
 Aaisha Opel1
Aaisha Opel1

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 24 April 2012
Sec. Cardiac Electrophysiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00096
This article is part of the Research Topic Autonomic regulation of the heart - emerging regulators and arrhythmogenic mechanisms View all 8 articles
Inhibitory heterotrimeric G proteins and the control of heart rate. The activation of cell signaling pathways involving inhibitory heterotrimeric G proteins acts to slow the heart rate via modulation of ion channels. A large number of Regulators of G protein signalings (RGSs) can act as GTPase accelerating proteins to inhibitory G proteins and thus it is important to understand the network of RGS\G-protein interaction. We will review our recent findings on in vivo heart rate control in mice with global genetic deletion of various inhibitory G protein alpha subunits. We will discuss potential central and peripheral contributions to the phenotype and the controversies in the literature.
The pacemaker in the sinoatrial node (SAN) of the heart has an intrinsic rate independent of autonomic innervation and this leads to a measurement of “intrinsic” heart rate that is determined in practice by pharmacologically inhibiting the sympathetic and parasympathetic systems. At the molecular level this self-regenerating electrical activity arises from a membrane clock and\or a calcium clock dependent on intracellular calcium release events (Lakatta and DiFrancesco, 2009). This pacemaker activity can be modulated: increased sympathetic activity leads to an increase in rate and an increase in vagal tone to a decrease. The release of noradrenaline from sympathetic nerve endings or the release of adrenaline from the adrenal medulla into the circulation modulates SAN cells by binding to β-adrenoreceptors. The stimulatory G protein is activated and this leads to stimulation of adenylate cyclase and the generation of cyclic adenosine monophosphate (cAMP). Increased cAMP can directly modulate the hyperpolarization-activated cation currents (Ih/If) leading to increased pacemaker depolarization and rate. However, increased cAMP can also activate protein kinase A (PKA), which can modulate intracellular calcium-handling proteins such as phospholamban and the ryanodine receptor to modulate rate.
However, the main focus of this review is on the inhibitory events typified by the negative chronotropic response in the SAN to vagal nerve efferent activation. Acetylcholine (ACh) released from the vagus nerve binds to the muscarinic (M2) receptor, leading to the activation, and dissociation of inhibitory G protein heterotrimers. The resulting βγ-dimer directly activates the G protein coupled inward rectifying potassium (GIRK) channel to cause membrane hyperpolarization, slowing pacemaker depolarization, and sinus rate. Furthermore, the activation of the inhibitory Gα subunit will inhibit adenylate cyclase and thus modulate Ih/If as detailed above. Regulators of G protein signaling (RGS) proteins interact with the inhibitory Gα subunit to switch off this pathway through GTPase accelerating protein (GAP) activity and allow the heterotrimer to reform. This process is summarized in Figure 1.
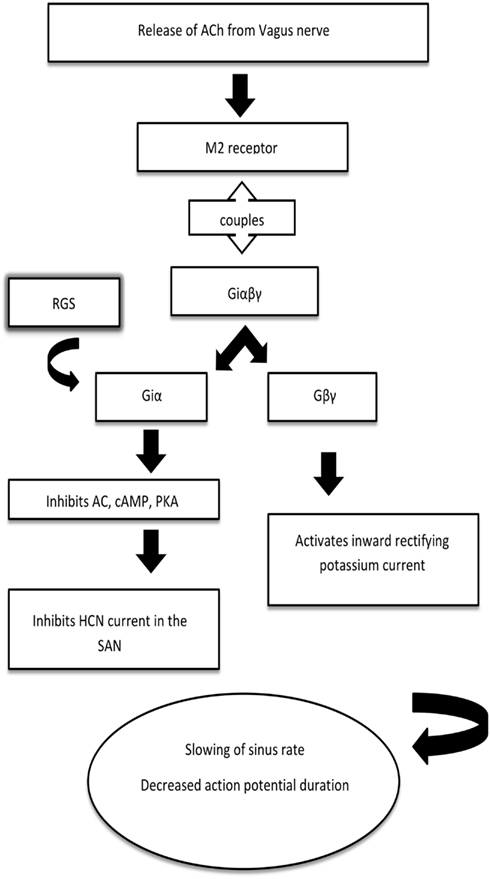
Figure 1. A simplified schematic for the mechanism of heart rate regulation by inhibitory G proteins. Acetylcholine released from the vagus nerve binds to the muscarinic (M2) receptor, which is coupled to the inhibitory G protein. The βγ-dimer activates the G protein coupled inward rectifying potassium (GIRK) channel to slow the sinus rate, decrease the action potential duration and allow arrhythmia generation. Regulators of G protein signaling (RGS) interact with the α-subunit to switch off this pathway through GTPase accelerating protein activity and allow the heterotrimer to reform. AC, adenylyl cyclase; cAMP, cyclic AMP; PKA, protein kinase A; HCN, hyperpolarization-activated cyclic nucleotide-gated channel.
It has emerged from the SHIFT trial using ivabradine, an inhibitor of Ih/If, in addition to standard therapy that heart rate per se may be an important risk factor in chronic heart failure (Swedberg et al., 2010). Furthermore, autonomic precipitants are well known for common arrhythmias such as atrial fibrillation (AF) and ventricular tachyarrhythmia (see below). Thus the focus in this review is an attempt to link physiological and pathophysiological function in the control of heart rate and rhythm with the known molecular isoforms of the inhibitory G proteins and RGSs. These proteins are widely distributed in the heart thus we will consider not only their effects on heart rate but also what electrical processes they might modulate in the atria, atrioventricular node (AVN), and ventricles. One of the key approaches is the use of genetically modified mice. However, these often have global knockout of the relevant gene and this will mean deletion of the signaling molecule in central circuits and peripheral nerves of the autonomic nervous system. Thus we review the role of inhibitory G proteins in the central circuits and peripheral limbs of the autonomic nervous system, consider the limited information available and experimental approaches by which this might be investigated.
We initially discuss this from a cardiac perspective but the role of the nervous system is obviously important and this is addressed in another section.
There are four families of G proteins namely Gαi/o, Gαs, Gαq/11, Gα12/13. The inhibitory G proteins (Gαi/o) are the most highly expressed and predominant class followed by Gαs, Gαq/11 then Gα12/13. The inhibitory G proteins themselves have multiple isoforms: Gαi1, Gαi2, Gαi3, and Gαo. The Gαo (“other”) isoform has two splice variants GαoA and GαoB. Gαo is the predominant isoform in the brain whereas Gαi1, Gαi2, Gαi3 are highly homologous and widely distributed in many tissues (Wettschureck and Offermanns, 2005). The inhibitory G proteins characteristically inhibit adenylate cyclase activity and lower the concentration of cAMP (Wong et al., 1991; Rudolph et al., 1996), however they also activate PI-3 kinase activity and directly regulate ion channel activity (see below).
Regulators of G protein signaling proteins act to effectively inhibit G protein signaling; they interact with the α-subunit and accelerate GTPase activity. Characteristically this family have a 120 amino acid conserved RGS domain, flanked by variable length N- and C-terminals. Six main subfamilies of mammalian RGSs are distinguished: R4, R7, R12, RA, RL, and RZ. The vast majority of RGSs are GAPs for inhibitory G proteins but some have activity to the Gq/11 family as well. They may also have other roles in signaling (Hollinger and Hepler, 2002). There are more than 20 mammalian RGS isoforms and many of these are expressed to some extent in the heart (Kardestuncer et al., 1998; Doupnik et al., 2001; Owen et al., 2001; Wieland and Mittmann, 2003). For both Gαi/o and RGSs the pattern of expression in conducting tissues, atria, and ventricles and in specific cell types such as myocytes and fibroblasts is not well delineated.
