTI and PRSS1 (Figure 1). However, human pancreas does not have BPTI. The gene encoding human PSTI was named as serine protease inhibitor, Kazal type 1 (SPINK1). Mouse gene homologous to human SPINK1 is Spink3. Hydra gene homologous to human SPINK1 is Kazal1. The human SPINK1 gene encodes mRNA of 237 bp, which is translated to a 79 amino acid peptide including a 23 amino acid signal peptide with three intramolecular disulfide bridges (Cys9–Cys33, Cys16–Cys35, and Cys24–Cys56; Bartelt et al., 1977; Horii et al., 1987), and the molecular weight is estimated to be 6240 based on the amino acid composition. SPINK1 (PSTI) is secreted by the acinar cells of exocrine pancreas into the pancreatic duct. It binds rapidly to trypsin and inhibits its activity both intracellularly and extracellularly. Hence, it is an important protective factor in the onset of pancreatitis (Hirota et al., 2006). The liver also secretes SPINK1 protein in the systemic circulation as one of acute phase proteins to inhibit trypsin activity in tissues such as the pancreas. Although it is known that SPINK1 molecule is widely expressed in extrapancreatic tissues, especially in the gastrointestinal and urinary tract (Marchbank et al., 1998), its roles in these tissues are not known.
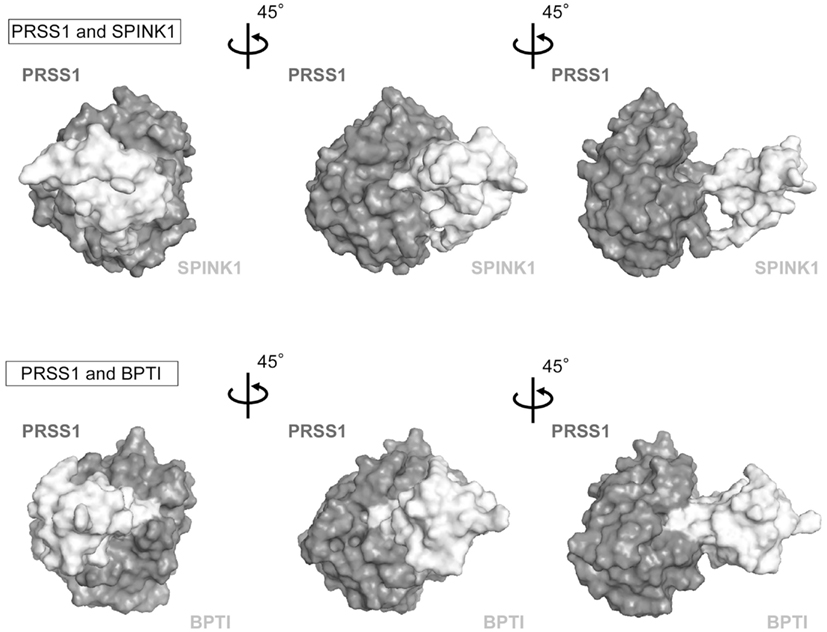
Figure 1. PRSS1 and trypsin inhibitor binding. The 3-D structures of SPINK1, BPTI, and PRSS1 were downloaded from Protein Data Bank (accession number: SPINK1, 1cgj; BPTI, 2ptc, and PRSS1, 2ra3). Hydrogen atoms were added to these structures by using PyMOL software. Molecular docking simulations for PRSS1 and SPINK1 were the carried out by using ZDOCK.
At second discovery, SPINK1 was isolated by Stenman et al. (1982) from urine of ovarian cancer patients and was reported as tumor-associated trypsin inhibitor (TATI). TATI was later shown to be identical to SPINK1 (Huhtala et al., 1982). Increased expression of SPINK1 protein was reported in various cancers, such as lung cancer (Tomita et al., 1987; Higashiyama et al., 1992), colon cancer (Higashiyama et al., 1990; Tomita et al., 1990), liver cancer (Ohmachi et al., 1993), breast cancer (Ogawa et al., 1987), prostate cancer (Tomlins et al., 2008), and pancreas cancer (Ogawa et al., 1987; Ozaki et al., 2009). Clinically, measurement of SPINK1 protein is most useful for monitoring of patients with mucinous ovarian cancer. Increased serum concentration ofSPINK1 protein may occur in most types of cancer (Paju and Stenman, 2006). And, increased expression of SPINK1 protein in tumor tissues has been reported to be associated with the poor survival in various cancers. Although this appears to be explained by the coexpression of SPINK1 molecule and tumor-associated trypsin, which is thought to participate in tumor-associated protease cascades mediating tumor invasion (Stenman et al., 1991), but it has not been established yet. Tomlins et al. (2008) reported that SPINK1 outlier expression is exclusively in a subset of ETS rearrangement-negative cancer (∼10% of total cases). ETS rearrangement is that the translocation of an ETS (E26 transformation specific) transcription factor (ERG or ETV1) to the TMPRSS2 promoter region, which contains androgen responsive elements, and it results in the aberrant androgen-regulated expression of ERG (Tomlins et al., 2005). They found that SPINK1 outlier expression is an independent predictor of biochemical recurrence after resection, and observed that SPINK1 outlier expression is an independent predictor of biochemical recurrence after resection. Tonouchi et al. (2006) reported that by the DNA microarray analysis and quantitative RT-PCR reaction, SPINK1 molecule is a candidate suggesting early recurrence of intrahepatic cholangiocarcinoma after resection. They described that the patients with higher levels of SPINK1 mRNA expression had significantly shorter recurrence-free survival. Gouyer et al. (2008) reported that from the conditioned medium of HT-29 5M21 human colon cancer cells, which are expressing a spontaneous invasive phenotype, SPINK1 molecule was identified and characterized as the major proinvasive secreted factor. SPINK1, which has a signal peptide, is secreted from not only pancreatic acinar cells, but also, colon cancer cells. SPINK1 protein may work as an autocrine and/or paracrine transforming factor, which is potentially involved in cancer progression, including local invasion of the primary tumor and its metastatic spread.
