




94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol., 29 May 2012
Sec. Cardiac Electrophysiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00122
This article is part of the Research TopicTransgenic models of cardiac arrhythmias and sudden deathView all 13 articles
Ankyrin polypeptides are cellular adapter proteins that tether integral membrane proteins to the cytoskeleton in a host of human organs. Initially identified as integral components of the cytoskeleton in erythrocytes, a recent explosion in ankyrin research has demonstrated that these proteins play prominent roles in cytoskeletal signaling pathways and membrane protein trafficking/regulation in a variety of excitable and non-excitable cells including heart and brain. Importantly, ankyrin research has translated from bench to bedside with the discovery of human gene variants associated with ventricular arrhythmias that alter ankyrin–based pathways. Ankyrin polypeptides have also been found to play an instrumental role in various forms of sinus node disease and atrial fibrillation (AF). Mouse models of ankyrin-deficiency have played fundamental roles in the translation of ankyrin-based research to new clinical understanding of human sinus node disease, AF, and ventricular tachycardia.
Ankyrin polypeptides are a family of adapter proteins that link integral membrane proteins with the submembranous actin/spectrin-based cytoskeleton. Ankyrin-R was identified in the late 1970s as a critical link between several anion exchanger isoforms and β-spectrin in red blood cells (Bennett, 1979). Over 30 years later, ankyrins are now widely regarded as key players in the formation of protein complexes including ion channels and transporters, cell adhesion molecules (CAMs), signaling proteins, and cytoskeletal elements. In heart, ankyrin-associated protein complexes organize specialized membrane-domains with distinct electrical and structural properties in cardiac sinus node, atrial, and ventricular cardiomyocytes. With particular emphasis given to ankyrin-B and ankyrin-G, this review provides important insight into the role of ankyrins in cardiac physiology as well as providing information on the link between ankyrin dysfunction and human arrhythmias based primarily on data garnered from mouse models.
ANK1, ANK2, and ANK3 genes encode three classes of ankyrin polypeptides (ankyrin-R, ankyrin-B, and ankyrin-G, respectively). While ankyrin-R expression is primarily limited to erythrocytes, neurons, and skeletal muscle, both ankyrin-B and ankyrin-G are widely expressed (Bennett and Chen, 2001; Cunha and Mohler, 2006). Although each gene is alternatively spliced to produce protein products of variable size, canonical ankyrin gene products include 210 kD ankyrin-R, 220 kD ankyrin-B, and 190 kD ankyrin-G (Figure 1; Bennett and Chen, 2001; Cunha and Mohler, 2006). Ankyrin genes are large, and complex splicing events determine cell-specific binding partners and function. For example, ANK1 contains over 40 exons resulting in gene products ranging from −30 to +200kD (Bennett and Chen, 2001). The ANK2 gene spans over 560 kb with >50 exons and diverse splice forms ranging from −50 to 440 kD in brain, heart, skeletal muscle and thymus (Bennett and Chen, 2001). ANK3 encodes numerous ankyrin-G isoforms broadly expressed in epithelial tissue, kidney, skeletal and cardiac muscle, and brain (Bennett and Chen, 2001). While the specific role of alternatively spliced ankyrin isoforms is unknown, it is likely that variability in expression contributes to organization and maintenance of distinct subcellular domains in eukaryotic cells.
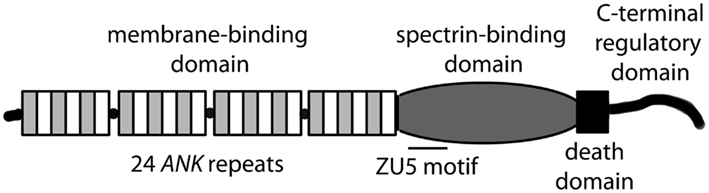
Figure 1. Canonical ankyrin (R, B, G) domain organization.
Canonical ankyrins are comprised of four functional domains: the membrane-binding domain (MBD), the spectrin-binding domain (SBD), the death domain (DD), and the C-terminal domain (CTD; Figure 1; Hashemi et al., 2009). The MBD mediates the majority of ankyrin interactions with integral membrane proteins including voltage-gated Na+ and K+ channels, the Na/Ca (NCX) exchanger, the Na+/K+ ATPase (NKA), the ammonium transporter, the KATP channel, the inositol 1,4,5 trisphosphate receptor (IP3R), and the anion exchanger (Table 1; Bennett and Healy, 2009; Kline et al., 2009). Furthermore, the MBD interacts with CAMs including CD44, E-cadherin, and L1-CAMs. Ankyrin membrane-domains are multivalent for integral membrane protein binding, and therefore have the ability to coordinate large membrane-associated protein complexes. For example, in cardiomyocytes, a single ankyrin-B polypeptide can form a ternary membrane protein complex including the NCX, NKA, and IP3R (Mohler et al., 2005). While there is a homology in the MBD across members of the ankyrin family, each ankyrin possesses a unique set of binding partners (e.g., ankyrin-G and voltage-gated Na+ and K+ channels, ankyrin-B and NCX, NKA, IP3R). The ankyrin SBD links ankyrin to the actin-based cytoskeleton via β-spectrin isoforms (Mohler et al., 2004c) thereby conferring structural stability to larger protein networks. Apart from structural capacities, the SBD has also been demonstrated to bind other proteins including the regulatory subunit (B56alpha) of the PP2A family (Bhasin et al., 2007). Lastly, the DD and CTD together comprise the ankyrin “regulatory domain.” This domain regulates the binding of ankyrin isoforms to large muscle proteins including obscurin (Kontrogianni-Konstantopoulos et al., 2003; Cunha and Mohler, 2008). However, beyond intermolecular interactions, this domain associates with the ankyrin-BMBD (Abdi et al., 2006). Relevant for this review, the ankyrin-B “regulatory domain” is the site of the majority of human gene variants linked with arrhythmia phenotypes.