The current paradigm for the mechanism underlying SAN automaticity involves a complex interaction between activity of the voltage gated ion channels/exchangers in the plasma membrane and the sarcoplasmic reticulum (SR) (Lakatta and DiFrancesco, 2009). The key membrane ion channel thought to be involved in pacemaker setting is the Ih/If channel (DiFrancesco, 2010), and pacemaker activity is further modulated by K+, Na+, and Ca2+ currents. In addition, recent studies suggest that rhythmic local calcium releases from the SR modulated by PKA also contribute to pacemaker activity by activating inward Na+/Ca2+ exchanger currents during late diastolic depolarization (Maltsev and Lakatta, 2008).
The hyperpolarization-activated cyclic nucleotide (HCN) gated cation current (Ih), also known as the “funny” current (If), is a non-specific cation conductance. Hyperpolarization and the binding of cAMP mediate activation of the channel. Thus inhibition of adenylate cyclase can reduce the current and slow pacemaker depolarization and heart rate. HCN4 is the major isoform present in the heart and it is present solely in conducting tissues in the healthy heart. There is not complete consensus on the relative roles of Ih/If and GIRK, but it is thought about half the negative chronotropic effect of muscarinic receptor activation is due to each mechanism (Wickman et al., 1998; Gehrmann et al., 2002).
GIRK channels (Kir3.x) are part of the family of inwardly rectifying K+ channels. The subfamily has five members (Kir3.1–3.5) with a heterotetramer of Kir3.1 and 3.4 constituting the cardiac channel and is present in the atria and conducting tissues but has little if any expression in the ventricle (Karschin et al., 1994; Krapivinsky et al., 1995). The channel is characteristically activated by ACh binding to muscarinic (M2) receptors leading to the common designation as IKACh. The increase in channel activity occurs because of a direct interaction of the Gβγ dimer, released from the inhibitory G protein heterotrimer, with specific domains in the N- and C-termini of the channel and as such is a paradigm for βγ subunit mediated signaling (Logothetis et al., 1987). The current is also stimulated via adenosine, sphingosine-1-phosphate, and endothelin-A receptors in the heart (Ono et al., 1994, 2001; Bösche et al., 2003; Ochi et al., 2006).
The L-type calcium current is the most prominent calcium current in mammalian ventricular heart cells. The alpha subunit is CaV1.2 (also known as α1C). It is classically regulated by phosphorylation by PKA and thus will be modulated by variations in G protein coupled receptor pathways involving Gαs and Gαi/o. For example, the activation of β1-adrenoreceptors by agonists leads to up to a fourfold increase in current and shifts the voltage dependence of activation and inactivation to more hyperpolarized potentials (McCleskey et al., 1986; Hirayama and Hartzell, 1997). Although ACh has no effect upon basal L-type calcium currents, it antagonizes their increase on application of a catecholamine (Fischmeister and Hartzell, 1986).
Other membrane and sarcolemmal ion currents modulated via the cAMP/PKA pathway can also play a role in modulation of pacemaker activity. For example, isoprenaline reduced action potential duration (APD) and increased spontaneous action potential firing rate in rabbit SAN cells by increasing the amplitude and causing a negative shift in the activation of the total delayed rectifier K+ current (IK) in a PKA-dependent manner (Lei et al., 2000).
It is clear then that there are a number of isoforms of inhibitory G proteins and RGSs expressed in the heart and other tissues, which have potentially overlapping functions. Thus the question is do specific isoforms carry out unique physiological roles or does the system have significant redundancy? For example, inhibitory G proteins are characterized by their sensitivity to pertussis toxin, which ADP ribosylates the proteins and uncouples them from receptors. It is possible to engineer Gαi/o isoforms that are resistant to the toxin and thus design experimental strategies in which the isoforms can be selectively investigated. In our own work using in vitro cellular systems, we were able to show that only rarely would there be an absolute preference of a receptor for an inhibitory G protein isoform: most G protein coupled receptors couple well with a number of isoforms (Leaney and Tinker, 2000).
We studied the question of which inhibitory G protein isoform in vivo governs heart rate modulation and dynamics using mice with global KO of Gαi2, Gαi1, and Gαi3 combined and Gαo (Zuberi et al., 2008). We analyzed heart rate dynamics in the time and frequency domain with implanted telemetry probes in awake and mobile mice. The influence of the autonomic system can be measured indirectly by the beat-to-beat variation of heart rate (“heart rate variability,” HRV). In man R–R interval shows characteristic patterns of variability with a low frequency (LF) component (>6 s cycle length) determined by sympathetic and parasympathetic drive and a high frequency (HF) component (2.5–6 s cycle length) governed by parasympathetic input (Stauss, 2003). Such analysis has also been applied to the mouse but there is less agreement on the contribution of the different arms of the autonomic nervous system to the frequency components (Thireau et al., 2007). We found that Gαi2 deficient mice were generally tachycardic with preservation of diurnal variation, had loss of HF power in HRV analysis similar to that seen with GIRK channel blocker TertiapinQ and with the muscarinic receptor blocker atropine and an attenuated bradycardic response to the muscarinic agonist carbachol. Mice with global deficiency in Gαi1,3 were similar to WT but Gαo showed a different phenotype that is discussed below. There is one conflicting study suggesting Gαo and not Gαi2 is necessary in muscarinic mediated parasympathetic activity which may be due to methodological issues as the mice were studied very early after probe implantation (Duan et al., 2007). In keeping with our observations, mice with a G184S knockin point mutation in the switch I region of Gαi2, preventing the binding of RGS proteins and deactivation of Gαi2, had marked enhancement of muscarinic agonist mediated bradycardia (Fu et al., 2007).
Gαi2 and Gαo signaling has also been implicated in the muscarinic inhibition of β-adrenoreceptor activated voltage gated L-type calcium channels in the heart. Using ventricular myocytes isolated from global Gαi2 and Gαi3 knockout mice, Nagata et al. (2000) demonstrated reduced calcium channel currents with carbachol following isoproterenol stimulation in Gαi3 knockout mice and littermate controls but not in Gαi2 knockout mice. Using a similar experimental strategy in Gαo knockout mice, Valenzuela et al. (1997) demonstrated similar impaired muscarinic regulation of L-type calcium channels in ventricular myocytes lacking Gαo but no difference in potassium current compared to wild type controls.
Differential coupling of G protein subtypes to adenosine A1 and muscarinic M2 receptor mediated heart rate slowing was also demonstrated in vitro using RGS insensitive Gαo and Gαi2 ES derived cardiomyocytes (Fu et al., 2006). The RGS insensitive Gαo homozygous knockin cells demonstrated enhanced adenosine A1 and muscarinic M2 receptor mediated bradycardic responses. In contrast, RGS insensitive Gαi2 homozygous knockin cells showed enhanced responses to M2 but not A1 receptors. Blocking GIRK channels largely abolished the mutation-induced enhancement of the M2 receptor mediated response but had a minimal effect on A1 responses.
Taken together, the evidence suggests that M2 mediated GIRK channel activation is coupled preferentially to the Gαi2 subunit whereas coupling to voltage gated L-type calcium channel appears to be less specific with both Gαo and Gαi2 implicated.
