 Ihsan Ekin Demir
Ihsan Ekin Demir Helmut Friess and Güralp O. Ceyhan*
Helmut Friess and Güralp O. Ceyhan*- Department of Surgery, Klinikum rechts der Isar, Technische Universität München, Munich, Germany
Interaction of cancer cells with diverse cell types in the tumor stroma is today recognized to have a fate-determining role for the progression and outcome of human cancers. Despite the well-described interactions of cancer cells with several stromal components, i.e., inflammatory cells, cancer-associated fibroblasts, endothelial cells, and pericytes, the investigation of their peculiar relationship with neural cells is still at its first footsteps. Pancreatic cancer (PCa) with its abundant stroma represents one of the best-studied examples of a malignant tumor with a mutually trophic interaction between cancer cells and the intratumoral nerves embedded in the desmoplastic stroma. Nerves in PCa are a rich source of neurotrophic factors like nerve growth factor (NGF), glial-cell-derived neurotrophic factor (GDNF), artemin; of neuronal chemokines like fractalkine; and of autonomic neurotransmitters like norepinephrine which can all enhance the invasiveness of PCa cells via matrix-metalloproteinase (MMP) upregulation, trigger neural invasion (NI), and activate pro-survival signaling pathways. Similarly, PCa cells themselves provide intrapancreatic nerves with abundant trophic agents which entail a remarkable neuroplasticity, leading to emergence of more routes for NI and cancer spread, to augmented local neuro-surveillance, neural sensitization, and neuropathic pain. The strong correlation of NI with PCa-associated desmoplasia suggests the potential presence of a triangular relationship between nerves, PCa cells, and other stromal partners like myofibroblasts and pancreatic stellate cells which generate tumor desmoplasia. Hence, although not a classical hallmark of human cancers, nerve-cancer interactions can be considered as an indispensable sub-class of cancer-stroma interactions in PCa. The present article provides an overview of the so far known nerve-cancer interactions in PCa and illustrates their ominous role in the stromal biology of human PCa.
Introduction
Lessons from cancer studies of the past decade have made an important contribution to our understanding of the general features of cancer by pointing out to the role of “tumor microenvironment” in cancer progression (Hanahan and Weinberg, 2000). In the “next generation” of hallmarks of cancer, what is considered a new hallmark is the interaction of cancer cells with apparently normal cells in their surroundings which potentiate their own tumorigenic capacity (Hanahan and Weinberg, 2011). These non-neoplastic cells acquire an activated state during cancer progression and maintain the continuous growth and spread of the malignant tumor (Dvorak, 1986; Riss et al., 2006; Egeblad et al., 2010). In most cases, these cells of the tumor microenvironment belong to the tumor-associated inflammatory cells which secrete growth factors, pro-angiogenic factors, and extracellular matrix-degrading enzymes which sustain invasion and metastasis (de Visser et al., 2006; de Visser, 2008). Hence, the logical role of the immune system in the eradication of malignant cells seems to be accompanied by a concomitant tumor-promoting property of inflammation, especially for those cancer cells that can escape from immune destruction (de Visser et al., 2006; de Visser, 2008; DeNardo et al., 2010).
However, cancer cells undergo an exploitative interaction not only with immune cells, but also with several other cell types which are present in their direct environment, termed stroma, i.e., interstitium, of the organ of tumor origin (Dvorak, 1986; Pietras and Ostman, 2010). Stroma, both in normal and neoplastic tissues, is composed of non-cellular and cellular components (Dvorak, 1986; Pietras and Ostman, 2010; Hanahan and Weinberg, 2011). The non-cellular part consists of deposits of large extracellular matrix proteins like collagen, fibronectin, laminin, etc. The cellular compartment comprises immune cells, fibroblasts, vascular and lymphatic endothelial cells, pericytes, and cells of peripheral nerves (Dvorak, 1986; Pietras and Ostman, 2010; Hanahan and Weinberg, 2011). All these cells have been the focus of investigation in the past 10 years due to their ability to promote tumor growth and metastasis (Dvorak, 1986; Pietras and Ostman, 2010). In addition to the above-mentioned role of immune cells, cancer-associated fibroblasts – or stellate cells as they are commonly referred to in the pancreas and the liver – have been shown to provide cancer cells with numerous growth factors, cytokines, and matrix-degrading enzymes which enhance the proliferation, invasion, and metastasis of cancer cells (Vonlaufen et al., 2008b; Rasanen and Vaheri, 2010). Similarly, endothelial cells are increasingly recruited during tumor angiogenesis, display distinct surface expression patterns for growth factor receptors and allow for tumor nourishment and dissemination (Nagy et al., 2010). As a novel niche of cancer research, pericytes have gained attention as potential “gatekeepers” in tumor metastasis due to their role as cells regulating vessel permeability and integrity (Raza et al., 2010; Hanahan and Weinberg, 2011).
Looking at the diversity of cells which have been studied in the tumor stroma, an emerging cell type is represented by cells of peripheral nerves. Although the study of their interaction with cancer is still at its very beginning, the human cancer which probably reflects the highest degree of nerve-cancer interactions and has thus been subject to most intense investigation is pancreatic cancer (PCa; Ceyhan et al., 2009b, 2010; Demir et al., 2010a). Termed “ductal adenocarcinoma of the pancreas,” PCa as one of the most aggressive human malignancies is characterized by pronounced tumor desmoplasia, and the associated reduction in intratumoral vessel density is considered the leading cause for the limited local delivery of chemotherapeutics into the tumor (Olive et al., 2009). In this “enlarged” microenvironment of PCa cells, what is a frequent striking observation are the local nerve alterations in the intra- and peritumoral tissue (Ceyhan et al., 2009a; Demir et al., 2010a). Research from the past 10 years uncovered several crucial features of nerve-cancer interactions in PCa, including neural plasticity, neural invasion (NI), and the associated neuropathic pain syndrome in PCa (Ceyhan et al., 2009a; Demir et al., 2010a). Just like increased appreciation of the role of the members of tumor stroma, neural cells are increasingly perceived as major modulators of tumor growth and spread (Entschladen et al., 2008). The present review aims at providing an overview of the so far elucidated interactions of intrapancreatic nerves with PCa cells and with other members of the pancreatic tumor stroma and raises several important questions related to these exciting interactions.
Nerve-Derived Factors Enhance Pancreatic Cancer Cell Invasiveness
A possibility for nerves to influence the growth of PCa was considered in conjunction with the frequently observed invasion of nerves by PCa cells (Zhu et al., 1999, 2001). This NI – or often denoted as perineural invasion due to the frequent alignment of PCa cells around the perineurium – is encountered in nearly 100% of PCa cases upon targeted histopathological inspection of surgical specimens (Hirai et al., 2002; Liu and Lu, 2002; Ceyhan et al., 2009a; Demir et al., 2010a; Bapat et al., 2011). In addition to being another route of cancer spread, NI bears a major clinical relevance: it is considered the foremost reason for local tumor recurrence after curative resection and correlates to the abdominal pain sensation of PCa patients (Kayahara et al., 1991, 1993, 1995; Ozaki et al., 1999). In an effort to address the pathogenesis of NI in PCa, Zhu and Friess et al. investigated the expression of the best known nerve-derived growth factor, i.e., nerve growth factor (NGF), in human PCa, motivated by the previously shown proliferative effect of NGF upon lung cancer, prostate cancer, and glioblastoma cells (Zhu et al., 1999, 2002). They could identify a close correlation with intratumoral NGF expression, the frequency of NI, and the degree of pain (Zhu et al., 1999). Subsequent studies showed that NGF and its receptors TrkA and p75NTR are expressed not only by nerves, but also by PCa cells themselves, making them responsive to autocrine NGF signaling and paracrine NGF effects upon its release from intrapancreatic nerves (Zhu et al., 2002; Ceyhan et al., 2010). Indeed, supplementation of NGF into the growth medium of PCa cells or stable NGF-transfection into PCa cells exerts a prominent enhancer effect upon proliferation and invasiveness of PCa cells (Zhu et al., 2001, 2002).
This demonstration of tumor-supporting effects of nerve-derived factors like NGF was followed by analysis of further neural factors in terms of their similar capabilities. Particularly, members of the glial-cell-derived neurotrophic factor (GDNF) family like GDNF and artemin have also been shown to increase the invasive capabilities of PCa cells (Figure 1; Funahashi et al., 2003, 2005; Okada et al., 2003; Takahashi et al., 2004; Zeng et al., 2008). For GDNF, the mechanisms of this tumorigenicity have been addressed in several studies. Especially, GDNF has been shown to activate phosphatidylinositol 3-kinase (PI3-K)/Akt, extracellular signal-regulated kinase (ERK; Veit et al., 2004), and nuclear factor kappa B (Takahashi et al., 2004) over RET activation and to increase the expression of matrix-metalloproteinases type 2 and 9 as direct mediators of invasion (Okada et al., 2003, 2004). The common observation in these studies is the prominent impact of neurotrophic factors upon the invasiveness of PCa cells, with an occasionally detected enhancement of proliferation. However, the even more important deduction is the capability of nerves to influence and actually enhance PCa spread, just like other components of the tumor stroma.
