
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 02 April 2012
Sec. Gastrointestinal Sciences
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00079
This article is part of the Research Topic Molecular mechanisms and therapeutic interventions in acute liver injury View all 11 articles
 Heike Bantel1* and Klaus Schulze-Osthoff1
Heike Bantel1* and Klaus Schulze-Osthoff1
Acute liver failure (ALF) can be the consequence of various etiologies, that might vary between different geographic regions. Most frequent are intoxications with acetaminophen, viral hepatitis, or liver damage of unknown origin. ALF occurs when the extent of hepatocyte death exceeds the regenerative capacity of the liver. The mode of liver cell death that is predominantly induced in ALF, i.e., apoptosis or necrosis, is still controversial and presumably determined by the etiology, duration, and magnitude of liver injury. Severe liver damage involves oxidative stress and depletion of ATP resulting in necrosis. In contrast, maintenance of ATP stores is required for the execution of apoptosis. Recent data suggest that necrosis resulting from severe liver damage is associated with poor outcome of ALF patients. Discrimination between apoptosis and necrosis might be therefore useful for the identification of ALF patients requiring liver transplantation. Identification of the molecular cell death mechanisms remains an important issue not only for early prediction of ALF outcome, but also for therapeutic interventions. In view of the pleiotropic functions of critical mediators of cell death and tissue regeneration, a particular challenge will be to reduce hepatocellular death without inhibiting the regenerative capacity of the liver. Here, we review the molecular mechanisms of hepatocyte injury and the pathways leading to apoptosis and necrosis, which might represent potential diagnostic and therapeutic targets in ALF.
Acute liver failure (ALF) can occur as a result of various etiologies including hepatic injury by drugs and poison, viral hepatitis, ischemia, or other causes. The mechanisms by which liver cells are destroyed as well as the processes mediating liver regeneration, remain largely unknown. It has become evident that liver cell death can occur via distinct biochemical pathways and morphological alterations, including apoptosis, autophagic cell death, and necrosis. Apoptosis is defined by chromatin condensation, nuclear fragmentation, cell shrinkage, blebbing of the plasma membrane, and formation of apoptotic bodies that contain nuclear or cytoplasmic material (Kerr et al., 1972; Russell et al., 1972). Autophagic cell death, on the other hand, is characterized by a massive accumulation of double-membrane containing vacuoles, called autophagosomes, that subsequently fuse with lysosomes. Necrotic cell death is often negatively defined as a form of cell death that lacks signs of apoptosis or autophagy. Typically, necrotic cells show cytoplasmic swelling, dilation of organelles, and mechanical rupture of the plasma membrane. Although necrosis has been deemed to be a mainly passive process, the initiation, and modulation of necrotic cell death are currently under intense investigation at the molecular level.
The relative contribution of apoptosis or necrosis to organ dysfunction in ALF remains controversial (Schulze-Osthoff and Bantel, 2011). Necrosis is typically the consequence of acute metabolic perturbation with ATP depletion, whereas apoptosis represents an ATP-dependent cell death program. Furthermore, in several cases, the nature and duration of cellular injury determine if cells die by apoptosis, necrosis, or other mechanisms. At low doses, a variety of injurious stimuli often induce apoptosis, but the same stimuli can result in necrosis at higher doses. Therefore, in many situations cell death might be not executed as a clear-cut form of cell death, but as a continuum with intermediate features of both apoptosis and necrosis. A distinction of different cell death forms is therefore not only relevant for semantical reasons, but has important clinical implications when considering the therapeutic targeting of cell death processes. Thus, an understanding of the cell death processes is most important for development of effective interventions to prevent hepatocellular death in acute liver damage (Fischer and Schulze-Osthoff, 2005).
Apoptosis represents a programmed form of cell death that is required for the maintenance of tissue homeostasis by counterbalancing cell proliferation and eliminating damaged, infected, or transformed cells. This process is particularly important in the liver as an organ that is naturally exposed to toxins, drugs, and viruses. However, excessive apoptosis can result in tissue destruction and organ failure.
Apoptosis results from a collapse of cellular infrastructure through internal proteolytic digestion, which leads to cytoskeletal disintegration, metabolic derangement, and genomic fragmentation. Members of the caspase family of proteases form the core engine of apoptosis and are involved in initiation, execution, and regulatory phases of the pathway. Caspases are cysteine proteases that cleave substrates after aspartate residues within specific peptide recognition sequences. To preclude unwarranted cell death, caspases are expressed as inactive zymogens consisting of a prodomain followed by two subunits with the catalytic domain. Caspases operate in hierarchical cascades that serve to amplify the apoptotic signal (Los et al., 1999). Based on their structure and order in cell death pathways, caspases can be divided into upstream initiators and downstream effectors of apoptosis. Effector caspases such as caspase-3, -6, and -7 cleave diverse cellular substrates including structural proteins such as cytokeratin-18 and many others (Fischer et al., 1998; Leers et al., 1999; Bantel et al., 2000, 2001a). In contrast, initiator caspases, such as caspase-8, -9, and -10, exert regulatory roles by activating downstream effector caspases.
Caspases are activated by two major signaling routes, namely the extrinsic death receptor and the intrinsic mitochondrial pathway, that both depend on the formation of large multi-protein complexes (Schulze-Osthoff et al., 1998; Yoon and Gores, 2002). Initiator caspase-8 is the key mediator of the extrinsic pathway. In a simplified model, binding of death ligands such as TRAIL or CD95L or tumor necrosis factor (TNF)-α to their respective death receptors leads to receptor oligomerization. This event then results in the recruitment of the adapter protein FADD and the initiator caspase-8 into a death-inducing signaling complex (DISC), wherein caspase-8 becomes activated by dimerization and autoproteolytic cleavage (Figure 1). Subsequently, caspase-8 cleaves and activates the effector caspase-3, culminating in the demise of so-called type I cells. In most cells including hepatocytes, however, only low amounts of initiator caspases are activated at the DISC, which is insufficient for cell death. In those type II cells, the extrinsic receptor pathway must be amplified by the intrinsic mitochondrial apoptotic pathway through the caspase-8-mediated cleavage of Bid, a pro-apoptotic Bcl-2 family protein, which subsequently initiates together with the Bcl-2 family members Bak and Bax the release of mitochondrial pro-apoptotic mediators (Schwerk and Schulze-Osthoff, 2005). Interestingly, CD95-induced hepatocyte apoptosis is delayed but not inhibited in Bak/Bax-deficient mice indicating that hepatocytes can act as type I cells in the absence of pro-apoptotic Bcl-2 proteins (Hikita et al., 2011).