Interestingly, there are some structural similarities between SPINK1 and epidermal growth factor (EGF); both have similar numbers of amino acid residues (56 and 53, respectively), molecular weights (about 6 kD), and three intra-chain disulfide bridges (Fukuoka et al., 1987; Marchbank et al., 1998). There is 50% gene sequence homology between SPINK1 and EGF (Hunt et al., 1974; Scheving, 1983; Yamamoto et al., 1985). Ogawa et al. (1985) reported that SPINK1 protein was mitogenic for human fibroblasts. Some studies support the concept that SPINK1 protein binds to the EGF receptor (EGFR). Rat monitor peptide (rat homolog of human SPINK1) has been reported to compete with mouse EGF for binding to EGFR of Swiss 3T3 fibroblasts (Fukuoka et al., 1987) and an EGFR-blocking antibody removed the promigratory effects of SPINK1 on human HT-29 cells (Marchbank et al., 1996).
Recently we elucidated several functions of SPINK1 molecule. These include trypsin inhibition in vivo, autophagy regulation, and growth stimulation.
Pancreatic digestive enzymes are stored as inactivated precursors in pancreatic acinar cells. Under normal conditions, digestive enzyme activation is strictly controlled to prevent autodigestion of the pancreas, which is called pancreatitis. However, in certain circumstances, excessive amounts of pancreatic trypsinogen are activated to trypsin intracellularly, resulting in activation of other zymogens and autodigestion of the pancreas. SPINK1 molecule is synthesized in acinar cells of the pancreas and is thought to inhibit the trypsin activity in the pancreas.
Intrapancreatic balance between trypsin and SPINK1 activities is important for pancreatitis development. It is hypothesized that mutations in SPINK1 gene that affect SPINK1 binding with trypsin will contribute to the onset of pancreatitis. There have been many reports of mutations in SPINK1 genes in patients with pancreatitis, and several hypothetical roles of these mutant proteins in pancreatitis (Pfutzer et al., 2000; Witt et al., 2000; Chen et al., 2001; Kaneko et al., 2001; Kuwata et al., 2001, 2003).
To analyze the importance of trypsinogen activation (trypsin production) and its regulation by SPINK1 molecule in the onset of pancreatitis, we generated Spink3 gene knockout mice by gene targeting and analyzed their phenotypes (Ohmuraya et al., 2005). The pancreatic acinar cells in knockout mouse showed excessive autophagy (Figure 2) and enhanced tryptic activity was detected in pancreatic acini prepared at 1 day after birth (Ohmuraya et al., 2006). All acinar cells disappeared completely after birth, indicating that Spink3 is not only a trypsin inhibitor within pancreatic acinar cells, but also important in maintaining the integrity of these cells.

Figure 2. The pancreatic acinar cells of Spink3 deficient mice. Many vacuoles, autophagosomes, are appeared in Spink3 deficient pancreatic acinar cells (Right panel). N, nucleus of the acinar cell.
The general mechanism to deliver cytoplasmic components to the lysosomes is called autophagy (Figure 3). The best understood role of autophagy is cellular housekeeping, a function that extends beyond the simple removal of damaged or unwanted products (Seglen and Bohley, 1992; Kim and Klionsky, 2000; Meijer, 2003). In fact, along with other proteolytic systems, lysosome participates in the continuous turnover of intracellular constituents. Not only soluble cytosolic proteins but also organelles, such as mitochondria and peroxisomes, can be removed by autophagy (Lemasters et al., 2002; Bellu and Kiel, 2003; Roberts et al., 2003). In addition to maintaining cellular homeostasis, there is growing evidence for the participation of autophagy in processes such as cellular differentiation, tissue remodeling, growth control, cell defense, and adaptation to adverse environments (Hennig and Neufeld, 2002; Jacinto and Hall, 2003; Melendez et al., 2003; Otto et al., 2003).
