

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 12 April 2012
Sec. Cardiac Electrophysiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00093
This article is part of the Research TopicOxidant Stress in Atrial Fibrillation: Sources, impact and therapeutic significanceView all 8 articles
Postoperative atrial fibrillation (AF) is the most common arrhythmia following cardiac surgery with extracorporeal circulation. The pathogenesis of postoperative AF is multifactorial. Oxidative stress, caused by the unavoidable ischemia–reperfusion event occurring in this setting, is a major contributory factor. Reactive oxygen species (ROS)-derived effects could result in lipid peroxidation, protein carbonylation, or DNA oxidation of cardiac tissue, thus leading to functional and structural myocardial remodeling. The vulnerability of myocardial tissue to the oxidative challenge is also dependent on the activity of the antioxidant system. High ROS levels, overwhelming this system, should result in deleterious cellular effects, such as the induction of necrosis, apoptosis, or autophagy. Nevertheless, tissue exposure to low to moderate ROS levels could trigger a survival response with a trend to reinforce the antioxidant defense system. Administration of n−3 polyunsaturated fatty acids (PUFA), known to involve a moderate ROS production, is consistent with a diminished vulnerability to the development of postoperative AF. Accordingly, supplementation of n−3 PUFA successfully reduced the incidence of postoperative AF after coronary bypass grafting. This response is due to an up-regulation of antioxidant enzymes, as shown in experimental models. In turn, non-enzymatic antioxidant reinforcement through vitamin C administration prior to cardiac surgery has also reduced the postoperative AF incidence. Therefore, it should be expected that a mixed therapy result in an improvement of the cardioprotective effect by modulating both components of the antioxidant system. We present novel available evidence supporting the hypothesis of an effective prevention of postoperative AF including a two-step therapeutic strategy: n−3 PUFA followed by vitamin C supplementation to patients scheduled for cardiac surgery with extracorporeal circulation. The present study should encourage the design of clinical trials aimed to test the efficacy of this strategy to offer new therapeutic opportunities to patients challenged by ischemia–reperfusion events not solely in heart, but also in other organs such as kidney or liver in transplantation surgeries.
Atrial fibrillation (AF) is the most common arrhythmia affecting patients after general thoracic surgery. It is associated with increased risk of stroke and death and constitutes a substantial use of healthcare resources, including increased duration of hospitalization and hospital costs (Zimmer et al., 2003; Gillespie et al., 2005, 2006). Together with flutter, it is one of the two most the common arrhythmias that develop in patients postoperatively following coronary artery bypass graft surgery with extracorporeal circulation. Age is the only risk factor most consistently identified as a major independent predictor for the development of postoperative AF. Among other risk factors even a genetic predisposition is plausible since there is influence of race (Rader et al., 2011). Despite the available resources of all the modern antiarrhythmic drugs for effective prophylaxis and advances in surgical techniques, the incidence of postoperative AF remains unchanged, ranging from 15 to 40% (Maisel et al., 2001; Elahi et al., 2003; Mathew et al., 2004). Postoperative AF is associated with significant morbidity and mortality (Villareal et al., 2004; El-Chami et al., 2010). Moreover, it has been recognized as a risk factor for short-term morbidity and decreased long-term survival (Saxena et al., 2012). It was reported that intensive care unit stay and hospital stay as well as in-hospital mortality were also significantly higher among the patients with AF compared with the sinus rhythm group (Attaran et al., 2011). The precise pathophysiology of postoperative AF is unknown, however most of the evidence suggests it is multifactorial (Alqahtani, 2010). The problem of AF is now widely appreciated, but the underlying mechanisms that lead to onset and persistence of arrhythmia have been difficult to elucidate (Van Wagoner, 2007). It is of interest that despite the documented clinical impact of postoperative AF, the lack of mechanistic understanding of its occurrence has contributed in part to explain why there are no uniformly accepted treatments for its prevention. The pathophysiological mechanisms of postoperative AF are influenced by preoperative, intraoperative, and postoperative factors. In on-pump cardiac surgery, the unavoidable occurrence of an ischemia–reperfusion event leads to the formation of reactive oxygen species (ROS), causing oxidative stress, and a systemic inflammatory response (Matata et al., 2000; Elahi et al., 2008; Van Wagoner, 2008). ROS react with cell molecules such as lipids, proteins, and nucleic acids, leading to alterations of a variety of metabolic pathways and abnormal function of cellular structures and macromolecules. Thus, ROS-mediated oxidative damage of cardiomyocyte is characterized by processes such as lipid peroxidation, protein carbonylation and/or nitration, DNA oxidation, among others, accounting for structural modifications in cardiac tissue. Accumulating evidence suggests a link between AF and myocardial oxidative processes, since the latter may contribute to atrial remodeling (Korantzopoulos et al., 2003; Van Wagoner, 2003). These changes are associated with breakdown of cell membrane, impairment of mitochondrial function, calcium overload, and apoptosis. In turn, there has been emerging data to support the association between inflammation and AF (Boos et al., 2006). Oxidative stress triggers proinflammatory signaling pathways that activate nuclear factor kappaB (NF-κB) and AP-1 transcription factors (Bowie and O’Neill, 2000). The deleterious consequences of high ROS exposure, such as inflammation, cell death (apoptosis/autophagy/necrosis), or fibrosis, may be abrogated by myocardial preconditioning caused by previous exposure to moderate ROS concentration known to trigger survival response mechanisms (Fruehauf and Meyskens, 2007). Marked inflammatory infiltrates, myocyte necrosis, and fibrosis have been demonstrated in the atrial biopsies of patients with lone AF refractory to antiarrhythmic drug therapy, but not in control patients (Frustaci et al., 1997). Consequently, the structural cardiomyocyte alterations may give rise to aberrations in impulse generation, propagation, or the duration and configuration of individual cardiac action potentials, thus accounting for the basis of disorders of cardiac rhythm. The Na+ channel plays a central role in the generation of sinus rhythm and its alteration is associated with AF (Grant, 2001; Olson et al., 2005). Therefore, the myocardial oxidative injury can lead to increased susceptibility to postoperative AF. On the basis of this paradigm, it should be expected that a reinforcement of the antioxidant defense system, prior to the oxidative challenge, results in a cardioprotective effect expressed by a decreased incidence of postoperative AF. Up to date, to our knowledge, the largely empirical prophylactic therapies have not taken into account this point of view. This study presents the molecular basis to achieve a decreased vulnerability of myocardial tissue to the oxidative challenge created by its exposure to the ischemia–reperfusion occurring during cardiac surgery with extracorporeal circulation (on-pump). The novel pharmacological design is based on a two-step strategy aimed to exert a positive modulation of the antioxidant system.
Oxidative stress constitutes a unifying mechanism of injury of many types of disease processes (Rodrigo, 2009), it occurs when there is an imbalance between the generation of ROS and the antioxidant defense systems in the body so that the latter become overwhelmed (Juránek and Bezek, 2005). In the cellular metabolism, the oxygen molecule itself is reduced to water after forming, as successive intermediates, superoxide, hydrogen peroxide (H2O2), and hydroxyl radical. It is estimated that between 0.15 and 2% of cellular O2 consumption results in superoxide formation (Tahara et al., 2009). In turn, RNS mainly include nitric oxide (NO), peroxynitrite anion (ONOO−) that is the product of the reaction between NO and superoxide, and nitrogen dioxide radical (NOO·) formed during the homolytic decomposition of peroxynitrous acid. Ischemic heart disease is perhaps the human condition in which the role of oxidative stress has been investigated in more detail. There is major evidence on the contribution of oxidative stress to myocardial damage in clinical settings related to ischemia–reperfusion events, such as acute myocardial infarction, percutaneous coronary interventions following myocardial infarction or postoperative AF, among others. ROS and RNS and consequent expression of oxidative damage have been demonstrated both in ischemia and during post-ischemic reperfusion in humans. However, they may also participate as important signaling species in the normal physiology of healthy cells. Moreover, available evidence suggests that these molecules can trigger defensive mechanisms essential for cell survival. The role of intermediate metabolism during myocardial ischemia together with the cellular redox state might represent a promising interpretative key (Ceconi et al., 2003). The mitochondrial electron transport chain is involved in oxygen sensing and would therefore need to respond to changes in oxygen levels (Debreceni and Debreceni, 2011).
Cardiac myocytes are able to generate ROS in a variety of ways. The greatest ROS source is from electrons lost during transfer between electron transport chain complexes in mitochondrial oxidative phosphorylation processes. Mitochondrial ROS are produced during oxidative metabolism through the one electron reduction of oxygen (O2) to form superoxide; the mitochondrial electron transport chain generates superoxide predominantly at complexes I and III (Murphy, 2009). In addition, ROS can be generated from enzymes, such as NADPH oxidase (from heart and phagocytes), uncoupled endothelial nitric oxide synthase (eNOS), xanthine oxidase (XO), and lipoxygenase/cyclooxygenase, aside from the oxidation of catecholamines. NADPH oxidase activity occurs in cardiomyocytes and heart endothelial cells. Furthermore, the phagocytic NADPH oxidase NOX2 (gp91) produces superoxide anions by reducing O2 (Brown and Griendling, 2009); local non-phagocytic NADPH oxidases have been implicated in ROS production following growth factor and cytokine stimulation (Ushio-Fukai, 2009). The cellular effects of ROS are partially mediated by NF-κB activation. NF-κB mainly exists in the cytosol as a pre-formed trimeric complex that consists of the inhibitory protein IκB and the P50/P65 protein dimer. ROS induce redox changes that result in phosphorylation of IκB subunit, thereby activating its proteolytic digestion. When the inhibitor subunit is dislodged from the P60/P65 heterodimer, NF-κB can translocate to the nucleus, bind DNA, and initiate transcription. In addition to being a major mediator of cytokine effects in the heart, NF-κB regulates cardiac gene expression programs downstream of multiple signal transduction cascades in a variety of physiological and pathophysiological states. Blocking NF-κB reduces infarct size in the murine heart after ischemia–reperfusion (Jones et al., 2003), implicating NF-κB as a major determinant of cell death in this setting, thus supporting the concept that it may be an important therapeutic target for specific cardiovascular disease. Antioxidants, including vitamin E, can abrogate NF-κB activation. Oxidation of P50 on its DNA-binding domain has been shown to act as a protective mechanism by preventing NF-κB binding (Wang et al., 2011a). Finally, a novel zinc-finger protein, monocyte chemoattractant protein-1 (MCP-1)-induced protein (MCPIP), is thought to have NF-κB inhibitory activity in certain cell cultures, but its pathophysiological consequence in vivo remains undefined. Compared with lipopolysaccharide (LPS)-treated wild-type mice, MCPIP transgenic mice have markedly reduced levels of myocardial inflammatory cytokines, less iNOS expression and peroxynitrite formation, decreased caspase-3/7 activities, and apoptotic cell death. This novel protein might protect the heart from inflammatory pathologies, possibly through inhibition of the IκB kinase complex, which prevents NF-κB activation, and attenuates the proinflammatory state and nitrosative stress in the myocardium (Niu et al., 2011).
Enzymatic antioxidant defenses include superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and catalase (CAT). Non-enzymatic antioxidants include a variety of mostly exogenous biological molecules, such as ascorbic acid (vitamin C), α-tocopherol (vitamin E), reduced glutathione (GSH), carotenoids, flavonoids, polyphenols, and other exogenous antioxidants (Gasparetto et al., 2005). Oxidative stress develops when ROS production is sufficient to overwhelm the antioxidant defense system, which leads to increased ROS and RNS steady-state concentrations thereby damaging biomolecules. Examples of pathophysiological cardiac states associated with oxidative stress derived from ischemia and reperfusion are acute myocardial infarction, cardiac surgery with extracorporeal circulation and heart transplantation, among others. The vulnerability of myocardial tissue to oxidative challenges caused by reperfusion is dependent on antioxidant system activity. The first line of cellular defense against oxidative injury include the antioxidant enzymes CAT, SOD, and GSH-Px. MnSOD overexpression protects the murine myocardium from post-ischemic injury; however, neither GSH-Px nor Cu/ZnSOD appear to be important determinants of the extent of myocardial injury in this in vivo model (Jones et al., 2003). Consistent with this view, it has been reported that infarct size is markedly reduced in transgenic mice that over-express SOD (Chen et al., 1998). In contrast, in a tamoxifen-inducible cardiomyocyte-specific catalase-over-expressing mouse model, there were no acute functional improvements with physiological CAT overexpression before myocardial infarction. However, hydrogen peroxide scavenging reduced proinflammatory cytokines and altered cardiac collagen isoforms, associated with an improvement in cardiac function after 21 days. These data suggest that sustained rather than acute H2O2 levels may be relevant in the myocardial remodeling and cardiac function (Pendergrass et al., 2011). Mitochondrial and cell cytosolic components of the antioxidant system can neutralize excess mitochondrial ROS under most conditions.
Reactive oxygen species are not just by-products of mitochondrial respiration, but also play a key role in cell signaling. Cardiac tissue exposure to low to moderate ROS levels should trigger a survival response and reinforce ROS scavengers of the antioxidant defense system to elicit a cardioprotective effect for myocardial reperfusion. In fact, the molecular mechanism responsible for this adaptive change involves enhanced antioxidant activity achieved by up-regulating several housekeeping genes partly under the control of nuclear factor erythroid 2-related factor-2 (Nrf2). Nrf2 is normally sequestered in the cytosol by Kelch-like ECH-associated protein 1 (Keap1). Upon oxidative stimulation, Nrf2 oxidizes or covalently modifies Keap1 thiol groups, dissociates from Keap1 and undergoes nuclear translocation. In the nucleus, Nrf2 binds to antioxidant response elements (ARE) in target gene promoters (Kobayashi and Yamamoto, 2005), which increase the expression of antioxidant enzymes. It was demonstrated that the constitutive levels/activities of a number of important antioxidants and phase 2 enzymes, such as CAT, GSH-Px, glutathione reductase, glutathione S-transferase, NAD(P)H:quinone oxidoreductase 1, and heme oxygenase-1 in primary cardiomyocytes are dependent on Nrf2 status. Therefore, Nrf2 should be expected to contribute to diminish the susceptibility of cardiomyocytes to injury elicited by oxidants and electrophilic species (Zhu et al., 2008), making the Nrf2 signaling pathway an important mechanism for myocardial cytoprotection.