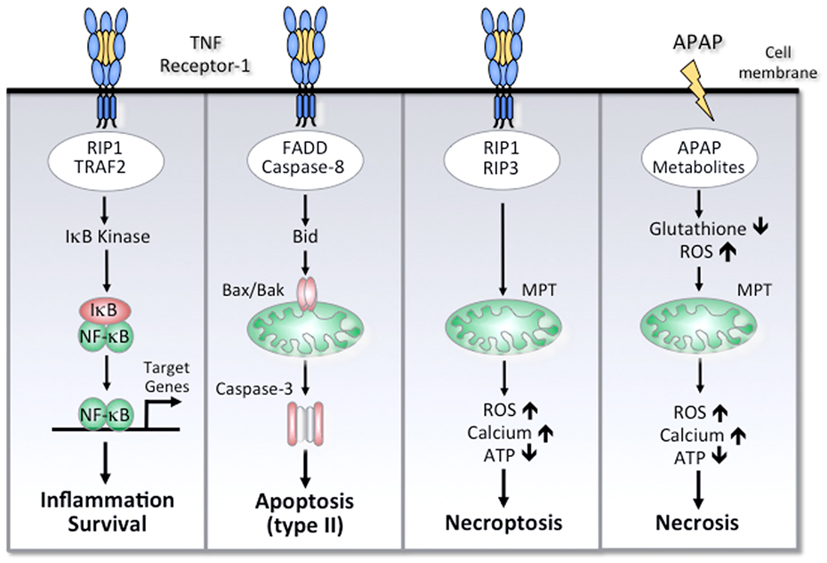
Figure 1. Simplified scheme of cell death and survival pathways involved in ALF. Activation of TNF receptor-1 can mediate NF-κB activation, apoptosis, or necroptosis. The different outcomes are determined by distinct TNF receptor-associated signaling complexes. Activation of NF-κB is mediated by TRAF-2, RIP-1, and other signaling molecules that lead to activation of IκB kinase and subsequent activation of NF-κB target genes. FADD and caspase-8 are the essential adapter proteins involved in apoptosis, which in hepatocytes requires a mitochondrial amplification loop through caspase-8-mediated cleavage of Bid. The subsequent translocation of Bax and Bak results in mitochondrial outer membrane permeabilization, cytochrome c release, and effector caspase-3 activation. Under conditions of impaired apoptosis, TNF receptor-1 can induce necroptosis, which involves RIP-1 and RIP-3 kinases. Among other effects, RIP-3 can increase the production of reactive oxygen species (ROS) due to increased oxidative phosphorylation, resulting in intracellular calcium overload, mitochondrial membrane permeability transition (MPT), depletion of ATP, and necrosis. APAP-induced necrosis is essentially mediated by a toxic metabolite, which depletes glutathione and forms APAP protein adducts, triggering oxidative stress, compromised respiratory function, and ATP depletion. Although APAP treatment can instigate the mitochondrial pathway of apoptosis, high doses of APAP will ultimately mediate liver cell death by necrosis.
Inappropriate activation of death receptors might lead to ALF. This has been impressively demonstrated in mice that died rapidly of liver failure with massive hepatocyte apoptosis when agonistic anti-CD95 antibody was injected (Ogasawara et al., 1993). Similarly, treatment of mice with TNF-α in combination with a transcription-blocking agent, such as D-galactosamine (D-GalN) or actinomycin D, induces lethal hepatitis (Leist et al., 1994, 1995; Libert et al., 1994). Another well established mouse model of ALF consists of a combined treatment with D-GalN and lipopolysaccharide (LPS), which induces TNF-α expression and an inflammatory response that is predominantly directed toward the liver (Galanos et al., 1979).
It has been suggested that the toxicity in the murine TNF-α and anti-CD95 models resembles viral forms of acute hepatic failure in patients (Keppler et al., 1968; El-Mofty et al., 1975). CD95/CD95L expression has been shown to be upregulated in viral hepatitis and to correlate with disease activity and hepatocyte apoptosis (Hiramatsu et al., 1994; Mita et al., 1994; Pianko et al., 2001; Lee et al., 2004). We have recently demonstrated that diseased, e.g., HCV-infected, livers show an upregulation of TRAIL receptors and increased susceptibility toward TRAIL-induced apoptosis (Volkmann et al., 2007). These data implicate that viral forms of ALF are associated with death receptor-induced cell death. Recent data show that the CD95 system is involved in human ALF caused not only by viral hepatitis but also by Wilson’s disease (Strand et al., 1998; Rivero et al., 2002). Increased levels of death ligands or receptors such as CD95L, TNF-α, or TNF receptors (TNF-R) were found in blood of patients with ALF (Ryo et al., 2000; Streetz et al., 2000; Tokushige et al., 2000; Nakae et al., 2001; Volkmann et al., 2008). Particularly, high serum levels of soluble death receptor CD95 have also been found in drug-induced ALF (Tagami et al., 2003; Rutherford et al., 2007).
Silencing of CD95 or caspase-8 protected mice from ALF or fulminant hepatitis induced by agonistic CD95 antibody or concanavalin A, respectively (Song et al., 2003; Zender et al., 2003). However, CD95 and caspase-8 also promotes liver regeneration by inducing differentiation of stellate cells and possibly of other non-parenchymal liver cells (Desbarats and Newell, 2000; Canbay et al., 2003; Ben Moshe et al., 2007). Additionally, TNF-α plays a pivotal role in liver regeneration by activation of transcription factors such as NF-κB, which induces the transcription of a huge number of cytokines and growth-promoting target genes (Wullaert et al., 2007). The activation of NF-κB by TNF-α is mediated by distinct adapter proteins that are recruited to TNF-R-1 upon ligand binding (Figure 1). Whether increased levels of circulating death receptors or ligands in human ALF mirror apoptotic cell death or liver regeneration and whether death receptor-induced cell death depends on special ALF etiologies remains unknown.