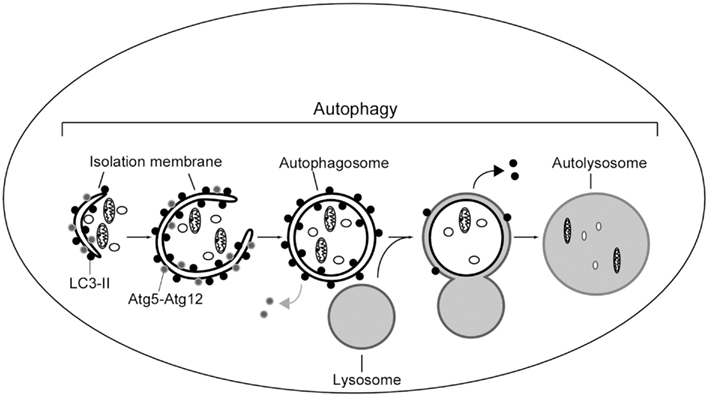
Figure 3. Scheme of autophagy in mammalian cells. A portion of cytoplasm is enclosed by isolation membrane to form an autophagosome. Autophagosome fuses with lysosome to degrade the inside materials. The Atg5–Atg12 conjugate localizes to the isolation membrane throughout its elongation process. LC3 is recruited to the membrane in the Atg5-dependent manner. Atg5–Atg12 dissociate from the membrane upon completion of autophagosome formation, while LC3 (-II) remains on the autophagosome membrane. Atg5 is required for elongation of the isolation membrane.
The vacuoles were observed by histological examination correspond to autophagosomes using autophagosome specific probe, microtubule-associated protein 1 light chain 3 (LC3-II), which are the hallmark of autophagy (Hashimoto et al., 2008). We reported that conversion of trypsinogen to trypsin within the pancreatic acinar cell was greatly suppressed in autophagy-related gene 5 (Atg5) deficient acinar cells, suggesting involvement of autophagy in trypsinogen activation by lysosomal hydrolase such as cathepsin B (Figure 4; Hashimoto et al., 2008; Ohmuraya and Yamamura, 2008). In previous study, the results indicate that all known mutations of SPINK1 gene are functionally innocuous for the trypsin inhibitory activity (Kuwata et al., 2002; Kiraly et al., 2007; Ohmuraya et al., 2009). Hence, it is possible that HP caused by mutation of SPINK1 gene is due to autophagy induction, but not to loss of binding to trypsin.
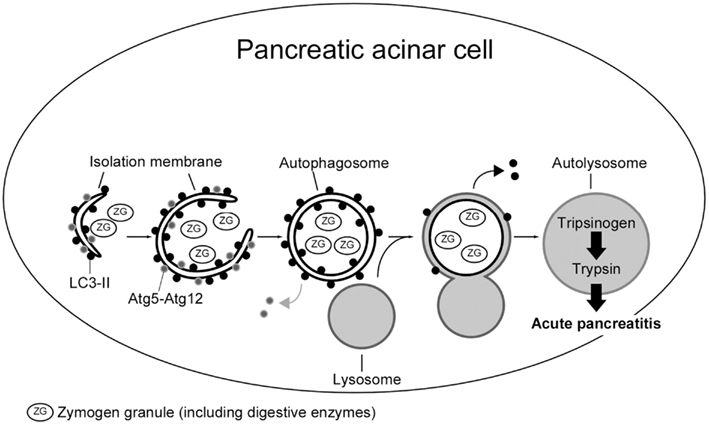
Figure 4. Schematic representation of autophagy within a pancreatic acinar cell in acute pancreatitis. After autophagic vacuole containing zymogen granules fuses lysosome, the trypsinogen is activated by lysosomal hydrolases in autophagic process, and release of activated digestive enzymes inside the cell.
Interestingly, Chera et al. (2006) reported that similar phenotypes parallel in the endodermal epithelial cells observed upon silencing of Kazal1 gene, which is a homolog of SPINK1 gene in hydra. In hydra, the endodermal epithelial cells carry out the digestive function together with the gland cells that produce zymogens and express the evolutionarily conserved gene Kazal1. A progressive Kazal1 silencing induced excessive autophagy in the cytoplasm of digestive cells, and dramatic disorganization followed by a massive death of these cells. These data suggests that SPINK1 activity is required to prevent excessive autophagy in food digestive systems.