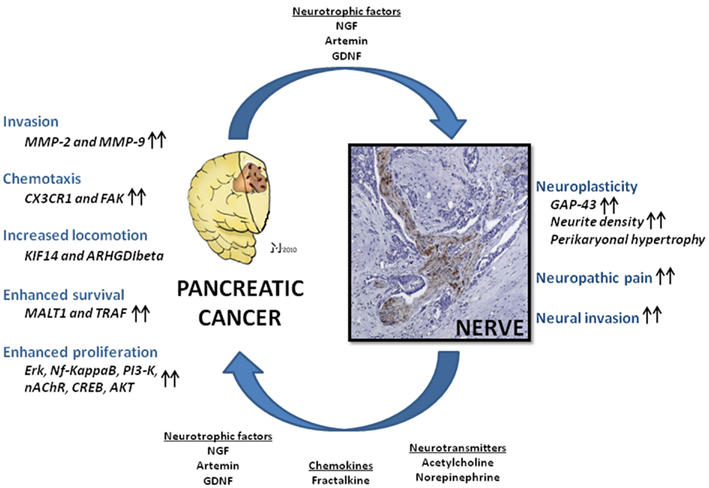
Figure 1. Nerve-cancer interactions in human pancreatic cancer (PCa). Neural cells and PCa cells undergo a mutually growth-supporting interaction. Nerve-derived molecules like neurotrophic factors, neurotransmitters, and chemokines are capable of enhancing PCa cell tumorigenicity in several aspects, including invasiveness, cancer cell chemotaxis, locomotion, proliferation, and cell survival via upregulation of numerous signaling pathways. In analogy, PCa cells secrete neuromodulatory agents which induce neuroplasticity, augment neural invasion, and neuropathic pain sensation. NGF, nerve growth factor; GDNF, glial-cell-derived neurotrophic factor; Nf-kappaB, nuclear factor kappa B; PI3-K, phosphoinositide-3-kinase; nAChR, nicotinic acetylcholine receptor; CREB, cAMP response element-binding; AKT, protein kinase B; MMP-2 and -9, matrix-metalloproteinase-2 and -9; FAK, focal adhesion kinase; Erk, extracellular signal-regulated kinase; MALT1, mucosa-associated lymphoid tissue lymphoma translocation gene 1; TRAF, tumor-necrosis-factor-receptor-associated factor; KIF14, kinesin family member 14 (KIF14); ARHGDIbeta, RHO-GDP dissociation inhibitor-β; GAP-43, growth-associated protein 43. Please refer to the manuscript for the respective references.
Nerves and Pancreatic Cancer Cells Undergo a Mutually Trophic Interaction
The expression of neurotrophic factors by PCa cells and nerves at the same time implies that PCa cells may make use of the same repertoire of trophic signals as nerves do. Hence, once in proximity, these two cell types would potentially establish a mutually beneficial relationship. Indeed, this relationship is best seen in invasion of nerves by PCa cells (Ceyhan et al., 2008a). Although PCa cells have been postulated to damage nerves during this peculiar invasion process (Bockman et al., 1994), surgical pathologists have steadily observed extension of PCa cell colonies along intrapancreatic nerves toward extrapancreatic neural plexus (Kayahara et al., 1991, 2007). Therefore, a true “destruction” of nerves by PCa cells has not been conclusively confirmed, and actually, several in vitro studies provide evidence for the exact opposite: in the past 10 years, several groups aimed at establishing novel in vitro and in vivo models with the aim of simulating NI in PCa (Eibl and Reber, 2005; Dai et al., 2007; Ceyhan et al., 2008a; Gil et al., 2008, 2010). These studies typically harbor heterotypic cell culture settings where PCa cells and neurons, usually derived from rodent dorsal root ganglia (DRG), are confronted with each other and observed for their interaction (Gil et al., 2010). In all these settings, researchers repeatedly detected targeted migration of PCa cells toward neurons, which extend their neurites toward the migrating PCa cells. Once together, PCa cells continue their migration along the DRG neurites, resembling a cell–cell adhesion (Dai et al., 2007; Ceyhan et al., 2008a; Gil et al., 2010). In one of these studies, a clear growth advantage, i.e., increased proliferation and colony of formation, was shown for PCa cells which built contacts with DRG neurites (Dai et al., 2007). Similarly, the density of DRG neurites significantly increased toward migrating PCa cell colonies (Dai et al., 2007; Ceyhan et al., 2008a). Further evidence for this mutual growth support was found in the upregulation of pro-survival genes, i.e. mucosa-associated lymphoid tissue lymphoma translocation gene 1 (MALT1) and tumor-necrosis-factor-receptor-associated factor (TRAF), in PCa cells which were confronted with DRG neurons (Figure 1; Dai et al., 2007). Thus, neural cells are today considered to be a further cancer-supporting cell in PCa stroma and microenvironment (Demir et al., 2010a). Importantly, nerves benefit from this effect also for their own growth advantage since PCa cells can also provide them with the same trophic signals (Demir et al., 2010a). Hence, nerve-cancer interactions in PCa stroma represent another major example of an “unholy alliance” in PCa, as previously shown for other stromal cells (Vonlaufen et al., 2008b).
Nerves Mediate Enhanced Pancreatic Cancer Cell Chemoattraction and Motility
For the mutually trophic interaction between nerves and cancer cells to occur, nerves and PCa cells must exchange signals to ensure their proximity. These signals would typically constitute agents with chemoattractive attributes. In this context, the abundance of neurotrophic signals in nerves and in PCa cells themselves represents not only a trophic support, but actually also a major mechanism of mutual chemoattraction due to the intrinsic chemoattractive properties of neurotrophins and GDNF family of neurotrophic factors (Tang et al., 1998; Paratcha et al., 2006; Zhou et al., 2007; Cornejo et al., 2010; Mwizerwa et al., 2011). These factors are known to be chemotactic upon neuronal precursors, cerebellar cell precursors, glia cells, and enteric neural crest precursors (Tang et al., 1998; Paratcha et al., 2006; Zhou et al., 2007; Cornejo et al., 2010; Mwizerwa et al., 2011). Furthermore, neurons from GDNF-knockout mice have reduced ability to attract PCa cells (Gil et al., 2010). Hence, neurotrophic factor secretion from intrapancreatic nerves and the associated chemoattraction of PCa cells toward nerves is assumed to be one of the initiating events in the nerve-cancer interactions and the entailing NI (Ceyhan et al., 2006, 2008a; Demir et al., 2010a).
Besides neurotrophic factors, there is recent evidence for PCa cell recruitment toward nerves via more classical chemoattractants like chemokines. The chemokine profile of nerves is still subject to further investigation. However, one of the well-characterized neuronal chemokines, fractalkine, bears the unique feature of possessing a single and specific receptor, i.e., CX3CR1, which has recently been demonstrated to be expressed on PCa cells (Marchesi et al., 2010). In detail, PCa cell lines which express CX3CR1 migrate and adhere to neuroblastoma cells, a process that involves beta1-integrin receptors and activation of focal adhesion kinase (Marchesi et al., 2010). In vivo, transfection of MiaPaCa2–PCa cells with CX3CR1 results in increased subcutaneous NI in nude mice (Marchesi et al., 2010). Moreover, increased CX3CR1 expression in PCa tissues is strongly associated with more prominent NI and earlier local tumor recurrence (Marchesi et al., 2010). Overall, these findings provide strong evidence for a key role of chemokine signaling in the mutual attraction of nerves and PCa cells and their resulting interaction (Figure 1). Further studies on the role of other chemokines in the nerve-cancer attraction in PCa are still lacking.
In accordance with the tumor-attractive properties of nerves, PCa cells respond to these neural signals by upregulation of their locomotor machinery (Abiatari et al., 2009a,b,c). In an ex vivo NI model, Abiatari et al. (2009a) determined the transcriptome profile of PCa cells which were co-cultivated with explanted rat vagal nerves. Under these conditions, PCa cell clones which showed NI were compared to non-invasive clones in terms of their differentially regulated genes. Here, the investigators identified increased expression of kinesin family member 14 (KIF14) and RHO-GDP dissociation inhibitor-β (ARHGDIbeta), and the siRNA-mediated specific knockdown of these cytoskeletal molecules significantly inhibited NI (Abiatari et al., 2009a). In a subsequent study, the same group identified microtubule-associated protein RP/EB family member 2 (MAPRE2) as another factor which is overexpressed by neural invasive PCa cell clones and associated with worse survival rates in PCa patients (Figure 1; Abiatari et al., 2009b). Overall, these studies underline the importance of PCa motility factors in the generation of NI and clearly show that intrapancreatic nerves are capable of strongly contributing to the local tumor progression in PCa by chemo-attracting PCa cells and activating their locomotion. Therefore, based on our knowledge to date, nerves solely have tumor-promoting effects in PCa and are therefore one of the “best partners in crime” for PCa cells (Legg, 2011).