There are two studies asking which individual RGS might be important for heart rate control. Cifelli et al. (2008) suggested RGS4 is likely to be the predominant RGS in the SAN. RGS4 mRNA levels were found to be higher in the SAN compared to the atrium, conscious RGS4 null mice showed an increased bradycardic response to carbachol and isolated sinus node myocytes from these mice have modified M2 receptor mediated KACh currents. More recently, RGS6 has been found to be highly expressed in the heart, in particular in the SAN and AVN. Conscious and anesthetized RGS6 null mice also show an increased bradycardic response to carbachol. Perfused hearts demonstrated a similar phenomenon, together with the inhibition of spontaneous action potential firing rates in the SAN, as well as atrioventricular block (Yang et al., 2010). RGS6 deficient mice show impaired desensitization and slower deactivation of the GIRK current in atrial myocytes in response to agonist application (Posokhova et al., 2010), confirming that RGS6 has GAP activity. Taken together, the results indicate that RGS4 and RGS6 have an important parasympathetic modulatory role in the SAN, preventing severe bradycardia and that there is very little redundancy. It would be interesting to know if the combined knockout has a more severe phenotype.
Atrial fibrillation is the most common cardiac arrhythmia and affects at least 10% of the octogenarian population (Ho et al., 1993; Benjamin et al., 1998). It is characterized by rapid, irregular, and chaotic electrical excitation of the atria, where these can beat 300 times per minute in the human. The AVN has decremental properties and prevents every beat from being conducted to the ventricle. Despite this, the ventricular rate can still be rapid resulting in symptoms of palpitations, shortness of breath, and dizziness and it can precipitate overt heart failure. There have been several proposed mechanisms of AF. Classically, it has been suggested that AF arises due to an atrial ectopic focus, a single re-entry circuit, or multiple re-entrant circuits (Nattel, 2002). A more modern variant of these mechanisms for AF involves the concept of rotors and spiral waves (Jalife, 2003). A rotor is a stable rotating pattern around a pivot point. This gives rise to a spiraling wavefront (spiral waves) into the surrounding tissue. Such a rotor is thought to be present at the pulmonary vein-left atrial junction, with spiral waves emanating into the atria, giving fibrillation. Catheter ablation producing anatomical block between the pulmonary veins and left atrium can terminate AF supporting this hypothesis (Haissaguerre et al., 1998).
It has been said that “AF begets AF” (Wijffels et al., 1995). This statement is an illustration of the remodeling that occurs once AF has commenced. Indeed permanent AF is often preceded by paroxysmal AF. Remodeling can take two forms: electrical, which occurs as a result of AF itself and structural, which is associated with congestive cardiac failure and other cardiac and extracardiac diseases predisposing to fibrosis. An increase in rate caused by AF results in increased cellular calcium loading, which threatens cell viability. Subsequently, the density of the L-type calcium channel is reduced with a corresponding increase in the inward rectifying potassium currents, leading to a decreased APD and effective refractory period (ERP). This, in turn, promotes and perpetuates the induction and maintenance of AF through multiple re-entrant circuits. These calcium-handling abnormalities impair atrial contractility and can cause atrial dilatation, which also promotes AF (Nattel et al., 2008). In contrast, patients with congestive cardiac failure are thought to develop AF secondary to a change in atrial architecture due to fibrosis, which interferes with electrical conductance. Profibrotic signaling pathways have been identified and these include angiotensin II, transforming growth factor-β1 and platelet derived growth factor. Extracardiac disease, such as diabetes, obesity, and sleep apnea, may also be responsible for an increased risk of AF through fibrosis. Atrial stretch, which can occur secondary to mitral valve disease, is also a known trigger. Genetic factors may contribute to remodeling; gain-of-function mutations in the potassium channels and loss-of-function mutations in the L-type calcium channel can promote AF. Moreover, single nucleotide polymorphisms alongside genes contributing to atrial integrity have been associated with AF, for example, angiotensin-converting enzyme and PITX2 (Wakili et al., 2011).
An imbalance in the autonomic system may increase the propensity to AF. In 18 subjects with paroxysmal AF, vagal overactivity was described as the precipitating factor. Here, each paroxysm was preceded by atrial coupling followed by a pause and accompanied by slowing of the sinus rate (Coumel et al., 1978). Changes in autonomic tone through the study of HRV has been assessed in 77 patients with paroxysmal AF (Bettoni and Zimmermann, 2002). This study showed that at least 20 min prior to the onset of paroxysmal AF, there was an initial increase in adrenergic drive followed by a shift in autonomic tone such that increased vagal tone dominated immediately before the onset of paroxysmal AF. The effects of both cholinergic and β-adrenergic stimulation and blockade were studied in the intact dog heart (Sharifov et al., 2004). ACh and catecholamines, individually and in combination, were infused into the heart via the sinus node artery. No electrical stimulation was performed. Cholinergic stimulation was found to be the main factor responsible for spontaneous AF initiation. Adrenergic tone, however, was involved in modulating the initiation and maintenance of cholinergically mediated AF. Further evidence for the role of a vagal mechanism in paroxysmal AF was demonstrated in patients undergoing circumferential pulmonary vein ablation in whom complete vagal denervation was also performed (Pappone et al., 2004). These patients had greater freedom from AF.
Activation of GIRK current results in membrane hyperpolarization, and shortening of APD and ERP. The change in atrial ERP may predispose to the induction and maintenance of AF. Thus any increased activity in inhibitory G protein signaling may predispose to AF. There is surprisingly little data mapping specific genetic modifications in this pathway to experimental supraventricular tachyarrhythmia. It is possible to induce non-sustained AF in the mouse using programmed electrical stimulation (Kovoor et al., 2001). Patch clamp studies utilizing atrial cardiomyocytes from humans (Dobrev et al., 2005) and dogs (Voigt et al., 2008) illustrate that the GIRK channel is constitutively active in AF. Furthermore, the Kir3.4 knockout mouse is resistant to AF (Kovoor et al., 2001). Overexpression of Gαi2 using a viral vector has also been tried as a therapeutic strategy (Bauer et al., 2004). Injection of the construct into the AVN of animals slowed the heart rate in AF (Bauer et al., 2004). The delivery of cell penetrating peptides to the posterior left atrium to disrupt M2-Gαi coupling prolonged refractoriness in the left atrium and suppressed vagally induced AF (Aistrup et al., 2009). It is interesting to note that it was necessary to target both Gαi2 and Gαi3 here. Furthermore, it has been shown that in Gαi2 knockout mice, where the hearts are studied ex vivo, pacemaking occurs from the atria not the SAN and there is no impairment of muscarinic receptor mediated heart rate slowing (Boknik et al., 2009). This suggests that there may be differences in the coupling profile between the M2 receptor and GIRK channel in the atria and SAN. Once again there is little information on which RGSs might be important in the atria. Of those studied functionally, RGS6 but not RGS4 is expressed there and RGS10 mediates β-adrenergic receptor effects on the GIRK current in rat atrial cells (Bender et al., 2008). There is one report showing inducible AF in the RGS2 knockout mouse (Tuomi et al., 2010).
Ventricular cardiac arrhythmias, namely ventricular tachycardia (VT) and ventricular fibrillation (VF), cause sudden cardiac death (SCD). SCD poses a significant clinical burden with an estimated 300 000 cases per year recorded in the USA (Noseworthy and Newton-Cheh, 2008). This highlights the importance in predicting risk of SCD, although precise calculation of such a risk remains challenging. Drug therapy, as in AF, can be limited by modest efficacy, significant toxicity, and can be proarrhythmic. ICDs have proven benefit but are complicated by inappropriate shocks, the need for a generator and possible lead changes and complications at the time of insertion, for example infection. Catheter ablation is invasive and is suitable for only modest numbers of patients. Therefore, there is a need to understand the molecular mechanisms responsible for VT initiation and maintenance, in order to develop successful treatments.