In contrast to viral infection, drug-induced liver injury is mainly associated with signaling pathways triggered by mitochondrial damage (Chan et al., 2005). In the intrinsic pathway, apoptosis is mediated by translocation of pro-apoptotic Bcl-2 molecules, such as Bax and Bak, from the cytosol to mitochondria to form pores in the outer mitochondrial membrane (Los et al., 1999). This process is followed by the mitochondrial release of cytochrome c and other pro-apoptotic factors. Cytochrome c normally functions in electron transport processes of the respiratory chain to generate ATP. In the cytosol of apoptotic cells, however, it serves as a cofactor for the adapter protein Apaf-1. Upon binding of cytochrome c and dATP, Apaf-1 oligomerizes, and recruits the initiator caspase-9 to trigger the formation of the apoptosome. Thus, similar to the DISC, the apoptosome is a high-molecular weight complex that serves as a caspase activation platform. Once assembled in the apoptosome, caspase-9 becomes activated and subsequently triggers the caspase cascade (Schulze-Osthoff et al., 1998). As mentioned above, there is also considerable crosstalk between the extrinsic and intrinsic pathways. For example, during death receptor-mediated apoptosis caspase-8 can proteolytically activate the Bcl-2 protein Bid, which facilitates cytochrome c release and amplifies the apoptotic signal following death receptor activation.
In contrast to apoptosis, necrosis is mediated by opening of the mitochondrial membrane permeability transition (MPT) pore, which triggers the collapse of the membrane potential and cessation of ATP formation. The resulting mitochondrial swelling leads to the rupture of the outer mitochondrial membrane with the release of intermembrane proteins and subsequent nuclear DNA fragmentation. Other prominent features include massive energy depletion, formation of reactive oxygen species (ROS), and activation of non-apoptotic proteases. Furthermore, during necrosis a strong increase of intracellular calcium is observed. The elevated calcium levels in the cytosol trigger mitochondrial calcium overload, leading to depolarization of the inner mitochondrial membrane and a shut-down of ATP production. While depletion of ATP impedes the function of membrane channels, increased calcium activates calcium-dependent proteases, such as calpains. Calcium fluxes, ATP depletion, and oxidative stress involve complex and interactive feedback loops, which self-amplify and potentiate each other leading to exaggerated cell death. The relative amount of ATP might be an important factor that determines whether hepatocytes die by apoptosis or necrosis (Ferrari et al., 1998; Hinson et al., 2010). Another important distinguishing feature of apoptotic versus necrotic cell death relates to inflammation. When the necrotic cell ruptures, an inflammatory response follows due to the release of intracellular contents. In contrast, inflammation is not typical of apoptosis, because phagocytic cells rapidly engulf apoptotic cells and thereby prevent the release of noxious intracellular compounds.
Acetaminophen (paracetamol, N-acetyl-p-aminophenol; APAP) overdose represents one of the most common causes of ALF in developed countries (Larson et al., 2005). APAP-induced hepatotoxicity is due to the formation of the toxic metabolite N-acetyl-p-benzoquinone imine by the cytochrome P450 system, which causes glutathione depletion, oxidative stress, alterations of calcium homeostasis, resulting in MPT, loss of mitochondrial membrane potential, and ATP depletion (Hinson et al., 2010). Although necrosis has been thought to be the predominant mode of cell death in APAP-induced liver injury, conflicting in vitro and animal data have emerged suggesting a potential role of apoptosis in acetaminophen-induced hepatotoxicity (El-Hassan et al., 2003; Kon et al., 2004). Mice treated with a toxic dose of acetaminophen showed 40% apoptotic and 60% necrotic hepatocytes (Ray et al., 1996). It was demonstrated that mice with defective CD95 receptor (lpr mice) or CD95 ligand (gld mice) were partially protected from APAP-induced liver injury (Liu et al., 2004). Moreover, increased circulating levels of CD95 have been found in humans with APAP intoxication (Tagami et al., 2003).
It has been recently suggested that APAP hepatotoxicity is caused by the mitochondrial apoptosis pathway and facilitated by chemokine (C-X-C motif) receptor 2 (CXCR2) signaling. In this study caspase inhibition prevented DNA fragmentation, although the authors did not investigate whether caspase inhibition was also associated with cell survival (Hu and Colletti, 2010; Schulze-Osthoff and Bantel, 2011). In contrast, in another study no caspase activation was observed and, accordingly, caspase inhibition did not protect from liver injury in APAP-treated mice (Jaeschke et al., 2006). Further data from animal models revealed also no evidence that apoptotic cell death contributes to APAP-induced liver injury. For instance, following the application of an APAP overdose in mice, less than 1% of the parenchymal cells revealed an apoptotic morphology (Gujral et al., 2002). Another study in mice showed that knockdown of the CD95 receptor protected against 300 mg/kg APAP overdose but not against 700 mg/kg overdose (Zhang et al., 2000), indicating that the mode of cell death might at least partially depend on the APAP dose. Indeed, several studies demonstrated that APAP induces mitochondrial dysfunction with ATP depletion which even interrupts initial CD95-induced mitochondrial signaling pathways (Lawson et al., 1999; Knight and Jaeschke, 2002). On the other hand, subliminal CD95 activation can also increase APAP-induced liver injury (Tinel et al., 2004), indicating that death receptor signaling might influence the extent of APAP-induced necrotic liver injury.
Conflicting data exist also about the role of TNF-α in APAP-induced liver injury. Increased TNF-α expression in liver and circulating TNF-α levels have been observed after APAP poisoning (Blazka et al., 1995, 1996). However, the role of TNF-α in APAP-induced necrosis remains controversial, as TNF-α inhibitors exerted either protection or no effect (Blazka et al., 1995; Simpson et al., 2000). In addition, TNF-α knockout mice showed similar sensitivity to acetaminophen compared to wildtype mice (Boess et al., 1998). It has been recently shown that inhibition of c-jun N-terminal kinase (JNK), a member of the mitogen-activated protein kinase family, reduced paracetamol-induced toxicity in mice by inhibiting hepatic TNF-α production (Henderson et al., 2007). Furthermore, APAP-induced JNK activation has been linked to activation of the pro-apoptotic Bcl-2 protein Bim. In line, APAP-induced necrotic liver injury was shown to be reduced in Bim knockout mice (Badmann et al., 2011). However, neither JNK1 nor JNK2 knockdown did protect mice from APAP-induced liver toxicity, raising concerns about a major role of JNK in APAP-induced liver injury (Gunawan et al., 2006; Henderson et al., 2007; Bourdi et al., 2008). Altogether, although apoptotic alterations can occur, profound energy depletion, and mitochondrial failure presumably divert cell death to necrosis as the principal mode of APAP-induced liver toxicity.