Nerve-Cancer Interactions in Pancreatic Cancer Induce Neuroplasticity and Neuropathic Pain
The tumor-promoting effects of nerves in PCa and the trophic support of PCa cells for nerves result in complex neuroplastic alterations in the PCa stroma. When analyzed systematically, PCa specimens frequently harbor increased neural density and intrapancreatic neural hypertrophy (Figure 2; Ceyhan et al., 2009a; Demir et al., 2010a). Furthermore, this neuroplasticity is encountered in no other pancreatic tumor, either benign or malignant, at this extent as seen in PCa (Ceyhan et al., 2009a; Demir et al., 2010a). Growth-associated-protein-43, which is known to be overexpressed following neural damage or after long-term potentiation in the brain, is much more prevalent in PCa tissue than in the normal pancreas or other pancreatic tumor entities (Ceyhan et al., 2009a; Demir et al., 2010a). Recently, these alterations have been demonstrated to be present in the apparently normal pancreatic tissue adjacent to the pancreatic tumor, where a specific upregulation of NGF and artemin have been observed within intrapancreatic nerves (Ceyhan et al., 2010).
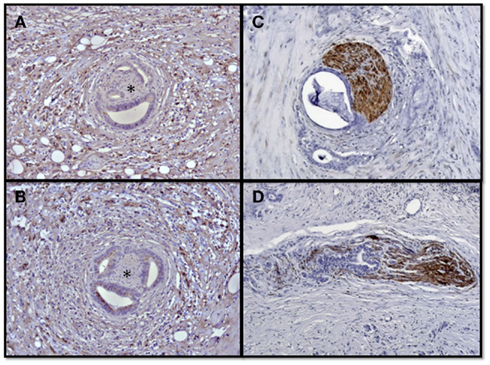
Figure 2. Neural invasion and neuroplasticity in the desmoplastic PCa stroma. Human PCa specimens contain hypertrophic nerves which are victims of neural invasion and frequently encountered in the desmoplastic tumor stroma. The asterisks demonstrate nerves surrounded by invading PCa cells in the activated perineural, immunolabeled tumor desmoplasia (A,B). This neural invasion is either present as perineural invasion (C) along their epi- or perineurium, or as endoneural invasion which destructs the neural integrity (D).
In order to enable functional studies on the pathomechanism of this neuroplasticity, a recent novel in vitro neuroplasticity assay was established (Demir et al., 2010b). Here, neurons from different sources, i.e., sensory neurons from DRG and myenteric neurons of the Auerbach’s plexus, have been cultivated under conditions resembling PCa tissue, particularly within PCa tissue extracts or within supernatants of human PCa cell lines (Demir et al., 2010b). Interestingly, these media exert a potent neurotrophic effect upon neurons as they significantly increase their neurite density, axonal branching capacity, and entail a perikaryonal hypertrophy (Demir et al., 2010b). Hence, novel functional studies help to recapitulate the major characteristics of pancreatic neuroplasticity in human PCa.
Besides being a remarkable example of disease-associated visceral neuroplasticity, these alterations seem to bear a unique association with the generation of the pain in PCa. Indeed, neural hypertrophy is particularly detected in patients with increasing severity of pain sensation (Ceyhan et al., 2009a). This association becomes further interesting when considered in the context of above-described, mutually trophic nerve-cancer cell interactions. As seen under in vitro co-culture conditions, NI and pain sensation of PCa patients correlate to neural hypertrophy and increased neural density in the PCa tissue (Ceyhan et al., 2009a). Therefore, it seems that the mutually trophic interaction between PCa cells and nerves has a major role in the generation of the pain in PCa. Based on these multifold interactions, but also due to the previously reported nerve damage which can be detected upon ultrastructural analysis of PCa tissues, pain in PCa is increasingly considered to be of neuropathic origin (Ceyhan et al., 2008b, 2009a, 2010; Demir et al., 2010a). While neuropathic pain medications are not in common use to treat PCa-associated pain, minimally invasive, interventional techniques like celiac plexus neurolysis or thoracic splanchnicectomy which intervene with the transmission of ongoing pain stimuli from the periphery, i.e., the cancerous pancreatic tissue, have been proven to be effective to treat neuropathic pain in advanced PCa (Stefaniak et al., 2005; Yan and Myers, 2007).
Intriguingly, emergence of pain as a result of nerve-cancer interactions in PCa raises important questions related to the biological significance of pain sensation in these patients. Normally, pain serves as a natural warning signal for the central nervous system, alarming the presence of a noxious process in the organ of origin (Fregni et al., 2007). Hence, the reactive response of the organism to this stimulus would be to activate noxi-clearing surveillance agents like inflammation (Fregni et al., 2007). However, in fact, PCa cells do not seem to be subject to any disadvantage out of this signaling: indeed, neuropathic pain sensation in PCa is associated with much worse survival when compared to its absence (Ceyhan et al., 2009a). This observation suggests that the pain-generating events which seem to be initiated by PCa cells eventually yield further support for PCa cells in their local and systemic spread. Hence, an “unintended” tumor-promoting character may not be unique to the direct microenvironment of the tumor within the pancreatic tissue, but may also be a feature of distant homeostatic organ systems like the central nervous system. This possibility as clearly observed in the context of PCa deserves future investigation to elucidate the contribution of “pain” to the biology of cancer.
Neurotransmitters Modulate Pancreatic Cancer Cell Biology
To what extent the nervous system can influence disease states has long been an issue of debate. Interestingly, early studies already demonstrated that antagonism of the sympathetic nervous system and parasympathomimetic agents are capable of ameliorating the course of experimental cancer, including chemically induced gastric cancer and colon cancer (Tatsuta et al., 1988, 1989a,b, 1990, 1992). In the era of research on oncogenic pathways and genetic deciphering, the modulatory role of nervous system upon cancer biology seems to have been somewhat neglected. Interestingly, there has been a revival of interest in cancer neuromodulation, owing to the discovery of neurotransmitter receptors on cancer cells (Schuller et al., 2001, 2008a,b; Weddle et al., 2001; Al-Wadei and Schuller, 2009; Al-Wadei et al., 2009b). In particular, cancer cells of the lung, colon, and pancreas have been shown to express nicotinic and muscarinic acetylcholine receptors of the parasympathetic nervous system, and the beta-adrenergic receptors of the sympathetic nervous system (Schuller et al., 2001; Wong et al., 2007; Schuller, 2008; Pettersson et al., 2009; Wu et al., 2011). In this context, the ability of nitrosamines which are constituents of tobacco to activate nicotinic acetylcholine receptors (nAChRs) and induce enhanced cancer cell proliferation and invasiveness has been crucial for our understanding of the association between tobacco smoking and carcinogenesis (Schuller, 2008). These studies demonstrated that activation of α3, α5, and α7-nAChRs leads to intracellular phosphorylation of growth-supporting pathways like CREB, ERK, Src, and AKT (Al-Wadei et al., 2011). Hence, in contrast with earlier studies where systemic administration of parasympathomimetics inhibited cancer progression, these studies showed that activation of cholinergic receptors may actually also lead to enhanced cancer cell growth (Schuller and Al-Wadei, 2010; Al-Wadei et al., 2011). These at first glance paradoxical findings reached recent harmony in the studies by Al-Wadei et al. (2011) who showed the cross-talk of beta-adrenergic signaling with nicotine-induced activation of nAChRs in PCa. Indeed, activation of these receptors results in the release of cellular stress-neurotransmitters which bind to beta-adrenergic receptors in an autocrine fashion and activate tumorigenic signaling pathways (Al-Wadei et al., 2011). In accordance, administration of propanolol, a non-selective beta-adrenergic receptor blocker, results in attenuation of experimental PCa (Al-Wadei et al., 2009a), whereas chronic neuropsychological stress exacerbated the tumor via increased systemic cathecholamine levels and subsequent activation of nAChRs (Schuller et al., 2011). Similar growth-promoting effects have also been demonstrated for acetylcholine which can significantly increase the proliferation and invasive ability of, e.g., colon cancer cells (Cheng et al., 2008; Pettersson et al., 2009; Xie et al., 2009). Moreover, there is increasing evidence that the autonomic nervous system may also influence tumor angiogenesis. Increased stress was demonstrated to lead to increased angiogenesis in a murine model of ovarian carcinoma through vascular endothelial growth factor (VEGF) upregulation (Thaker et al., 2006; Lee et al., 2009; Tilan and Kitlinska, 2010). Conversely, the norepinephrine precursor dopamine was shown inhibit angiogenesis by prevention of VEGF receptor phosphorylation and mitogen-activated protein kinase phosphorylation (Asada et al., 2008). In conjunction with the tumor-promoting properties of tobacco smoking, nicotine was shown to increase neo-vascularization over nAChRs in, e.g., lung cancer and gastric cancer (Natori et al., 2003; Shin et al., 2004; Jarzynka et al., 2006). In colon cancer, it was shown that 5-HT can support tumor angiogenesis by reduction of MMP-12 expression in tumor-associated macrophages, decreasing the levels of the anti-angiogenetic mediator angiostatin (Nocito et al., 2008). So far, similar data from the angiogenetic properties of neurotransmitters in PCa are lacking. Still, it was previously demonstrated that the naturally occurring perivascular sympathetic nerve fibers are lost around intratumoral vessels in PCa, suggesting a potential neural dysregulation around tumor neo-vascularization (Ceyhan et al., 2009b). Overall, it becomes increasingly clear that neurotransmitters, whether from the sympathetic or parasympathetic nervous system, can remarkably enhance PCa cell growth, angiogenesis, and metastasis by augmenting the release of growth factors, angiogenetic factors, and pro-inflammatory cytokines (Schuller, 2008; Schuller and Al-Wadei, 2010). Therefore, owing to all these recent studies, neurotransmitters are likely to be an ongoing focus of investigation in future studies on the interaction of PCa with the primary source of these neurotransmitters in its stroma, i.e., intrapancreatic nerves. However, what deserves particular attention in this regard is the recently demonstrated remodeling of the pancreatic autonomic innervation in PCa (Ceyhan et al., 2009b). Analysis of nerve fiber qualities in PCa revealed that both sympathetic and cholinergic nerve fibers are prominently suppressed as part of the neuroplastic alterations in the pancreas (Ceyhan et al., 2009b). This “neural remodeling” implies that the local activity of the autonomic nervous system may be subject to an inhibition, which is why studies on the effect of (autonomic) neurotransmitters on PCa cells and in murine PCa models which do not reflect pancreatic neuroplasticity should be interpreted and translated cautiously.