The etiology of VT/VF can be broadly divided into those occurring in patients with structurally normal or abnormal hearts. Normal heart VT/VF has traditionally been considered a primary electrical problem, caused by “channelopathies” including the long QT (LQT) syndromes and Brugada syndrome. In some cases, the cause of VT/VF is unknown and hence diagnosed as idiopathic VT. Structurally abnormal hearts causing VT/VF can be due to inherited conditions such as hypertrophic cardiomyopathy (HCM) and arrhythmogenic right ventricular cardiomyopathy (ARVC) or acquired cardiomyopathies, the most common of which is ischemic cardiomyopathy.
Autonomic precipitants to VT/VF is well recognized clinically. Both increased sympathetic and parasympathetic tone can lead to VT/VF depending on etiology and the underlying electrical abnormalities. For example, LQT 1 and 2 are precipitated by an increase in sympathetic tone whereas in LQT 3, it is protective (Moss and Kass, 2005). In Brugada syndrome, vagotonic agents and β adrenergic blockers can precipitate VT/VF (Kasanuki et al., 1997). In patients with idiopathic VT/VF and short coupled variant of torsades des pointes, HRV was significantly depressed prior to the onset of ventricular tachyarrhythmia suggesting vagal precipitant (Leenhardt et al., 1994). The arrhythmogenic substrate is thought to be due to the amplification of spatial dispersion of repolarization by the underlying autonomic tone, causing abnormal re-entry, or triggered activity (Verrier and Antzelevitch, 2004).
Bradycardia itself per se may precipitate polymorphic VT by causing prolongation of APD and development of early afterdepolarizations, leading to triggered activity in the form of ventricular premature beats (El-Sherif et al., 1988). Hence any perturbation of Gαi/o signaling in the conduction system which leads to bradycardia could in theory increase susceptibility to ventricular arrhythmia.
In patients with ischemic cardiomyopathy induced VT/VF and heart failure, the picture is somewhat different. Sympathetic nervous system activation is beneficial acutely in heart failure but detrimental in the long term. Such a mechanism promotes apoptosis of cardiac myocytes, ion channel remodeling, and is proarrhythmic (DeGeorge et al., 2008). Cardiac Gαi/o signaling pathways may counteract this process (Zheng et al., 2005). Indeed, in chronic heart failure, β1-adrenoceptors are downregulated whereas Gαi2 is significantly increased (Eschenhagen et al., 1992). Overexpression of β1-adrenoceptors in transgenic mice results in a dilated cardiomyopathy but a more modest phenotype is seen with β2-adrenoceptor overexpression. β2-adrenoceptors are coupled to Gαi/o and Gαs, suggesting a cardioprotective role for Gαi/o (Foerster et al., 2003; DeGeorge et al., 2008).
In heart failure patients with ICDs, a loss in HRV was noted immediately before an episode of VT and this was found to be an independent predictor of SCD (Pruvot et al., 2000). Parasympathetic activation can terminate VT and increase VF threshold whereas increases in sympathetic activation can precipitate ventricular arrhythmias (Kolman et al., 1975; Waxman and Wald, 1977; Brack et al., 2007). Taken together, there is good evidence that increased vagal tone protects against the development of heart failure and VT/VF precipitated by ischemic cardiomyopathy. Certainly β-blocker therapy improves prognosis in those with heart failure (Poole-Wilson et al., 2003).
To explore the role of Gαi2 and predisposition to cardiac ventricular arrhythmias, we performed in vivo cardiac electrophysiological studies on global Gαi2 knockout mice and compared them with wild type littermates and combined Gαi1 and Gαi3 knockout controls (Zuberi et al., 2010). Mice with global genetic deletion of Gαi2 were found to have a reduced ventricular effective refractive period and propensity for VT induced with programmed electrical stimulation compared to controls. Conscious Gαi2 knockout mice had a prolonged QT interval and single ventricular myocytes studied with patch clamping showed a steep restitution curve but prolonged APD. Increased levels of message for the L-type calcium channel was found on gene expression studies, with confirmation of an increase in this current on patch clamping. No structural heart disease was noted on histology or echocardiography. These findings suggest that the absence of Gαi2 is a substrate for VT. Interestingly, Dizayee et al. (2011) studied ventricular myocytes isolated from Gαi2 and Gαi3 knockout mice and found reduced L-type calcium channel current density in Gαi2 knockouts compared to littermate controls but increased in Gαi3 knockouts where there was a compensatory increase in Gαi2 expression. The differences between our results and theirs may be accounted for by strain differences in the mice (129\Sv versus C57\Black) Clinically, pravastatin, given to patients in order to lower cholesterol, selectively upregulates Gαi2 and increases HF power in HRV analysis (Welzig et al., 2003).
The autonomic nervous system maintains homeostasis and coordinates physiological responses in the body via its two functional limbs: the sympathetic and parasympathetic nervous systems. The heart is regulated in this manner within the cardio-respiratory system (Figure 2). Afferent nerves convey baroreceptor and chemoreceptor inputs to the cardio-respiratory centers within the brainstem and generally opposing reflex responses are mediated by the sympathetic and parasympathetic efferent to the heart, blood vessels, and other effector organs such as the adrenal glands. Both sympathetic and parasympathetic systems are made up of central and peripheral components. The central component consists of cell groups in the brain and spinal cord, and myelinated preganglionic neurons. It is responsible for maintenance of autonomic tone, and integration of afferent inputs to generate appropriate autonomic reflexes. Much of the basic structure involved in the reflex control of the cardiovascular system is contained within the medulla. Integrated and behaviorally appropriate responses also require reciprocal connections between the medulla, pons, midbrain, and hypothalamus (Spyer, 1994). The peripheral component consists of ganglia and unmyelinated postganglionic afferent and efferent fibers innervating the heart and associated effector organs (e.g., blood vessels). It is increasingly recognized that the pattern of cardiac innervation peripherally may be important in cardiac arrhythmogenesis (Verrier and Antzelevitch, 2004).
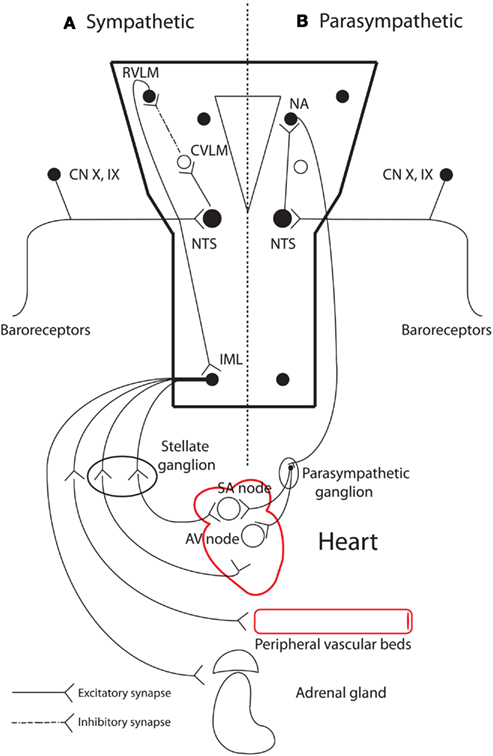
Figure 2. The main peripheral and neural centers involved in autonomic cardiovascular regulation. (A) Sympathetic baroreflex arm: sensory neurons in the periphery transmit baroreceptor information via a glutamatergic synapse to the nucleus tractus solitaries (NTS) in the medulla. NTS neurons project to and excite neurons in the caudal ventrolateral medulla (CVLM). CVLM neurons in turn project and inhibit (via a GABAergic synapse) presympathetic neurons in the rostral ventrolateral medulla (RVLM). Spinally projecting presympathetic RVLM neurons make direct connections with sympathetic preganglionic neurons in the intermediolateral (IML) spinal column, which projects sympathetic outflow to effector organs. (B) Parasympathetic control of heart rate: NTS neurons receive baroreceptor and chemoreceptor information and relay via a glutamatergic pathway to the nucleus ambiguous (NA). Cardioinhibitory neurons in NA then project to the heart via the vagus nerve, which mediates its effect via muscarinic receptors in the heart. Adapted from Pilowsky and Goodchild (2002). GABA, gamma-Aminobutyric acid; SA, sinoatrial; AV, atrioventricular.