It is of interest to note that the magnitude of ROS levels could determine the activation of NF-κB and/or Nrf2 pathways.
The pathophysiological effects of ROS depend upon the type, concentration, and specific site of production and involve three broad types of action. A wide mechanistic survey of AF is beyond the scope of the present review, but it has been previously discussed in detail (Van Wagoner, 2007). This section is devoted to present the experimental and clinical evidence to account for the role of ROS in the pathogenesis of AF following cardiac surgery.
Multiple lines of evidence have strongly suggested a link between oxidative stress and cardiac arrhythmias, especially AF (Neuman et al., 2007; Negi et al., 2010). Interestingly, cardiac surgery has also been reported to increase oxidative stress as measured by thiol ratios in the plasma and myocardium (De Vecchi et al., 1998). Moreover, these data suggest that oxidative stress markers may have predictive value in AF management. Interestingly, recent studies have demonstrated the implication of oxidative stress within the atrial tissue during AF suggesting a potential role in the remodeling phenomenon (Van Wagoner, 2003; Korantzopoulos et al., 2007).
Further evidence of the role of ROS and RNS in the development of AF has been provided. Thus, AF induced by rapid atrial pacing in pigs is characterized by increased nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity and superoxide production in the left atrium (Dudley et al., 2005). Right human atrial appendages of patients with AF exhibit higher levels of the nitrosative and oxidative stress markers 3-nitrotyrosine and protein carbonyls, respectively, compared with patients with sinus rhythm (Mihm et al., 2001). In mice, chronic cardiac overexpression of Rac1 represents a novel model for AF. Rac1 GTPase contributes to the pathogenesis of AF and might represent a target for the prevention and its treatment (Adam et al., 2007).
The myocardium can tolerate brief periods (up to 15 min) of severe and even total myocardial ischemia without resultant cardiomyocyte death. This is observed in clinical settings like coronary vasospasm, angina, and balloon angioplasty, and is therefore not associated with concomitant myocyte death (Kloner and Jennings, 2001a,b). It was suggested that endothelial dysfunction occurs early during reperfusion of a previously ischemic tissue and that it will be present for long time (Lefer and Lefer, 1996). The hypoxia and reoxygenation significantly increased pulmonary veins arrhythmogenesis and induces different electrophysiological responses in the right and left atria, which may play a role in the pathophysiology of AF (Lin et al., 2012). In a clinical setting, reperfusion injury after revascularization of the ischemia-related artery is manifested by myocardial stunning, reperfusion arrhythmia, myocyte death, and endothelial- and microvascular-dysfunction including the no-reflow phenomenon (Moens et al., 2005). Early reperfusion is an absolute prerequisite for the survival of ischemic myocardium. However, this process has been referred as the double edged sword because reperfusion may itself lead to accelerated and additional myocardial injury beyond that generated by ischemia alone (Braunwald and Kloner, 1985). This paradoxical effect results in a spectrum of reperfusion-associated pathologies, collectively called reperfusion injury (Yellon and Hausenloy, 2007). These observations are consistent with the notion that reintroduction of abundant oxygen at the onset of reperfusion evokes a burst of ROS within the first few minutes of reflow as demonstrated experimentally and in patients with acute myocardial infarction undergoing thrombolysis (Béard et al., 1994) or percutaneous coronary intervention (Babu et al., 2011), and patients undergoing open heart on-pump surgery (Akila et al., 2007).
The ability of tissues to tolerate ROS is partly conditioned by the activity of their antioxidant defense system to scavenge and degrade ROS to non-toxic molecules. Antioxidant enzymes are mainly located in the intracellular space, and, the antioxidant molecules both within the intra- and extra-cellular spaces. The setting of an ischemia–reperfusion event alters this homeostasis giving rise to a prevalence of prooxidants over antioxidant activity (oxidative stress). Consequently, the increased ROS concentration at the steady-state causes direct damage to lipids, proteins, and DNA. A large body of experimental evidence supports the notion that ROS-generated reperfusion injury occurs when oxygen is reintroduced to the ischemic tissue (Hess and Manson, 1984; Weisfeldt et al., 1988; Opie, 1991; Kilgore and Lucchesi, 1993; Zughaib et al., 1994; Ambrosio and Tritto, 1999; Park and Lucchesi, 1999). Previously, it has been proposed the beneficial effects of ROS occur at moderate concentrations and involve physiological roles in cellular responses (Martindale and Holbrook, 2002; Valko et al., 2007). It is of interest to mention that superoxide is able to react with nitric oxide (NO) via a high rate constant non-enzymatic reaction to form peroxynitrite, a highly peroxidant molecule responsible for additional structural changes, such as nitrotyrosine derivatives formation, nitrosylation, and oxidation of thiol groups. Redox signaling processes are involved in the activation of many signal transduction protein kinases and transcription factors, the stimulation of DNA synthesis and expression of growth-related genes, and the regulation of myocardial excitation–contraction coupling. Interestingly, while high levels of peroxynitrite may induce non-specific toxic effects, at lower levels it is capable of modulating signaling events in vivo indicating an additional level of complexity. NO synthase normally generate NO but may instead generate superoxide if it becomes “uncoupled,” a state that may occur from deficiency of NOS cofactor tetrahydrobiopterin (BH4) or the NOS substrate L-arginine (Verhaar et al., 2004). Interestingly, ROS produced by NADPH oxidases can promote ROS generation by other sources thereby amplifying total levels of ROS. Superoxide may oxidize and degrade BH4, thereby leading to eNOS uncoupling. Similarly, NADPH oxidase-derived ROS may also activate xanthine oxidase (Li and Shah, 2004). The evidence indicates that the primary source of superoxide production in the human atrial myocardium is an NADPH oxidase, whose activity is increased in AF patients compared with matched patients in sinus rhythm (Kim et al., 2005), what is accompanied by increased mRNA expression of NOX2 and P22 phox. In addition, it was reported that AF is associated with a 6.9-fold increase in active Rac1 (Adam et al., 2007). Possible mechanisms of Rac1 activation include increased Ang II receptor activation, or increased cytosolic Ca2+ (Wassmann et al., 2001; Cook-Mills et al., 2004; Li et al., 2006).
Compelling evidence shows that AF development and perpetuation depends on the electrophysiological and structural substrates of the atria (Allessie et al., 2002; Nattel et al., 2007). The structural substrates refer to abnormalities in atrial architecture such as atrial dilatation (Kojodjojo et al., 2007), fibrosis (Everett and Olgin, 2007), apoptosis phenomena, tissue dedifferentiation, etc. (Shiroshita-Takeshita et al., 2005). The cardiomyocytes have to cope with new circumstances and consequently change the expression and organization pattern of proteins involved in activation, conduction, and contraction. Ions like Ca2+, Na+, and K+ are important players in the excitation–contraction cycle, and the expression patterns of their channels and other proteins involved in ion homeostasis have been extensively studied. Oxidative membrane damage has been found to be of great pathological importance in ischemia–reperfusion of myocardial tissue injury. At the molecular level, membrane functions may be modified by direct attack of ROS on its related components. ROS exposure influences the physical properties of membranes, and hence ion transport mainly due to changes in the dielectric constant, which is enhanced by lipid peroxidation (Killig and Stark, 2002). This effect is caused by the accumulation of polar products of lipid peroxidation, or secondary reactive species, in the membrane interior. Consequently, membrane barrier, experienced by the charged species throughout their translocation across the membrane, would be further reduced (Stark, 2005) giving rise to many of the activation phenomena of membrane transport described in the literature. Ion channels (contrary to ion carriers) provide polar pathways for ions across the hydrophobic membrane barrier. For their proper functioning, the structural element forming the polar pathway must be inserted into the membrane, a process which is substantially facilitated by increased dielectric constant. Membrane proteins have a natural lipid environment and are therefore particularly exposed to an eventual attack of secondary reactive species.
The oxidative modification of (Na + K)-ATPase has been studied considering the direct and the indirect inactivation pathways (Shao et al., 1995). In agreement with this view it was reported that erythrocyte (Na + K)-ATPase activity correlated negatively with 8-isoprostane (in vivo index of lipid peroxidation) in hypertensive and normotensive subjects (Rodrigo et al., 2007a). Several authors have found altered mRNA and/or protein levels of ion channels during AF (Van Wagoner et al., 1997; Van Gelder et al., 1999; Brundel et al., 2001). In this setting, downregulation has been described for the L-type calcium channel (Lai et al., 1999), the sarcoplasmic reticulum calcium ATPase (Brundel et al., 1999), and several potassium channel subunits (Brundel et al., 1999; Lai et al., 1999), whereas the expression of other proteins involved in calcium handling like the sodium/calcium exchanger, phospholamban, calsequestrin, and the ryanodine receptor were not altered (Lai et al., 1999; Van Gelder et al., 1999; Brundel et al., 2001). It is of interest to mention that electrical remodeling is capable to alter ion channels being the base of AF, such as L-type calcium channel (Cav1.2), inward rectifier potassium channel (Kir2.1), transient outward potassium channel (Kv4.3), ultrarapid delayed rectifier potassium channel (Kv1.5), acetylcholine-activated potassium channel (Kir3.1), ATP-sensitive potassium channel (Kir6.2), fast sodium channel (Nav1.5). For example, it has been shown that in AF patients with a reduced density of outward potassium current, the expression of the potassium channel Kv1.5 subunit decreases (Van Wagoner et al., 1997). This indicates that the current densities are influenced by alterations in channel protein expression level, an observation that was also described in a dog model of chronic AF (Gaspo et al., 1997; Yue et al., 1997, 1999). ROS production occurs in the heart sarcolemmal and t-tubule membranes where NOX2 is located and sensitizes nearby ryanodine receptors in the sarcoplasmic reticulum. This triggers a burst of Ca2+ sparks and, consequently, arrhythmogenic Ca2+ waves (Prosser et al., 2011). These effects were prevented by the antioxidant N-(2-mercaptopropionyl) glycine in isolated cardiomyocytes, after pretreatment with digitoxin, known to increases the incidence of arrhythmogenic spontaneous Ca2+ waves, decreases the sarcoplasmic reticulum Ca2+ load, and increases both ROS and ryanodine thiol oxidation (Ho et al., 2011). In addition, inhibitors of NADPH oxidase, mitochondrial ATP-dependent channels, or permeability transition pore, but not inhibitors of xanthine oxidase, also were able to inhibit these effects. Furthermore, it was demonstrated that H2O2 enhances the late, slowly inactivating current through cardiac Na channels, thereby leading to action potential prolongation and arrhythmias (Song et al., 2006), being Ca2+/calmodulin-dependent protein kinase II (CaMKII)δ suggested as an important factor for ROS-induced arrhythmias. This is supported by the finding that the expression and activity of CaMKII are increased in arrhythmias (Wagner et al., 2006), although ROS-induced sarcoplasmic reticulum Ca2+ release may be a prerequisite for ROS-dependent CaMKII activation. It is of interest to mention that it was identified a ROS-dependent, Ca2+ independent CaMKII activation by oxidative modification of methionine 281/282, thereby showing that direct activation of CaMKII by ROS engenders Ca2+ autonomous activity, a clear but previously molecular mechanism by which CaMKII can integrate Ca2+ and ROS signals (Erickson et al., 2008).
There is considerable evidence supporting that the function of the L-type Ca2+-channel is influenced by the redox state of the cell, leading to intracellular Ca2+ overload. Ischemia–reperfusion causes an increased in the production of 20-hydroxyeicosatetraenoic acid (20-HETE), a potent vasoconstrictor that activates L-type Ca2+ channels. Inhibition of 20-HETE production has been shown to reduce infarct size caused by ischemia. In isolated crat cardiomyocytes, it was shown that this effect includes 20-HETE stimulation of NADPH oxidase-derived superoxide production, which activates L-type Ca2+ channels via a PKC-dependent mechanism (Zeng et al., 2010). The effect explaining the action of ROS on the heart was provided by the first time relating the concomitant disruption of calcium and energy metabolism. It was concluded that ROS-induced glycolytic inhibition and Ca2+ overload (Corretti et al., 1991). More recently, it was reported that a related mechanism for the development of atrial dysrhythmias is Ca2+ alternans, a subcellular phenomenon caused by modulation of sarcoplasmic reticulum Ca2+ release, which is mediated, at least in part, by local inhibition of energy metabolism (Kockskämper and Blatter, 2002). Furthermore, atrial myocytes glycolysis regulates Ca2+ release from the sarcoplasmic reticulum by direct modulation of ryanodine receptors activity by intermediates and products of glycolysis and modulation of sarcoplasmic reticulum Ca2+-ATPase activity through local changes of glycolytically derived ATP (Kockskämper et al., 2005). In support of these postulated mechanisms, it has been demonstrated that glycolytic inhibition interacts with the aged and fibrotic atria to amplify intracellular Ca2+handling abnormalities that facilitate AF development (Ono et al., 2007). Furthermore, in aged fibrotic unlike in adult rat hearts, glycolytic inhibition promotes ventricular fibrillation by activation of ATP-sensitive K channels, CaMKII, and Na+–Ca2+-exchanger (Morita et al., 2011), but these arrhythmias were suppressed by the reducing agent N-acetylcysteine and CaMKII inhibitor KN-93 (Morita et al., 2009).
Gap junctions are involved in cell–cell coupling; they form electrical conduits between adjacent myocardial cells, permitting rapid spatial passage of the excitation current essential to each heartbeat. Arrhythmogenic decreases in gap junction coupling are a characteristic of stressed, failing, and aging myocardium, but the mechanisms of decreased coupling are poorly understood. Intercellular gap junction channels are formed by homomeric or heteromeric connexin subunits (Köhler and Hoyer, 2007), thus supporting the concept that local intercellular electrical communication between the cardiomyocytes occurs through these structures (Félétou and Vanhoutte, 2006). This provides an electrical continuity allowing a uniform membrane potential among coupled cells. Nevertheless, connexins functions are beyond facilitating the exchange of metabolites and electrical signals between cells and contributing to cardiomyocyte volume regulation (Burra and Jiang, 2011). The role of gap junction channels on cardiac impulse propagation is complex. Atrial muscle preferentially expresses connexins 40 and 43. Microtubules have a well-studied role in the trafficking of connexin 43 to the plasma membrane. EB1 is a protein involved in the targeted delivery of connexin 43 to adherens junctions (Shaw et al., 2007). Oxidative stress perturbs connexon forward trafficking along microtubules to gap junctions (Smyth et al., 2010) and EB1 levels are reduced at intercalated disks in end-stage human ischemic cardiomyopathy and in adult mouse hearts exposed to acute ischemia–reperfusion injury or acute oxidative stress alone. Furthermore, mutational disruption of the EB1–tubulin interaction mimicked the effects of oxidant stress, including a reduction in surface Cx43 expression (Tomaselli, 2010). In contrast, dietary selenium, an antioxidant compound, exerts a cardioprotective effect that might be mediated by a reduced oxidative stress, a lower connexin 43 dephosphorylation, and a decreased TNF-α expression (Tanguy et al., 2011). Furthermore, in endothelial cells challenged by hypoxia-reoxygenation it was recently found that p47phox rather than NADPH oxidase-derived ROS affects the modulation of intercellular coupling, suggesting a contribution of a cross-talk between connexin 40 and NADPH oxidase (Yu et al., 2010).