Autophagy represents another process that might influence the outcome of APAP-induced liver toxicity. Autophagy is a catabolic mechanism by which long-lived proteins and organelles are recycled in order to maintain energy and protein synthesis. It is characterized by the appearance of numerous cytosolic vacuole-like structures, called autophagosomes, which encapsulate cytosolic materials and fuse with lysosomes. Although the role of the autophagy in protection during nutrient starvation is accepted, its function in programmed cell death remains controversial. Under normal physiological conditions autophagy occurs at low basal levels, contributing to the turnover of cytoplasmic components and promoting cell survival during stress conditions. Excess autophagy, on the other hand, leads to autophagic cell death. Interestingly, it has been recently demonstrated that APAP induces autophagy in mouse liver and primary human hepatocytes and that activation of autophagy protects against APAP-induced hepatotoxicity (Ni et al., 2012). In this study it was suggested that the induction of oxidative stress might play an important role in APAP-induced autophagy. Moreover, it was shown that pharmacological inhibition of autophagy increased APAP-induced liver injury. Clearly, more work is needed to elucidate the role of autophagy in APAP-induced hepatotoxicity and the complex crosstalk with other cell death pathways.
In addition to measuring death ligands, several other cell death biomarkers have been proposed for monitoring the clinical outcome of ALF and other liver diseases (Volkmann et al., 2006; Rutherford et al., 2007; Bechmann et al., 2008; Joka et al., 2012). Caspases cleave the intermediate filament protein cytokeratin (CK)-18 into specific fragments that are released into circulating blood and can be detected by the M30 ELISA (Bantel et al., 2001b, 2004; Seidel et al., 2005). Moreover, when this assay is combined with a second ELISA that detects the total release of caspase-cleaved and uncleaved CK-18 (M65 ELISA), even different forms of cell death, such as necrosis and apoptosis, can be discriminated. Using these serological assays, it has been recently demonstrated that the predictive sensitivity of total CK-18 for lethal outcome was comparable to the model for end-stage liver disease (MELD) score at time of admission of ALF patients (Bechmann et al., 2010). Moreover, modification of the MELD score by substitution of bilirubin for total CK-18 significantly increased the prediction of ALF outcome.
Interestingly, we have demonstrated that ALF patients display considerable caspase activity and high levels of caspase-cleaved CK-18 in the serum, which was unexpectedly higher in spontaneous survivors than in patients that required transplantation or died (Volkmann et al., 2008). Nevertheless, despite a weaker activation of caspases, liver biopsies of patients without spontaneous recovery revealed extensive TUNEL reactivity, which detects both apoptotic and necrotic cell death. Moreover, sera from those patients contained increased levels of total CK-18, but reduced levels of its caspase-generated fragments as compared to patients with spontaneous recovery. These findings therefore indicate that necrosis but not apoptosis is the predominant cell death in those critically ill ALF patients, whereas in ALF patients with spontaneous recovery apoptotic cell death predominates. In contrast, in another study, detection of caspase-cleaved CK-18 could not adequately predict ALF outcome (Rutherford et al., 2007). However, unlike in our study in which APAP-induced ALF only played a minor role, in this study a relevant number of patients with APAP-induced ALF was included. As mentioned above, in those patients necrotic cell death might predominate irrespective of the outcome. In line with this observation, patients with APAP-induced ALF showed higher levels of total CK-18 compared to caspase-cleaved CK-18 levels (Bechmann et al., 2008; Volkmann et al., 2008; Craig et al., 2011; Bantel and Schulze-Osthoff, 2012).
Caspase activation might also play a role in liver regeneration and this might explain our observation of higher caspase activity in spontaneous survivors. Caspases can cleave different cytokine precursors to generate active cytokines and thus create an environment that could be essential for liver regeneration. Experiments in mice showed that caspase activation is associated with chemokine production and inflammation in the liver, whereas caspase-3 inhibition strongly reduced activity of pro-inflammatory transcription factors and chemokines (Faouzi et al., 2001). A role of caspases in liver regeneration is best exemplified by a recent report showing that a hepatocyte-specific knockout of caspase-8 attenuates hepatocyte proliferation after partial hepatectomy (Ben Moshe et al., 2007).
In addition to cell death biomarkers, circulating levels of nucleosomes and high-mobility group box 1 (HMBG1) protein, both of which are released during hepatocyte death, have been shown to be elevated in patients with ALF (Roth et al., 2009; Craig et al., 2011). However, those biomarkers were not able to predict ALF outcome, and no significant differences in HMBG1 or nucleosome levels between paracetamol- and non-paracetamol-induced liver injury were found (Craig et al., 2011). One explanation for the lacking correlation of those biomarkers with ALF outcome might be that the release of HMBG1 and nucleosomes are not strictly related to necrotic cell death, but also occur during apoptosis (Bell et al., 2006; Jaeschke et al., 2012). Certainly, further larger cohort studies are required to evaluate the predictive value of cell death markers in patients with paracetamol- or non-paracetamol-induced ALF.
An admixture of necrosis and apoptosis occurs in ALF and might therefore open up novel strategies for therapeutic intervention (Fischer and Schulze-Osthoff, 2005). More extreme injury leads to necrotic killing, whereas milder injury may result in apoptosis. Whether inhibition of apoptosis using available pharmacological caspase inhibitors can indeed prevent liver cell death or will just simply shift the mode of cell death to necrosis, remains to be shown (Los et al., 2002). As caspases not only play a role in apoptosis but also in processes of liver regeneration, the possibility of adverse effects should not be ignored when considering the therapeutic use of caspase inhibitors in ALF treatment. Death receptor-blocking agents, such as CD95-Fc or TRAIL-R-Fc proteins, might be promising candidates in virus-induced ALF (Bantel and Schulze-Osthoff, 2003). MicroRNAs (miRNAs) are non-coding RNAs which have been implicated in the posttranscriptional regulation of various cellular pathways. It was recently demonstrated that overexpression of miRNA-221 delays CD95-induced fulminant liver failure in mice (Sharma et al., 2011). Thus, miRNAs involved in cell death signaling pathways might serve as potential therapeutic targets in ALF.