As described above, the murine models used to study (patho)physiology are often global knockouts or knock-ins. It is conceivable (and probably very likely) that there are contributions to the phenotype from the effects on inhibitory G protein signaling in the nervous system. Gαi1, Gαi2, and Gαi3 are ubiquitously expressed but Gαo protein expression is restricted mainly to the central and peripheral nervous systems, endocrine cells, and cardiomyocytes (Wettschureck and Offermanns, 2005). Expression of Gαo is greatest in the brain where it is estimated to constitute around 1% of all membrane proteins (Sternweis and Robishaw, 1984). Mice lacking the α subunit of Go are smaller and weaker than their littermates, have greatly reduced life expectancy and display numerous neurological abnormalities including hyperalgesia, tremors, seizures, and increased motor activity with an extreme turning behavior (Jiang and Bajpayee, 2009).
GIRK channels are widely distributed in neuronal and endocrine tissues and play key roles in generating late inhibitory postsynaptic potentials and modulating hormone release in addition to their cardiac functions. In the central nervous system, GIRK channels hyperpolarize neurons in response to activation by neurotransmitters including adenosine, somatostatin, 5-HT, and opioids, and is composed of heterotetramers of Kir3.1, Kir3.2, and Kir3.3 and homotetramers of Kir3.2 (Lüscher et al., 1997; Lüscher and Slesinger, 2010). Pharmacological investigations of GIRK channels and studies in animal models suggest that GIRK activity has an important role in physiological responses, including pain perception and memory modulation. Abnormal GIRK function has been implicated in altering neuronal excitability and cell death, which may be important in the pathophysiology of diseases such as epilepsy, Down’s syndrome, Parkinson’s disease, and drug addiction (Lüscher and Slesinger, 2010). There is limited study of GIRK’s role in autonomic traffic. Feldman and colleagues found reduced BP depressor effects of the I1-imidazoline receptor (I1R) selective ligands administered intracisternally in rabbits pretreated with TertiapinQ suggesting that the central sympatho-inhibitory effects of I1R ligands are mediated by GIRK channels (Yoro Sy et al., 2008).
The regulation of neurotransmitter release at presynaptic terminals is an important mechanism underlying the modulation of synaptic transmission in the nervous system. Inhibitory regulation of neurotransmitter release via inhibitory G proteins is mediated by various G protein coupled receptors like α2-adrenoceptors, μ- and δ-opioid receptors, GABA-B receptors, adenosine A1, or endocannabinoid CB1-receptors. A major mechanism by which these G proteins mediate the inhibition of transmitter release is the direct inhibitory modulation of the action potential-evoked Ca2+ entry to the presynaptic terminal by the Gβγ subunit, which is required to trigger neurotransmitter release. N- and P/Q-type calcium channels that are concentrated at nerve terminals as well as R-type calcium channels have been shown to be inhibited via Gαi/o-coupled receptors (Dolphin, 2003).
There is evidence showing selectivity of inhibitory G protein coupling to voltage gated Ca2+ channels. Neuropeptide Y (NPY) and bradykinin both inhibit N-type Ca2+ channels in vitro, and pertussis toxin treatment could completely abolish this inhibition. Addition of purified bovine Gαo subunits effectively restored the NPY modulation of Ca2+ currents while purified Gαi1 or Gαi2 had little or no effect. Interestingly, the bradykinin inhibitory effects on Ca2+ currents in pertussis toxin-treated cells was only partially restored by the addition of either purified Gαo or Gαi2, even at high concentrations. Complete restoration required a combination of both Gαo and Gαi2 proteins suggesting that Gαo and Gαi2 proteins may couple to distinct subpopulations of bradykinin receptors since Gαo nor Gαi2 protein alone cannot compensate the loss of the other (Ewald et al., 1989). Gαo antibodies in the superior cervical ganglion has also been shown to reduce calcium current inhibition by noradrenaline in superior cervical ganglion neurons (Caulfield et al., 1994). Signaling via Gαo has also been implicated in vesicular glutamate transporter within the central nervous system (Winter et al., 2005) and GAP43 receptor signaling for neuronal pathfinding (Strittmatter et al., 1990).
In vivo experiments manipulating the expression of inhibitory G proteins and its subtypes either directly with knockout models or indirectly by manipulation of their regulatory proteins have shed limited light on their physiological importance and relative contribution to the peripheral and central control of autonomic heart rate regulation. Methodologically, autonomic tone in vivo has been quantified by indirect methods such as catecholamine sampling and analysis of HRV. The baroreceptor reflex is the prototypical cardiovascular reflex studied and is commonly quantified by computation of baroreflex set point and gain from ambulatory BP recordings, although more direct methods have been described (Ma et al., 2002; Young and Davisson, 2011).
Interestingly, mice overexpressing RGS insensitive Gαi2 were tachycardic during the daytime, have an enlarged heart and hyperdynamic echo profile. Along with reduced viability, growth retardation, and multiple neurological deficit, the cardiovascular phenotype observed was attributed to increased central sympathetic tone (Huang et al., 2006). To study the cardiac specific effect of enhanced Gαi2 signaling, isoproterenol-stimulated beating isolated perfused hearts were studied and showed the expected enhancement of muscarinic mediated bradycardia (Fu et al., 2007).
Mice deficient in RGS2 were found to be hypertensive but with normal HR suggestive of resetting of baroreceptor sensitivity. There was an associated increase in urinary catecholamine secretion and reduction in both LF and HF power of HRV suggesting increased central sympathetic tone (Gross et al., 2005). This along with data on the RGS insensitive Gαi2 mice described above implies that overactivity of inhibitory G proteins may contribute to increased central sympathetic tone. In our study of global Gαo knockout mice (Zuberi et al., 2008), in addition to the neurological abnormalities previously described, we found the mice to be tachycardic with loss of diurnal rhythm and fairly selective loss of the LF component of HRV with preserved total power. However, carbachol still had a negative chronotropic effect. This is suggestive of increased sympathetic tone, likely to be of central origin.
Significant progress has been made in our understanding of the autonomic nervous system and the effects upon cardiac function. Nonetheless, further study of the chronic alterations in the autonomic nervous system and their cardiac consequences may prove useful in the prevention of cardiac disease and provide valuable treatment modalities (Brodde and Michel, 1999).
It is clear that inhibitory G proteins and their associated RGS proteins play a critical role in autonomic signaling at multiple levels. Their role in mediating the parasympathetic tone to the heart is well established. What is unclear is their role in autonomic traffic in the central autonomic circuits. This will need further elucidation to dissect the role of autonomic tone in cardiac pathophysiology. What is also unclear is what role the autonomic nervous system and the specific signaling molecules have in generating cardiac arrhythmia. Mice with global G protein deletion often have reduced viability and are difficult to work with and it can be unclear at which level signaling is contributing to the phenotype. Use of more refined temporal and spatial conditional deletion strategies would circumvent some of the problems and help define the relative role of inhibitory G proteins and its subtypes in the central and peripheral autonomic control of the heart.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aistrup, G. L., Villuendas, R., Ng, J., Gilchrist, A., Lynch, T. W., Gordon, D., Cokic, I., Mottl, S., Zhou, R., Dean, D. A., Wasserstrom, J. A., Goldberger, J. J., Kadish, A. H., and Arora, R. (2009). Targeted G-protein inhibition as a novel approach to decrease vagal atrial fibrillation by selective parasympathetic attenuation. Cardiovasc. Res. 83, 481–492.