The modulation by gap junction channels is prominent in cardiac tissues such as sinoatrial node-atrial muscles, atrioventricular node-His bundle, His bundle–bundle branch, and Purkinje fibers-ventricular muscles (Desplantez et al., 2007). Under conditions of oxidative stress connexin 43 distribution pattern is heterogeneous giving rise to aberrant electrical conduction, facilitating self-perpetuation of re-entry pathways thereby causing the development of AF (Tansey et al., 2006; Wilhelm et al., 2006). However, further studies are still lacking to determine the precise molecular basis linking oxidative stress with these complex phenomena occurring in postoperative AF.
Inflammation is another heavily studied pathophysiological process implicated in the pathophysiology of postoperative AF (Aviles et al., 2003; Korantzopoulos et al., 2003; Boos et al., 2006). Aside from increased ROS and calcium overload, inflammatory cell infiltration into the myocardium may promote oxidative injury, directly affecting atrial myocyte contraction (Mihm et al., 2001), and electrical conduction (Kumagai et al., 1997). Accordingly, patients with persistent AF have increased white cell count (Abdelhadi et al., 2004), as well as plasma levels of the biomarker C-reactive protein (CRP), relative to patients with paroxysmal AF, and both of these patient groups have levels greater than those with no AF (Chung et al., 2001). Thus, cardiac surgery with extracorporeal circulation leads to elevation of CRP, interleukin-6, and the complement system (Gaudino et al., 2003; Canbaz et al., 2008; Echahidi et al., 2008). The temporal course of AF occurring after cardiac surgery closely follows the activation of the complement system and release of proinflammatory cytokines. It was demonstrated that the levels of CRP on postoperative day 2 were more elevated in patients that experienced postoperative AF than in those that did not (Bruins et al., 1997). It was reported that a more pronounced increase in postoperative white cell count independently predicts development of postoperative AF (Lamm et al., 2006). During the first hour of reperfusion, superoxide triggers neutrophil infiltration, which increases cardiac damage by further release of ROS, inflammatory mediators, and proteases (Yellon and Hausenloy, 2007). Chemokines are known to induce leukocyte migration, growth, and activation. Specifically, the chemokine interleukin-8 appears to have a fundamental role in regulating neutrophil localization in ischemic myocardium. In mice, CXCL2, the homolog of human interleukin-8, is up-regulated in reperfused myocardium (Vandervelde et al., 2007). The chemokine response in ischemic tissues may be induced by various factors, including ROS, cytokines (e.g., TNF-α and complement), and NF-κB activation (Chandrasekar et al., 2001). Prospective epidemiological studies have shown that serum levels of CRP is a strong predictor of cardiovascular ischemia–reperfusion injury events, such as myocardial infarction, postoperative AF, and stroke (Kinoshita et al., 2011). Several studies revealed an independent association of high plasma CRP levels with adverse prognosis in acute myocardial infarction patients. Interestingly, preconditioning was found to inhibit post-ischemic CRP increases in a rat model of acute myocardial infarction (Valtchanova-Matchouganska et al., 2004).
Recently, it has been discussed the possible role of microRNAs in AF initiation and maintenance, as they influence cardiac excitability and other processes pertinent to AF (Wang et al., 2011b). Enhanced expression of miR-200, a family of microRNAs affecting tumorigenesis and chemosensitivity, can modulate the oxidative stress response. It was reported that miR-200a also sensitizes tumors to treatment, which could account for the limited success of antioxidants in clinical trials (Mateescu et al., 2011).
As a consequence of inflammation, impairment on the electrical properties of atrial tissue, such as inhomogeneity in atrial conduction may result (Ishii et al., 2005; Tselentakis et al., 2006).
Indeed the role of inflammation response in the intra- and extra-cellular mechanisms implicated in this disorder cannot be separated from that of oxidative stress. The biochemical nexus between these two processes represents an essential piece in the puzzle. It is of interest to mention that the transcriptional factor NF-κB was found to play a pivotal role in this connection (Pavlovic et al., 2002). It has been found that this transcriptional factor responds to changes of the cellular oxidative status (Chandra et al., 2000). When NF-κB is activated, by phosphorylation of its inhibitory cofactor, it bonds to a DNA response element (ARE) and promotes the transcription of genes coding to inflammation mediators such as CRP, interleukin-6, and fibrinogen, among others (Liakopoulos et al., 2007). NF-κB activation has been associated with cardiac dysfunction, ventricular hypertrophy, and maladaptive cardiac growth (Opie et al., 2006). Increased levels of several inflammation markers were found in serum and atria biopsies of AF and postoperative AF patients (Korantzopoulos et al., 2003; Neuman et al., 2007; Yu et al., 2010). Therefore it is reasonable to assume that oxidative stress and inflammation response act in a synergic way in the underlying pathophysiological mechanisms of postoperative AF, giving the foundation for studies involving anti-inflammatory AF therapy (Halonen et al., 2007; Goldstein et al., 2008).
Necrosis is a catastrophic type of cell death that is uncontrollable and does not involve any specific mechanisms. Necrosis is classically characterized by early plasma membrane rupture and swelling of cytoplasmic organelles, in particular mitochondria. It only refers to an irreversible stage of cell death, even though dying cells generally progress from a reversible to an irreversible stage. ROS is involved in necrotic cardiomyocyte death through MPT pore opening. Apoptosis has been implicated in the pathogenesis of several acute and chronic conditions affecting the cardiovascular system in the last decade (Gale et al., 2011). The loss of endothelial cells precedes and may predispose cardiomyocytes to undergo apoptosis (Dallabrida et al., 2005), indicating that salvaging endothelial cells is of paramount importance. It is likely that myocyte apoptosis is initiated during ischemia but dependent on reperfusion injury. Reperfusion appears to accelerate apoptosis when compared with permanent occlusion (Whelan et al., 2010). Active caspases cleave vital substrates in the cell, such as actin, actinin, β-myosin heavy chain, myosin light chain, tropomyosin, and cardiac troponins, leading to cellular demise (Communal et al., 2002). The “intrinsic” pathway utilizes mitochondria to induce cell death by opening the mitochondrial permeability transition pore (MPTP) or rupturing the outer mitochondrial membrane, both of which trigger the sudden and complete release of cytochrome c and other proteins from the intermembrane mitochondrial space into other cellular compartments. The “intrinsic” pathway is primarily activated in cardiac myocytes by cellular stimuli, such as hypoxia, ischemia–reperfusion, and oxidative stress, which perturb the MPTP and increase the permeability of the outer and inner mitochondrial membranes (Singh et al., 2011). Once released, cytochrome c binds to the cytosolic protein apaf1 and facilitates formation of the “apoptosome” complex, which results in caspase-9 activation that provokes caspase-3 activation (Adrain and Martin, 2001). Smac/DIABLO indirectly activates caspases by sequestering caspase-inhibitory proteins, while the mitochondrial release of endonuclease-G and apoptosis-inducing factor results in their translocation into the nucleus where they directly or indirectly facilitate DNA fragmentation (Cande et al., 2002). Experimental studies in mice demonstrated that the activation of Fas can induce apoptosis in cardiac myocytes and that Fas is a critical mediator of myocardial infarction due to ischemia–reperfusion in vivo (Lee et al., 2003).
Autophagy – in contrast to necrosis and apoptosis, autophagy is primarily a survival mechanism. Cellular oxidative stress and ROS have been reported to serve as important autophagic stimuli during periods of ischemia–reperfusion (Hariharan et al., 2011). Autophagic degradation and removal of damaged oxidized proteins in response to low to moderate oxidative stress is reportedly beneficial for cells. Conversely, severe oxidative stress and increasing amounts of ROS may activate signaling pathways that lead to autophagy-induced cell death. In addition, ultrastructural atrial remodeling characterized by an impaired cardiac autophagy was found in patients developing postoperative AF after coronary artery bypass surgery (García et al., 2011). Whether autophagy promotes cell survival or death depends upon the severity and degree of stress in the cellular environment (Essick and Sam, 2010). During the initial period of ischemia, enzyme XO is formed, and substrates for XO (hypoxanthine and xanthine) accumulate. Upon reperfusion, the reintroduction of O2 leads to XO-mediated superoxide generation due to the presence of xanthine and hypoxanthine (Ushio-Fukai, 2009). During this period, ATP generation decreases and results in the phosphorylation of 5′ adenosine monophosphate-activated protein kinase (AMPK), which leads to autophagosome formation through inhibition of mammalian target of rapamycin (mTOR; Matsui et al., 2008). Meanwhile, ROS damage organelles and cytosolic proteins and cause mitochondrial lipid peroxidation, all of which exacerbate autophagy (Juhaszova et al., 2004). Additionally, antioxidant enzymes, such as CAT and SOD, are targeted by autophagosomes. This ultimately leads to the induction of cell death and is thus detrimental to tissue function. Furthermore, AMPK activity decreases during reperfusion, thus increasing autophagic death and up-regulating beclin-1 (Zhu et al., 2007). Autophagy has been reported to be involved in cardioprotection against lethal ischemic injury; thus, repetitive ischemia by coronary stenosis or occlusion enhances autophagy and subsequent cardioprotection when compared with classical ischemia–reperfusion insult (Yan et al., 2009). Aside from the key role that the chemokine MCP-1 plays in cardiac damage following ischemia, it also mediates autophagy through MCP-1-induced protein (MCPIP), a novel zinc-finger protein that has transcription factor-like activity (Zhou et al., 2006). MCPI stimulates inducible NO synthase, translocation of the NADPH oxidase subunit phox47 from the cytoplasm to the membrane, ROS production, induction of endoplasmic reticulum stress markers HSP40 and autophagy, as indicated by beclin-1 induction, cleavage of microtubule-associated protein 1 light chain 3 and autophagolysosome formation, and apoptosis, respectively (Younce and Kolattukudy, 2010).
A wide survey of AF is beyond the scope of the present chapter. Nevertheless, a mechanistic hypothesis representing the effects of ischemia, as well as those derived from the re-establishment of blood flow in the ischemic myocardium, together with the inflammation caused by the surgical procedure and enhanced by ROS is presented in Figure 1.
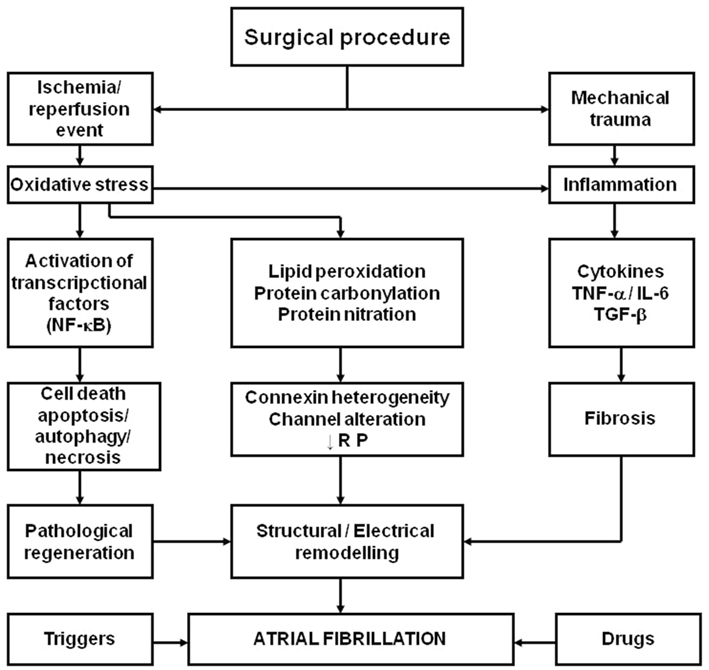
Figure 1. Schema representing the hypothesis for the role of oxidative stress and inflammation in the pathophysiology of postoperative atrial fibrillation in patients subjected to cardiac surgery with extracorporeal circulation. NF-κB, nuclear factor kappaB; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; TGF-β, transforming growth factor-β; RP, refractory period. Adapted from Rodrigo et al. (2008) with permission.
The high incidence of postoperative AF has led to the development of therapies that target the sympathetic nervous system, refractory period, or alter cardiac conduction. In summary, available evidence strongly supports the use of β-blockers as standard prophylaxis of postoperative AF (Koniari et al., 2010). However, while these agents reduce the incidence of the arrhythmia, they do not eliminate it. Moreover, β-blockers should not be used in intolerant patients or as additive therapy in high risk patients. To study the evidence supporting the use of traditional pharmacologic therapies, other than β-blockers, for the prevention of postoperative AF, the reader could be directed to a recent review (Davis et al., 2010).
At present, preventive strategies against postoperative AF offer only suboptimal benefits. Despite improvements in anesthesia, surgical technique, and medical therapy, the incidence of this complication of surgery remains high. Likely, this could be the result of a limited knowledge of the mechanisms for the development of postoperative AF. Therefore, there is a need of alternative therapies. Accordingly, newly identified pathways have given rise to novel strategies under investigation, including renin–angiotensin–aldosterone modulators, statins, non-steroidal anti-inflammatory drugs, corticosteroids, thiazolidinediones, n−3 polyunsaturated fatty acids (n−3 PUFA), ascorbic acid, N-acetylcysteine, and sodium nitroprusside. Our interest will be focused in the reinforcement of the antioxidant defense system, since ROS could contribute to the mechanism accounting for structural remodeling and inflammation of myocardial tissue through the occurrence of ischemia–reperfusion events. Therefore, it should be expected a decreased risk of myocardial tissue for the development postoperative AF in the presence of increased antioxidant activity.