The “Third Party” in Nerve-Cancer Interactions: Desmoplasia and Pancreatic Stellate Cells
The above-described neural alterations in PCa suggest that the direct interaction of PCa cells with nerves can bring about increased neural density, neural hypertrophy, and NI by PCa cells via neurotrophic factor and chemokine signaling. The possibility of other agents or even other cell types which may play a mediator role in the interaction between nerves and PCa cells has so far not been genuinely considered. This possibility rises out of recent observations on the actual localization of such alterations within the PCa tissue. In detail, there is a firm correlation between the degree of tumor desmoplasia and the severity of NI in PCa (Ceyhan et al., 2009a; Figure 2). This observation underlines that desmoplasia may be a factor triggering increased PCa cell invasiveness and thus NI. Desmoplasia, i.e. increased production of dense extracellular matrix around tumor cells, is a hallmark of PCa (Erkan et al., 2008, 2010; Olive et al., 2009; Pandol et al., 2009). It is estimated that over 80% of the actual tumor mass in PCa consists of tumor desmoplasia (Erkan et al., 2010). The key role of tumor desmoplasia on the biology of PCa is best represented by findings from recent studies targeting signaling pathways which contribute to the generation of tumor desmoplasia. These studies convincingly demonstrated enhanced chemotherapeutic agent delivery into the tumor and improved survival upon depletion of PCa-associated desmoplasia upon Hedgehog inhibition in transgenic mouse models of PCa (Olive et al., 2009). In the past decade, research on PCa-associated desmoplasia turned its focus onto the role of cancer-associated myofibroblasts in PCa, i.e., pancreatic stellate cells (PSCs), which have been shown to be the foremost producer of tumor desmoplasia in their activated state (Apte et al., 1998; Haber et al., 1999; Apte and Wilson, 2004). Besides producing extracellular matrix components like type I collagen, fibronectin and periostin, these cells represent another major tumor growth-promoting cell type in the PCa microenvironment (Omary et al., 2007; Vonlaufen et al., 2008b). Particularly, PSCs produce growth factors like PDGF which can increase the proliferation of PCa cells, and induce epithelial–mesenchymal transition (EMT) in PCa cells and thereby enhance their invasiveness via transforming growth factor-β (Omary et al., 2007). Moreover, PSCs have also been shown to influence tumor angiogenesis by augmenting the endostatin production of PCa cells under co-culture and by increasing their own VEGF secretion under hypoxia (Erkan et al., 2009). In accordance with these tumorigenic properties of PSCs, Vonlaufen et al. (2008a) demonstrated increased tumor size, metastasis, and desmoplasia in athymic mice which were co-injected with human PSCs and PCa cells as opposed to solitary injection of PCa cells. Furthermore, co-injected PSCs were also observed to readily extravasate during the metastatic spread of the pancreatic tumor and the associated angiogenesis (Xu et al., 2010). There is further evidence that PCa cells which are treated with PSCs-conditioned medium are more resistant to chemotherapy with gemcitabine or to radiotherapy (Hwang et al., 2008). Interestingly, PSCs can also secrete high levels of matrix-metalloproteinase-2, even much higher than the level produced by PCa cells, thereby potentially enhancing their in-tissue invasiveness (Schneiderhan et al., 2007). Overall, the tumor-promoting effects of PSCs represent one of the leading “unholy” interactions in PCa microenvironment (Vonlaufen et al., 2008b).
As cells which are particularly important for the aggressive behavior of PCa cells, PSCs have recently been subject to investigation in terms of their interaction with stromal nerves (Figure 3). Particularly, there is increasing evidence for the presence of intrinsic neurotrophic attributes of PSCs (Demir et al., 2010b). In an in vitro neuroplasticity model, PSCs derived from human chronic pancreatitis tissues could prominently enhance the neurite outgrowth of isolated DRG neurons and myenteric plexus cells. Even though this neurotrophic effect was not achieved by supernatants of PSCs from PCa, PSCs derived from PCa could noticeably induce dense neural networks among isolated DRG neurons and increase their neurite density (Demir et al., 2010b). This important effect could not be seen in PSCs derived from chronic pancreatitis patients. However, studies using PSCs derived from human pathological tissues bear the assumption that these cells preserve their in-tissue expression and secretory profiles after their isolation and in vitro cultivation. Therefore, in vitro studies on isolated PSCs may not allow a conclusive statement on the actual neurotrophic potential of PSCs within PCa tissue. The possibility of PSCs as a producer of neurotrophic agents has recently been demonstrated by Haas et al. (2009) who could show increased NGF production over ALK5-activiation following stimulation of PSCs with TGF-β. Similarly, PSCs from chronic pancreatitis tissues also upregulate their expression of the neurotrophic factor artemin when exposed to TGF-β (Ceyhan et al., 2007). While such a neurotrophic factor upregulation may well influence PCa cell invasiveness, proliferation, and contribute to local repair/regenerative processes, its exact contribution to intrapancreatic neuroplastic alterations remains to be determined, especially in the absence of any major correlation of desmoplasia to neural density, size, and pancreatic neuro-inflammation (pancreatic neuritis) in PCa (Ceyhan et al., 2009a). In fact, desmoplastic tissue areas in PCa have even been shown to exhibit a loss of the normal parenchymal nerve fiber network within the dense tumor stroma (Samkharadze et al., 2011). Still, this loss of small-caliber nerves fibers was accompanied by the emergence of hypertrophic, GAP-43-immunoreactive nerves embedded in the tumor stroma (Samkharadze et al., 2011). At this point, it should be considered that PSCs, in particular the abundant ECM components that they produce in PCa stroma, may exert a strong trophic effect upon neurons and their nerve fibers. It is well established that ECM components like collagen, laminin, and fibronectin can considerably enhance neurite formation by neurons (Fujii et al., 1982). For example, on type I collagen or heparan sulfate-collagen surfaces, neurons build dense neuronal networks as opposed to dermatan sulfate (DS), chondroitin-6-sulfate (C6S), or hyaluronic acid (HA)-bound collagen substrates. Interestingly, it was shown that neurites avoid surfaces coated with DS, C6S, or HA in order to remain in contact with collagen (Fujii et al., 1982). In chick embryo explants, collagen types I and III supported neuritogenesis at least as efficiently as laminin, whereas this effect was less pronounced for collagen type V and absent for types II, IV, and VI (Carri et al., 1992). Considering the abundant production of collagen type I by activated PSCs, it is conceivable that this particular stroma in PCa may be one of the leading factors in the neural sprouting and neural hypertrophy in PCa. The exact phenotype of these “novel” nerve fibers is still subject to investigation. Nevertheless, these sprouting large nerve trunks were shown to undergo the above-mentioned remodeling, characterized by suppression of sympathetic and cholinergic nerve fibers, especially in nerves which are invaded PCa cells (Ceyhan et al., 2009b). Moreover, this suppression was also identified to be even more prominent in tissues derived from PCa patients who reported to have abdominal pain (Ceyhan et al., 2009b). Hence, in this remodeling process where neurons alter their chemical code, it seems that at the interface of the interaction between PSCs and neurons, there is a suppression of autonomic nerve fibers. The functional implications of this neural remodeling in the desmoplastic tumor stroma in PCa remain to be elucidated.
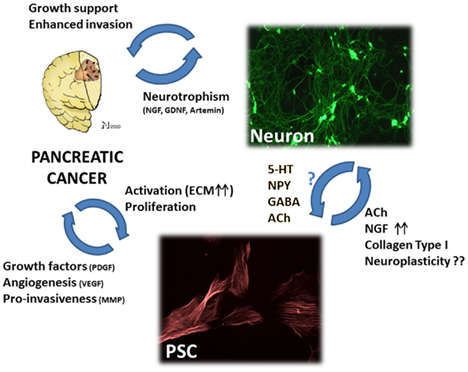
Figure 3. The hypothetic triangular relationship between nerves, PCa cells, and pancreatic stellate cells (PSC). PCa cells benefit not only from nerves, but also from PSCs which can – in their activated state – increase PCa cell growth, invasiveness, and tumor angiogenesis. Moreover, activated PSCs can provide neural cells with neurotrophic factors like NGF and neurotransmitters which can modulate neuronal activity. Conversely, there is yet insufficient evidence for a modulating activity of nerve-derived molecules upon PSC activity. Still, studies on hepatic stellate cells demonstrated that neurotransmitters including 5-hydroxytryptophan (5-HT), neuropeptide Y, gamma-amino butyric acid (GABA), and acetylcholine (ACh) can significantly increase the fibrogenic activity, proliferation, and calcium currents of stellate cells. Right upper image: myenteric plexus neurons treated with neurotrophic PCa tissue extracts, immunostained against the neuronal marker beta-III-tubulin. Lower image: rat pancreatic stellate cells immunostained against alpha smooth muscle actin. PDGF, platelet-derived growth factor; VEGF, vascular endothelial growth factor; MMP, matrix-metalloproteinase; ACh, acetylcholine; NGF, nerve growth factor; 5-HT, 5-hydroxytryptophan; NPY, neuropeptide Y; GABA, gamma-amino butyric acid; ECM, extracellular matrix; GDNF, glial-cell-derived neurotrophic factor. Please refer to the manuscript for the respective references.