In mammalian cells, autophagy continuously occurs at basal level, but can be induced in response to environmental signals including nutrients and hormones (Mizushima, 2005; Mizushima et al., 2008). The molecular machinery of autophagy is regulated by a class I PI3K and mammalian target of rapamycin (mTOR), which act to inhibit autophagy (Klionsky, 2005; Mizushima et al., 2008). It is interesting to note that SPINK1 and EGF have structural similarities, including the number of amino acid residues and the presence of three intrachain disulfide bridges (Scheving, 1983). Can SPINK1 bind to EGFR and stimulate its, as a growth factor?
Spink3 deficient pancreas shows no sign of regeneration of acinar cells (Ohmuraya et al., 2005), and Kazal1 silencing decreases budding rate in hydra (Chera et al., 2006). SPINK1 protein was previously shown to stimulate the growth of non-neoplastic (Ogawa et al., 1985; McKeehan et al., 1986) and neoplastic cells (Freeman et al., 1990; Niinobu et al., 1990).
Marchbank et al. (1996) reported that SPINK1 molecule involved in the regenerative process at the ulcer edge. The cell migration induced by SPINK1 protein was inhibited by adding an EGFR-blocking antibody, which suggests that SPINK1 protein mediated this effect through binding to the EGFR. This idea is supported by a previous report that rat PSTI competed with mouse EGF for binding to mouse Swiss 3T3 cells (Fukuoka et al., 1987), although Niinobu et al. (1990) reported that the binding of human 125I-labeled SPINK1 protein to the same cells was displaced by cold SPINK1 protein but not EGF, suggesting a separate receptor.
Recently, we showed that SPINK1 protein binds to EGFR to activate its downstream signaling; resulting in proliferation of pancreatic and breast cancer cells (Ozaki et al., 2009). We showed that SPINK1 protein coprecipitated with EGFR in an immunoprecipitation experiment and that the binding affinity of SPINK1 to EGFR is about half of that of EGF using quartz-crystal microbalance technique. In addition, we performed molecular docking simulations using ZDOCK for the following pairs: EGF and EGFR; SPINK1 and EGFR. In 2000 docking runs, each binding pairs bound 795 and 287 times at the same position, respectively. These data indicate that SPINK1 binds to EGFR at the same position of EGF–EGFR binding site, but binding affinity is about half compared to EGF–EGFR affinity.
Although binding affinity of SPINK1–EGFR is lower than that of EGF–EGFR, EGFR, and its downstream molecules, signal transducer and activator of transcription 3 (STAT3), v-Akt murine thymoma viral oncogene homolog (AKT), and extracellular signal-regulated kinase 1/2 (ERK1/2), were phosphorylated by SPINK1 as well as EGF. These results suggest that SPINK1 protein stimulates the proliferation of pancreatic cancer cells through EGFR (Ozaki et al., 2009). Autophagy is regulated by PI3K–AKT–mTOR pathway (Klionsky, 2005; Mizushima et al., 2008). Secreted SPINK1 protein may activate this pathway via EGFR to prevent insufficient or excessive autophagy, which induces cell death.
In summary, SPINK1 is a multifunctional fascinating molecule. It is important not only in pancreatitis pathogenesis, but also in cellular growth and death. Although generally favoring a central role for trypsin activity during pancreatitis, none of the hereditary variants provides direct evidence that intracellular trypsin activity alone is sufficient to initiate the CP. We proved new roles of SPINK1 molecule, namely autophagy regulation and growth stimulation via EGFR. These new functions of SPINK1 molecule may be related to the development of CP. It is possible that HP caused by mutation of the SPINK1 gene is due to autophagy induction, but not to loss of binding to trypsin (Ohmuraya and Yamamura, 2008). As next steps, human SPINK1 replacement models in Spink3 knockout mice are in progress.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Archer, H., Jura, N., Keller, J., Jacobson, M., and Bar-Sagi, D. (2006). A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology 131, 1844–1855.
Bartelt, D. C., Shapanka, R., and Greene, L. J. (1977). The primary structure of the human pancreatic secretory trypsin inhibitor. Amino acid sequence of the reduced S-aminoethylated protein. Arch. Biochem. Biophys. 179, 189–199.
Bellu, A. R., and Kiel, J. A. (2003). Selective degradation of peroxisomes in yeasts. Microsc. Res. Tech. 61, 161–170.
Braganza, J. M., Lee, S. H., Mccloy, R. F., and Mcmahon, M. J. (2011). Chronic pancreatitis. Lancet 377, 1184–1197.
Chen, J. M., Mercier, B., Audrezet, M. P., Raguenes, O., Quere, I., and Ferec, C. (2001). Mutations of the pancreatic secretory trypsin inhibitor (PSTI) gene in idiopathic chronic pancreatitis. Gastroenterology 120, 1061–1064.