Bauer, A., McDonald, A. D., Nasir, K., Peller, L., Rade, J. J., Miller, J. M., Heldman, A. W., and Donahue, J. K. (2004). Inhibitory G protein overexpression provides physiologically relevant heart rate control in persistent atrial fibrillation. Circulation 110, 3115–3120.
Bender, K., Nasrollahzadeh, P., Timpert, M., Liu, B., Pott, L., and Kienitz, M. C. (2008). A role for RGS10 in adrenergic modulation of G-protein-activated K+ (GIRK) channel current in rat atrial myocytes. J. Physiol. (Lond.) 586, 2049–2060.
Benjamin, E. J., Wolf, P. A., D’Agostino, R. B., Silbershatz, H., Kannel, W. B., and Levy, D. (1998). Impact of atrial fibrillation on the risk of death: the Framingham Heart Study. Circulation 98, 946–952.
Bettoni, M., and Zimmermann, M. (2002). Autonomic tone variations before the onset of paroxysmal atrial fibrillation. Circulation 105, 2753–2759.
Boknik, P., Grote-Wessels, S., Barteska, G., Jiang, M., Müller, F. U., Schmitz, W., Neumann, J., and Birnbaumer, L. (2009). Genetic disruption of G proteins, G(i2)alpha or G(o)alpha, does not abolish inotropic and chronotropic effects of stimulating muscarinic cholinoceptors in atrium. Br. J. Pharmacol. 158, 1557–1564.
Bösche, L. I., Wellner-Kienitz, M.-C., Bender, K., and Pott, L. (2003). G protein-independent inhibition of GIRK current by adenosine in rat atrial myocytes overexpressing A1 receptors after adenovirus-mediated gene transfer. J. Physiol. (Lond.) 550, 707–717.
Brack, K. E., Patel, V. H., Coote, J. H., and Ng, G. A. (2007). Nitric oxide mediates the vagal protective effect on ventricular fibrillation via effects on action potential duration restitution in the rabbit heart. J. Physiol. (Lond.) 583, 695–704.
Brodde, O. E., and Michel, M. C. (1999). Adrenergic and muscarinic receptors in the human heart. Pharmacol. Rev. 51, 651–690.
Caulfield, M. P., Jones, S., Vallis, Y., Buckley, N. J., Kim, G. D., Milligan, G., and Brown, D. A. (1994). Muscarinic M-current inhibition via G alpha q/11 and alpha-adrenoceptor inhibition of Ca2+ current via G alpha o in rat sympathetic neurones. J. Physiol. (Lond.) 477(Pt 3), 415–422.
Cifelli, C., Rose, R. A., Zhang, H., Voigtlaender-Bolz, J., Bolz, S.-S., Backx, P. H., and Heximer, S. P. (2008). RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ. Res. 103, 527–535.
Coumel, P., Attuel, P., Lavallee, J., Flammang, D., Leclercq, J. F., and Slama, R. (1978). The atrial arrhythmia syndrome of vagal origin. Arch. Mal. Coeur. Vaiss. 71, 645–656.
DeGeorge, B. R. J., Gao, E., Boucher, M., Vinge, L. E., Martini, J. S., Raake, P. W., Chuprun, J. K., Harris, D. M., Kim, G. W., Soltys, S., Eckhart, A. D., and Koch, W. J. (2008). Targeted inhibition of cardiomyocyte Gi signaling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation 117, 1378–1387.
DiFrancesco, D. (2010). The role of the funny current in pacemaker activity. Circ. Res. 106, 434–446.
Dizayee, S., Kaestner, S., Kuck, F., Hein, P., Klein, C., Piekorz, R. P., Meszaros, J., Matthes, J., Nurnberg, B., and Herzig, S. (2011). Galphai2- and Galphai3-specific regulation of voltage-dependent L-type calcium channels in cardiomyocytes. PLoS ONE 6, e24979. doi: 10.1371/journal.pone.0024979
Dobrev, D., Friedrich, A., Voigt, N., Jost, N., Wettwer, E., Christ, T., Knaut, M., and Ravens, U. (2005). The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112, 3697–3706.
Dolphin, A. C. (2003). G protein modulation of voltage-gated calcium channels. Pharmacol. Rev. 55, 607–627.
Doupnik, C. A., Xu, T., and Shinaman, J. M. (2001). Profile of RGS expression in single rat atrial myocytes. Biochim. Biophys. Acta 1522, 97–107.
Duan, S. Z., Christe, M., Milstone, D. S., and Mortensen, R. M. (2007). Go but not Gi2 or Gi3 is required for muscarinic regulation of heart rate and heart rate variability in mice. Biochem. Biophys. Res. Commun. 357, 139–143.
El-Sherif, N., Zeiler, R. H., Craelius, W., Gough, W. B., and Henkin, R. (1988). QTU prolongation and polymorphic ventricular tachyarrhythmias due to bradycardia-dependent early afterdepolarizations. Afterdepolarizations and ventricular arrhythmias. Circ. Res. 63, 286–305.
Eschenhagen, T., Mende, U., Nose, M., Schmitz, W., Scholz, H., Haverich, A., Hirt, S., Doring, V., Kalmar, P., and Hoppner, W. (1992). Increased messenger RNA level of the inhibitory G protein alpha sub- unit Gi alpha-2 in human end-stage heart failure. Circ. Res. 70, 688–696.
Ewald, D. A., Pang, I. H., Sternweis, P. C., and Miller, R. J. (1989). Differential G protein-mediated coupling of neurotransmitter receptors to Ca2+ channels in rat dorsal root ganglion neurons in vitro. Neuron 2, 1185–1193.
Fischmeister, R., and Hartzell, H. C. (1986). Mechanism of action of acetylcholine on calcium current in single cells from frog ventricle. J. Physiol. (Lond.) 376, 183–202.
Foerster, K., Groner, F., Matthes, J., Koch, W. J., Birnbaumer, L., and Herzig, S. (2003). Cardioprotection specific for the G protein Gi2 in chronic adrenergic signaling through beta 2-adrenoceptors. Proc. Natl. Acad. Sci. U.S.A. 100, 14475–14480.
Fu, Y., Huang, X., Piao, L., Lopatin, A. N., and Neubig, R. R. (2007). Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: implications for sick sinus syndrome and AV block. Am. J. Physiol. Heart Circ. Physiol. 292, H2532–H2539.
Fu, Y., Huang, X., Zhong, H., Mortensen, R. M., D’Alecy, L. G., and Neubig, R. R. (2006). Endogenous RGS proteins and G alpha subtypes differentially control muscarinic and adenosine-mediated chronotropic effects. Circ. Res. 98, 659–666.
Gehrmann, J., Meister, M., Maguire, C. T., Martins, D. C., Hammer, P. E., Neer, E. J., Berul, C. I., and Mende, U. (2002). Impaired parasympathetic heart rate control in mice with a reduction of functional G protein betagamma-subunits. Am. J. Physiol. Heart Circ. Physiol. 282, H445–56.