While these findings suggest a potential, at least indirect influence of desmoplasia upon pancreatic neuropathy in PCa, there is recent evidence for a direct cross-signaling between PSCs and nerves, as PSCs have been demonstrated to release acetylcholine (ACh), the main parasympathetic neurotransmitter (Figure 3; Phillips et al., 2010). Strikingly, PSCs possess the complete cellular ACh synthesis and release machinery which can get activated by cholecystokinin (CCK; Phillips et al., 2010). Owing to this neurotransmitter, PSCs can induce amylase secretion from pancreatic acini (Phillips et al., 2010). Besides demonstrating a novel mechanism for CCK-induced exocrine secretion in pancreas, this study points out to the profound ability of PSCs to directly interact with intrapancreatic nerves (Phillips et al., 2010).
The extent of interactions of PSCs with neural cells, in particular with neurons has not yet been to subject to detailed investigation. On a theoretical basis, seeing the ACh production by PSCs, one can imagine that PSCs may similarly possess receptors for nerve-derived molecules, in particular for a large battery of neurotransmitters which are produced by intrinsic pancreatic neurons as derivatives of myenteric neurons (Figure 3). These neurotransmitters include calcitonin-gene-related-peptide (CGRP), vasoactive intestinal peptide (VIP), substance P, 5-hydroxytrytophan (5-HT), acetylcholine, and norepinephrine. It has to be underlined that the presence of the respective receptors for these neurotransmitters has not yet been demonstrated in PSCs. However, research on liver fibrogenesis has delineated the presence of several of these receptors on hepatic stellate cells (HSCs). It is known that HSCs can build calcium currents and thus get activated by 5-HT via the 5HT2 receptor (Park et al., 2011). Furthermore, rat and human HSCs possess the 5-HT1B, 5-HT1F 5-HT2A 5-HT2B, and 5-HT7 receptors, where 5-HT2B was shown to closely correlate to the degree of fibrosis in experimental liver fibrosis induced by carbon tetrachloride (Ruddell et al., 2006). Conversely, the GABA(B) receptor agonist baclofen can inhibit the myofibroblastic differentiation, ECM production, and TGFβ1-secretion by HSCs (Xiao et al., 2010). For adrenergic signaling, it was shown that activated human HSC express alpha(1A)-adrenoceptors which can lead to rapid calcium currents following norepinephrine-induced activation (Sancho-Bru et al., 2006). Moreover, norepinephrine can induce phosphorylation of myosin light chain II, contraction of HSCs and promote the secretion of pro-inflammatory cytokines from HSCs (Oben and Diehl, 2004; Oben et al., 2004; Sancho-Bru et al., 2006). A similar support of proliferation for HSCs was also reported for neuropeptide Y and acetylcholine (Oben et al., 2003a,b). Overall, even though similar data from PSCs are missing, the multitude of previously demonstrated effects of neurotransmitters upon HSCs suggests that a similar neuromodulation may also well exist for PSCs. Moreover, a very recent study on the interconvertibility of fibroblast-like cells and neurons (Yoo et al., 2011) adds another dimension to this exciting interaction, implying a potentially direct contribution of PSCs to pancreatic neuroplasticity in PCa. However, whether such microRNA signals are indeed present in PCa tissue (Yoo et al., 2011), and whether intrapancreatic myofibroblast-like cells can generate neurons of extrinsic origin (i.e., residing in DRG) which represent the most likely source of neuroplasticity, remain an issue of debate. Regarding all these novel findings and as suggested by the consensus report from a recent consensus conference on PSC research (StellaTUM), future studies are awaited that shall elucidate the interactions of PSCs with the peripheral nervous system in the pancreas (Erkan et al., 2011).
Summary and Conclusion
Tumor stroma in PCa harbors a wide spectrum of cells which, in their adaptive response to carcinogenesis and tumor growth, activate signaling mechanisms that paradoxically promote tumor invasion and metastasis. Despite remarkable advances in our understanding of the interaction of PCa cells with stromal fibroblasts, endothelial cells, pericytes, and inflammatory cells, nerve-cancer interactions in PCa still deserve much further and intensive investigation in order to elucidate the impact of nerves as an important stromal partner to PCa cells. Like several other stromal cells, neural cells potentiate PCa aggressiveness, particularly via (1) neurotrophic factor and autonomic neurotransmitter signaling which can be proliferative, pro-angiogenetic, and pro-invasive, (2) release of chemoattractive agents, and (3) serving as a protective, shelter path of tumor dissemination. Certainly, these features are in part similarly observed attributes of other stromal cell types and compartments. However, nerves and their inhabitants bear the unique ability of combining all these features as a single partner of interaction. Therefore, future research should not neglect the contribution of nerves to PCa cell and stromal biology. The interaction of nerves with PCa, but also with other stromal partners including angiogenesis, desmoplasia, and cancer-associated fibroblasts, is a fascinating niche of research, and not only their abundant electric potentials, but also the major therapeutic potential imbued in nerves in PCa deserves considerable attention.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Matthias Maak for his assistance with the generation of the figures in the manuscript.
References
Abiatari, I., Deoliveira, T., Kerkadze, V., Schwager, C., Esposito, I., Giese, N. A., Huber, P., Bergman, F., Abdollahi, A., Friess, H., and Kleeff, J. (2009a). Consensus transcriptome signature of perineural invasion in pancreatic carcinoma. Mol. Cancer Ther. 8, 1494–1504.
Abiatari, I., Gillen, S., Deoliveira, T., Klose, T., Bo, K., Giese, N. A., Friess, H., and Kleeff, J. (2009b). The microtubule-associated protein MAPRE2 is involved in perineural invasion of pancreatic cancer cells. Int. J. Oncol. 35, 1111–1116.
Abiatari, I., Kiladze, M., Kerkadze, V., Friess, H., and Kleeff, J. (2009c). Expression of YPEL1 in pancreatic cancer cell lines and tissues. Georgian Med. News 175, 60–62.
Al-Wadei, H. A., Al-Wadei, M. H., and Schuller, H. M. (2009a). Prevention of pancreatic cancer by the beta-blocker propranolol. Anticancer Drugs 20, 477–482.
Al-Wadei, H. A., Plummer, H. K. III, and Schuller, H. M. (2009b). Nicotine stimulates pancreatic cancer xenografts by systemic increase in stress neurotransmitters and suppression of the inhibitory neurotransmitter gamma-aminobutyric acid. Carcinogenesis 30, 506–511.
Al-Wadei, H. A., and Schuller, H. M. (2009). Nicotinic receptor-associated modulation of stimulatory and inhibitory neurotransmitters in NNK-induced adenocarcinoma of the lungs and pancreas. J. Pathol. 218, 437–445.
Al-Wadei, M. H., Al-Wadei, H. A., and Schuller, H. M. (2011). Pancreatic cancer cells and normal pancreatic duct epithelial cells express an autocrine catecholamine loop that is activated by nicotinic acetylcholine receptors alpha3, alpha5, and alpha7. Mol. Cancer Res. 10, 239–249.
Apte, M. V., Haber, P. S., Applegate, T. L., Norton, I. D., Mccaughan, G. W., Korsten, M. A., Pirola, R. C., and Wilson, J. S. (1998). Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 43, 128–133.
Asada, M., Ebihara, S., Numachi, Y., Okazaki, T., Yamanda, S., Ikeda, K., Yasuda, H., Sora, I., and Arai, H. (2008). Reduced tumor growth in a mouse model of schizophrenia, lacking the dopamine transporter. Int. J. Cancer 123, 511–518.
Bapat, A. A., Hostetter, G., Von Hoff, D. D., and Han, H. (2011). Perineural invasion and associated pain in pancreatic cancer. Nat. Rev. Cancer 11, 695–707.
Bockman, D. E., Buchler, M., and Beger, H. G. (1994). Interaction of pancreatic ductal carcinoma with nerves leads to nerve damage. Gastroenterology 107, 219–230.
Carri, N. G., Rubin, K., Gullberg, D., and Ebendal, T. (1992). Neuritogenesis on collagen substrates. Involvement of integrin-like matrix receptors in retinal fibre outgrowth on collagen. Int. J. Dev. Neurosci. 10, 393–405.