Chera, S., De Rosa, R., Miljkovic-Licina, M., Dobretz, K., Ghila, L., Kaloulis, K., and Galliot, B. (2006). Silencing of the hydra serine protease inhibitor Kazal1 gene mimics the human SPINK1 pancreatic phenotype. J. Cell. Sci. 119, 846–857.
Etemad, B., and Whitcomb, D. C. (2001). Chronic pancreatitis: diagnosis, classification, and new genetic developments. Gastroenterology 120, 682–707.
Freeman, T. C., Curry, B. J., Calam, J., and Woodburn, J. R. (1990). Pancreatic secretory trypsin inhibitor stimulates the growth of rat pancreatic carcinoma cells. Gastroenterology 99, 1414–1420.
Fukuoka, S., Fushiki, T., Kitagawa, Y., Sugimoto, E., and Iwai, K. (1987). Competition of a growth stimulating-/cholecystokinin (CCK) releasing-peptide (monitor peptide) with epidermal growth factor for binding to 3T3 fibroblasts. Biochem. Biophys. Res. Commun. 145, 646–650.
Gaiser, S., Daniluk, J., Liu, Y., Tsou, L., Chu, J., Lee, W., Longnecker, D. S., Logsdon, C. D., and Ji, B. (2011). Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut 60, 1379–1388.
Gouyer, V., Fontaine, D., Dumont, P., De Wever, O., Fontayne-Devaud, H., Leteurtre, E., Truant, S., Delacour, D., Drobecq, H., Kerckaert, J. P., De Launoit, Y., Bracke, M., Gespach, C., Desseyn, J. L., and Huet, G. (2008). Autocrine induction of invasion and metastasis by tumor-associated trypsin inhibitor in human colon cancer cells. Oncogene 27, 4024–4033.
Gullo, L., Barbara, L., and Labo, G. (1988). Effect of cessation of alcohol use on the course of pancreatic dysfunction in alcoholic pancreatitis. Gastroenterology 95, 1063–1068.
Hashimoto, D., Ohmuraya, M., Hirota, M., Yamamoto, A., Suyama, K., Ida, S., Okumura, Y., Takahashi, E., Kido, H., Araki, K., Baba, H., Mizushima, N., and Yamamura, K. (2008). Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J. Cell Biol. 181, 1065–1072.
Hennig, K. M., and Neufeld, T. P. (2002). Inhibition of cellular growth and proliferation by dTOR overexpression in Drosophila. Genesis 34, 107–110.
Higashiyama, M., Doi, O., Kodama, K., Yokouchi, H., Tateishi, R., Matsuura, N., Murata, A., Tomita, N., Monden, T., and Ogawa, M. (1992). Immunohistochemical analysis of pancreatic secretory trypsin inhibitor expression in pulmonary adenocarcinoma: its possible participation in scar formation of the tumor tissues. Tumour Biol. 13, 299–307.
Higashiyama, M., Monden, T., Tomita, N., Murotani, M., Kawasaki, Y., Morimoto, H., Murata, A., Shimano, T., Ogawa, M., and Mori, T. (1990). Expression of pancreatic secretory trypsin inhibitor (PSTI) in colorectal cancer. Br. J. Cancer 62, 954–958.
Hirota, M., Ohmuraya, M., and Baba, H. (2006). Genetic background of pancreatitis. Postgrad. Med. J. 82, 775–778.
Horii, A., Kobayashi, T., Tomita, N., Yamamoto, T., Fukushige, S., Murotsu, T., Ogawa, M., Mori, T., and Matsubara, K. (1987). Primary structure of human pancreatic secretory trypsin inhibitor (PSTI) gene. Biochem. Biophys. Res. Commun. 149, 635–641.
Huhtala, M. L., Pesonen, K., Kalkkinen, N., and Stenman, U. H. (1982). Purification and characterization of a tumor-associated trypsin inhibitor from the urine of a patient with ovarian cancer. J. Biol. Chem. 257, 13713–13716.
Hunt, L. T., Barker, W. C., and Dayhoff, M. O. (1974). Epidermal growth factor: internal duplication and probable relationship to pancreatic secretory trypsin inhibitor. Biochem. Biophys. Res. Commun. 60, 1020–1028.
Ida, S., Ohmuraya, M., Hirota, M., Ozaki, N., Hiramatsu, S., Uehara, H., Takamori, H., Araki, K., Baba, H., and Yamamura, K. (2010). Chronic pancreatitis in mice by treatment with choline-deficient ethionine-supplemented diet. Exp. Anim. 59, 421–429.
Jacinto, E., and Hall, M. N. (2003). Tor signalling in bugs, brain and brawn. Nat. Rev. Mol. Cell Biol. 4, 117–126.