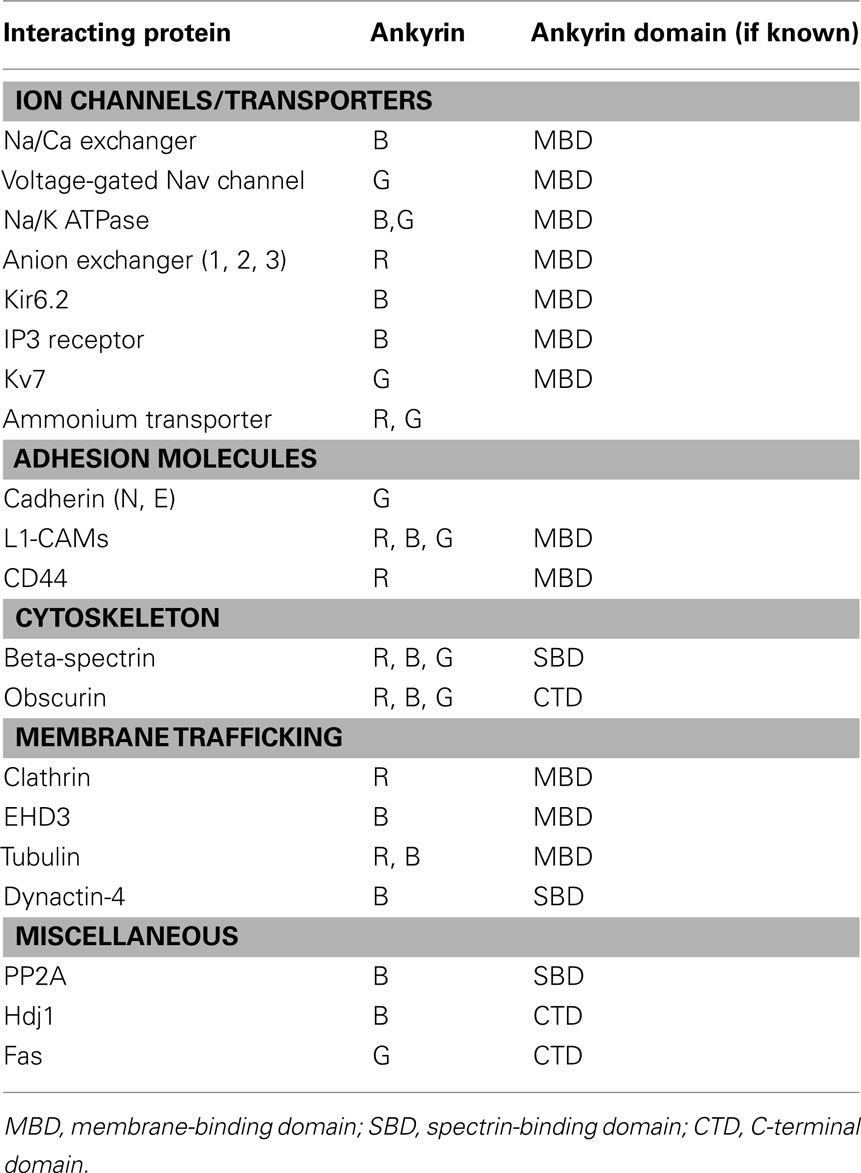
Table 1. Select ankyrin (R, B, G) interacting proteins and site of protein binding.
Defects in ankyrin have been linked to a variety of human diseases, and play an integral role in cardiovascular disease and arrhythmia. In heart, a growing number of inherited arrhythmia syndromes have been linked to ankyrin dysfunction with mounting data that ankyrin defects may also play a role in common forms of acquired heart disease. The first such disorder linked to ankyrin dysfunction was inherited long-QT syndrome (LQTS), characterized by abnormalities on the electrocardiogram (ECG), and increased susceptibility to polymorphic ventricular arrhythmias and sudden death. Mutations within ion channels that are involved in the generation and termination of action potentials make up the majority of inherited LQTS (Splawski et al., 2000). However, work nearly a decade ago identified the first loss-of-function gene variant in ANK2 in large pedigrees with LQTS (Mohler et al., 2003). Since this initial discovery, ANK2 loss-of-function variants are now well documented to cause dominantly inherited type 4 LQTS (LQT4) and have been identified throughout the world in cohorts with phenotypes linked with LQT4 (Mohler et al., 2004b, 2007). Beyond loss-of-function mutations, analysis of large human populations has linked ANK2 single-nucleotide polymorphisms with alterations in the cardiac QT interval (Sedlacek et al., 2008) further supporting the role of ankyrin-B in cardiac electrical regulation.