Ceyhan, G. O., Bergmann, F., Kadihasanoglu, M., Altintas, B., Demir, I. E., Hinz, U., Muller, M. W., Giese, T., Buchler, M. W., Giese, N. A., and Friess, H. (2009a). Pancreatic neuropathy and neuropathic pain – a comprehensive pathomorphological study of 546 cases. Gastroenterology 136, 177–186.e1.
Ceyhan, G. O., Demir, I. E., Rauch, U., Bergmann, F., Muller, M. W., Buchler, M. W., Friess, H., and Schafer, K. H. (2009b). Pancreatic neuropathy results in “neural remodeling” and altered pancreatic innervation in chronic pancreatitis and pancreatic cancer. Am. J. Gastroenterol. 104, 2555–2565.
Ceyhan, G. O., Bergmann, F., Kadihasanoglu, M., Erkan, M., Park, W., Hinz, U., Giese, T., Muller, M. W., Buchler, M. W., Giese, N. A., and Friess, H. (2007). The neurotrophic factor artemin influences the extent of neural damage and growth in chronic pancreatitis. Gut 56, 534–544.
Ceyhan, G. O., Demir, I. E., Altintas, B., Rauch, U., Thiel, G., Muller, M. W., Giese, N. A., Friess, H., and Schafer, K. H. (2008a). Neural invasion in pancreatic cancer: a mutual tropism between neurons and cancer cells. Biochem. Biophys. Res. Commun. 374, 442–447.
Ceyhan, G. O., Michalski, C. W., Demir, I. E., Muller, M. W., and Friess, H. (2008b). Pancreatic pain. Best Pract. Res. Clin. Gastroenterol. 22, 31–44.
Ceyhan, G. O., Giese, N. A., Erkan, M., Kerscher, A. G., Wente, M. N., Giese, T., Buchler, M. W., and Friess, H. (2006). The neurotrophic factor artemin promotes pancreatic cancer invasion. Ann. Surg. 244, 274–281.
Ceyhan, G. O., Schafer, K. H., Kerscher, A. G., Rauch, U., Demir, I. E., Kadihasanoglu, M., Bohm, C., Muller, M. W., Buchler, M. W., Giese, N. A., Erkan, M., and Friess, H. (2010). Nerve growth factor and artemin are paracrine mediators of pancreatic neuropathy in pancreatic adenocarcinoma. Ann. Surg. 251, 923–931.
Cheng, K., Samimi, R., Xie, G., Shant, J., Drachenberg, C., Wade, M., Davis, R. J., Nomikos, G., and Raufman, J. P. (2008). Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G591–G597.
Cornejo, M., Nambi, D., Walheim, C., Somerville, M., Walker, J., Kim, L., Ollison, L., Diamante, G., Vyawahare, S., and De Bellard, M. E. (2010). Effect of NRG1, GDNF, EGF and NGF in the migration of a Schwann cell precursor line. Neurochem. Res. 35, 1643–1651.
Dai, H., Li, R., Wheeler, T., Ozen, M., Ittmann, M., Anderson, M., Wang, Y., Rowley, D., Younes, M., and Ayala, G. E. (2007). Enhanced survival in perineural invasion of pancreatic cancer: an in vitro approach. Hum. Pathol. 38, 299–307.
de Visser, K. E. (2008). Spontaneous immune responses to sporadic tumors: tumor-promoting, tumor-protective or both? Cancer Immunol. Immunother. 57, 1531–1539.
de Visser, K. E., Eichten, A., and Coussens, L. M. (2006). Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 6, 24–37.
Demir, I. E., Ceyhan, G. O., Liebl, F., D’Haese, J. G., Maak, M., and Friess, H. (2010a). Neural invasion in pancreatic cancer: the past, present and future. Cancers 2, 1513–1527.
Demir, I. E., Ceyhan, G. O., Rauch, U., Altintas, B., Klotz, M., Muller, M. W., Buchler, M. W., Friess, H., and Schafer, K. H. (2010b). The microenvironment in chronic pancreatitis and pancreatic cancer induces neuronal plasticity. Neurogastroenterol. Motil. 22, 480–490.
DeNardo, D. G., Andreu, P., and Coussens, L. M. (2010). Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 29, 309–316.
Dvorak, H. F. (1986). Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659.
Egeblad, M., Nakasone, E. S., and Werb, Z. (2010). Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell 18, 884–901.
Eibl, G., and Reber, H. A. (2005). A xenograft nude mouse model for perineural invasion and recurrence in pancreatic cancer. Pancreas 31, 258–262.
Entschladen, F., Palm, D., Niggemann, B., and Zaenker, K. S. (2008). The cancer’s nervous tooth: considering the neuronal crosstalk within tumors. Semin. Cancer Biol. 18, 171–175.
Erkan, M., Adler, G., Apte, M. V., Bachem, M. G., Buchholz, M., Detlefsen, S., Esposito, I., Friess, H., Gress, T. M., Habisch, H. J., Hwang, R. F., Jaster, R., Kleeff, J., Kloppel, G., Kordes, C., Logsdon, C. D., Masamune, A., Michalski, C. W., Oh, J., Phillips, P. A., Pinzani, M., Reiser-Erkan, C., Tsukamoto, H., and Wilson, J. (2011). StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 61, 172–178.
Erkan, M., Michalski, C. W., Rieder, S., Reiser-Erkan, C., Abiatari, I., Kolb, A., Giese, N. A., Esposito, I., Friess, H., and Kleeff, J. (2008). The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 6, 1155–1161.
Erkan, M., Reiser-Erkan, C., Michalski, C. W., Deucker, S., Sauliunaite, D., Streit, S., Esposito, I., Friess, H., and Kleeff, J. (2009). Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 11, 497–508.
Erkan, M., Reiser-Erkan, C., Michalski, C. W., and Kleeff, J. (2010). Tumor microenvironment and progression of pancreatic cancer. Exp. Oncol. 32, 128–131.
Fregni, F., Pascual-Leone, A., and Freedman, S. D. (2007). Pain in chronic pancreatitis: a salutogenic mechanism or a maladaptive brain response? Pancreatology 7, 411–422.
Fujii, D. K., Massoglia, S. L., Savion, N., and Gospodarowicz, D. (1982). Neurite outgrowth and protein synthesis by PC12 cells as a function of substratum and nerve growth factor. J. Neurosci. 2, 1157–1175.
Funahashi, H., Okada, Y., Sawai, H., Takahashi, H., Matsuo, Y., Takeyama, H., and Manabe, T. (2005). The role of glial cell line-derived neurotrophic factor (GDNF) and integrins for invasion and metastasis in human pancreatic cancer cells. J. Surg. Oncol. 91, 77–83.
Funahashi, H., Takeyama, H., Sawai, H., Furuta, A., Sato, M., Okada, Y., Hayakawa, T., Tanaka, M., and Manabe, T. (2003). Alteration of integrin expression by glial cell line-derived neurotrophic factor (GDNF) in human pancreatic cancer cells. Pancreas 27, 190–196.
Gil, Z., Cavel, O., Kelly, K., Brader, P., Rein, A., Gao, S. P., Carlson, D. L., Shah, J. P., Fong, Y., and Wong, R. J. (2010). Paracrine regulation of pancreatic cancer cell invasion by peripheral nerves. J. Natl. Cancer Inst. 102, 107–118.
Gil, Z., Kelly, K. J., Brader, P., Shah, J. P., Fong, Y., and Wong, R. J. (2008). Utility of a herpes oncolytic virus for the detection of neural invasion by cancer. Neoplasia 10, 347–353.
Haas, S. L., Fitzner, B., Jaster, R., Wiercinska, E., Gaitantzi, H., Jesnowski, R., Lohr, J. M., Singer, M. V., Dooley, S., and Breitkopf, K. (2009). Transforming growth factor-beta induces nerve growth factor expression in pancreatic stellate cells by activation of the ALK-5 pathway. Growth Factors 27, 289–299.
Haber, P. S., Keogh, G. W., Apte, M. V., Moran, C. S., Stewart, N. L., Crawford, D. H., Pirola, R. C., Mccaughan, G. W., Ramm, G. A., and Wilson, J. S. (1999). Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am. J. Pathol. 155, 1087–1095.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Hirai, I., Kimura, W., Ozawa, K., Kudo, S., Suto, K., Kuzu, H., and Fuse, A. (2002). Perineural invasion in pancreatic cancer. Pancreas 24, 15–25.
Hwang, R. F., Moore, T., Arumugam, T., Ramachandran, V., Amos, K. D., Rivera, A., Ji, B., Evans, D. B., and Logsdon, C. D. (2008). Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 68, 918–926.
Jarzynka, M. J., Guo, P., Bar-Joseph, I., Hu, B., and Cheng, S. Y. (2006). Estradiol and nicotine exposure enhances A549 bronchioloalveolar carcinoma xenograft growth in mice through the stimulation of angiogenesis. Int. J. Oncol. 28, 337–344.
Kayahara, M., Nagakawa, T., Konishi, I., Ueno, K., Ohta, T., and Miyazaki, I. (1991). Clinicopathological study of pancreatic carcinoma with particular reference to the invasion of the extrapancreatic neural plexus. Int. J. Pancreatol. 10, 105–111.
Kayahara, M., Nagakawa, T., Ueno, K., Ohta, T., Takeda, T., and Miyazaki, I. (1993). An evaluation of radical resection for pancreatic cancer based on the mode of recurrence as determined by autopsy and diagnostic imaging. Cancer 72, 2118–2123.