The available evidence for oxidative stress in the pathophysiology of postoperative AF has stimulated the prophylactic use of antioxidants. Based on the pathogenic role of ROS and RNS, it should be expected that treatments with exogenous antioxidant agents protect against reperfusion injury. Clinical trials designed to study cardioprotection by long-term administration of vitamins C and E have failed to demonstrate beneficial effects (Flaherty et al., 1994; Stephens et al., 1996; Rapola et al., 1997; GISSI-Prevenzione Trial, 1999; Heart Protection Study Collaborative Group, 2000; Yusuf et al., 2000; de Gaetano, 2001; Guan et al., 2003; Tsujita et al., 2006; Cook et al., 2007; Sesso et al., 2008). This disappointed result could be due to the lack of consideration of the pharmacodynamics and/or the biochemical properties of these compounds. Thus, the efficacy of vitamin C as ROS scavenger cannot be reached by oral doses. In turn, high doses of vitamin E (more than 400 IU) could exert a prooxidant effect and should be avoided. Some studies have suggested that antioxidant agents attenuate left ventricular remodeling following acute myocardial infarction. When these patients had undergone primary percutaneous transluminal coronary angioplasty, pretreatment with allopurinol, an XO inhibitor, resulted in effective inhibition of oxygen-derived radical generation during reperfusion therapy and left ventricular function recovery (Guan et al., 2003). Accordingly, in a model of AF induced by rapid atrial pacing in pigs, it was demonstrated that increased activity of NADPH and xanthine oxidases in atrial tissue could account for increased superoxide anion, what was prevented by the inhibitors apocinin and oxypurinol, respectively (Dudley et al., 2005). More recently, administering the free radical scavenger edaravone to patients with acute myocardial infarction immediately prior to reperfusion significantly reduced infarct size and reperfusion arrhythmia (Tsujita et al., 2006). A prospective, randomized, placebo-controlled pilot study reported a significant reduction of the incidence of postoperative AF in patients undergoing coronary artery bypass and/or valve surgery who were treated with N-acetylcysteine (Ozaydin et al., 2011). However, other attempts, such as intravenous bolus of SOD (Yusuf et al., 2000), showed no beneficial effect on patient outcome.
Concerning the role of antioxidant vitamins in the prevention of postoperative AF, although the scientific rationale, epidemiologic data, and retrospective studies have been persuasive, prospective, randomized, placebo-controlled trials have not verified the actual benefit of antioxidant vitamins in human diseases (Rodrigo et al., 2007b). There are several possible contributory factors that could explain this discrepancy, and the lack of consideration of basic aspects, such as the pharmacokinetic properties and biochemical effects of antioxidant vitamins at the cell level. Consistent with this view, previous attempts to reduce free radical production with oral vitamin C following primary coronary angioplasty for AMI failed to attenuate increased F2-isoprostane production (Guan et al., 1999). Jaxa-Chamiec et al. (2005) performed a randomized, double-blind, placebo-controlled multicentre study in 800 patients and analyzed the combined effects of vitamins C and E through infusion and capsules but could not demonstrate a major effect of this antioxidant treatment on the clinical outcome, although diabetic patients showed a reduction in 30-day cardiac mortality (Jaxa-Chamiec et al., 2009). It should be noted that the authors acknowledged that the dose of vitamin C used only raised plasma levels to 0.1 mmol/L, a concentration 100 times lower than that required for scavenging superoxide anion. Recently, a meta-analysis of randomized controlled trials concluded that antioxidant vitamins C and E may have prophylactic effect to reduce the incidence of postoperative AF and all-cause arrhythmia following cardiac surgery; however, the overall quality of current studies is poor and further research is needed (Harling et al., 2011; Rasoli et al., 2011). Most investigations on the protective effects of vitamins C and E have merely focused on their antioxidant power. However, these compounds have the potential to influence a broad range of mechanisms underlying human health and disease; the biological properties of α-tocopherol and ascorbic acid overwhelm their antioxidant effects. Despite the enormous interest in antioxidant vitamins as potential protective agents against human disease development, the actual contributions, and mechanisms of such compounds remain unclear. The biological properties and beneficial effects of vitamins C and E against postoperative AF have been reviewed (Rodrigo et al., 2010) and will be analyzed separately.
This antioxidant, primarily α-tocopherol, is the major peroxyl radical scavenger in biological lipid phases, such as membranes or LDL (Upston et al., 2001; Terentis et al., 2002). The antioxidant action of vitamin E has been ascribed to its ability to act chemically as a lipid-based free radical chain-breaking molecule, thereby inhibiting lipid peroxidation through its own conversion into an oxidized product, α-tocopheroxyl radical. α-Tocopherol can be restored by reducing α-tocopheroxyl radicals with redox-active reagents, such as vitamin C or ubiquinol (Shi et al., 1999). The discovery of α-tocopheryl phosphate, a novel and natural water-soluble form of vitamin E, should expand the knowledge of the roles of this vitamin in biological systems (Gianello et al., 2005) and facilitate its intravenous administration. In fact, vitamin E oral supplementation hours before acute episodes of oxidative stress may be not effective, but short-term parenteral administration was shown to enrich vitamin E in endothelial cells (Engelhart et al., 1998). In clinical studies of ischemia–reperfusion injury, positive effects of a multivitamin antioxidant solution, including preoperative administration of vitamin E, were described for revascularization of the lower extremities (Rabl et al., 1995), kidney transplantation (Rabl et al., 1993), liver surgery, and aortic aneurysm repair (Bartels et al., 2004). However, homologous studies in the myocardium have not been performed. Although it was reported that long-term vitamin E supplements given at high dosage (≥400 IU/day) may increase all-cause mortality (Miller et al., 2005), this was not the case for short-term administration of conventional doses required for the prevention of myocardial reperfusion damage.
Intraarterial administration of high doses of ascorbate has demonstrated to abolish the in vivo effects of superoxide anion in the impairment of vascular endothelial function in subjects with essential hypertension (Schneider et al., 2005). In addition, recent in vitro studies have also been successful to study the effects in vitro of oxidative stress with and without this vitamin C concentration, thus validating ascorbate to counteract the effects of oxidative stress (Virdis et al., 2009). It should be noted that most human cells have vitamin C transporters, coupled with sodium transport at a ratio Na:ascorbate = 2:1, having a K0.5 by 70 μmol/L, being distributed about 90% in the cytosol (Mackenzie et al., 2008). When vitamin C is given by mouth, the relationship between oral dose and plasma concentration is sigmoidal. Its concentration in plasma is tightly controlled and excess of vitamin C is excreted as a function of dose, being completely saturated at doses of 400 mg daily and higher, producing a steady-state plasma concentration of approximately 80 μmol/L (Graumlich et al., 1997). Unfortunately, while this concentration approaches to that required for enzyme modulation, it is not enough to scavenge superoxide anion. Therefore, in settings accompanied by oxidative stress, such as the acute myocardial infarction, or percutaneous coronary intervention following acute myocardial infarction, it should not be expected a beneficial effect of oral administration of vitamin C in the prevention of oxidative damage, and intravenous infusion could be considered with this purpose. Indeed, superoxide reacts with NO at a rate 105-fold greater than the rate at which superoxide reacts with ascorbic acid (Jackson et al., 1998). As a consequence, 10 mmol/L ascorbate is needed to support its competition with NO for superoxide. It should be expected a high rate of cellular transport at 10 mmol/L ascorbate plasma concentration, being saturated the transporters, as this concentration is much higher than Vmax. Short-term infusion of high vitamin C doses make possible to reach peak concentrations higher than 20 mmol/L, but being over 10 mm/L for 3 h (Duconge et al., 2008). Interestingly, plasma levels of such magnitude, or three times higher, have not demonstrated adverse events, except in the case of known complications such as renal failure or glucose 6 phosphate dehydrogenase deficiency (Padayatty et al., 2010). Therefore, these data are consistent with the fact that vitamin C high doses offer an unexplored therapeutic opportunity to counteract the myocardial reperfusion damage caused by percutaneous coronary intervention. Despite oral doses of vitamin C fail to protect the heart against the effect of peroxynitrite, some beneficial effects other than that of superoxide scavenging may be observed when ascorbate is administered following acute myocardial infarction. Accordingly, in patients undergoing elective percutaneous coronary intervention, impaired microcirculatory reperfusion is improved by vitamin C infusion (Basili et al., 2010). Furthermore, in patients subjected to thrombolysis, SOD in the blood was found significantly reduced; whereas the activity of the oxidant enzyme XO, and malondialdehyde levels were found significantly increased. However, oral supplementation of vitamin C to the post reperfusion patients restored these parameters back to normal or near normal levels (Bhakuni et al., 2006). These effects could be related to the modulation of ROS production by vitamin C. A major enzymatic source of ROS is given by their production via NADPH oxidase, an enzyme subjected to downregulation by vitamin C. In addition, vitamin C prevents the oxidation of tetrahydrobiopterin, a cofactor of eNOS that is highly sensitive to oxidation. When tetrahydrobiopterin is oxidized, eNOS activity becomes uncoupled, resulting in the production of superoxide rather than NO, thus enhancing the consequences of oxidative damage (Yang and Ming, 2006). Therefore, it should be expected a beneficial effect of vitamin C in the outcome of patients, even at oral doses, when it is administered before cardiac surgery with extracorporeal circulation. On this line, another clinical model of oxidative stress likely to have beneficial effects of oral doses of vitamin C is postoperative AF. This is supported by previous studies demonstrating a major reduction in the incidence of postoperative AF in patients subjected to coronary artery bypass who were given 2 g of vitamin C the day before the surgery, followed by 500 mg daily until the fifth postoperative day (Carnes et al., 2001). In agreement with these data, more recent studies demonstrated that the combination of ascorbic acid and β-blockers may be more effective in reducing postoperative AF than β-blockers alone (Eslami et al., 2007), and that supplementation of vitamin C reduces the incidence of postoperative AF, the time needed for rhythm restoration and length of hospital stay (Papoulidis et al., 2011). However, available data are few and more large-scale clinical trials are still lacking to confirm this beneficial effect of vitamin C (Rasoli et al., 2011). It is noteworthy that vitamin C could also abrogate the beneficial effects of ischemic preconditioning, a phenomenon induced by a series of brief sub-lethal episodes of ischemia and reperfusion prior to a potentially lethal episode of ischemia that renders the heart more resistant to myocardial infarction (Tsovolas et al., 2008). Likely, the preconditioning is abolished due to an abrogation of ROS production, a phenomenon that otherwise could give rise to a survival response by the antioxidant defense system. Ascorbate preloading, among its many effects, has been shown to down-regulate p47phox and thus attenuate NADPH oxidase-derived ROS production (Wu et al., 2007).
It has been demonstrated the electrophysiological effects of n−3 PUFA account for their acute antiarrhythmic potential, being more remarkable with DHA and EPA as compared with α-linolenic acid (Milberg et al., 2011). However, while the anti-inflammatory or antiarrhythmic effects of n−3 PUFA may decrease the risk of postoperative AF, interventional studies have yielded conflicting results. Thus, at present there exists a controversy respect the beneficial effects of n−3 PUFA in the prevention of AF recurrence after direct cardioversion (Kumar et al., 2011; Nodari et al., 2011; Ozaydin et al., 2011). Although the mechanism of postoperative AF is based on acute factors, such as inflammation and oxidative stress, the vulnerability of myocardial tissue will be dependent on the tissue redox status at the time of receiving the oxidative challenge. Likely, vitamins C and E supplementation contributes with a reinforcement of the non-enzymatic antioxidant system. However, the antioxidant enzymes also could be a target of interventions aimed to induce up-regulation of their activities, thus further enhancing the antioxidant potential against the unavoidable ROS burst during the tissue reperfusion. Low to moderate ROS production is thought to lead to a survival response mediated by Nrf2. In addition to repeated short-term ischemia–reperfusion cycles, or through pharmacological agents, this response could be induced by n−3 PUFA supplementation. Due to their highly unsaturated chemical structure, n−3 PUFA are highly prone to peroxidation, which proceeds through both enzymatic and non-enzymatic pathways. Therefore, these compounds suitable to create a condition of increased ROS production regulated by the administered dose. An appropriate dose to cause increase of ROS concentration enough to activate Nrf2 pathway, but not cell death pathways, will elicit a survival response rendering the myocardial tissue more resistant to a subsequent ROS exposure and derived consequences, such as postoperative AF. In support of this view, experimental studies performed in heart of rats supplemented with n−3 PUFA rich fish oil demonstrated a significant enhancement of the expression of antioxidant enzymes (Jahangiri et al., 2006). Some studies aimed to the prevention of postoperative AF have demonstrated a significant reduction of its incidence in patients subjected to cardiac surgery with extracorporeal circulation (Calò et al., 2005; Mariscalco et al., 2010; Castillo et al., 2011). However, other studies found no evidence of a beneficial effect of treatment with n−3 PUFA on these patients (Heidarsdottir et al., 2010; Saravanan et al., 2010). It should be mentioned a meta-analysis that did not demonstrate significant effects of n−3 PUFA supplementation on AF prevention (Liu et al., 2011), although these compounds may favorably affect atrial remodeling in AF (Korantzopoulos et al., 2005). Likely, this controversy arises from the nature of n−3 PUFA administered to patients, having a key role the 1/2 = EPA/DHA ratio. In fact, the three successful studies in diminishing the incidence of the arrhythmia administered n−3 PUFA nearly in this ratio. In turn, the unsuccessful studies showed a higher EPA/DHA ratio. This view is supported by a recent study reporting a trend toward a benefit from n−3 PUFA supplementation when the EPA/DHA ratio is 1/2 (Benedetto et al., 2011). However, more mechanistic studies to ascertain the molecular basis of these effects are still lacking.