Due to the pleiotropic effects of TNF-α, inhibitors of this cytokine might not only influence liver cell death but also immune response and liver regeneration. Treatment of APAP intoxicated mice with anti-TNF-α partially prevented hepatotoxicity (Blazka et al., 1995). Other studies showed no alterations of APAP toxicity in mice treated either with anti-TNF-α antibody or soluble TNF receptor (Simpson et al., 2000). Whether the observed discrepancy is due to the variant experimental conditions, such as different mouse strains, remains to be elucidated (Jaeschke et al., 2012). There is increasing evidence suggesting that TNF-α triggers cell death not only by apoptosis, but also by a necrosis-like process, which has been recently called necroptosis (Schulze-Osthoff et al., 1994; Vandenabeele et al., 2010). Whereas TNF-α-induced apoptosis involves caspase-8, TNF-α-induced necroptosis is essentially mediated by the kinases RIP-1 and RIP-3 that are recruited to TNF-R1 (Figure 1). Interestingly, specific inhibitors of RIP-1, called necrostatins, have been recently designed as novel cytoprotective agents that might be a promising tool to positively influence ALF outcome, at least in conditions in which death receptor-mediated necrosis predominates.
Therapeutic targets to improve APAP-induced ALF outcome might be JNK or other members of the mitogen-activated protein kinase family, such as p38. JNK inhibition by SP600125, a small-molecule reversible ATP-competitive inhibitor, or by D-JNKII, a peptide inhibitor that inhibits the interaction of JNK with substrates, markedly reduced mortality in murine paracetamol-induced hepatotoxicity, with a significant reduction of hepatic apoptosis and necrosis (Bennett et al., 2001; Borsello et al., 2003; Henderson et al., 2007). Since glutathione depletion and subsequent oxidative stress formation are key pathogenic mechanisms of APAP-induced hepatocyte death, application of N-acetylcysteine, a pro-drug for glutathione synthesis, which has been shown to reduce APAP-induced liver necrosis, is the current standard therapy in APAP-induced ALF (Corcoran et al., 1985; Saito et al., 2010). In addition, other drugs with anti-oxidant effects might improve ALF outcome. In this respect, application of cyclooxygenase inhibitors exerted protective effects in an experimental ALF mouse model with decreased oxidative stress formation and marked reduction of hepatic necrosis (Chang et al., 2011; Liong et al., 2012). A novel therapeutic target in ALF that connects the immune system with cell death might be cyclophilin A. This intracellular protein exerts pro-inflammatory and hepatotoxic activity when released from necrotic liver cells in APAP-induced liver injury. Conversely, inhibition of cyclophilin reduced inflammatory response to necrotic liver (Dear et al., 2011). Thus, targeting of cyclophilin might represent an opportunity for a novel therapeutic approach in acetaminophen poisoning. As mentioned above, pharmacological inhibition of autophagy exacerbated APAP-induced liver toxicity. Vice versa, induction of autophagy by rapamycin was shown to inhibit APAP-induced hepatotoxicity (Ni et al., 2012). Thus, autophagy induction might be a further strategy to improve ALF outcome in APAP intoxicated patients.
Taken together, there are many promising therapeutic approaches for inhibition of APAP- and non-APAP-induced ALF. However, it should be kept in mind that most of the described targets exert a pleiotropic role and might interfere not only with cell death but also with survival pathways.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Badmann, A., Keough, A., Kaufmann, T., Bouillet, P., Brunner, T., and Corazza, N. (2011). Role of TRAIL and pro-apoptotic Bcl2 homolog Bim in acetaminophen-induced liver damage. Cell Death Dis. 2, e171.
Bantel, H., Lügering, A., Heidemann, J., Volkmann, X., Poremba, C., Strassburg, C. P., Manns, M. P., and Schulze-Osthoff, K. (2004). Detection of apoptotic caspase activation in sera from patients with chronic HCV infection is associated with fibrotic liver injury. Hepatology 40, 1078–1087.
Bantel, H., Lügering, A., Poremba, C., Lügering, N., Held, J., Domschke, W., and Schulze-Osthoff, K. (2001a). Caspase activation correlates with the degree of inflammatory liver injury in chronic hepatitis C virus infection. Hepatology 34, 758–767.
Bantel, H., Ruck, P., Gregor, M., and Schulze-Osthoff, K. (2001b). Detection of elevated caspase activation and early apoptosis in liver diseases. Eur. J. Cell Biol. 80, 230–239.
Bantel, H., Ruck, P., and Schulze-Osthoff, K. (2000). In situ monitoring of caspase activation in hepatobiliary diseases. Cell Death Differ. 7, 504–505.
Bantel, H., and Schulze-Osthoff, K. (2003). Apoptosis in hepatitis C virus infection. Cell Death Differ. 10, 48–58.
Bantel, H., and Schulze-Osthoff, K. (2012). Can cell death biomarkers predict the outcome of acute liver failure? Liver Int. 32, 346.
Bechmann, L. P., Jochum, C., Kocabayoglu, P., Sowa, J. P., Kassalik, M., Gieseler, R. K., Saner, F., Paul, A., Trautwein, C., Gerken, G., and Canbay, A. (2010). Cytokeratin 18-based modification of the MELD score improves prediction of spontaneous survival after acute liver injury. J. Hepatol. 53, 639–647.
Bechmann, L. P., Marquitan, G., Jochum, C., Saner, F., Gerken, G., and Canbay, A. (2008). Apoptosis versus necrosis rate as a predictor in acute liver failure following acetaminophen intoxication compared with acute-on-chronic liver failure. Liver Int. 28, 713–716.