Gross, V., Tank, J., Obst, M., Plehm, R., Blumer, K. J., Diedrich, A., Jordan, J., and Luft, F. C. (2005). Autonomic nervous system and blood pressure regulation in RGS2-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R1134–R1142.
Haissaguerre, M., Jais, P., Shah, D. C., Takahashi, A., Hocini, M., Quiniou, G., Garrigue, S., Le Mouroux, A., Le Metayer, P., and Clementy, J. (1998). Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659–666.
Hirayama, Y., and Hartzell, H. C. (1997). Effects of protein phosphatase and kinase inhibitors on Ca2+ and Cl− currents in guinea pig ventricular myocytes. Mol. Pharmacol. 52, 725–734.
Ho, K. K., Pinsky, J. L., Kannel, W. B., and Levy, D. (1993). The epidemiology of heart failure: the Framingham Study. J. Am. Coll. Cardiol. 22, 6A–13A.
Hollinger, S., and Hepler, J. R. (2002). Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol. Rev. 54, 527–559.
Huang, X., Fu, Y., Charbeneau, R. A., Saunders, T. L., Taylor, D. K., Hankenson, K. D., Russell, M. W., D’Alecy, L. G., and Neubig, R. R. (2006). Pleiotropic phenotype of a genomic knock-in of an RGS-insensitive G184S Gnai2 allele. Mol. Cell. Biol. 26, 6870–6879.
Jalife, J. (2003). Rotors and spiral waves in atrial fibrillation. J. Cardiovasc. Electrophysiol. 14, 776–780.
Jiang, M., and Bajpayee, N. S. (2009). Molecular mechanisms of go signaling. Neurosignals 17, 23–41.
Kardestuncer, T., Wu, H., Lim, A. L., and Neer, E. J. (1998). Cardiac myocytes express mRNA for ten RGS proteins: changes in RGS mRNA expression in ventricular myocytes and cultured atria. FEBS Lett. 438, 285–288.
Karschin, C., Schreibmayer, W., Dascal, N., Lester, H., Davidson, N., and Karschin, A. (1994). Distribution and localization of a G-protein-coupled inwardly rectifying K+ channel in the rat. FEBS Lett. 348, 139–144.
Kasanuki, H., Ohnishi, S., Ohtuka, M., Matsuda, N., Nirei, T., Isogai, R., Shoda, M., Toyoshima, Y., and Hosoda, S. (1997). Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease. Circulation 95, 2277–2285.
Kolman, B. S., Verrier, R. L., and Lown, B. (1975). The effect of vagus nerve stimulation upon vulnerability of the canine ventricle: role of sympathetic-parasympathetic interactions. Circulation 52, 578–585.
Kovoor, P., Wickman, K., Maguire, C. T., Pu, W., Gehrmann, J., Berul, C. I., and Clapham, D. E. (2001). Evaluation of the role of I(KACh) in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 37, 2136–2143.
Krapivinsky, G., Gordon, E. A., Wickman, K., Velimirovic, B., Krapivinsky, L., and Clapham, D. E. (1995). The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K(+)-channel proteins. Nature 374, 135–141.
Lakatta, E. G., and DiFrancesco, D. (2009). What keeps us ticking: a funny current, a calcium clock, or both? J. Mol. Cell. Cardiol. 47, 157–170.
Leaney, J. L., and Tinker, A. (2000). The role of members of the pertussis toxin-sensitive family of G proteins in coupling receptors to the activation of the G protein-gated inwardly rectifying potassium channel. Proc. Natl. Acad. Sci. U.S.A. 97, 5651–5656.
Leenhardt, A., Glaser, E., Burguera, M., Nürnberg, M., Maison-Blanche, P., and Coumel, P. (1994). Short-coupled variant of torsade de pointes. A new electrocardiographic entity in the spectrum of idiopathic ventricular tachyarrhythmias. Circulation 89, 206–215.
Lei, M., Brown, H. F., and Terrar, D. A. (2000). Modulation of delayed rectifier potassium current, iK, by isoprenaline in rabbit isolated pacemaker cells. Exp. Physiol. 85, 27–35.
Logothetis, D. E., Kurachi, Y., Galper, J., Neer, E. J., and Clapham, D. E. (1987). The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature 325, 321–326.
Lüscher, C., Jan, L. Y., Stoffel, M., Malenka, R. C., and Nicoll, R. A. (1997). G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19, 687–695.
Lüscher, C., and Slesinger, P. A. (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11, 301–315.
Ma, X., Abboud, F. M., and Chapleau, M. W. (2002). Analysis of afferent, central, and efferent components of the baroreceptor reflex in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283, R1033–R1040.
Maltsev, V. A., and Lakatta, E. G. (2008). Dynamic interactions of an intracellular Ca2+ clock and membrane ion channel clock underlie robust initiation and regulation of cardiac pacemaker function. Cardiovasc. Res. 77, 274–284.
McCleskey, E. W., Fox, A. P., Feldman, D., and Tsien, R. W. (1986). Different types of calcium channels. J. Exp. Biol. 124, 177–190.
Moss, A. J., and Kass, R. S. (2005). Long QT syndrome: from channels to cardiac arrhythmias. J. Clin. Invest. 115, 2018–2024.
Nagata, K., Ye, C., Jain, M., Milstone, D. S., Liao, R., and Mortensen, R. M. (2000). Galpha(i2) but not Galpha(i3) is required for muscarinic inhibition of contractility and calcium currents in adult cardiomyocytes. Circ. Res. 87, 903–909.
Nattel, S., Burstein, B., and Dobrev, D. (2008). Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ. Arrhythm. Electrophysiol. 1, 62–73.
Noseworthy, P. A., and Newton-Cheh, C. (2008). Genetic determinants of sudden cardiac death. Circulation 118, 1854–1863.
Ochi, R., Momose, Y., Oyama, K., and Giles, W. R. (2006). Sphingosine-1-phosphate effects on guinea pig atrial myocytes: alterations in action potentials and K+ currents. Cardiovasc. Res. 70, 88–96.
Ono, K., Masumiya, H., Sakamoto, A., Christé, G., Shijuku, T., Tanaka, H., Shigenobu, K., and Ozaki, Y. (2001). Electrophysiological analysis of the negative chronotropic effect of endothelin-1 in rabbit sinoatrial node cells. J. Physiol. (Lond.) 537, 467–488.
Ono, K., Tsujimoto, G., Sakamoto, A., Eto, K., Masaki, T., Ozaki, Y., and Satake, M. (1994). Endothelin-A receptor mediates cardiac inhibition by regulating calcium and potassium currents. Nature 370, 301–304.
Owen, V. J., Burton, P. B., Mullen, A. J., Birks, E. J., Barton, P., and Yacoub, M. H. (2001). Expression of RGS3, RGS4 and Gi alpha 2 in acutely failing donor hearts and end-stage heart failure. Eur. Heart J. 22, 1015–1020.
Pappone, C., Santinelli, V., Manguso, F., Vicedomini, G., Gugliotta, F., Augello, G., Mazzone, P., Tortoriello, V., Landoni, G., Zangrillo, A., Lang, C., Tomita, T., Mesas, C., Mastella, E., and Alfieri, O. (2004). Pulmonary vein denervation enhances long-term benefit after circumferential ablation for paroxysmal atrial fibrillation. Circulation 109, 327–334.
Pilowsky, P. M., and Goodchild, A. K. (2002). Baroreceptor reflex pathways and neurotransmitters: 10 years on. J. Hypertens. 20, 1675–1688.
Poole-Wilson, P. A., Swedberg, K., Cleland, J. G., Di Lenarda, A., Hanrath, P., Komajda, M., Lubsen, J., Lutiger, B., Metra, M., Remme, W. J., Torp-Pedersen, C., Scherhag, A., and Skene, A. (2003). Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet 362, 7–13.