Cumulated evidence, previously discussed, leads to support the view for a potential role of n−3 PUFA and vitamins C and E in protecting the myocardial damage derived from reperfusion following extracorporeal circulation. This hypothesis is based on two-step regime applied from a week before surgery, composed of (Zimmer et al., 2003) an oxidative stress stimulus to promote up-regulation of the endogenous enzymatic antioxidant defense system, through n−3 PUFA supplementation, followed by (Gillespie et al., 2005) a reinforcement of the non-enzymatic antioxidant component through supplements of vitamins C and E (Rodrigo et al., 2007b, 2008). Under these conditions, it should be expected a decreased vulnerability of cardiac tissue to the oxidative challenge occurring because of the ischemia–reperfusion event proper of this surgery. Consequently, taking into account the role of ROS and RNS in the development of postoperative AF, this antioxidant supplementation could reduce the incidence of the arrhythmia. Although the available clinical trial are controversial to support this hypothesis, the latter should be complemented with the need to administer n−3 PUFA in a 1/2 = EPA/DHA ratio, since all the studies using this protocol found a successful prevention of postoperative AF. In addition, an independent effect of ascorbate could enhance this cardioprotection. This hypothesis is summarized in Figure 2.
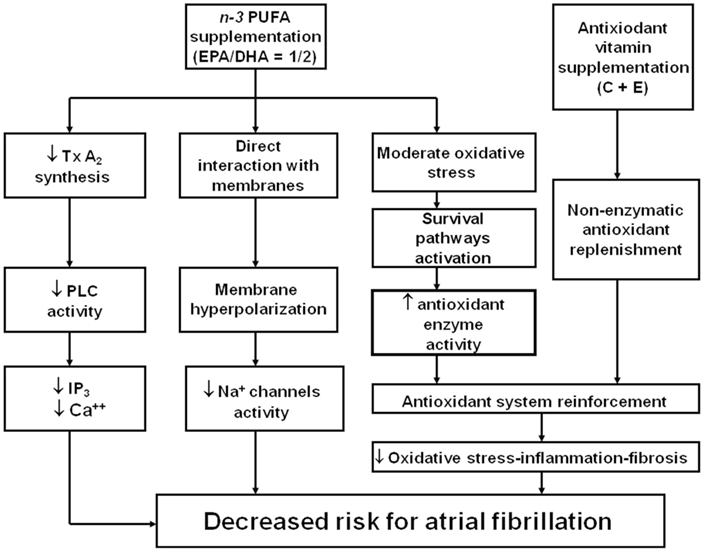
Figure 2. Mechanism supporting a novel strategy for the protective effect of n− 3 PUFA and antioxidant vitamin administration against postoperative atrial fibrillation in patients subjected to cardiac surgery with extracorporeal circulation. EPA, eicosapentaenoic acid; DHA, docosahexaenoic acid; PLC, Phospholipase C; Tx A2, Thromboxane A2; IP3, Inositol trisphosphate; Ca++, cytosolic calcium. Adapted from Rodrigo et al. (2008) with permission.
Postoperative AF is a common complication, particularly following cardiac surgery with extracorporeal circulation. It increases the incidence of other complications, hospital, and healthcare costs. Although the precise pathophysiological mechanisms of this arrhythmia related to cardiac surgery are poorly understood, oxidative stress and inflammation in atrial tissue may be causally related to AF. High ROS levels could give rise to pathological effects of oxidative stress characterized by oxidative damage, resulting in cell dysfunction, apoptosis, and/or necrosis. Conversely, at lower concentrations, local targeted production of ROS serves as a second messenger system that transmits biological information by highly specific modulation of intracellular signaling molecules, enzymes, and proteins. The latter process includes an adaptive response to enhance antioxidant defenses, mainly by up-regulating antioxidant enzymes expression and/or anti-apoptotic pathways. The reduction of ROS, with hydroxyl radical scavengers, in the heart attenuates the rate changes induced by hypoxia and reoxygenation, and also decreases the burst firing incidence. To date, prophylactic therapies for postoperative AF are largely empirical and suboptimal. Despite the uncertainty of the beneficial effects provided by clinical studies, optimal EPA/DHA ratio has been recently argued to explain the preventive effects of n−3 PUFA against postoperative AF. The hypothesis for a novel therapeutic strategy based on the paradigm that a reinforcement of the antioxidant system before cardiac surgery should decrease the vulnerability of myocardium to the oxidative challenges is presented. Low to moderate ROS exposure can be created by n−3 PUFA supplementation, giving rise to up-regulation of antioxidant enzymes in cardiac tissue. Additionally, vitamins C and E are capable of the reinforcement of non-enzymatic antioxidant defenses. The intervention supported by this hypothesis would be safe, low cost, and readily available, and the systemic effects of this treatment make possible its use in other clinical settings related to ischemia–reperfusion, such as percutaneous coronary interventions following acute myocardial infarction or transplantation of live organ donation. The present study should encourage the design of clinical trials aimed to offer new therapeutic opportunities to all these patients.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This study was supported by the Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT), grant 1120594.
Abdelhadi, R. H., Gurm, H. S., VanWagoner, D. R., and Chung, M. K. (2004). Relation of an exaggerated rise in white blood cells after coronary bypass or cardiac valve surgery to development of atrial fibrillation postoperatively. Am. J. Cardiol. 93, 1176–1178.
Adam, O., Frost, G., Custodis, F., Sussman, M. A., Schäfers, H. J., Böhm, M., and Laufs, U. (2007). Role of Rac1 GTPase activation in atrial fibrillation. J. Am. Coll. Cardiol. 50, 359–367.
Adrain, C., and Martin, S. J. (2001). The mitochondrial apoptosome: a killer unleashed by the cytochrome seas. Trends Biochem. Sci. 26, 390–397.
Akila, D’souza, B., Vishwanath, P., and D’souza, V. (2007). Oxidative injury and antioxidants in coronary artery bypass graft surgery: off-pump CABG significantly reduces oxidative stress. Clin. Chim. Acta 375, 147–152.
Allessie, M., Ausma, J., and Schotten, U. (2002). Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 54, 230–246.
Alqahtani, A. A. (2010). Atrial fibrillation post cardiac surgery trends toward management. Heart Views 11, 57–63.
Ambrosio, G., and Tritto, I. (1999). Reperfusion injury: experimental evidence and clinical implications. Am. Heart J. 138, S69–S75.
Attaran, S., Shaw, M., Bond, L., Pullan, M. D., and Fabri, B. M. (2011). A comparison of outcome in patients with preoperative atrial fibrillation and patients in sinus rhythm. Ann. Thorac. Surg. 92, 1391–1395.
Aviles, R. J., Martin, D. O., Apperson-Hansen, C., Houghtaling, P. L., Rautaharju, P., Kronmal, R. A., Tracy, R. P., Van Wagoner, D. R., Psaty, B. M., Lauer, M. S., and Chung, M. K. (2003). Inflammation as a risk factor for atrial fibrillation. Circulation 108, 3006–3010.
Babu, G. G., Walker, J. M., Yellon, D. M., and Hausenloy, D. J. (2011). Peri-procedural myocardial injury during percutaneous coronary intervention: an important target for cardioprotection. Eur. Heart J. 32, 23–31.
Bartels, M., Biesalski, H. K., Engelhart, K., Sendlhofer, G., Rehak, P., and Nagel, E. (2004). Pilot study on the effect of parenteral vitamin E on ischemia and reperfusion induced liver injury: a double blind, randomized, placebo-controlled trial. Clin. Nutr. 23, 1360–1370.
Basili, S., Tanzilli, G., Mangieri, E., Raparelli, V., Di Santo, S., Pignatelli, P., and Violi, F. (2010). Intravenous ascorbic acid infusion improves myocardial perfusion grade during elective percutaneous coronary intervention: relationship with oxidative stress markers. JACC Cardiovasc. Interv. 3, 221–229.
Béard, T., Carrié, D., Boyer, M. J., Boudjemaa, B., Ferrières, J., Delay, M., Bernadet, P., and Thouvenot, J. P. (1994). Production of oxygen free radicals in myocardial infarction treated by thrombolysis. Analysis of glutathione peroxidase, superoxide dismutase and malondialdehyde. Arch. Mal. Coeur Vaiss. 87, 1289–1296.
Benedetto, U., Angeloni, E., Melina, G., Danesi, T. H., Di Bartolomeo, R., Lechiancole, A., Refice, S., Roscitano, A., Comito, C., and Sinatra, R. (2011). n-3 Polyunsaturated fatty acids for the prevention of postoperative atrial fibrillation: a meta-analysis of randomized controlled trials. J. Cardiovasc. Med. (Hagerstown). doi: 10.2459/JCM.0b013e32834a13c1. [Epub ahead of print].
Bhakuni, P., Chandra, M., and Misra, M. K. (2006). Effect of ascorbic acid supplementation on certain oxidative stress parameters in the post reperfusion patients of myocardial infarction. Mol. Cell. Biochem. 290, 153–158.
Boos, C. J., Anderson, R. A., and Lip, G. Y. (2006). Is atrial fibrillation an inflammatory disorder? Eur. Heart J. 27, 136–149.
Bowie, A., and O’Neill, L. A. (2000). Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem. Pharmacol. 59, 13–23.
Braunwald, E., and Kloner, R. A. (1985). Myocardial reperfusion: a double-edged sword? J. Clin. Invest. 76, 1713–1719.
Brown, D. I., and Griendling, K. K. (2009). Nox proteins in signal transduction. Free Radic. Biol. Med. 47, 1239–1253.
Bruins, P., te Velthuis, H., Yazdanbakhsh, A. P., Jansen, P. G., van Hardevelt, F. W., de Beaumont, E. M., Wildevuur, C. R., Eijsman, L., Trouwborst, A., and Hack, C. E. (1997). Activation of the complement system during and after cardiopulmonary bypass surgery: postsurgery activation involves C-reactive protein and is associated with postoperative arrhythmia. Circulation 96, 3542–3548.
Brundel, B. J., van Gelder, I. C., Henning, R. H., Tuinenburg, A. E., Deelman, L. E., Tieleman, R. G., Grandjean, J. G., van Gilst, W. H., and Crijns, H. J. (1999). Gene expression of proteins influencing the calcium homeostasis in patients with persistent and paroxysmal atrial fibrillation. Cardiovasc. Res. 42, 443–454.
Brundel, B. J., Van Gelder, I. C., Henning, R. H., Tuinenburg, A. E., Wietses, M., Grandjean, J. G., Wilde, A. A., Van Gilst, W. H., and Crijns, H. J. (2001). Alterations in potassium channel gene expression in atria of patients with persistent and paroxysmal atrial fibrillation: differential regulation of protein and mRNA levels for K+ channels. J. Am. Coll. Cardiol. 37, 926–932.
Burra, S., and Jiang, J. X. (2011). Regulation of cellular function by connexin hemichannels. Int. J. Biochem. Mol. Biol. 2, 119–128.
Calò, L., Bianconi, L., Colivicchi, F., Lamberti, F., Loricchio, M. L., de Ruvo, E., Meo, A., Pandozi, C., Staibano, M., and Santini, M. (2005). N-3 Fatty acids for the prevention of atrial fibrillation after coronary artery bypass surgery: a randomized, controlled trial. J. Am. Coll. Cardiol. 45, 1723–1728.
Canbaz, S., Erbas, H., Huseyin, S., and Duran, E. (2008). The role of inflammation in atrial fibrillation following open heart surgery. J. Int. Med. Res. 36, 1070–1076.
Cande, C., Cohen, I., Daugas, E., Ravagnan, L., Larochette, N., Zamzami, N., and Kroemer, G. (2002). Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie 84, 215–222.
Carnes, C. A., Chung, M. K., Nakayama, T., Nakayama, H., Baliga, R. S., Piao, S., Kanderian, A., Pavia, S., Hamlin, R. L., McCarthy, P. M., Bauer, J. A., Van, and Wagoner, D. R. (2001). Ascorbate attenuates atrial pacing-induced peroxynitrite formation and electrical remodeling and decreases the incidence of postoperative atrial fibrillation. Circ. Res. 89, E32–E38.
Castillo, R., Rodrigo, R., Perez, F., Cereceda, M., Asenjo, R., Zamorano, J., Navarrete, R., Villalabeitia, E., Sanz, J., Baeza, C., and Aguayo, R. (2011). Antioxidant therapy reduces oxidative and inflammatory tissue damage in patients subjected to cardiac surgery with extracorporeal circulation. Basic Clin. Pharmacol. Toxicol. 108, 256–262.
Ceconi, C., Boraso, A., Cargnoni, A., and Ferrari, R. (2003). Oxidative stress in cardiovascular disease: myth or fact? Arch. Biochem. Biophys. 420, 217–221.
Chandra, J., Samali, A., and Orrenius, S. (2000). Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 29, 323–333.
Chandrasekar, B., Smith, J. B., and Freeman, G. L. (2001). Ischemia-reperfusion of rat myocardium activates nuclear factor-kappaB and induces neutrophil infiltration via lipopolysaccharide-induced CXC chemokine. Circulation 103, 2296–2302.
Chen, Z., Siu, B., Ho, Y. S., Vincent, R., Chua, C. C., Hamdy, R. C., and Chua, B. H. (1998). Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J. Mol. Cell. Cardiol. 30, 2281–2289.
Chung, M. K., Martin, D. O., Sprecher, D., Wazni, O., Kanderian, A., Carnes, C. A., Bauer, J. A., Tchou, P. J., Niebauer, M. J., Natale, A., and Van Wagoner, D. R. (2001). C-reactive protein elevation in patients with atrial arrhythmias: inflammatory mechanisms and persistence of atrial fibrillation. Circulation 104, 2886–2891.
Communal, C., Sumandea, M., de Tombe, P., Narula, J., Solaro, R. J., and Hajjar, R. J. (2002). Functional consequences of caspase activation in cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 99, 6252–6256.
Cook, N. R., Albert, C. M., Gaziano, J. M., Zaharris, E., MacFadyen, J., Danielson, E., Buring, J. E., and Manson, J. E. (2007). A randomized factorial trial of vitamins C, E, and beta-carotene in the secondary prevention of cardiovascular events in women: results from the Women’s Antioxidant Cardiovascular Study (WACS). Arch. Intern. Med. 167, 1610–1618.
Cook-Mills, J. M., Johnson, J. D., Deem, T. L., Ochi, A., Wang, L., and Zheng, Y. (2004). Calcium mobilization and Rac1 activation are required for VCAM-1 (vascular cell adhesion molecule-1) stimulation of NADPH oxidase activity. Biochem. J. 378, 539–547.
Corretti, M. C., Koretsune, Y., Kusuoka, H., Chacko, V. P., Zweier, J. L., and Marban, E. (1991). Glycolytic inhibition and calcium overload as consequences of exogenously generated free radicals in rabbit hearts. J. Clin. Invest. 88, 1014–1025.