Bell, C. W., Jiang, W., Reich, C. F. III, and Pisetsky, D. S. (2006). The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 291, 1318–1325.
Ben Moshe, T., Barash, H., Kang, T. B., Kim, J. C., Kovalenko, A., Gross, E., Schuchmann, M., Abramovitch, R., Galun, E., and Wallach, D. (2007). Role of caspase-8 in hepatocyte response to infection and injury in mice. Hepatology 45, 1014–1024.
Bennett, B. L., Sasaki, D. T., Murray, B. W., O’Leary, E. C., Sakata, S. T., Xu, W., Leisten, J. C., Motiwala, A., Pierce, S., Satoh, Y., Bhagwat, S. S., Manning, A. M., and Anderson, D. W. (2001). SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. U.S.A. 98, 13681–11386.
Blazka, M. E., Elwell, M. R., Holladay, S. D., Wilson, R. E., and Luster, M. I. (1996). Histopathology of acetaminophen-induced liver changes: role interleukin 1 alpha and tumor necrosis factor alpha. Toxicol. Pathol. 24, 181–189.
Blazka, M. E., Wilmer, J. L., Holladay, S. D., Wilson, R. E., and Luster, M. I. (1995). Role of proinflammatory cytokines in acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 133, 43–52.
Boess, F., Bopst, M., Althaus, R., Polsky, S., Cohen, S. D., Euster, H. P., and Boelsterli, U. A. (1998). Acetaminophen hepatotoxicity in tumor necrosis factor/lymphotoxin-alpha gene knockout mice. Hepatology 27, 1021–1029.
Borsello, T., Clarke, P. G., Hirt, L., Vercelli, A., Repici, M., Schorderet, D. F., Bogousslavsky, J., and Bonny, C. (2003). A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 9, 1180–1186.
Bourdi, M., Korrapati, M. C., Chakraborty, M., Yee, S. B., and Pohl, L. R. (2008). Protective role of c-Jun N-terminal kinase 2 in acetaminophen-induced liver injury. Biochem. Biophys. Res. Commun. 374, 6–10.
Canbay, A., Higuchi, H., Bronk, S. F., Taniai, M., Sebo, T. J., and Gores, G. J. (2003). Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology 123, 1323–1330.
Chan, K., Truong, D., Shangari, N., and O’Brien, P. J. (2005). Drug-induced mitochondrial toxicity. Expert Opin. Drug Metab. Toxicol. 4, 655–669.
Chang, C. C., Wang, S. S., Huang, H. C., Chan, C. Y., Lee, F. Y., Lin, H. C., Nong, J. Y., Chuang, C. L., and Lee, S. D. (2011). Selective cyclooxygenase inhibition improves hepatic encephalopathy in fulminant hepatic failure of rat. Eur. J. Pharmacol. 666, 226–232.
Corcoran, G. B., Racz, W. J., Smith, C. V., and Mitchell, J. R. (1985). Effects of N-acetylcysteine on acetaminophen covalent binding and hepatic necrosis in mice. J. Pharmacol. Exp. Ther. 232, 864–872.
Craig, D. G., Lee, P., Pryde, E. A., Masterton, G. S., Hayes, P. C., and Simpson, K. J. (2011). Circulating apoptotic and necrotic cell death markers in patients with acute liver injury. Liver Int. 31, 1127–1136.
Dear, J. W., Simpson, K. J., Nicolai, M. P., Catterson, J. H., Street, J., Huizinga, T., Craig, D. G., Dhaliwal, K., Webb, S., Bateman, D. N., and Webb, D. J. (2011). Cyclophilin A is a damage-associated molecular pattern molecule that mediates acetaminophen-induced liver injury. J. Immunol. 187, 3347–3352.
Desbarats, J., and Newell, M. K. (2000). Fas engagement accelerates liver regeneration after partial hepatectomy. Nat. Med. 6, 920–923.
El-Hassan, H., Anwar, K., Macanas-Pirard, P., Crabtree, M., Chow, S. C., Johnson, V. L., Lee, P. C., Hinton, R. H., Price, S. C., and Kass, G. E. (2003). Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: roles of cytochrome c, bax, bid, and caspases. Toxicol. Appl. Pharmacol. 191, 118–129.
El-Mofty, S. K., Scrutton, M. C., Serroni, A., Nicolini, C., and Farber, J. L. (1975). Early, reversible plasma membrane injury in galactosamine-induced liver cell death. Am. J. Pathol. 79, 579–596.
Faouzi, S., Burckhardt, B. E., Hanson, J. C., Campe, C. B., Schrum, L. W., Rippe, R. A., and Maher, J. J. (2001). Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-kappa B-independent, caspase-3-dependent pathway. J. Biol. Chem. 276, 49077–49082.
Ferrari, D., Stepzynska, A., Los, M., Wesselborg, S., and Schulze-Osthoff, K. (1998). Differential regulation and ATP requirement for caspase-8 and -3 activation during CD95- and anticancer drug-induced apoptosis. J. Exp. Med. 188, 979–984.
Fischer, U., Jänicke, R. U., and Schulze-Osthoff, K. (1998). Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 10, 76–100.
Fischer, U., and Schulze-Osthoff, K. (2005). New approaches and therapeutics targeting apoptosis in disease. Pharmacol. Rev. 57, 187–215.
Galanos, C., Freudenberg, M. A., and Reutter, W. (1979). Galactosamine-induced sensitization to the lethal effects of endotoxin. Proc. Natl. Acad. Sci. U.S.A. 76, 5939–5943.
Gujral, J. S., Knight, T. R., Farhood, A., Bajt, M. L., and Jaeschke, H. (2002). Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol. Sci. 67, 322–328.
Gunawan, B. K., Liu, Z. X., Han, D., Hanawa, N., Gaarde, W. A., and Kaplowitz, N. (2006). C-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 131, 165–178.
Henderson, N. C., Pollock, K. J., Frew, J., Mackinnon, A. C., Flavell, R. A., Davis, R. J., Sethi, T., and Simpson, K. J. (2007). Critical role of c-jun (NH2) terminal kinase in paracetamol-induced acute liver failure. Gut 56, 982–990.