The development a mouse model heterozygous for a null mutation in ankyrin-B (ankyrin-B+/−) was critical for understanding the pathophysiology of the human disease. In fact, ankyrin-B+/− mice phenocopy human LQT4. It is also imperative to highlight that mice homozygous for null mutations in ankyrin-B (ankyrin-B−/−) also demonstrate abnormalities on ECG that mirrors those of LQT4 with abnormal QT rate adaptation resulting in greater QT prolongation with heart rate deceleration (Chauhan et al., 2000 #2). In regards to the heterozygous model, ankyrin-B+/−mice and human ANK2 E1425G carriers display bradycardia, prolonged QT interval, heart rate variability, and episodes of isorhythmic atrio-ventricular dissociation (Mohler et al., 2003; Le Scouarnec et al., 2008). Furthermore, similar to their human counterparts, ankyrin-B+/− mice display polymorphic ventricular tachycardia, syncope, and sudden cardiac death in response to exercise and increased circulating catecholamines. In light of the complex phenotype presented by human ANK2 variant carriers that includes ventricular and supraventricular defects, the disease name was revised to the moniker “Ankyrin-B Syndrome.”
Great strides have been made over the past 10 years to establish the link between ankyrin-deficiency and increased susceptibility to cardiac arrhythmia. At the level of the single myocyte, loss of ankyrin-B results in aberrant membrane targeting and regulation of the NKA, NCX, KATP, and IP3R. Loss of these proteins ultimately produces defects in intracellular Na+ and Ca2+ regulation, that in the cardiomyocyte leads to increased sarcoplasmic reticulum calcium load, inappropriate calcium release, and early and delayed after depolarizations in the presence of high levels of circulating catecholamines. Subsequent computational modeling has further supported the link between ankyrin-deficiency, loss of membrane function of NCX and NKA, intracellular Na+ and Ca2+ accumulation, and increased susceptibility of myocytes to develop SR Ca2+ overload (Wolf et al., 2010). In particular, the ankyrin-deficient cellular phenotype was illustrated to produce frequent spontaneous Ca2+ release events and increased the incidence of pro-arrhythmic after depolarizations during rapid pacing and/or elevated catecholamines (Wolf et al., 2010). This biophysical phenotype was independently confirmed in a recent report investigating Ca2+ handling in intact cardiomyocytes isolated from ankyrin-B+/− mice (Camors et al., 2012).
Beyond ventricular arrhythmias, loss-of-function variants in ankyrin-B have more recently been linked with sinus node dysfunction (Le Scouarnec et al., 2008). Similar to the discovery of the link between ANK2 and human ventricular phenotypes, large human pedigrees were initially utilized to provide genetic linkage between sinus node disease and the ANK2 locus. Related directly to sinus node disease, two large European cohorts mutant ANK2 alleles were shown to display highly penetrant bradycardia and exaggerated heart rate variability. Immunoblotting of muscle biopsies in affected individuals confirmed loss of ankyrin-B expression associated with the phenotype (Le Scouarnec et al., 2008). The degree of penetrance and the age of onset of sinus node dysfunction were truly remarkable in affected individuals. Sinus node dysfunction is largely a disease of the elderly population (Bernstein and Parsonnet, 1992). However, carriers of the ANK2 mutation received pacemakers (devices implanted underneath the skin of the clavicle with leads attached to the heart designed to pace the heart at a safe heart rate if the native conduction system falters) at a mean age of 30-extraordinarily rare in clinical practice. Akin to these patients with ankyrin-B-associated sinus node disease, mice lacking ankyrin-B display severe sinus node dysfunction, with reduced expression of NKA, NCX, and IP3R (Le Scouarnec et al., 2008). However, unlike ventricular cardiomyocytes, ICa,L was also significantly reduced in isolated ankyrin-B+/− sinoatrial node cells, further altering calcium handling and sinoatrial node automaticity (Le Scouarnec et al., 2008). Together these data link ankyrin-B dysfunction to sinus node disease and provide key mechanistic insights into molecular basis for the disease at the cellular level. Subsequent studies have provided new insight into the role of ankyrin-B in cardiac pacing at the level of the whole heart. This past year, regulation of automaticity in wild-type and ankyrin-B+/− mouse hearts was examined using optical mapping (Glukhov et al., 2010). As expected in wild-type mice, isoproterenol accelerated the sinoatrial rate and shifted the leading pacemaker site whereas acetylcholine had the opposite effect. In contrast, following isoproterenol, ankyrin-B+/− mice exhibited significant beat-to-beat variability (bradycardia followed by abrupt bursts of tachycardia), a disorganized shift of the leading pacemaker, and multiple competing pacemaker foci- phenotypes consistent with the human sinus node disease. Ankyrin-B+/− mouse hearts also displayed reduced sensitivity to acetylcholine. Collectively these data highlight the importance of the functional anatomy of the entire sinoatrial node pacemaker complex, and clearly demonstrate the role of ankyrin-B in cardiac automaticity in humans and in mice.