Dallabrida, S. M., Ismail, N., Oberle, J. R., Himes, B. E., and Rupnick, M. A. (2005). Angiopoietin-1 promotes cardiac and skeletal myocyte survival through integrins. Circ. Res. 96, e8–e24.
Davis, E. M., Packard, K. A., and Hilleman, D. E. (2010). Pharmacologic prophylaxis of postoperative atrial fibrillation in patients undergoing cardiac surgery: beyond beta-blockers. Pharmacotherapy 30, 274e–318e.
de Gaetano, G. (2001). Collaborative Group of the Primary Prevention Project. Low-dose aspirin and vitamin E in people at cardiovascular risk: a randomised trial in general practice. Collaborative Group of the Primary Prevention Project. Lancet 357, 89–95.
De Vecchi, E., Pala, M. G., Di Credico, G., Agape, V., Paolini, G., Bonini, P. A., Grossi, A., and Paroni, R. (1998). Relation between left ventricular function and oxidative stress in patients undergoing bypass surgery. Heart 79, 242–247.
Debreceni, B., and Debreceni, L. (2011). Why do homocysteine-lowering B vitamin and antioxidant E vitamin supplementations appear to be ineffective in the prevention of cardiovascular diseases? Cardiovasc. Ther. doi:10.1111/j.1755-5922.2011.00266.x
Desplantez, T., Dupont, E., Severs, N. J., and Weingart, R. (2007). Gap junction channels and cardiac impulse propagation. J. Membr. Biol. 218, 13–28.
Duconge, J., Miranda-Massari, J. R., Gonzalez, M. J., Jackson, J. A., Warnock, W., and Riordan, N. H. (2008). Pharmacokinetics of vitamin C: insights into the oral and intravenous administration of ascorbate. P. R. Health Sci. J. 27, 7–19.
Dudley, S. C. Jr., Hoch, N. E., McCann, L. A., Honeycutt, C., Diamandopoulos, L., Fukai, T., Harrison, D. G., Dikalov, S. I., and Langberg, J. (2005). Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation 112, 1266–1273.
Echahidi, N., Pibarot, P., O’Hara, G., and Mathieu, P. (2008). Mechanisms, prevention, and treatment of atrial fibrillation after cardiac surgery. J. Am. Coll. Cardiol. 51, 793–801.
Elahi, M., Hadjinikolaou, L., and Galinanes, M. (2003). Incidence and clinical consequences of atrial fibrillation within 1 year of first-time isolated coronary bypass surgery. Circulation 108(Suppl. 1), II207–II212.
Elahi, M. M., Flatman, S., and Matata, B. M. (2008). Tracing the origins of postoperative atrial fibrillation: the concept of oxidative stress-mediated myocardial injury phenomenon. Eur. J. Cardiovasc. Prev. Rehabil. 15, 735–741.
El-Chami, M. F., Kilgo, P., Thourani, V., Lattouf, O. M., Delurgio, D. B., Guyton, R. A., Leon, A. R., and Puskas, J. D. (2010). New-onset atrial fibrillation predicts long-term mortality after coronary artery bypass graft. J. Am. Coll. Cardiol. 55, 1370–1376.
Engelhart, K., Jentzsch, A. M., Fürst, P., and Biesalski, H. K. (1998). Short-term parenteral application of α-tocopherol leads to increased concentration in plasma and tissue of the rat. Free Radic. Res. 29, 421–426.
Erickson, J. R., Joiner, M. L., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., Bartlett, R. K., Lowe, J. S., O’Donnell, S. E., Aykin-Burns, N., Zimmerman, M. C., Zimmerman, K., Ham, A. J., Weiss, R. M., Spitz, D. R., Shea, M. A., Colbran, R. J., Mohler, P. J., and Anderson, M. E. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474.
Eslami, M., Badkoubeh, R. S., Mousavi, M., Radmehr, H., Salehi, M., Tavakoli, N., and Avadi, M. R. (2007). Oral ascorbic acid in combination with beta-blockers is more effective than beta-blockers alone in the prevention of atrial fibrillation after coronary artery bypass grafting. Tex. Heart Inst. J. 34, 268–274.
Essick, E. E., and Sam, F. (2010). Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid. Med. Cell. Longev. 3, 168–177.
Everett, T. H. 4th, and Olgin, J. E. (2007). Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. 4(Suppl. 3), S24–S27.
Félétou, M., and Vanhoutte, P. M. (2006). Endothelium-derived hyperpolarizing factor: where are we now? Arterioscler. Thromb. Vasc. Biol. 26, 1215–1225.
Flaherty, J. T., Pitt, B., Gruber, J. W., Heuser, R. R., Rothbaum, D. A., Burwell, L. R., George, B. S., Kereiakes, D. J., Deitchman, D., and Gustafson, N. (1994). Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation 89, 1982–1991.
Fruehauf, J. P., and Meyskens, F. L. (2007). Reactive oxygen species: a breath of life or death? Clin. Cancer Res. 13, 789–794.
Frustaci, A., Chimenti, C., Bellocci, F., Morgante, E., Russo, M. A., and Maseri, A. (1997). Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation 96, 1180–1184.
Gale, C. P., Metcalfe, E., West, R. M., Das, R., Kilcullen, N., Morrell, C., Crook, R., Batin, P. D., Hall, A. S., and Barth, J. H. (2011). An assessment of the concentration-related prognostic value of cardiac troponin I following acute coronary syndrome. Am. J. Cardiol. 108, 1259–1265.
García, A., Eiras, S., Parguiña, A. F., Alonso, J., Rosa, I., Salgado-Somoza, A., Rico, T. Y., Teijeira-Fernández, E., and González-Juanatey, J. R. (2011). High-resolution two-dimensional gel electrophoresis analysis of atrial tissue proteome reveals down-regulation of fibulin-1 in atrial fibrillation. Int. J. Cardiol. 150, 283–290.
Gasparetto, C., Malinverno, A., Gulacciati, D., Gritti, D., and Prosperini, P. G. (2005). Specchia G, ricevuti G. Antioxidant vitamins reduce oxidative stress and ventricular remodeling in patients with acute myocardial infarction. Int. J. Immunopathol. Pharmacol. 18, 487–496.
Gaspo, R., Bosch, R. F., Bou-Abboud, E., and Nattel, S. (1997). Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ. Res. 81, 1045–1052.
Gaudino, M., Andreotti, F., Zamparelli, R., Di Castelnuovo, A., Nasso, G., Burzotta, F., Iacoviello, L., Donati, M. B., Schiavello, R., Maseri, A., and Possati, G. (2003). The -174G/C interleukin-6 polymorphism influences postoperative interleukin-6 levels and postoperative atrial fibrillation. Is atrial fibrillation an inflammatory complication? Circulation 108(Suppl. 1), II195–II199.
Gianello, R., Libinaki, R., Azzi, A., Gavin, P. D., Negis, Y., Zingg, J. M., Holt, P., Keah, H. H., Griffey, A., Smallridge, A., West, S. M., and Ogru, E. (2005). Alpha-tocopheryl phosphate: a novel, natural form of vitamin E. Free Radic. Biol. Med. 39, 970–976.
Gillespie, E. L., White, C. M., Kluger, J., Rancourt, J. A., Gallagher, R., and Coleman, C. I. (2006). Cost-effectiveness of amiodarone for prophylaxis of atrial fibrillation after cardiothoracic surgery. Pharmacotherapy 26, 499–504.
Gillespie, E. L., White, M., Kluger, J., Sahni, J., Gallagher, R., and Coleman, C. (2005). A hospital perspective on the cost-effectiveness of beta-blockade for prophylaxis of atrial fibrillation after cardiothoracic surgery. Clin. Ther. 27, 1963–1969.
GISSI-Prevenzione Trial. (1999). Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione Trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet 354, 447–455.
Goldstein, R. N., Ryu, K., Khrestian, C., van Wagoner, D. R., and Waldo, A. L. (2008). Prednisone prevents inducible atrial flutter in the canine sterile pericarditis model. J. Cardiovasc. Electrophysiol. 19, 74–81.
Grant, A. O. (2001). Molecular biology of sodium channels and their role in cardiac arrhythmias. Am. J. Med. 110, 296–305.
Graumlich, J. F., Ludden, T. M., Conry-Cantilena, C., Cantilena, L. R. Jr., Wang, Y., and Levine, M. (1997). Pharmacokinetic model of ascorbic acid in healthy male volunteers during depletion and repletion. Pharm. Res. 14, 1133–1139.
Guan, W., Osanai, T., Kamada, T., Hanada, H., Ishizaka, H., Onodera, H., Iwasa, A., Fujita, N., Kudo, S., Ohkubo, T., and Okumura, K. (2003). Effect of allopurinol pretreatment on free radical generation after primary coronary angioplasty for acute myocardial infarction. J. Cardiovasc. Pharmacol. 41, 699–705.
Guan, W., Osanai, T., Kamada, T., Ishizaka, H., Hanada, H., and Okumura, K. (1999). Time course of free radical production after primary coronary angioplasty for acute myocardial infarction and the effect of vitamin C. Jpn. Circ. J. 63, 924–928.
Halonen, J., Halonen, P., Järvinen, O., Taskinen, P., Auvinen, T., Tarkka, M., Hippeläinen, M., Juvonen, T., and Hartikainen, J. (2007). Corticosteroids for the prevention of atrial fibrillation after cardiac surgery: a randomized controlled trial. JAMA 297, 1562–1567.
Hariharan, N., Zhai, P., and Sadoshima, J. (2011). Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 14, 2179–2190.
Harling, L., Rasoli, S., Vecht, J. A., Ashrafian, H., Kourliouros, A., and Athanasiou, T. (2011). Do antioxidant vitamins have an anti-arrhythmic effect following cardiac surgery? A meta-analysis of randomised controlled trials. Heart 97, 1636–1642.
Heart Protection Study Collaborative Group. (2000). MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 360, 23–33.
Heidarsdottir, R., Arnar, D. O., Skuladottir, G. V., Torfason, B., Edvardsson, V., Gottskalksson, G., Palsson, R., and Indridason, O. S. (2010). Does treatment with n-3 polyunsaturated fatty acids prevent atrial fibrillation after open heart surgery? Europace 12, 356–363.
Hess, M. L., and Manson, N. H. (1984). Molecular oxygen: friend and foe. The role of the oxygen free radical system in the calcium paradox, the oxygen paradox and ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 16, 969–985.
Ho, H. T., Stevens, S. C., Terentyeva, R., Carnes, C. A., Terentyev, D., and Györke, S. (2011). Arrhythmogenic adverse effects of cardiac glycosides are mediated by redox modification of ryanodine receptors. J. Physiol. (Lond.) 589(Pt 19), 4697–4708.
Ishii, Y., Schuessler, R. B., Gaynor, S. L., Yamada, K., Fu, A. S., Boineau, J. P., and Damiano, R. J. Jr. (2005). Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation. Circulation 111, 2881–2888.
Jackson, T. S., Xu, A., Vita, J. A., and Keaney, J. F. Jr. (1998). Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ. Res. 83, 916–922.
Jahangiri, A., Leifert, W. R., Kind, K. L., and McMurchie, E. J. (2006). Dietary fish oil alters cardiomyocyte Ca2+ dynamics and antioxidant status. Free Radic. Biol. Med. 40, 1592–1602.
Jaxa-Chamiec, T., Bednarz, B., Drozdowska, D., Gessek, J., Gniot, J., Janik, K., Kawka-Urbanek, T., Maciejewski, P., Ogórek, M., Szpajer, M., MIVIT Trial Group. (2005). Antioxidant effects of combined vitamins C and E in acute myocardial infarction. The randomized, double-blind, placebo controlled, multicenter pilot Myocardial Infarction and VITamins (MIVIT) trial. Kardiol. Pol. 62, 344–350.
Jaxa-Chamiec, T., Bednarz, B., Herbaczynska-Cedro, K., Maciejewski, P., Ceremuzynski, L., MIVIT Trial Group. (2009). Effects of vitamins C and E on the outcome after acute myocardial infarction in diabetics: a retrospective, hypothesis-generating analysis from the MIVIT study. Cardiology 112, 219–223.
Jones, W. K., Brown, M., Ren, X., He, S., and McGuinness, M. (2003). NF-kappaB as an integrator of diverse signaling pathways: the heart of myocardial signaling? Cardiovasc. Toxicol. 3, 229–254.
Juhaszova, M., Zorov, D. B., Kim, S. H., Pepe, S., Fu, Q., Fishbein, K. W., Ziman, B. D., Wang, S., Ytrehus, K., Antos, C. L., Olson, E. N., and Sollott, S. J. (2004). Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 113, 1535–1549.
Juránek, I., and Bezek, S. (2005). Controversy of free radical hypothesis: reactive oxygen species – cause or consequence of tissue injury? Gen. Physiol. Biophys. 24, 263–278.
Kilgore, K. S., and Lucchesi, B. R. (1993). Reperfusion injury after myocardial infarction: the role of free radicals and the inflammatory response. Clin. Biochem. 26, 359–370.
Killig, F., and Stark, G. (2002). Photodynamic activation of ion transport through lipid membranes and its correlation with an increased dielectric constant of the membrane. Biochim. Biophys. Acta 1564, 207–213.
Kim, Y. M., Guzik, T. J., Zhang, Y. H., Zhang, M. H., Kattach, H., Ratnatunga, C., Pillai, R., Channon, K. M., and Casadei, B. (2005). A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 97, 629–636.
Kinoshita, T., Asai, T., Takashima, N., Hosoba, S., Suzuki, T., Kambara, A., and Matsubayashi, K. (2011). Preoperative C-reactive protein and atrial fibrillation after off-pump coronary bypass surgery. Eur. J. Cardiothorac. Surg. 40, 1298–1303.
Kloner, R. A., and Jennings, R. B. (2001a). Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 1. Circulation 104, 2981–2989.
Kloner, R. A., and Jennings, R. B. (2001b). Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2. Circulation 104, 3158–3167.
Kobayashi, M., and Yamamoto, M. (2005). Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 7, 385–394.
Kockskämper, J., and Blatter, L. A. (2002). Subcellular Ca2+ alternans represents a novel mechanism for the generation of arrhythmogenic Ca2+ waves in cat atrial myocytes. J. Physiol. (Lond.) 545(Pt 1), 65–79.