Kayahara, M., Nagakawa, T., Ueno, K., Ohta, T., Tsukioka, Y., and Miyazaki, I. (1995). Surgical strategy for carcinoma of the pancreas head area based on clinicopathologic analysis of nodal involvement and plexus invasion. Surgery 117, 616–623.
Kayahara, M., Nakagawara, H., Kitagawa, H., and Ohta, T. (2007). The nature of neural invasion by pancreatic cancer. Pancreas 35, 218–223.
Lee, J. W., Shahzad, M. M., Lin, Y. G., Armaiz-Pena, G., Mangala, L. S., Han, H. D., Kim, H. S., Nam, E. J., Jennings, N. B., Halder, J., Nick, A. M., Stone, R. L., Lu, C., Lutgendorf, S. K., Cole, S. W., Lokshin, A. E., and Sood, A. K. (2009). Surgical stress promotes tumor growth in ovarian carcinoma. Clin. Cancer Res. 15, 2695–2702.
Marchesi, F., Piemonti, L., Mantovani, A., and Allavena, P. (2010). Molecular mechanisms of perineural invasion, a forgotten pathway of dissemination and metastasis. Cytokine Growth Factor Rev. 21, 77–82.
Mwizerwa, O., Das, P., Nagy, N., Akbareian, S. E., Mably, J. D., and Goldstein, A. M. (2011). Gdnf is mitogenic, neurotrophic, and chemoattractive to enteric neural crest cells in the embryonic colon. Dev. Dyn. 240, 1402–1411.
Nagy, J. A., Chang, S. H., Shih, S. C., Dvorak, A. M., and Dvorak, H. F. (2010). Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 36, 321–331.
Natori, T., Sata, M., Washida, M., Hirata, Y., Nagai, R., and Makuuchi, M. (2003). Nicotine enhances neovascularization and promotes tumor growth. Mol. Cells 16, 143–146.
Nocito, A., Dahm, F., Jochum, W., Jang, J. H., Georgiev, P., Bader, M., Graf, R., and Clavien, P. A. (2008). Serotonin regulates macrophage-mediated angiogenesis in a mouse model of colon cancer allografts. Cancer Res. 68, 5152–5158.
Oben, J. A., and Diehl, A. M. (2004). Sympathetic nervous system regulation of liver repair. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 280, 874–883.
Oben, J. A., Roskams, T., Yang, S., Lin, H., Sinelli, N., Torbenson, M., Smedh, U., Moran, T. H., Li, Z., Huang, J., Thomas, S. A., and Diehl, A. M. (2004). Hepatic fibrogenesis requires sympathetic neurotransmitters. Gut 53, 438–445.
Oben, J. A., Yang, S., Lin, H., Ono, M., and Diehl, A. M. (2003a). Acetylcholine promotes the proliferation and collagen gene expression of myofibroblastic hepatic stellate cells. Biochem. Biophys. Res. Commun. 300, 172–177.
Oben, J. A., Yang, S., Lin, H., Ono, M., and Diehl, A. M. (2003b). Norepinephrine and neuropeptide Y promote proliferation and collagen gene expression of hepatic myofibroblastic stellate cells. Biochem. Biophys. Res. Commun. 302, 685–690.
Okada, Y., Eibl, G., Duffy, J. P., Reber, H. A., and Hines, O. J. (2003). Glial cell-derived neurotrophic factor upregulates the expression and activation of matrix metalloproteinase-9 in human pancreatic cancer. Surgery 134, 293–299.
Okada, Y., Eibl, G., Guha, S., Duffy, J. P., Reber, H. A., and Hines, O. J. (2004). Nerve growth factor stimulates MMP-2 expression and activity and increases invasion by human pancreatic cancer cells. Clin. Exp. Metastasis 21, 285–292.
Olive, K. P., Jacobetz, M. A., Davidson, C. J., Gopinathan, A., Mcintyre, D., Honess, D., Madhu, B., Goldgraben, M. A., Caldwell, M. E., Allard, D., Frese, K. K., Denicola, G., Feig, C., Combs, C., Winter, S. P., Ireland-Zecchini, H., Reichelt, S., Howat, W. J., Chang, A., Dhara, M., Wang, L., Ruckert, F., Grutzmann, R., Pilarsky, C., Izeradjene, K., Hingorani, S. R., Huang, P., Davies, S. E., Plunkett, W., Egorin, M., Hruban, R. H., Whitebread, N., Mcgovern, K., Adams, J., Iacobuzio-Donahue, C., Griffiths, J., and Tuveson, D. A. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461.
Omary, M. B., Lugea, A., Lowe, A. W., and Pandol, S. J. (2007). The pancreatic stellate cell: a star on the rise in pancreatic diseases. J. Clin. Invest. 117, 50–59.
Ozaki, H., Hiraoka, T., Mizumoto, R., Matsuno, S., Matsumoto, Y., Nakayama, T., Tsunoda, T., Suzuki, T., Monden, M., Saitoh, Y., Yamauchi, H., and Ogata, Y. (1999). The prognostic significance of lymph node metastasis and intrapancreatic perineural invasion in pancreatic cancer after curative resection. Surg. Today 29, 16–22.
Pandol, S., Edderkaoui, M., Gukovsky, I., Lugea, A., and Gukovskaya, A. (2009). Desmoplasia of pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 7, S44–S47.
Paratcha, G., Ibanez, C. F., and Ledda, F. (2006). GDNF is a chemoattractant factor for neuronal precursor cells in the rostral migratory stream. Mol. Cell. Neurosci. 31, 505–514.
Park, K. S., Sin, P. J., Lee, D. H., Cha, S. K., Kim, M. J., Kim, N. H., Baik, S. K., Jeong, S. W., and Kong, I. D. (2011). Switching-on of serotonergic calcium signaling in activated hepatic stellate cells. World J. Gastroenterol. 17, 164–173.
Pettersson, A., Nilsson, L., Nylund, G., Khorram-Manesh, A., Nordgren, S., and Delbro, D. S. (2009). Is acetylcholine an autocrine/paracrine growth factor via the nicotinic alpha7-receptor subtype in the human colon cancer cell line HT-29? Eur. J. Pharmacol. 609, 27–33.
Phillips, P. A., Yang, L., Shulkes, A., Vonlaufen, A., Poljak, A., Bustamante, S., Warren, A., Xu, Z., Guilhaus, M., Pirola, R., Apte, M. V., and Wilson, J. S. (2010). Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc. Natl. Acad. Sci. U.S.A. 107, 17397–17402.
Pietras, K., and Ostman, A. (2010). Hallmarks of cancer: interactions with the tumor stroma. Exp. Cell Res. 316, 1324–1331.
Rasanen, K., and Vaheri, A. (2010). Activation of fibroblasts in cancer stroma. Exp. Cell Res. 316, 2713–2722.
Raza, A., Franklin, M. J., and Dudek, A. Z. (2010). Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am. J. Hematol. 85, 593–598.
Riss, J., Khanna, C., Koo, S., Chandramouli, G. V., Yang, H. H., Hu, Y., Kleiner, D. E., Rosenwald, A., Schaefer, C. F., Ben-Sasson, S. A., Yang, L., Powell, J., Kane, D. W., Star, R. A., Aprelikova, O., Bauer, K., Vasselli, J. R., Maranchie, J. K., Kohn, K. W., Buetow, K. H., Linehan, W. M., Weinstein, J. N., Lee, M. P., Klausner, R. D., and Barrett, J. C. (2006). Cancers as wounds that do not heal: differences and similarities between renal regeneration/repair and renal cell carcinoma. Cancer Res. 66, 7216–7224.
Ruddell, R. G., Oakley, F., Hussain, Z., Yeung, I., Bryan-Lluka, L. J., Ramm, G. A., and Mann, D. A. (2006). A role for serotonin (5-HT) in hepatic stellate cell function and liver fibrosis. Am. J. Pathol. 169, 861–876.
Samkharadze, T., Erkan, M., Reiser-Erkan, C., Demir, I. E., Kong, B., Ceyhan, G. O., Michalski, C. W., Esposito, I., Friess, H., and Kleeff, J. (2011). Pigment epithelium-derived factor associates with neuropathy and fibrosis in pancreatic cancer. Am. J. Gastroenterol. 106, 968–980.
Sancho-Bru, P., Bataller, R., Colmenero, J., Gasull, X., Moreno, M., Arroyo, V., Brenner, D. A., and Gines, P. (2006). Norepinephrine induces calcium spikes and proinflammatory actions in human hepatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G877–G884.
Schneiderhan, W., Diaz, F., Fundel, M., Zhou, S., Siech, M., Hasel, C., Moller, P., Gschwend, J. E., Seufferlein, T., Gress, T., Adler, G., and Bachem, M. G. (2007). Pancreatic stellate cells are an important source of MMP-2 in human pancreatic cancer and accelerate tumor progression in a murine xenograft model and CAM assay. J. Cell. Sci. 120, 512–519.
Schuller, H. M. (2008). Neurotransmission and cancer: implications for prevention and therapy. Anticancer Drugs 19, 655–671.