This past year, the role of ankyrin-B in cardiovascular electrophysiology and disease has continued to expand by the linkage of ankyrin-B dysfunction in human atrial fibrillation (AF; Cunha et al., 2011). Specifically, individuals harboring ANK2 loss-of-function alleles presented with a high incidence of early-onset AF, commonly progressing to permanent AF. In line with this finding, continuous telemetry ECG recordings from ankyrin-B-deficient mice revealed atrial arrhythmias similar to those observed in human carriers of ANK2 variants, including bradycardia with erratic atrial activity, lack of discrete P waves, and variable ventricular response (Cunha et al., 2011). Ankyrin-B+/− mice also showed increased susceptibility to induction of AFin response to atrial burst pacing. Primary atrial myocytes from ankyrin-B+/− mice displayed shortened action potentials, a hallmark of AF, and a sharp decrease of L-type Ca2+ current. Subsequent molecular studies determined that loss of L-type calcium current was due to loss of Cav1.3, but not Cav1.2 in atrial cardiomyocytes also explaining previously discovered reductions in ICa,L in sinoatrial node myocytes (Le Scouarnec et al., 2008). Moreover, Cav1.3 was identified as a novel ankyrin-B-interacting protein, and this direct interaction was shown to be mediated by a short motif in the C-terminus. As a final link between ankyrin-B and AF, analysis of human paroxysmal AF showed decreased ankyrin-B, as well as Cav1.3 expression. Together, these data provided a new molecular mechanism underlying congenital AF, a new binding partner for ankyrin-B in heart, and implicate ankyrin-B in the pathogenesis of common acquired forms of AF. These studies also provide insight into how defects in a single protein can result in a broad spectrum clinical disease with unique regional manifestations. In other words, the complexity of “ankyrin-B syndrome” arises from the fact that ankyrin-B is both broadly expressed in the heart and forms distinct, region-specific macromolecular complexes.
In addition to roles in congenital arrhythmias, ankyrin-B has more recently been linked with more common forms of heart disease. Electrical and structural remodeling in the peri-infarct zone creates a substrate favorable for the initiation and maintenance of reentrant arrhythmias following myocardial infarction, in particular, ventricular fibrillation and tachycardia. An essential component of this remodeling process involves the redistribution of ion channels and transporters in surviving myocytes near the infarct (Baba et al., 2005). Recently, ankyrin-B mRNA and protein levels, along with downstream ankyrin-B-associated channels were identified as differentially regulated following coronary artery occlusion in the canine (Hund et al., 2009). Specifically, a marked increase in ankyrin-B mRNA levels 5 days post-occlusion were seen, presumably in response to the observed decrease in protein levels of ankyrin-B at the same time. Protein levels and/or membrane expression of NCX, NKA, and IP3R were also decreased at 5 days post-occlusion, in line with their known role as ankyrin-B-binding partners (Hund et al., 2009). These changes were observed in absence of any changes to β2-spectrin. By 14 days post-occlusion, ankyrin-B protein levels had returned to baseline, and the recovery of NCX, NKA, and IP3R expression were, also, underway, consistent with the normalization of electrical remodeling observed in this model. While these studies focus on a chronic stage of myocardial infarction, ankyrin-B likely is involved in heart’s response to acute ischemia. Specifically, ankyrin-B plays a pivotal role in ATP-sensitive K+ channel (KATP) regulation. Ankyrin-B associates with the KATP channel via the major subunit, Kir6.2, a key component of the cellular apparatus required for intrinsic cardioprotection from ischemia (Li et al., 2010). Hearts and isolated cardiomyocytes lacking ankyrin-B display a loss of Kir6.2 membrane expression resulting in decreased membrane IK,ATP (Li et al., 2010). KATP channels serve a cardioprotective role in ischemia via a KATP channel-mediated shortening of the action potential (Suzuki et al., 2001), hence decreased expression of KATP could increase the susceptibility to arrhythmias. More recently, we have observed similar changes in ankyrin and related proteins (e.g., EHD family of trafficking proteins) in a variety of animal models of ischemic and non-ischemic disease and in human heart failure (Gudmundsson et al., 2010, 2012). In summary, these data suggest potential new roles for ankyrin-B in the processes of electrical and structural remodeling in common forms of heart disease (myocardial infarction/heart failure).