Hikita, H., Takehara, T., Kodama, T., Shimizu, S., Shigekawa, M., Hosui, A., Miyagi, T., Tatsumi, T., Ishida, H., Li, W., Kanto, T., Hiramatsu, N., Shimizu, S., Tsujimoto, Y., and Shimizu, N. (2011). Delayed-onset caspase-dependent massive hepatocyte apoptosis upon Fas activation in Bak/Bax-deficient mice. Hepatology 54, 240–251.
Hinson, J. A., Roberts, D. W., and James, L. P. (2010). Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 196, 369–405.
Hiramatsu, N., Hayashi, N., Katayama, K., Mochizuki, K., Kawanishi, Y., Kasahara, A., Fusamoto, H., and Kamada, T. (1994). Immunohistochemical detection of Fas antigen in liver tissue of patients with chronic hepatitis C. Hepatology 19, 1354–1359.
Hu, B., and Colletti, L. M. (2010). CXC receptor-2 knockout genotype increases X-linked inhibitor of apoptosis protein and protects mice from acetaminophen hepatotoxicity. Hepatology 52, 691–702.
Jaeschke, H., Cover, C., and Bajt, M. L. (2006). Role of caspases in acetaminophen-induced liver injury. Life Sci. 78, 1670–1676.
Jaeschke, H., Williams, C. D., Ramachandran, A., and Bajt, M. L. (2012). Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity. Liver Int. 32, 8–20.
Joka, D., Wahl, K., Moeller, S., Schlue, J., Vaske, B., Bahr, M. J., Manns, M. P., Schulze-Osthoff, K., and Bantel, H. (2012). Prospective biopsy-controlled evaluation of cell death biomarkers for prediction of liver fibrosis and nonalcoholic steatohepatitis. Hepatology 55, 455–464.
Keppler, D., Lesch, R., Reutter, W., and Decker, K. (1968). Experimental hepatitis induced by D-galactosamine. Exp. Mol. Pathol. 9, 279–290.
Kerr, J. F. R., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257.
Knight, T. R., and Jaeschke, H. (2002). Acetaminophen-induced inhibition of Fas receptor-mediated liver cell apoptosis: mitochondrial dysfunction versus glutathione depletion. Toxicol. Appl. Pharmacol. 181, 133–141.
Kon, K., Kim, J. S., Jaeschke, H., and Lemasters, J. J. (2004). Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 40, 1170–1179.
Larson, A. M., Polson, J., Fontana, R. J., Davern, T. J., Lalani, E., Hynan, L. S., Reisch, J. S., Schiødt, F. V., Ostapowicz, G., Shakil, A. O., and Lee, W. M. (2005). Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42, 1364–1372.
Lawson, J. A., Fisher, M. A., Simmons, C. A., Farhood, A., and Jaeschke, H. (1999). Inhibition of Fas receptor (CD95)-induced hepatic caspase activation and apoptosis by acetaminophen in mice. Toxicol. Appl. Pharmacol. 156, 179–186.
Lee, J. Y., Chae, D. W., Kim, S. M., Nam, E. S., Jang, M. K., Lee, J. H., Kim, H. Y., and Yoo, J. Y. (2004). Expression of FasL and perforin/granzyme B mRNA in chronic hepatitis B virus infection. J. Viral Hepat. 11, 130–135.
Leers, M. P. G., Kölgen, W., Björklund, V., Bergmann, T., Gordon, T., Persson, B., Björklund, P., Ramaekers, F. C., Björklund, B., Nap, M., Jörnvall, H., and Schutte, B. (1999). Immunohistochemical detection and mapping of cytokeratin 18 neo-epitope exposed during early apoptosis. J. Pathol. 187, 567–572.
Leist, M., Gantner, F., Bohlinger, I., Germann, P. G., Tiegs, G., and Wendel, A. (1994). Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J. Immunol. 153, 1778–1788.
Leist, M., Gantner, F., Bohlinger, I., Tiegs, G., Germann, P. G., and Wendel, A. (1995). Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am. J. Pathol. 146, 1220–1234.
Libert, C., Brouckaert, P., and Fiers, W. (1994). Protection by alpha 1-acid glycoprotein against tumor necrosis factor-induced lethality. J. Exp. Med. 180, 1571–1575.
Liong, E. C., Xiao, J., Lau, T. Y., Nanji, A. A., and Tipoe, G. L. (2012). Cyclooxygenase inhibitors protect d-galactosamine/lipopolysaccharide induced acute hepatic injury in experimental mice model. Food Chem. Toxicol. 50, 861–866.
Liu, Z. X., Govindarajan, S., and Kaplowitz, N. (2004). Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology 127, 1760–1774.
Los, M., Mozoluk, M., Ferrari, D., Stepczynska, A., Stroh, C., Renz, A., Herceg, Z., Wang, Z. Q., and Schulze-Osthoff, K. (2002). Activation and caspase-mediated inhibition of PARP: a molecular switch between necrosis and apoptosis in death receptor signaling. Mol. Biol. Cell 13, 978–988.
Los, M., Wesselborg, S., and Schulze-Osthoff, K. (1999). The role of caspases in development, immunity and apoptotic signal transduction: lessons from knock-out mice. Immunity 10, 629–639.
Mita, E., Hayashi, N., Iio, S., Takehara, T., Hijioka, T., Kasahara, A., Fusamoto, H., and Kamada, T. (1994). Role of Fas ligand in apoptosis induced by hepatitis C virus infection. Biochem. Biophys. Res. Commun. 204, 468–474.
Nakae, H., Narita, K., and Endo, S. (2001). Soluble Fas and soluble Fas ligand levels in patients with acute hepatic failure. J. Crit. Care 16, 59–63.
Ni, H. M., Bockus, A., Boggess, N., Jaeschke, H., and Ding, W. X. (2012). Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 55, 222–232.
Ogasawara, J., Watanabe-Fukunaga, R., Adachi, M., Matsuzawa, A., Kasugai, T., Kitamura, Y., Itoh, N., Suda, T., and Nagata, S. (1993). Lethal effect of the anti-Fas antibody in mice. Nature 364, 806–809.
Pianko, S., Patella, S., Ostapowicz, G., Desmond, P., and Sievert, W. (2001). Fas-mediated hepatocyte apoptosis is increased by hepatitis C virus infection and alcohol consumption, and may be associated with hepatic fibrosis: mechanisms of liver cell injury in chronic hepatitis C virus infection. J. Viral Hepat. 8, 406–413.