Kaneko, K., Nagasaki, Y., Furukawa, T., Mizutamari, H., Sato, A., Masamune, A., Shimosegawa, T., and Horii, A. (2001). Analysis of the human pancreatic secretory trypsin inhibitor (PSTI) gene mutations in Japanese patients with chronic pancreatitis. J. Hum. Genet. 46, 293–297.
Kazal, L. A., Spicer, D. S., and Brahinsky, R. A. (1948). Isolation of a crystalline trypsin inhibitor-anticoagulant protein from pancreas. J. Am. Chem. Soc. 70, 304–340.
Kim, J., and Klionsky, D. J. (2000). Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu. Rev. Biochem. 69, 303–342.
Kiraly, O., Wartmann, T., and Sahin-Toth, M. (2007). Missense mutations in pancreatic secretory trypsin inhibitor (SPINK1) cause intracellular retention and degradation. Gut 56, 1433–1438.
Klionsky, D. J. (2005). The molecular machinery of autophagy: unanswered questions. J. Cell. Sci. 118, 7–18.
Kuwata, K., Hirota, M., Nishimori, I., Otsuki, M., and Ogawa, M. (2003). Mutational analysis of the pancreatic secretory trypsin inhibitor gene in familial and juvenile pancreatitis in Japan. J. Gastroenterol. 38, 365–370.
Kuwata, K., Hirota, M., Shimizu, H., Nakae, M., Nishihara, S., Takimoto, A., Mitsushima, K., Kikuchi, N., Endo, K., Inoue, M., and Ogawa, M. (2002). Functional analysis of recombinant pancreatic secretory trypsin inhibitor protein with amino-acid substitution. J. Gastroenterol. 37, 928–934.
Kuwata, K., Hirota, M., Sugita, H., Kai, M., Hayashi, N., Nakamura, M., Matsuura, T., Adachi, N., Nishimori, I., and Ogawa, M. (2001). Genetic mutations in exons 3 and 4 of the pancreatic secretory trypsin inhibitor in patients with pancreatitis. J. Gastroenterol. 36, 612–618.
Lemasters, J. J., Qian, T., He, L., Kim, J. S., Elmore, S. P., Cascio, W. E., and Brenner, D. A. (2002). Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid. Redox Signal. 4, 769–781.
Lowenfels, A. B., and Maisonneuve, P. (2005). Risk factors for pancreatic cancer. J. Cell. Biochem. 95, 649–656.
Lowenfels, A. B., Maisonneuve, P., Dimagno, E. P., Elitsur, Y., Gates, L. K. Jr., Perrault, J., and Whitcomb, D. C. (1997). Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J. Natl. Cancer Inst. 89, 442–446.
Marchbank, T., Chinery, R., Hanby, A. M., Poulsom, R., Elia, G., and Playford, R. J. (1996). Distribution and expression of pancreatic secretory trypsin inhibitor and its possible role in epithelial restitution. Am. J. Pathol. 148, 715–722.
Marchbank, T., Freeman, T. C., and Playford, R. J. (1998). Human pancreatic secretory trypsin inhibitor. Distribution, actions and possible role in mucosal integrity and repair. Digestion 59, 167–174.
McKeehan, W. L., Sakagami, Y., Hoshi, H., and Mckeehan, K. A. (1986). Two apparent human endothelial cell growth factors from human hepatoma cells are tumor-associated proteinase inhibitors. J. Biol. Chem. 261, 5378–5383.
Meijer, A. J. (2003). Amino acids as regulators and components of nonproteinogenic pathways. J. Nutr. 133, 2057S–2062S.
Melendez, A., Talloczy, Z., Seaman, M., Eskelinen, E. L., Hall, D. H., and Levine, B. (2003). Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391.
Mizushima, N. (2005). The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 12(Suppl. 2), 1535–1541.
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075.
Neuschwander-Tetri, B. A., Burton, F. R., Presti, M. E., Britton, R. S., Janney, C. G., Garvin, P. R., Brunt, E. M., Galvin, N. J., and Poulos, J. E. (2000). Repetitive self-limited acute pancreatitis induces pancreatic fibrogenesis in the mouse. Dig. Dis. Sci. 45, 665–674.
Niinobu, T., Ogawa, M., Murata, A., Nishijima, J., and Mori, T. (1990). Identification and characterization of receptors specific for human pancreatic secretory trypsin inhibitor. J. Exp. Med. 172, 1133–1142.
Nishimori, I., Kamakura, M., Fujikawa-Adachi, K., Morita, M., Onishi, S., Yokoyama, K., Makino, I., Ishida, H., Yamamoto, M., Watanabe, S., and Ogawa, M. (1999). Mutations in exons 2 and 3 of the cationic trypsinogen gene in Japanese families with hereditary pancreatitis. Gut 44, 259–263.
Ogawa, M., Matsuura, N., Higashiyama, K., and Mori, T. (1987). Expression of pancreatic secretory trypsin inhibitor in various cancer cells. Res. Commun. Chem. Pathol. Pharmacol. 55, 137–140.