Clearly, loss of membrane ion channels and transporters is an important link between ankyrin dysfunction and disease. However, recent findings suggest that ankyrin-B regulation extends beyond these integral membrane proteins to cardiac signaling in health and disease. For example, in ventricular myocytes, ankyrin-B has also been identified as a binding partner of B56α, the regulatory subunit of PP2A (Bhasin et al., 2007). PP2A is a multifunctional serine/threonine phosphatase known to regulate cardiac β-adrenergic signaling (Klein et al., 2003; De Arcangelis et al., 2008). Cardiomyocytes lacking ankyrin-B expression display a marked loss of B56α (Bhasin et al., 2007). As inhibition of B56α in myocytes results in RyR2 hyper-phosphorylation [by calcium/calmodulin-dependent kinase II (CaMKII)] and myocyte electrical instability, it is reasonable to speculate that defects in CaMKII-based signaling pathways may contribute to human ankyrin-B-linked arrhythmias (Terentyev et al., 2009). In summary, work from the past decade has illustrated complex roles for ankyrin-B in myocyte function and in disease. The multifactorial nature of ankyrin-B and its broad distribution present unique challenges and opportunities as we consider the development of therapies to treat patients with inherited or acquired ankyrin-deficiency and associated disease.
While much focus has been placed on the role of ankyrin-B in heart due in part to the identified cohorts of human patients with ankyrin-B mutations, the influence of ankyrin polypeptides on heart function extends beyond ankyrin-B. Specifically, ankyrin-G has been shown to play roles in heart health and disease. Importantly, similar to its role in neurons, ankyrin-G has been shown to target Nav1.5, for maintenance of normal cardiac cell excitability. Ankyrin-G and Na channels directly associate and are co-localized in heart at the intercalated disk, where neighboring cells are electrically and mechanically coupled. Silencing of ankyrin-G in myocytes results in loss of Nav1.5 expression, membrane targeting, and membrane current (Lowe et al., 2008).
Defects in ankyrin-G-based targeting pathway have been linked to excitable cell disease. Interestingly, ankyrin-G has emerged as a candidate in multiple GWAS studies as associated with bipolar disorder (Ferreira et al., 2008 #2309; Scott et al., 2009 #5597). In heart, disruption of ankyrin-G/Nav1.5 interaction has been linked to the Brugada Syndrome, an autosomal-dominant, cardiac arrhythmia syndrome characterized by ST segment elevation in the precordial leads associated with right bundle branch block and T-wave inversion (Antzelevitch et al., 2005). SCN5A encodes Nav1.5, and gene variants account for nearly a third of Brugada Syndrome cases. SCN5A variants largely lead to biophysical defects that disrupt the inward sodium current, which is a key current in cellular depolarization during the action potential. A SCN5A variant (E1053K) that is localized to the ankyrin-G-binding motif in Nav1.5 and disrupts ankyrin-G/Nav1.5 association results in Brugada Syndrome (Mohler et al., 2004a). Mechanistically, this variant disrupts normal membrane targeting of Nav1.5 resulting in loss of membrane sodium channel current density and impaired excitability (Mohler et al., 2004a).
Beyond Nav1.5 membrane targeting, ankyrin-G plays additional roles in Nav1.5 membrane regulation via recruitment of Nav1.5 regulatory proteins. Specifically, Hund et al. (2010) recently demonstrated that βIV-spectrin is co-localized and interacts with ankyrin-G and Nav1.5 at the cardiac intercalated disk. Moreover, βIV-spectrin through direct binding, targets CaMKII to the intercalated disk to directly phosphorylate Nav1.5 at a specific residue (571) in the DI–DII linker. This study revealed that the βIV-spectrin/CaMKII complex has a direct effect on membrane excitability, and mutant βIV-spectrin mice lacking CaMKII binding activity (qv3J) displayed defects in Nav1.5 phosphorylation, activity, excitability, and heart function (Hund et al., 2010). Based on these initial findings, it is interesting to consider the potential role of βIV-spectrin/ankyrin-G-based signaling platform in regulating excitable cell function in human disease (Hund et al., 2010).
Apart from regulating membrane excitability, ankyrin-G was recently proposed to play secondary roles in regulation of intercalated disk structural integrity. Delmar and colleagues identified ankyrin-G as member of a larger complex of Nav1.5 and gap junctions at the cardiac desmosome (Sato et al., 2011). Loss of ankyrin-G caused significant changes in the subcellular distribution and abundance of desmosomal proteins. This complex could potentially play key roles in arrhythmia susceptibility as human mutations in desmosomal genes have been linked with the development of arrhythmogenic right ventricular dysplasia (Pilichou et al., 2006), characterized by inflammation, fibrosis or adiposis, and susceptibility to cardiac arrhythmia and sudden cardiac death (Sen-Chowdhry et al., 2010). In summary, based on the ability to regulate both critical electrical and structural signaling, ankyrin-G-based pathways have been shown to play key roles in myocyte physiology and likely are a central key node for human structural and electrical heart disease.