Kockskämper, J., Zima, A. V., and Blatter, L. A. (2005). Modulation of sarcoplasmic reticulum Ca2+ release by glycolysis in cat atrial myocytes. J. Physiol. (Lond.) 564(Pt 3), 697–714.
Köhler, R., and Hoyer, J. (2007). The endothelium-derived hyperpolarizing factor: insights from genetic animal models. Kidney Int. 72, 145–150.
Kojodjojo, P., Peters, N. S., Davies, D. W., and Kanagaratnam, P. (2007). Characterization of the electroanatomical substrate in human atrial fibrillation: the relationship between changes in atrial volume, refractoriness, wavefront propagation velocities, and AF burden. J. Cardiovasc. Electrophysiol. 18, 269–275.
Koniari, I., Apostolakis, E., Rogkakou, C., Baikoussis, N. G., and Dougenis, D. (2010). Pharmacologic prophylaxis for atrial fibrillation following cardiac surgery: a systematic review. J. Cardiothorac. Surg. 5, 121.
Korantzopoulos, P., Kolettis, T., Siogas, K., and Goudevenos, J. (2003). Atrial fibrillation and electrical remodeling: the potential role of inflammation and oxidative stress. Med. Sci. Monit. 9, RA225–RA229.
Korantzopoulos, P., Kolettis, T. M., Galaris, D., and Goudevenos, J. A. (2007). The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation. Int. J. Cardiol. 115, 135–143.
Korantzopoulos, P., Kolettis, T. M., and Goudevenos, J. A. (2005). The anti-inflammatory and antioxidant effects of long-chain n-3 fatty acids or oil-rich fish may favorably affect atrial remodeling in atrial fibrillation. Med. Hypotheses 64, 1245–1246.
Kumagai, K., Khrestian, C., and Waldo, A. L. (1997). Simultaneous multisite mapping studies during induced atrial fibrillation in the sterile pericarditis model. Insights into the mechanism of its maintenance. Circulation 95, 511–521.
Kumar, S., Sutherland, F., Morton, J. B., Lee, G., Morgan, J., Wong, J., Eccleston, D. E., Voukelatos, J., Garg, M. L., and Sparks, P. B. (2011). Long-term omega-3 polyunsaturated fatty acid supplementation reduces the recurrence of persistent atrial fibrillation after electrical cardioversion. Heart Rhythm. 9, 483–491.
Lai, L. P., Su, M. J., Lin, J. L., Lin, F. Y., Tsai, C. H., Chen, Y. S., Huang, S. K., Tseng, Y. Z., and Lien, W. P. (1999). Down-regulation of L-type calcium channel and sarcoplasmic reticular Ca(2+)-ATPase mRNA in human atrial fibrillation without significant change in the mRNA of ryanodine receptor, calsequestrin and phospholamban: an insight into the mechanism of atrial electrical remodeling. J. Am. Coll. Cardiol. 33, 1231–1237.
Lamm, G., Auer, J., Weber, T., Berent, R., Ng, C., and Eber, B. (2006). Postoperative white blood cell count predicts atrial fibrillation after cardiac surgery. J. Cardiothorac. Vasc. Anesth. 20, 51–55.
Lee, P., Sata, M., Lefer, D. J., Factor, S. M., Walsh, K., and Kitsis, R. N. (2003). Fas pathway is a critical mediator of cardiac myocyte death and MI during ischemia–reperfusion in vivo. Am. J. Physiol. Heart Circ. Physiol. 284, H456–H463.
Lefer, A. M., and Lefer, D. J. (1996). The role of nitric oxide and cell adhesion molecules on the microcirculation in ischaemia-reperfusion. Cardiovasc. Res. 32, 743–751.
Li, J. M., and Shah, A. M. (2004). Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R1014–R1030.
Li, S. Y., Yang, X., Ceylan-Isik, A. F., Du, M., Sreejayan, N., and Ren, J. (2006). Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia 49, 1434–1446.
Liakopoulos, O. J., Schmitto, J. D., Kazmaier, S., Bräuer, A., Quintel, M., Schoendube, F. A., and Dörge, H. (2007). Cardiopulmonary and systemic effects of methylprednisolone in patients undergoing cardiac surgery. Ann. Thorac. Surg. 84, 110–118.
Lin, Y. K., Lai, M. S., Chen, Y. C., Cheng, C. C., Huang, J. H., Chen, S. A., Chen, Y. J., and Lin, C. I. (2012). Hypoxia and reoxygenation modulate the arrhythmogenic activity of the pulmonary vein and atrium. Clin. Sci. 122, 121–132.
Liu, T., Korantzopoulos, P., Shehata, M., Li, G., Wang, X., and Kaul, S. (2011). Prevention of atrial fibrillation with omega-3 fatty acids: a meta-analysis of randomised clinical trials. Heart 97, 1034–1040.
Mackenzie, B., Illing, A. C., and Hediger, M. A. (2008). Transport model of the human Na+-coupled L-ascorbic acid (vitamin C) transporter SVCT1. Am. J. Physiol. Cell Physiol. 294, C451–C459.
Maisel, W. H., Rawn, J. D., and Stevenson, W. G. (2001). Atrial fibrillation after cardiac surgery. Ann. Intern. Med. 135, 1061–1073.
Mariscalco, G., Sarzi Braga, S., Banach, M., Borsani, P., Bruno, V. D., Napoleone, M., Vitale, C., Piffaretti, G., Pedretti, R. F., and Sala, A. (2010). Preoperative n-3 polyunsatured fatty acids are associated with a decrease in the incidence of early atrial fibrillation following cardiac surgery. Angiology 61, 643–650.
Martindale, J. L., and Holbrook, N. J. (2002). Cellular response to oxidative stress: signaling for suicide and survival. J. Cell. Physiol. 192, 1–15.
Matata, B. M., Sosnowski, A. W., and Galiñanes, M. (2000). Off-pump bypass graft operation significantly reduces oxidative stress and inflammation. Ann. Thorac. Surg. 69, 785–791.
Mateescu, B., Batista, L., Cardon, M., Gruosso, T., de Feraudy, Y., Mariani, O., Nicolas, A., Meyniel, J. P., Cottu, P., Sastre-Garau, X., and Mechta-Grigoriou, F. (2011). miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med. 17, 1627–1235.
Mathew, J. P., Fontes, M. L., Tudor, I. C., Ramsay, J., Duke, P., Mazer, C. D., Barash, P. G., Hsu, P. H., Mangano, D. T., Investigators of the Ischemia Research, and Education Foundation, and Multicenter Study of Perioperative Ischemia Research Group. (2004). A multicenter risk index for atrial fibrillation after cardiac surgery. J. Am. Med. Assoc. 291, 1720–1729.
Matsui, Y., Kyoi, S., Takagi, H., Hsu, C. P., Hariharan, N., Ago, T., Vatner, S. F., and Sadoshima, J. (2008). Molecular mechanisms and physiological significance of autophagy during myocardial ischemia and reperfusion. Autophagy 4, 409–415.
Mihm, M. J., Yu, F., Carnes, C. A., Reiser, P. J., McCarthy, P. M., Van Wagoner, D. R., and Bauer, J. A. (2001). Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 104, 174–180.
Milberg, P., Frommeyer, G., Kleideiter, A., Fischer, A., Osada, N., Breithardt, G., Fehr, M., and Eckardt, L. (2011). Antiarrhythmic effects of free polyunsaturated fatty acids in an experimental model of LQT2 and LQT3 due to suppression of early afterdepolarizations and reduction of spatial and temporal dispersion of repolarization. Heart Rhythm 8, 1492–1500.
Miller, E. R. III, Pastor-Barriuso, R., Dalal, D., Riemersma, R. A., Appel, L. J., and Guallar, E. (2005). Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 142, 37–46.
Moens, A. L., Claeys, M. J., Timmermans, J. P., and Vrints, C. J. (2005). Myocardial ischemia/reperfusion-injury, a clinical view on a complex pathophysiological process. Int. J. Cardiol. 100, 179–190.
Morita, N., Lee, J. H., Bapat, A., Fishbein, M. C., Mandel, W. J., Chen, P. S., Weiss, J. N., and Karagueuzian, H. S. (2011). Glycolytic inhibition causes spontaneous ventricular fibrillation in aged hearts. Am. J. Physiol. Heart Circ. Physiol. 301, H180–H191.
Morita, N., Sovari, A. A., Xie, Y., Fishbein, M. C., Mandel, W. J., Garfinkel, A., Lin, S. F., Chen, P. S., Xie, L. H., Chen, F., Qu, Z., Weiss, J. N., and Karagueuzian, H. S. (2009). Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 297, H1594–H1605.
Nattel, S., Maguy, A., Le Bouter, S., and Yeh, Y. H. (2007). Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 87, 425–456.
Negi, S., Sovari, A. A., and Dudley, S. C. Jr. (2010). Atrial fibrillation: the emerging role of inflammation and oxidative stress. Cardiovasc. Hematol. Disord. Drug Targets 10, 262–268.
Neuman, R. B., Bloom, H. L., and Shukrullah, I. (2007). Oxidative stress markers are associated with persistent atrial fibrillation. Clin. Chem. 53, 1652–1657.
Niu, J., Wang, K., Graham, S., Azfer, A., and Kolattukudy, P. E. (2011). MCP-1-induced protein attenuates endotoxin-induced myocardial dysfunction by suppressing cardiac NF-κB activation via inhibition of IκB kinase activation. J. Mol. Cell. Cardiol. 51, 177–186.
Nodari, S., Triggiani, M., Campia, U., Manerba, A., Milesi, G., Cesana, B. M., Gheorghiade, M., and Dei Cas, L. (2011). n-3 Polyunsaturated fatty acids in the prevention of atrial fibrillation recurrences after electrical cardioversion: a prospective, randomized study. Circulation 124, 1100–1106.
Olson, T. M., Michels, V. V., Ballew, J. D., Reyna, S. P., Karst, M. L., Herron, K. J., Horton, S. C., Rodeheffer, R. J., and Anderson, J. L. (2005). Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. J. Am. Med. Assoc. 293, 447–454.
Ono, N., Hayashi, H., Kawase, A., Lin, S. F., Li, H., Weiss, J. N., Chen, P. S., and Karagueuzian, H. S. (2007). Spontaneous atrial fibrillation initiated by triggered activity near the pulmonary veins in aged rats subjected to glycolytic inhibition. Am. J. Physiol. Heart Circ. Physiol. 292, H639–H648.
Opie, L. H. (1991). Role of calcium and other ions in reperfusion injury. Cardiovasc. Drugs Ther. 5(Suppl. 2), 237–247.
Opie, L. H., Commerford, P. J., Gersh, B. J., and Pfeffer, M. A. (2006). Controversies in ventricular remodelling. Lancet 367, 356–367.
Ozaydin, M., Erdoğan, D., Tayyar, S., Uysal, B. A., Dogan, A., Içli, A., Ozkan, E., Varol, E., Türker, Y., and Arslan, A. (2011). N-3 polyunsaturated fatty acids administration does not reduce the recurrence rates of atrial fibrillation and inflammation after electrical cardioversion: a prospective randomized study. Anadolu Kardiyol. Derg. 11, 305–309.
Padayatty, S. J., Sun, A. Y., Chen, Q., Espey, M. G., Drisko, J., and Levine, M. (2010). Vitamin C: intravenous use by complementary and alternative medicine practitioners and adverse effects. PLoS ONE 5, e11414.
Papoulidis, P., Ananiadou, O., Chalvatzoulis, E., Ampatzidou, F., Koutsogiannidis, C., Karaiskos, T., Madesis, A., and Drossos, G. (2011). The role of ascorbic acid in the prevention of atrial fibrillation after elective on-pump myocardial revascularization surgery: a single-center experience – a pilot study. Interact. Cardiovasc. Thorac. Surg. 12, 121–124.
Park, J. L., and Lucchesi, B. R. (1999). Mechanisms of myocardial reperfusion injury. Ann. Thorac. Surg. 68, 1905–1912.
Pavlović, D., Đorđević, V., and Kocić, G. (2002). A “cross-talk” between oxidative stress and redox cell signalling. Med. Biol. 2, 131–137.
Pendergrass, K. D., Varghese, S. T., Maiellaro-Rafferty, K., Brown, M. E., Taylor, W. R., and Davis, M. E. (2011). Temporal effects of catalase overexpression on healing after myocardial infarction. Circ. Heart Fail. 4, 98–106.
Prosser, B. L., Ward, C. W., and Lederer, W. J. (2011). X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333, 1440–1445.
Rabl, H., Khoschsorur, G., Colombo, T., Petritsch, P., Rauchenwald, M., Költringer, P., Tatzber, F., and Esterbauer, H. (1993). A multivitamin infusion prevents lipid peroxidation and improves transplantation performance. Kidney Int. 43, 912–917.
Rabl, H., Koschsorur, G., and Petek, W. (1995). Antioxidant vitamin treatment: effect on lipid peroxidation and limb swelling after revascularisation operations. World J. Surg. 19, 738–744.
Rader, F., Van Wagoner, D. R., Ellinor, P. T., Gillinov, A. M., Chung, M. K., Costantini, O., and Blackstone, E. H. (2011). Influence of race on atrial fibrillation after cardiac surgery. Circ. Arrhythm. Electrophysiol. 4, 644–652.
Rapola, J. M., Virtamo, J., Ripatti, S., Huttunen, J. K., Albanes, D., Taylor, P. R., and Heinonen, O. P. (1997). Randomised trial of alpha-tocopherol and beta-carotene supplements on incidence of major coronary events in men with previous myocardial infarction. Lancet 349, 1715–1720.
Rasoli, S., Kourliouros, A., Harling, L., and Athanasiou, T. (2011). Does prophylactic therapy with antioxidant vitamins have an effect on atrial fibrillation following cardiac surgery? Interact. Cardiovasc. Thorac. Surg. 13, 82–85.
Rodrigo, R. (2009). Oxidative Stress and Antioxidants: Their Role in Human Disease. New York: Nova Science Publishers
Rodrigo, R., Bächler, J. P., Araya, J., Prat, H., and Passalacqua, W. (2007a). Relationship between (Na+K)-ATPase activity, lipid peroxidation and fatty acid profile in erythrocytes of hypertensive and normotensive subjects. Mol. Cell. Biochem. 303, 73–81.
Rodrigo, R., Guichard, C., and Charles, R. (2007b). Clinical pharmacology and therapeutic use of antioxidant vitamins. Fundam. Clin. Pharmacol. 21, 111–127.