Ogawa, M., Tsushima, T., Ohba, Y., Ogawa, N., Tanaka, S., Ishida, M., and Mori, T. (1985). Stimulation of DNA synthesis in human fibroblasts by human pancreatic secretory trypsin inhibitor. Res. Commun. Chem. Pathol. Pharmacol. 50, 155–158.
Ohmachi, Y., Murata, A., Matsuura, N., Yasuda, T., Monden, M., Mori, T., Ogawa, M., and Matsubara, K. (1993). Specific expression of the pancreatic-secretory-trypsin-inhibitor (PSTI) gene in hepatocellular carcinoma. Int. J. Cancer 55, 728–734.
Ohmuraya, M., Hirota, M., Araki, K., Baba, H., and Yamamura, K. (2006). Enhanced trypsin activity in pancreatic acinar cells deficient for serine protease inhibitor Kazal type 3. Pancreas 33, 104–106.
Ohmuraya, M., Hirota, M., Araki, M., Mizushima, N., Matsui, M., Mizumoto, T., Haruna, K., Kume, S., Takeya, M., Ogawa, M., Araki, K., and Yamamura, K. (2005). Autophagic cell death of pancreatic acinar cells in serine protease inhibitor Kazal type 3-deficient mice. Gastroenterology 129, 696–705.
Ohmuraya, M., Ozaki, N., Hirota, M., Baba, H., and Yamamura, K. (2009). Serine protease inhibitor Kazal type 1 (SPINK1): beyond the trypsin inhibitor. Curr. Enzym. Inhib. 5, 110–116.
Ohmuraya, M., and Yamamura, K. (2008). Autophagy and acute pancreatitis: a novel autophagy theory for trypsinogen activation. Autophagy 4, 1060–1062.
Otto, G. P., Wu, M. Y., Kazgan, N., Anderson, O. R., and Kessin, R. H. (2003). Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. J. Biol. Chem. 278, 17636–17645.
Ozaki, N., Ohmuraya, M., Hirota, M., Ida, S., Wang, J., Takamori, H., Higashiyama, S., Baba, H., and Yamamura, K. (2009). Serine protease inhibitor Kazal type 1 promotes proliferation of pancreatic cancer cells through the epidermal growth factor receptor. Mol. Cancer Res. 7, 1572–1581.
Paju, A., and Stenman, U. H. (2006). Biochemistry and clinical role of trypsinogens and pancreatic secretory trypsin inhibitor. Crit. Rev. Clin. Lab. Sci. 43, 103–142.
Pfutzer, R. H., Barmada, M. M., Brunskill, A. P., Finch, R., Hart, P. S., Neoptolemos, J., Furey, W. F., and Whitcomb, D. C. (2000). SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 119, 615–623.
Roberts, P., Moshitch-Moshkovitz, S., Kvam, E., O’toole, E., Winey, M., and Goldfarb, D. S. (2003). Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol. Biol. Cell 14, 129–141.
Sahin-Toth, M., and Toth, M. (2000). Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem. Biophys. Res. Commun. 278, 286–289.
Scheving, L. A. (1983). Primary amino acid sequence similarity between human epidermal growth factor-urogastrone, human pancreatic secretory trypsin inhibitor, and members of porcine secretin family. Arch. Biochem. Biophys. 226, 411–413.
Seglen, P. O., and Bohley, P. (1992). Autophagy and other vacuolar protein degradation mechanisms. Experientia 48, 158–172.
Steer, M. L., Waxman, I., and Freedman, S. (1995). Chronic pancreatitis. N. Engl. J. Med. 332, 1482–1490.
Stenman, U. H., Huhtala, M. L., Koistinen, R., and Seppala, M. (1982). Immunochemical demonstration of an ovarian cancer-associated urinary peptide. Int. J. Cancer 30, 53–57.
Stenman, U. H., Koivunen, E., and Itkonen, O. (1991). Biology and function of tumor-associated trypsin inhibitor, TATI. Scand. J. Clin. Lab. Invest. Suppl. 207, 5–9.
Teich, N., Rosendahl, J., Toth, M., Mossner, J., and Sahin-Toth, M. (2006). Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum. Mutat. 27, 721–730.
Tomita, N., Doi, S., Higashiyama, M., Morimoto, H., Murotani, M., Kawasaki, Y., Monden, T., Shimano, T., Horii, A., Yokouchi, H., Ogawa, M., Mori, T., and Matsubara, K. (1990). Expression of pancreatic secretory trypsin inhibitor gene in human colorectal tumor. Cancer 66, 2144–2149.
Tomita, N., Horii, A., Yamamoto, T., Ogawa, M., Mori, T., and Matsubara, K. (1987). Expression of pancreatic secretory trypsin inhibitor gene in neoplastic tissues. FEBS Lett. 225, 113–119.