The past decade has illustrated critical roles for ankyrins in cardiac physiology and in disease. Unlike their classic roles in the erythrocyte, ankyrins play dynamic roles in protein targeting and stability, and secondary unexpected roles in protein regulation and cardiac signaling. While ankyrins are linked with congenital ventricular and atrial arrhythmias, current data suggest that these proteins serve central nodal points of regulation for common forms of acute and chronic cardiovascular disease. Moreover, based on expression, it is not unreasonable that ankyrins likely play key roles in other types of human cardiovascular disease including inflammation and metabolic and vascular disease.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported in part by a grant from the Saving Tiny Hearts Society (Peter J. Mohler). This work was also supported by the National Institutes of Health (HL084583, HL083422 to Peter J Mohler; HL096805 to Thomas J. Hund); and American Heart Association Established Investigator Award (Peter J. Mohler).
Abdi, K. M., Mohler, P. J., Davis, J. Q., and Bennett, V. (2006). Isoform specificity of ankyrin-B: a site in the divergent C-terminal domain is required for intramolecular association. J. Biol. Chem. 281, 5741–5749.
Antzelevitch, C., Brugada, P., Borggrefe, M., Brugada, J., Brugada, R., Corrado, D., Gussak, I., Lemarec, H., Nademanee, K., Perez Riera, A. R., Shimizu, W., Schulze-Bahr, E., Tan, H., and Wilde, A. (2005). Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 111, 659–670.
Baba, S., Dun, W., Cabo, C., and Boyden, P. A. (2005). Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation 112, 2386–2396.
Bennett, V. (1979). Immunoreactive forms of human erythrocyte ankyrin are present in diverse cells and tissues. Nature 281, 597–599.
Bennett, V., and Chen, L. (2001). Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr. Opin. Cell Biol. 13, 61–67.
Bennett, V., and Healy, J. (2009). Membrane domains based on ankyrin and spectrin associated with cell-cell interactions. Cold Spring Harb. Perspect. Biol. 1, a003012.
Bernstein, A. D., and Parsonnet, V. (1992). Survey of cardiac pacing in the United States in 1989. Am. J. Cardiol. 69, 331–338.
Bhasin, N., Cunha, S. R., Mudannayake, M., Gigena, M. S., Rogers, T. B., and Mohler, P. J. (2007). Molecular basis for PP2A regulatory subunit B56alpha targeting in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 293, H109–H119.
Camors, E., Mohler, P. J., Bers, D. M., and Despa, S. (2012). Ankyrin-B reduction enhances Ca spark-mediated SR Ca release promoting cardiac myocyte arrhythmic activity. J. Mol. Cell. Cardiol. PMID:22406428. [Epub ahead of print].
Chauhan, V. S., Tuvia, S., Buhusi, M., Bennett, V., and Grant, A. O. (2000). Abnormal cardiac Na(+) channel properties and QT heart rate adaptation in neonatal ankyrin(B) knockout mice. Circ. Res. 86, 441–447.
Cunha, S. R., Hund, T. J., Hashemi, S., Voigt, N., Li, N., Wright, P., Koval, O., Li, J., Gudmundsson, H., Gumina, R. J., Karck, M., Schott, J. J., Probst, V., Le Marec, H., Anderson, M. E., Dobrev, D., Wehrens, X. H., and Mohler, P. J. (2011). Defects in ankyrin-based membrane protein targeting pathways underlie atrial fibrillation. Circulation 124, 1212–1222.
Cunha, S. R., and Mohler, P. J. (2006). Cardiac ankyrins: essential components for development and maintenance of excitable membrane domains in heart. Cardiovasc. Res. 71, 22–29.
Cunha, S. R., and Mohler, P. J. (2008). Obscurin targets ankyrin-B and protein phosphatase 2A to the cardiac M-line. J. Biol. Chem. 283, 31968–31980.
De Arcangelis, V., Soto, D., and Xiang, Y. (2008). Phosphodiesterase 4 and phosphatase 2A differentially regulate cAMP/protein kinase a signaling for cardiac myocyte contraction under stimulation of beta1 adrenergic receptor. Mol. Pharmacol. 74, 1453–1462.
Ferreira, M. A., O’Donovan, M. C., Meng, Y. A., Jones, I. R., Ruderfer, D. M., Jones, L., Fan, J., Kirov, G., Perlis, R. H., Green, E. K., Smoller, J. W., Grozeva, D., Stone, J., Nikolov, I., Chambert, K., Hamshere, M. L., Nimgaonkar, V. L., Moskvina, V., Thase, M. E., Caesar, S., Sachs, G. S., Franklin, J., Gordon-Smith, K., Ardlie, K. G., Gabriel, S. B., Fraser, C., Blumenstiel, B., Defelice, M., Breen, G., Gill, M., Morris, D. W., Elkin, A., Muir, W. J., McGhee, K. A., Williamson, R., MacIntyre, D. J., MacLean, A. W., St, C. D., Robinson, M., Van Beck, M., Pereira, A. C., Kandaswamy, R., McQuillin, A., Collier, D. A., Bass, N. J., Young, A. H., Lawrence, J., Ferrier, I. N., Anjorin, A., Farmer, A., Curtis, D., Scolnick, E. M., McGuffin, P., Daly, M. J., Corvin, A. P., Holmans, P. A., Blackwood, D. H., Gurling, H. M., Owen, M. J., Purcell, S. M., Sklar, P., Craddock, N., and Wellcome Trust Case Control Consortium. (2008). Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 40, 1056–1058.