Schuller, H. M., and Al-Wadei, H. A. (2010). Neurotransmitter receptors as central regulators of pancreatic cancer. Future Oncol. 6, 221–228.
Schuller, H. M., Al-Wadei, H. A., and Majidi, M. (2008a). GABA B receptor is a novel drug target for pancreatic cancer. Cancer 112, 767–778.
Schuller, H. M., Al-Wadei, H. A., and Majidi, M. (2008b). Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis 29, 1979–1985.
Schuller, H. M., Al-Wadei, H. A., Ullah, M. F., and Plummer, H. K. Iii (2011). Regulation of pancreatic cancer by neuropsychological stress responses: a novel target for intervention. Carcinogenesis 33, 191–196.
Schuller, H. M., Plummer, H. K. III, Bochsler, P. N., Dudric, P., Bell, J. L., and Harris, R. E. (2001). Co-expression of beta-adrenergic receptors and cyclooxygenase-2 in pulmonary adenocarcinoma. Int. J. Oncol. 19, 445–449.
Shin, V. Y., Wu, W. K., Ye, Y. N., So, W. H., Koo, M. W., Liu, E. S., Luo, J. C., and Cho, C. H. (2004). Nicotine promotes gastric tumor growth and neovascularization by activating extracellular signal-regulated kinase and cyclooxygenase-2. Carcinogenesis 25, 2487–2495.
Stefaniak, T., Basinski, A., Vingerhoets, A., Makarewicz, W., Connor, S., Kaska, L., Stanek, A., Kwiecinska, B., Lachinski, A. J., and Sledzinski, Z. (2005). A comparison of two invasive techniques in the management of intractable pain due to inoperable pancreatic cancer: neurolytic celiac plexus block and videothoracoscopic splanchnicectomy. Eur. J. Surg. Oncol. 31, 768–773.
Takahashi, H., Funahashi, H., Sawai, H., Sakamoto, M., Matsuo, Y., Yamamoto, M., Okada, Y., Hayakawa, T., and Manabe, T. (2004). Glial cell line-derived neurotrophic factor enhances nuclear factor-kappaB activity and invasive potential in human pancreatic cancer cells. Pancreas 29, 22–27.
Tang, M. J., Worley, D., Sanicola, M., and Dressler, G. R. (1998). The RET-glial cell-derived neurotrophic factor (GDNF) pathway stimulates migration and chemoattraction of epithelial cells. J. Cell Biol. 142, 1337–1345.
Tatsuta, M., Iishi, H., and Baba, M. (1989a). Inhibition by neostigmine and isoproterenol and promotion by atropine of experimental carcinogenesis in rat stomach by N-methyl-N′-nitro-N-nitrosoguanidine. Int. J. Cancer 44, 188–189.
Tatsuta, M., Iishi, H., Baba, M., and Taniguchi, H. (1989b). Effect of 6-hydroxydopamine on gastric carcinogenesis and tetragastrin inhibition of gastric carcinogenesis induced by N-methyl-N′-nitro-N-nitrosoguanidine in Wistar rats. Cancer Res. 49, 4199–4203.
Tatsuta, M., Iishi, H., Baba, M., and Taniguchi, H. (1992). Inhibitions by 6-hydroxydopamine and neostigmine singly or together of gastric carcinogenesis induced by N-methyl-N′-nitro-N-nitrosoguanidine in Wistar rats. Int. J. Cancer 51, 767–771.
Tatsuta, M., Iishi, H., Baba, M., Uehara, H., and Nakaizumi, A. (1990). Inhibition by neostigmine of hepatocarcinogenesis induced by N-nitrosomorpholine in Sprague-Dawley rats. Br. J. Cancer 62, 773–775.
Tatsuta, M., Iishi, H., Yamamura, H., Baba, M., and Taniguchi, H. (1988). Inhibition by isoproterenol and neostigmine of experimental carcinogenesis in rat colon by azoxymethane. Br. J. Cancer 58, 619–620.
Thaker, P. H., Han, L. Y., Kamat, A. A., Arevalo, J. M., Takahashi, R., Lu, C., Jennings, N. B., Armaiz-Pena, G., Bankson, J. A., Ravoori, M., Merritt, W. M., Lin, Y. G., Mangala, L. S., Kim, T. J., Coleman, R. L., Landen, C. N., Li, Y., Felix, E., Sanguino, A. M., Newman, R. A., Lloyd, M., Gershenson, D. M., Kundra, V., Lopez-Berestein, G., Lutgendorf, S. K., Cole, S. W., and Sood, A. K. (2006). Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 12, 939–944.
Tilan, J., and Kitlinska, J. (2010). Sympathetic neurotransmitters and tumor angiogenesis-link between stress and cancer progression. J. Oncol. 2010, 539706.
Veit, C., Genze, F., Menke, A., Hoeffert, S., Gress, T. M., Gierschik, P., and Giehl, K. (2004). Activation of phosphatidylinositol 3-kinase and extracellular signal-regulated kinase is required for glial cell line-derived neurotrophic factor-induced migration and invasion of pancreatic carcinoma cells. Cancer Res. 64, 5291–5300.
Vonlaufen, A., Joshi, S., Qu, C., Phillips, P. A., Xu, Z., Parker, N. R., Toi, C. S., Pirola, R. C., Wilson, J. S., Goldstein, D., and Apte, M. V. (2008a). Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 68, 2085–2093.
Vonlaufen, A., Phillips, P. A., Xu, Z., Goldstein, D., Pirola, R. C., Wilson, J. S., and Apte, M. V. (2008b). Pancreatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer Res. 68, 7707–7710.
Weddle, D. L., Tithoff, P., Williams, M., and Schuller, H. M. (2001). Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis 22, 473–479.
Wong, H. P., Yu, L., Lam, E. K., Tai, E. K., Wu, W. K., and Cho, C. H. (2007). Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol. Appl. Pharmacol. 221, 261–267.
Wu, C. H., Lee, C. H., and Ho, Y. S. (2011). Nicotinic acetylcholine receptor-based blockade: applications of molecular targets for cancer therapy. Clin. Cancer Res. 17, 3533–3541.
Xiao, F., Yu, K., Dong, F., Liang, Y., Jun, C., and Wei, H. (2010). The GABAB receptor inhibits activation of hepatic stellate cells. Dig. Dis. Sci. 55, 261–267.
Xie, G., Cheng, K., Shant, J., and Raufman, J. P. (2009). Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G755–G763.
Xu, Z., Vonlaufen, A., Phillips, P. A., Fiala-Beer, E., Zhang, X., Yang, L., Biankin, A. V., Goldstein, D., Pirola, R. C., Wilson, J. S., and Apte, M. V. (2010). Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 177, 2585–2596.
Yan, B. M., and Myers, R. P. (2007). Neurolytic celiac plexus block for pain control in unresectable pancreatic cancer. Am. J. Gastroenterol. 102, 430–438.
Yoo, A. S., Sun, A. X., Li, L., Shcheglovitov, A., Portmann, T., Li, Y., Lee-Messer, C., Dolmetsch, R. E., Tsien, R. W., and Crabtree, G. R. (2011). MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476, 228–231.
Zeng, Q., Cheng, Y., Zhu, Q., Yu, Z., Wu, X., Huang, K., Zhou, M., Han, S., and Zhang, Q. (2008). The relationship between overexpression of glial cell-derived neurotrophic factor and its RET receptor with progression and prognosis of human pancreatic cancer. J. Int. Med. Res. 36, 656–664.
Zhou, P., Porcionatto, M., Pilapil, M., Chen, Y., Choi, Y., Tolias, K. F., Bikoff, J. B., Hong, E. J., Greenberg, M. E., and Segal, R. A. (2007). Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron 55, 53–68.
Zhu, Z., Friess, H., Dimola, F. F., Zimmermann, A., Graber, H. U., Korc, M., and Buchler, M. W. (1999). Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. J. Clin. Oncol. 17, 2419–2428.
Zhu, Z., Kleeff, J., Kayed, H., Wang, L., Korc, M., Buchler, M. W., and Friess, H. (2002). Nerve growth factor and enhancement of proliferation, invasion, and tumorigenicity of pancreatic cancer cells. Mol. Carcinog. 35, 138–147.
Keywords: stroma, pancreatic cancer, neural invasion, neurotrophic factors, neurotransmitters, neuropathic pain, neuroplasticity, desmoplasia
Citation: Demir IE, Friess H and Ceyhan GO (2012) Nerve-cancer interactions in the stromal biology of pancreatic cancer. Front. Physio. 3:97. doi: 10.3389/fphys.2012.00097
Received: 31 December 2011; Paper pending published: 23 January 2012;
Accepted: 28 March 2012; Published online: 17 April 2012.
Edited by:
Phoebe Phillips, University of New South Wales, AustraliaReviewed by:
Shuping Vincent Wu, VA Greater Los Angeles Healthcare System, USAPhoebe Phillips, University of New South Wales, Australia
Alain Vonlaufen, University of Geneva Medical School, Switzerland
Copyright: © 2012 Demir, Friess and Ceyhan. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Güralp O. Ceyhan, Department of Surgery, Klinikum Rechts der Isar, Technische Universität München, Ismaninger Str. 22, D-81675 München, Germany. e-mail:Z3VlcmFscC5jZXloYW5AdHVtLmRl