Rodrigo, R., Cereceda, M., Castillo, R., Asenjo, R., Zamorano, J., Araya, J., Castillo-Koch, R., Espinoza, J., and Larraín, E. (2008). Prevention of atrial fibrillation following cardiac surgery: basis for a novel therapeutic strategy based on non-hypoxic myocardial preconditioning. Pharmacol. Ther. 118, 104–127.
Rodrigo, R., Vinay, J., Castillo, R., Cereceda, M., Asenjo, R., Zamorano, J., Araya, J., Castillo-Koch, R., Espinoza, J., and Larraín, E. (2010). Use of vitamins C and E as a prophylactic therapy to prevent postoperative atrial fibrillation. Int. J. Cardiol. 138, 221–228.
Saravanan, P., Bridgewater, B., West, A. L., O’Neill, S. C., Calder, P. C., and Davidson, N. C. (2010). Omega-3 fatty acid supplementation does not reduce risk of atrial fibrillation after coronary artery bypass surgery: a randomized, double-blind, placebo-controlled clinical trial. Circ. Arrhythm. Electrophysiol. 3, 46–53.
Saxena, A., Dinh, D. T., Smith, J. A., Shardey, G. C., Reid, C. M., and Newcomb, A. E. (2012). Usefulness of postoperative atrial fibrillation as an independent predictor for worse early and late outcomes after isolated coronary artery bypass grafting (Multicenter Australian Study of 19,497 Patients). Am. J. Cardiol. 109, 219–225.
Schneider, M. P., Delles, C., Schmidt, B. M., Oehmer, S., Schwarz, T. K., Schmieder, R. E., and John, S. (2005). Superoxide scavenging effects of N-acetylcysteine and vitamin C in subjects with essential hypertension. Am. J. Hypertens. 18, 1111–1117.
Sesso, H. D., Buring, J. E., Christen, W. G., Kurth, T., Belanger, C., MacFadyen, J., Bubes, V., Manson, J. E., Glynn, R. J., and Gaziano, J. M. (2008). Vitamins E and C in the prevention of cardiovascular disease in men: the Physicians’ Health Study II randomized controlled trial. JAMA 300, 2123–2133.
Shao, Q., Matsubara, T., Bhatt, S. K., and Dhalla, N. S. (1995). Inhibition of cardiac sarcolemma Na(+)-K+ ATPase by oxyradical generating systems. Mol. Cell. Biochem. 147, 139–144.
Shaw, R. M., Fay, A. J., Puthenveedu, M. A., von Zastrow, M., Jan, Y. N., and Jan, L. Y. (2007). Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128, 547–560.
Shi, H., Noguchi, N., and Niki, E. (1999). Comparative study on dynamics of antioxidative action of alpha-tocopheryl hydroquinone, ubiquinol, and alpha-tocopherol against lipid peroxidation. Free Radic. Biol. Med. 27, 334–336.
Shiroshita-Takeshita, A., Brundel, B. J., and Nattel, S. (2005). Atrial fibrillation: basic mechanisms, remodeling and triggers. J. Interv. Card. Electrophysiol. 13, 181–193.
Singh, T., Sharma, S. D., and Katiyar, S. K. (2011). Grape proanthocyanidins induce apoptosis by loss of mitochondrial membrane potential of human non-small cell lung cancer cells in vitro and in vivo. PLoS ONE 6, e27444. doi:10.1371/journal.pone.0027444
Smyth, J. W., Hong, T. T., Gao, D., Vogan, J. M., Jensen, B. C., Fong, T. S., Simpson, P. C., Stainier, D. Y., Chi, N. C., and Shaw, R. M. (2010). Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Invest. 120, 266–279.
Song, Y., Shryock, J., Wagner, S., Maier, L., and Belardinelli, L. (2006). Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J. Pharmacol. Exp. Ther. 318, 214–222.
Stephens, N. G., Parsons, A., Schofield, P. M., Kelly, F., Cheeseman, K., and Mitchinson, M. J. (1996). Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 347, 781–786.
Tahara, E. B., Navarete, F. D., and Kowaltowski, A. J. (2009). Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 46, 1283–1297.
Tanguy, S., Rakotovao, A., Jouan, M. G., Ghezzi, C., de Leiris, J., and Boucher, F. (2011). Dietary selenium intake influences Cx43 dephosphorylation, TNF-α expression and cardiac remodeling after reperfused infarction. Mol. Nutr. Food Res. 55, 522–529.
Tansey, E. E., Kwaku, K. F., Hammer, P. E., Cowan, D. B., Federman, M., Levitsky, S., and McCully, J. D. (2006). Reduction and redistribution of gap and adherens junction proteins after ischemia and reperfusion. Ann. Thorac. Surg. 82, 1472–1479.
Terentis, A. C., Thomas, S. R., Burr, J. A., Liebler, D. C., and Stocker, R. (2002). Vitamin E oxidation in human atherosclerotic lesions. Circ. Res. 90, 333–339.
Tomaselli, G. F. (2010). Oxidant stress derails the cardiac connexon connection. J. Clin. Invest. 120, 87–89.
Tselentakis, E. V., Woodford, E., Chandy, J., Gaudette, G. R., and Saltman, A. E. (2006). Inflammation effects on the electrical properties of atrial tissue and inducibility of postoperative atrial fibrillation. J. Surg. Res. 135, 68–75.
Tsovolas, K., Iliodromitis, E. K., Andreadou, I., Zoga, A., Demopoulou, M., Iliodromitis, K. E., Manolaki, T., Markantonis, S. L., and Kremastinos, D. T. (2008). Acute administration of vitamin C abrogates protection from ischemic preconditioning in rabbits. Pharmacol. Res. 57, 283–289.
Tsujita, K., Shimomura, H., Kaikita, K., Kawano, H., Hokamaki, J., Nagayoshi, Y., Yamashita, T., Fukuda, M., Nakamura, Y., Sakamoto, T., Yoshimura, M., and Ogawa, H. (2006). Long-term efficacy of edaravone in patients with acute myocardial infarction. Circ. J. 70, 832–837.
Upston, J. M., Witting, P. K., Brown, A. J., Stocker, R., and Keaney, J. F. (2001). Effect of vitamin E on aortic lipid oxidation and intimal proliferation after arterial injury in cholesterol-fed rabbits. Free Radic. Biol. Med. 31, 1245–1253.
Ushio-Fukai, M. (2009). Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal. 11, 1289–1299.
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84.
Valtchanova-Matchouganska, A., Gondwe, M., and Nadar, A. (2004). The role of C-reactive protein in ischaemia/reperfusion injury and preconditioning in a rat model of myocardial infarction. Life Sci. 75, 901–910.
Van Gelder, I. C., Brundel, B. J., Henning, R. H., Tuinenburg, A. E., Tieleman, R. G., Deelman, L., Grandjean, J. G., De Kam, P. J., Van Gilst, W. H., and Crijns, H. J. (1999). Alterations in gene expression of proteins involved in the calcium handling in patients with atrial fibrillation. J. Cardiovasc. Electrophysiol. 10, 552–560.
Van Wagoner, D. R. (2003). Molecular basis of atrial fibrillation: a dream or a reality? J. Cardiovasc. Electrophysiol. 14, 667–669.
Van Wagoner, D. R. (2007). Recent insights into the pathophysiology of atrial fibrillation. Semin. Thorac. Cardiovasc. Surg. 19, 9–15.
Van Wagoner, D. R. (2008). Oxidative stress and inflammation in atrial fibrillation: role in pathogenesis and potential as a therapeutic target. J. Cardiovasc. Pharmacol. 52, 306–313.
Van Wagoner, D. R., Pond, A. L., McCarthy, P. M., Trimmer, J. S., and Nerbonne, J. M. (1997). Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ. Res. 80, 772–781.
Vandervelde, S., van Luyn, M. J., Rozenbaum, M. H., Petersen, A. H., Tio, R. A., and Harmsen, M. C. (2007). Stem cell-related cardiac gene expression early after murine myocardial infarction. Cardiovasc. Res. 73, 783–793.
Verhaar, M. C., Westerweel, P. E., van Zonneveld, A. J., and Rabelink, T. J. (2004). Free radical production by dysfunctional eNOS. Heart 90, 494–495.
Villareal, R. P., Hariharan, R., Liu, B. C., Kar, B., Lee, V. V., Elayda, M., Lopez, J. A., Rasekh, A., Wilson, J. M., and Massumi, A. (2004). Postoperative atrial fibrillation and mortality after coronary artery bypass surgery. J. Am. Coll. Cardiol. 43, 742–748.
Virdis, A., Colucci, R., Fornai, M., Polini, A., Daghini, E., Duranti, E., Ghisu, N., Versari, D., Dardano, A., Blandizzi, C., Taddei, S., Del Tacca, M., and Monzani, F. (2009). Inducible nitric oxide synthase is involved in endothelial dysfunction of mesenteric small arteries from hypothyroid rats. Endocrinology 150, 1033–1042.
Wagner, S., Dybkova, N., Rasenack, E. C. L., Jacobshagen, C., Fabritz, L., Kirchhof, P., Maier, S. K. G., Zhang, T., Hasenfuss, G., Brown, J. H., Bers, D. M., and Maier, L. S. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138.
Wang, Y. Y., Liu, S., Lian, F., Yang, W. G., and Xue, S. (2011a). Toll-like receptor 7/8 agonist resiquimod induces late preconditioning in neonatal cardiac myocytes. Acta Pharmacol. Sin. 32, 565–572.
Wang, Z., Lu, Y., and Yang, B. (2011b). MicroRNAs and atrial fibrillation: new fundamentals. Cardiovasc. Res. 89, 710–721.
Wassmann, S., Laufs, U., Bäumer, A. T., Müller, K., Konkol, C., Sauer, H., Böhm, M., and Nickenig, G. (2001). Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol. Pharmacol. 59, 646–654.
Weisfeldt, M. L., Zweier, J., Ambrosio, G., Becker, L. C., and Flaherty, J. T. (1988). Evidence that free radicals result in reperfusion injury in heart muscle. Basic Life Sci. 49, 911–919.
Whelan, R. S., Kaplinskiy, V., and Kitsis, R. N. (2010). Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu. Rev. Physiol. 72, 19–44.
Wilhelm, M., Kirste, W., Kuly, S., Amann, K., Neuhuber, W., Weyand, M., Daniel, W. G., and Garlichs, C. (2006). Atrial distribution of connexin 40 and 43 in patients with intermittent, persistent, and postoperative atrial fibrillation. Heart Lung Circ. 15, 30–37.
Wu, F., Schuster, D. P., Tyml, K., and Wilson, J. X. (2007). Ascorbate inhibits NADPH oxidase subunit p47phox expression in microvascular endothelial cells. Free Radic. Biol. Med. 42, 124–231.
Yan, L., Sadoshima, J., Vatner, D. E., and Vatner, S. F. (2009). Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy 5, 709–712.
Yang, Z., and Ming, X. F. (2006). Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin. Med. Res. 4, 53–65.
Yellon, D. M., and Hausenloy, D. J. (2007). Myocardial reperfusion injury. N. Engl. J. Med. 357, 1121–1135.
Younce, C. W., and Kolattukudy, P. E. (2010). MCP-1 causes cardiomyoblast death via autophagy resulting from ER stress caused by oxidative stress generated by inducing a novel zinc-finger protein, MCPIP. Biochem. J. 426, 43–53.
Yu, G., Bolon, M., Laird, D. W., and Tyml, K. (2010). Hypoxia and reoxygenation-induced oxidant production increase in microvascular endothelial cells depends on connexin40. Free Radic. Biol. Med. 49, 1008–1013.
Yue, L., Feng, J., Gaspo, R., Li, G. R., Wang, Z., and Nattel, S. (1997). Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ. Res. 81, 512–525.
Yue, L., Melnyk, P., Gaspo, R., Wang, Z., and Nattel, S. (1999). Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ. Res. 84, 776–784.
Yusuf, S., Dagenais, G., Pogue, J., Bosch, J., and Sleight, P. (2000). Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N. Engl. J. Med. 342, 154–160.
Zeng, Q., Han, Y., Bao, Y., Li, W., Li, X., Shen, X., Wang, X., Yao, F., O’Rourke, S. T., and Sun, C. (2010). 20-HETE increases NADPH oxidase-derived ROS production and stimulates the L-type Ca2+ channel via a PKC-dependent mechanism in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 299, H1109–H1117.
Zhou, L., Azfer, A., Niu, J., Graham, S., Choudhury, M., Adamski, F. M., Younce, C., Binkley, P. F., and Kolattukudy, P. E. (2006). Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ. Res. 98, 1177–1185.
Zhu, H., Jia, Z., Misra, B. R., Zhang, L., Cao, Z., Yamamoto, M., Trush, M. A., Misra, H. P., and Li, Y. (2008). Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc. Toxicol. 8, 71–85.
Zhu, H., Tannous, P., Johnstone, J. L., Kong, Y., Shelton, J. M., Richardson, J. A., Le, V., Levine, B., Rothermel, B. A., and Hill, J. A. (2007). Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 117, 1782–1793.
Zimmer, J., Pezzullo, J., Choucair, W., Southard, J., Kokkinos, P., Karasik, P., Greenberg, M. D., and Singh, S. N. (2003). Meta-analysis of antiarrhythmic therapy in the prevention of postoperative atrial fibrillation and the effect on hospital length of stay, costs, cerebrovascular accidents, and mortality in patients undergoing cardiac surgery. Am. J. Cardiol. 91, 1137–1140.
Keywords: postoperative atrial fibrillation, oxidative stress, vitamin C, vitamin E, n−3 polyunsaturated fatty acids
Citation: Rodrigo R (2012) Prevention of postoperative atrial fibrillation: novel and safe strategy based on the modulation of the antioxidant system. Front. Physio. 3:93. doi: 10.3389/fphys.2012.00093
Received: 28 January 2012; Accepted: 26 March 2012;
Published online: 12 April 2012.
Edited by:
David R. Van Wagoner, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USAReviewed by:
Bao-Feng Yang, Harbin Medical University, ChinaCopyright: © 2012 Rodrigo. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ramón Rodrigo, Molecular and Clinical Pharmacology Program, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, Reyes Lavalle 3415, Depto. 71, Las Condes CP 7550164, Santiago, Chile. e-mail:cnJvZHJpZ29AbWVkLnVjaGlsZS5jbA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.