Glukhov, A. V., Fedorov, V. V., Anderson, M. E., Mohler, P. J., and Efimov, I. R. (2010). Functional anatomy of the murine sinus node: high-resolution optical mapping of ankyrin-B heterozygous mice. Am. J. Physiol. Heart Circ. Physiol. 299, H482–H491.
Gudmundsson, H., Curran, J., Kashef, F., Snyder, J. S., Smith, S. A., Vargas-Pinto, P., Bonilla, I. M., Weiss, R. M., Anderson, M. E., Binkley, P., Felder, R. B., Carnes, C. A., Band, H., Hund, T. J., and Mohler, P. J. (2012). Differential regulation of EHD3 in human and mammalian heart failure. J. Mol. Cell. Cardiol. 52, 1183–1190.
Gudmundsson, H., Hund, T. J., Wright, P. J., Kline, C. F., Snyder, J. S., Qian, L., Koval, O. M., Cunha, S. R., George, M., Rainey, M. A., Kashef, F. E., Dun, W., Boyden, P. A., Anderson, M. E., Band, H., and Mohler, P. J. (2010). EH domain proteins regulate cardiac membrane protein targeting. Circ. Res. 107, 84–95.
Hashemi, S. M., Hund, T. J., and Mohler, P. J. (2009). Cardiac ankyrins in health and disease. J. Mol. Cell. Cardiol. 47, 203–209.
Hund, T. J., Koval, O. M., Li, J., Wright, P. J., Qian, L., Snyder, J. S., Gudmundsson, H., Kline, C. F., Davidson, N. P., Cardona, N., Rasband, M. N., Anderson, M. E., and Mohler, P. J. (2010). A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 120, 3508–3519.
Hund, T. J., Wright, P. J., Dun, W., Snyder, J. S., Boyden, P. A., and Mohler, P. J. (2009). Regulation of the ankyrin-B-based targeting pathway following myocardial infarction. Cardiovasc. Res. 81, 742–749.
Klein, G., Schroder, F., Vogler, D., Schaefer, A., Haverich, A., Schieffer, B., Korte, T., and Drexler, H. (2003). Increased open probability of single cardiac L-type calcium channels in patients with chronic atrial fibrillation. Role of phosphatase 2A. Cardiovasc. Res. 59, 37–45.
Kline, C. F., Kurata, H. T., Hund, T. J., Cunha, S. R., Koval, O. M., Wright, P. J., Christensen, M., Anderson, M. E., Nichols, C. G., and Mohler, P. J. (2009). Dual role of K ATP channel C-terminal motif in membrane targeting and metabolic regulation. Proc. Natl. Acad. Sci. U.S.A. 106, 16669–16674.
Kontrogianni-Konstantopoulos, A., Jones, E. M., Van Rossum, D. B., and Bloch, R. J. (2003). Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol. Biol. Cell 14, 1138–1148.
Le Scouarnec, S., Bhasin, N., Vieyres, C., Hund, T. J., Cunha, S. R., Koval, O., Marionneau, C., Chen, B., Wu, Y., Demolombe, S., Song, L. S., Le Marec, H., Probst, V., Schott, J. J., Anderson, M. E., and Mohler, P. J. (2008). Dysfunction in ankyrin-B-dependent ion channel and transporter targeting causes human sinus node disease. Proc. Natl. Acad. Sci. U.S.A. 105, 15617–15622.
Li, J., Kline, C. F., Hund, T. J., Anderson, M. E., and Mohler, P. J. (2010). Ankyrin-B regulates Kir6.2 membrane expression and function in heart. J. Biol. Chem. 285, 28723–28730.
Lowe, J. S., Palygin, O., Bhasin, N., Hund, T. J., Boyden, P. A., Shibata, E., Anderson, M. E., and Mohler, P. J. (2008). Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J. Cell Biol. 180, 173–186.
Mohler, P. J., Davis, J. Q., and Bennett, V. (2005). Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP(3) receptor in a cardiac T-Tubule/SR microdomain. PLoS Biol. 3, e423. doi:10.1371/journal.pbio.0030423
Mohler, P. J., Le Scouarnec, S., Denjoy, I., Lowe, J. S., Guicheney, P., Caron, L., Driskell, I. M., Schott, J. J., Norris, K., Leenhardt, A., Kim, R. B., Escande, D., and Roden, D. M. (2007). Defining the cellular phenotype of “ankyrin-B syndrome” variants: human ANK2 variants associated with clinical phenotypes display a spectrum of activities in cardiomyocytes. Circulation 115, 432–441.