Tomlins, S. A., Rhodes, D. R., Perner, S., Dhanasekaran, S. M., Mehra, R., Sun, X. W., Varambally, S., Cao, X., Tchinda, J., Kuefer, R., Lee, C., Montie, J. E., Shah, R. B., Pienta, K. J., Rubin, M. A., and Chinnaiyan, A. M. (2005). Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648.
Tomlins, S. A., Rhodes, D. R., Yu, J., Varambally, S., Mehra, R., Perner, S., Demichelis, F., Helgeson, B. E., Laxman, B., Morris, D. S., Cao, Q., Cao, X., Andren, O., Fall, K., Johnson, L., Wei, J. T., Shah, R. B., Al-Ahmadie, H., Eastham, J. A., Eggener, S. E., Fine, S. W., Hotakainen, K., Stenman, U. H., Tsodikov, A., Gerald, W. L., Lilja, H., Reuter, V. E., Kantoff, P. W., Scardino, P. T., Rubin, M. A., Bjartell, A. S., and Chinnaiyan, A. M. (2008). The role of SPINK1 in ETS rearrangement-negative prostate cancers. Cancer Cell 13, 519–528.
Tonouchi, A., Ohtsuka, M., Ito, H., Kimura, F., Shimizu, H., Kato, M., Nimura, Y., Iwase, K., Hiwasa, T., Seki, N., Takiguchi, M., and Miyazaki, M. (2006). Relationship between pancreatic secretory trypsin inhibitor and early recurrence of intrahepatic cholangiocarcinoma following surgical resection. Am. J. Gastroenterol. 101, 1601–1610.
Whitcomb, D. C. (2004). Inflammation and cancer V. Chronic pancreatitis and pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G315–G319.
Whitcomb, D. C., Gorry, M. C., Preston, R. A., Furey, W., Sossenheimer, M. J., Ulrich, C. D., Martin, S. P., Gates, L. K. Jr., Amann, S. T., Toskes, P. P., Liddle, R., Mcgrath, K., Uomo, G., Post, J. C., and Ehrlich, G. D. (1996). Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 14, 141–145.
Witt, H., Luck, W., Hennies, H. C., Classen, M., Kage, A., Lass, U., Landt, O., and Becker, M. (2000). Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat. Genet. 25, 213–216.
Witt, H., Sahin-Toth, M., Landt, O., Chen, J. M., Kahne, T., Drenth, J. P., Kukor, Z., Szepessy, E., Halangk, W., Dahm, S., Rohde, K., Schulz, H. U., Le Marechal, C., Akar, N., Ammann, R. W., Truninger, K., Bargetzi, M., Bhatia, E., Castellani, C., Cavestro, G. M., Cerny, M., Destro-Bisol, G., Spedini, G., Eiberg, H., Jansen, J. B., Koudova, M., Rausova, E., Macek, M. Jr., Malats, N., Real, F. X., Menzel, H. J., Moral, P., Galavotti, R., Pignatti, P. F., Rickards, O., Spicak, J., Zarnescu, N. O., Bock, W., Gress, T. M., Friess, H., Ockenga, J., Schmidt, H., Pfutzer, R., Lohr, M., Simon, P., Weiss, F. U., Lerch, M. M., Teich, N., Keim, V., Berg, T., Wiedenmann, B., Luck, W., Groneberg, D. A., Becker, M., Keil, T., Kage, A., Bernardova, J., Braun, M., Guldner, C., Halangk, J., Rosendahl, J., Witt, U., Treiber, M., Nickel, R., and Ferec, C. (2006). A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat. Genet. 38, 668–673.
Keywords: chronic pancreatitis, hereditary pancreatitis, trypsinogen, SPINK1, Spink3, autophagy, EGF, EGFR
Citation: Ohmuraya M, Sugano A, Hirota M, Takaoka Y and Yamamura K (2012) Role of intrapancreatic SPINK1/Spink3 expression in the development of pancreatitis. Front. Physio. 3:126. doi: 10.3389/fphys.2012.00126
Received: 20 February 2012; Paper pending published: 01 March 2012;
Accepted: 16 April 2012; Published online: 07 May 2012.
Edited by:
Atsushi Masamune, Tohoku University Graduate School of Medicine, JapanReviewed by:
Hiroshi Ishiguro, Nagoya University Graduate School of Medicine, JapanCopyright: © 2012 Ohmuraya, Sugano, Hirota, Takaoka and Yamamura. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ken-ichi Yamamura, Institute of Resource Development and Analysis, Kumamoto University, 2-2-1 Honjo, Kumamoto 860-0811, Japan. e-mail:eWFtYW11cmFAZ3BvLmt1bWFtb3RvLXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.