Posokhova, E., Wydeven, N., Allen, K. L., Wickman, K., and Martemyanov, K. A. (2010). RGS6/Gss5 complex accelerates IKACh gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ. Res. 107, 1350–1354.
Pruvot, E., Thonet, G., Vesin, J. M., van-Melle, G., Seidl, K., Schmidinger, H., Brachmann, J., Jung, W., Hoffmann, E., Tavernier, R., Block, M., Podczeck, A., and Fromer, M. (2000). Heart rate dynamics at the onset of ventricular tachyarrhythmias as retrieved from implantable cardioverter-defibrillators in patients with coronary artery disease. Circulation 101, 2398–2404.
Rudolph, U., Spicher, K., and Birnbaumer, L. (1996). Adenylyl cyclase inhibition and altered G protein subunit expression and ADP-ribosylation patterns in tissues and cells from Gi2 alpha-/-mice. Proc. Natl. Acad. Sci. U.S.A. 93, 3209–3214.
Sharifov, O. F., Fedorov, V. V., Beloshapko, G. G., Glukhov, A. V., Yushmanova, A. V., and Rosenshtraukh, L. V. (2004). Roles of adrenergic and cholinergic stimulation in spontaneous atrial fibrillation in dogs. J. Am. Coll. Cardiol. 43, 483–490.
Spyer, K. M. (1994). Annual review prize lecture. Central nervous mechanisms contributing to cardiovascular control. J. Physiol. (Lond.) 474, 1–19.
Stauss, H. M. (2003). Heart rate variability. Am. J. Physiol. Regul. Integr. Comp. Physiol. 285, R927–R931.
Sternweis, P. C., and Robishaw, J. D. (1984). Isolation of two proteins with high affinity for guanine nucleotides from membranes of bovine brain. J. Biol. Chem. 259, 13806–13813.
Strittmatter, S. M., Valenzuela, D., Kennedy, T. E., Neer, E. J., and Fishman, M. C. (1990). G0 is a major growth cone protein subject to regulation by GAP-43. Nature 344, 836–841.
Swedberg, K., Komajda, M., Böhm, M., Borer, J. S., Ford, I., Dubost-Brama, A., Lerebours, G., Tavazzi, L., and SHIFT Investigators. (2010). Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet 376, 875–885.
Thireau, J., Zhang, B. L., Poisson, D., and Babuty, D. (2007). Heart rate variability in mice: a theoretical and practical guide. Exp. Physiol. 93, 83–94.
Tuomi, J. M., Chidiac, P., and Jones, D. L. (2010). Evidence for enhanced M3 muscarinic receptor function and sensitivity to atrial arrhythmia in the RGS2-deficient mouse. Am. J. Physiol. Heart Circ. Physiol. 298, H554–H561.
Valenzuela, D., Han, X., Mende, U., Fankhauser, C., Mashimo, H., Huang, P., Pfeffer, J., Neer, E. J., and Fishman, M. C. (1997). G alpha(o) is necessary for muscarinic regulation of Ca2+ channels in mouse heart. Proc. Natl. Acad. Sci. U.S.A. 94, 1727–1732.
Verrier, R. L., and Antzelevitch, C. (2004). Autonomic aspects of arrhythmogenesis: the enduring and the new. Curr. Opin. Cardiol. 19, 2–11.
Voigt, N., Maguy, A., Yeh, Y. H., Qi, X., Ravens, U., Dobrev, D., and Nattel, S. (2008). Changes in I K, ACh single-channel activity with atrial tachycardia remodelling in canine atrial cardiomyocytes. Cardiovasc. Res. 77, 35–43.
Wakili, R., Voigt, N., Kaab, S., Dobrev, D., and Nattel, S. (2011). Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 121, 2955–2968.
Waxman, M. B., and Wald, R. W. (1977). Termination of ventricular tachycardia by an increase in cardiac vagal drive. Circulation 56, 385–391.
Welzig, C. M., Shin, D. G., Park, H. J., Kim, Y. J., Saul, J. P., and Galper, J. B. (2003). Lipid lowering by pravastatin increases parasympathetic modulation of heart rate. Circulation 108, 2743–2746.
Wettschureck, N., and Offermanns, S. (2005). Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204.
Wickman, K., Nemec, J., Gendler, S. J., and Clapham, D. E. (1998). Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20, 103–114.
Wieland, T., and Mittmann, C. (2003). Regulators of G-protein signalling: multifunctional proteins with impact on signalling in the cardiovascular system. Pharmacol. Ther. 97, 95–115.
Wijffels, M. C., Kirchhof, C. J., Dorland, R., and Allessie, M. A. (1995). Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92, 1954–1968.
Winter, S., Brunk, I., Walther, D. J., Höltje, M., Jiang, M., Peter, J.-U., Takamori, S., Jahn, R., Birnbaumer, L., and Ahnert-Hilger, G. (2005). Galphao2 regulates vesicular glutamate transporter activity by changing its chloride dependence. J. Neurosci. 25, 4672–4680.
Wong, Y. H., Federman, A., Pace, A. M., Zachary, I., Evans, T., Pouysségur, J., and Bourne, H. R. (1991). Mutant alpha subunits of Gi2 inhibit cyclic AMP accumulation. Nature 351, 63–65.
Yang, J., Huang, J., Maity, B., Gao, Z., Lorca, R. A., Gudmundsson, H., Li, J., Stewart, A., Swaminathan, P. D., Ibeawuchi, S. R., Shepherd, A., Chen, C. K., Kutschke, W., Mohler, P. J., Mohapatra, D. P., Anderson, M. E., and Fisher, R. A. (2010). RGS6, a modulator of parasympathetic activation in heart. Circ. Res. 107, 1345–1349.
Yoro Sy, G., Urosevic, D., Fellmann, L., Greney, H., Bousquet, P., and Feldman, J. (2008). G-protein inwardly rectifying potassium channels are involved in the hypotensive effect of I1-imidazoline receptor selective ligands. J. Hypertens. 26, 1025–1032.
Young, C. N., and Davisson, R. L. (2011). In vivo assessment of neurocardiovascular regulation in the mouse: principles, progress, and prospects. Am. J. Physiol. Heart Circ. Physiol. 301, H654–H662.
Zheng, M., Zhu, W., Han, Q., and Xiao, R. P. (2005). Emerging concepts and therapeutic implications of beta-adrenergic receptor subtype signaling. Pharmacol. Ther. 108, 257–268.
Zuberi, Z., Birnbaumer, L., and Tinker, A. (2008). The role of inhibitory heterotrimeric G proteins in the control of in vivo heart rate dynamics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1822–R1830.
Keywords: inhibitory G protein, regulators of G protein signaling, heart, arrhythmia
Citation: Ang R, Opel A and Tinker A (2012) The role of inhibitory G proteins and regulators of G protein signaling in the in vivo control of heart rate and predisposition to cardiac arrhythmias. Front. Physio. 3:96. doi: 10.3389/fphys.2012.00096
Received: 02 February 2012; Paper pending published: 23 February 2012;
Accepted: 27 March 2012; Published online: 24 April 2012.
Edited by:
Craig Doupnik, University of South Florida College of Medicine, USAReviewed by:
Andrew F. James, University of Bristol, UKCopyright: © 2012 Ang, Opel and Tinker. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Andrew Tinker, William Harvey Heart Centre, Barts and The London School of Medicine and Dentistry, Room 1.02, Charterhouse Square, London EC1M 6BQ, UK. e-mail:YS50aW5rZXJAcW11bC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.