Mohler, P. J., Rivolta, I., Napolitano, C., Lemaillet, G., Lambert, S., Priori, S. G., and Bennett, V. (2004a). Nav1.5E. 1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 101, 17533–17538.
Mohler, P. J., Splawski, I., Napolitano, C., Bottelli, G., Sharpe, L., Timothy, K., Priori, S. G., Keating, M. T., and Bennett, V. (2004b). A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl. Acad. Sci. U.S.A. 101, 9137–9142.
Mohler, P. J., Yoon, W., and Bennett, V. (2004c). Ankyrin-B targets {beta}2-spectrin to an intracellular compartment in neonatal cardiomyocytes. J. Biol. Chem. 279, 40185–40193.
Mohler, P. J., Schott, J. J., Gramolini, A. O., Dilly, K. W., Guatimosim, S., Dubell, W. H., Song, L. S., Haurogne, K., Kyndt, F., Ali, M. E., Rogers, T. B., Lederer, W. J., Escande, D., Le Marec, H., and Bennett, V. (2003). Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421, 634–639.
Pilichou, K., Nava, A., Basso, C., Beffagna, G., Bauce, B., Lorenzon, A., Frigo, G., Vettori, A., Valente, M., Towbin, J., Thiene, G., Danieli, G. A., and Rampazzo, A. (2006). Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113, 1171–1179.
Sato, P. Y., Coombs, W., Lin, X., Nekrasova, O., Green, K. J., Isom, L. L., Taffet, S. M., and Delmar, M. (2011). Interactions between ankyrin-G, plakophilin-2, and connexin43 at the cardiac intercalated disc. Circ. Res. 109, 193–201.
Scott, L. J., Muglia, P., Kong, X. Q., Guan, W., Flickinger, M., Upmanyu, R., Tozzi, F., Li, J. Z., Burmeister, M., Absher, D., Thompson, R. C., Francks, C., Meng, F., Antoniades, A., Southwick, A. M., Schatzberg, A. F., Bunney, W. E., Barchas, J. D., Jones, E. G., Day, R., Matthews, K., McGuffin, P., Strauss, J. S., Kennedy, J. L., Middleton, L., Roses, A. D., Watson, S. J., Vincent, J. B., Myers, R. M., Farmer, A. E., Akil, H., Burns, D. K., and Boehnke, M. (2009). Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc. Natl. Acad. Sci. U. S. A. 106, 7501–7506.
Sedlacek, K., Stark, K., Cunha, S. R., Pfeufer, A., Weber, S., Berger, I., Perz, S., Kaab, S., Wichmann, H. E., Mohler, P. J., Hengstenberg, C., and Jeron, A. (2008). Common genetic variants in ANK2 modulate QT interval: results from the KORA Study. Circ. Cardiovasc. Genet. 1, 93–99.
Sen-Chowdhry, S., Syrris, P., Pantazis, A., Quarta, G., Mckenna, W. J., and Chambers, J. C. (2010). Mutational heterogeneity, modifier genes, and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ. Cardiovasc. Genet. 3, 323–330.
Splawski, I., Shen, J., Timothy, K. W., Lehmann, M. H., Priori, S., Robinson, J. L., Moss, A. J., Schwartz, P. J., Towbin, J. A., Vincent, G. M., and Keating, M. T. (2000). Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102, 1178–1185.
Suzuki, M., Li, R. A., Miki, T., Uemura, H., Sakamoto, N., Ohmoto-Sekine, Y., Tamagawa, M., Ogura, T., Seino, S., Marban, E., and Nakaya, H. (2001). Functional roles of cardiac and vascular ATP-sensitive potassium channels clarified by Kir6.2-knockout mice. Circ. Res. 88, 570–577.
Terentyev, D., Belevych, A. E., Terentyeva, R., Martin, M. M., Malana, G. E., Kuhn, D. E., Abdellatif, M., Feldman, D. S., Elton, T. S., and Gyorke, S. (2009). MiR-1 overexpression enhances Ca2+ release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56{alpha} and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ. Res. 104, 514–521.
Keywords: ankyrin, spectrin, arrhythmia, cytoskeleton, mouse model
Citation: Smith S, Curran J, Hund TJ and Mohler PJ (2012) Defects in cytoskeletal signaling pathways, arrhythmia, and sudden cardiac death. Front. Physio. 3:122. doi: 10.3389/fphys.2012.00122
Received: 18 March 2012; Paper pending published: 05 April 2012;
Accepted: 12 April 2012; Published online: 04 May 2012.
Edited by:
Carol Ann Remme, University of Amsterdam, NetherlandsReviewed by:
Hugues Abriel, University of Lausanne, SwitzerlandCopyright: © 2012 Smith, Curran, Hund and Mohler. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Peter J. Mohler, Dorothy M. Davis Heart and Lung Research Institute, The Ohio State University Medical Center, 473 W. 12th Avenue, 110G DHLRI, Columbus, OH 43210, USA. e-mail:cGV0ZXIubW9obGVyQG9zdW1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.