Ray, S. D., Mumaw, V. R., Raje, R. R., and Fariss, M. W. (1996). Protection of acetaminophen-induced hepatocellular apoptosis and necrosis by cholesteryl hemisuccinate pretreatment. J. Pharmacol. Exp. Ther. 279, 1470–1483.
Rivero, M., Crespo, J., Fabrega, E., Casafont, F., Mayorga, M., Gomez-Fleitas, M., and Pons-Romero, F. (2002). Apoptosis mediated by the Fas system in the fulminant hepatitis by hepatitis B virus. J. Viral Hepat. 9, 107–113.
Roth, G. A., Lubsczyk, B. A., Pilz, J., Faybik, P., Hetz, H., and Krenn, C. G. (2009). Nucleosome serum levels in acute hepatic failure and MARS treatment. Transplant. Proc. 41, 4207–4210.
Russell, S. W., Rosenau, W., and Lee, J. C. (1972). Cytolysis induced by human lymphotoxin. Am. J. Pathol. 69, 103–118.
Rutherford, A. E., Hynan, L. S., Borges, C. B., Forcione, D. G., Blackard, J. T., Lin, W., Gorman, A. R., Obaid, S. S., Reuben, A., Harrison, E., Reddy, K. R., Le, W. M., Chung, R. T., and for the ALF Study Group. (2007). Serum apoptosis markers in acute liver failure: a pilot study. Clin. Gastroenterol. Hepatol. 5, 1477–1483.
Ryo, K., Kamogawa, Y., Ikeda, I., Yamauchi, K., Yonehara, S., Nagat, S., and Hayashi, N. (2000). Significance of Fas antigen-mediated apoptosis in human fulminant hepatic failure. Am. J. Gastroenterol. 95, 2047–2055.
Saito, C., Zwingmann, C., and Jaeschke, H. (2010). Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 51, 246–254.
Schulze-Osthoff, K., and Bantel, H. (2011). Necrosis versus apoptosis in acetaminophen-induced hepatotoxicity. Hepatology 53, 1070.
Schulze-Osthoff, K., Ferrari, D., Los, M., Wesselborg, S., and Peter, M. E. (1998). Apoptosis signaling by death receptors. Eur. J. Biochem. 254, 439–459.
Schulze-Osthoff, K., Krammer, P. H., and Dröge, W. (1994). Divergent signaling via APO-1/Fas and the TNF receptor, two homologous molecules involved in physiological cell death. EMBO J. 13, 4587–4596.
Schwerk, C., and Schulze-Osthoff, K. (2005). Regulation of apoptosis by alternative pre-mRNA splicing. Mol. Cell 19, 1–13.
Seidel, N., Volkmann, X., Länger, F., Flemming, P., Manns, M. P., Schulze-Osthoff, K., and Bantel, H. (2005). The extent of liver steatosis in chronic hepatitis C virus infection is mirrored by caspase activity in serum. Hepatology 42, 113–120.
Sharma, A. D., Narain, N., Händel, E. M., Iken, M., Singhal, N., Cathomen, T., Manns, M. P., Schöler, H. R., Ott, M., and Cantz, T. (2011). MicroRNA-221 regulates FAS-induced fulminant liver failure. Hepatology 53, 1651–1661.
Simpson, K. J., Lukas, N. W., McGregor, A. H., Harrison, D. J., Strieter, R. M., and Kunkel, S. L. (2000). Inhibition of tumor necrosis factor alpha does not prevent experimental paracetamol-induced hepatic necrosis. J. Pathol. 190, 489–494.
Song, E., Lee, S. K., Wang, J., Ince, N., Ouyang, N., Min, J., Chen, J., Shankar, P., and Lieberman, J. (2003). RNA interference targeting Fas protects mice from fulminant hepatitis. Nat. Med. 3, 347–351.
Strand, S., Hofmann, W. J., Grambihler, A., Hug, H., Volkmann, M., Otto, G., Wesch, H., Mariani, S. M., Hack, V., Krammer, P. H., and Galle, P. R. (1998). Hepatic failure and liver cell damage in acute Wilson’s disease involve CD95 (APO-1/Fas) mediated apoptosis. Nat. Med. 4, 588–593.
Streetz, K., Leifeld, L., Grundmann, D., Ramakers, J., Eckert, K., Spengler, U., Brenner, D., Manns, M., and Trautwein, C. (2000). Tumor necrosis factor alpha in the pathogenesis of human and murine fulminant hepatic failure. Gastroenterology 119, 446–460.
Tagami, A., Ohnishi, H., and Huges, R. D. (2003). Increased serum soluble Fas in patients with acute liver failure due to paracetamol overdose. Hepatogastroenterology 5, 1477–1483.
Tinel, M., Berson, A., Vadrot, M., Descatoire, V., Grodet, A., Feldmann, G., Thénot, J. P., and Pessayre, D. (2004). Subliminal Fas stimulation increases the hepatotoxicity of acetaminophen and bromobenzene in mice. Hepatology 39, 655–666.
Tokushige, K., Yamaguchi, N., Ikeda, I., Hashimoto, E., Yamauchi, K., and Hayashi, N. (2000). Significance of soluble TNF receptor I in acute type fulminant heaptitis. Am. J. Gastroenterol. 95, 2040–2046.
Vandenabeele, P., Galluzzi, L., Berghe, T. V., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714.
Volkmann, X., Ansteatt, M., Hadem, J., Stiefel, P., Bahr, M. J., Lehner, F., Manns, M. P., Schulze-Osthoff, K., and Bantel, H. (2008). Caspase activation is associated with spontaneous recovery from acute liver failure. Hepatology 47, 1624–1633.
Volkmann, X., Cornberg, M., Wedemeyer, H., Lehner, F., Manns, M. P., Schulze-Osthoff, K., and Bantel, H. (2006). Caspase activation is required for antiviral treatment response in chronic hepatitis C virus infection. Hepatology 43, 1311–1316.
Volkmann, X., Fischer, U., Bahr, M. J., Ott, M., Lehner, F., Macfarlane, M., Cohen, G. M., Manns, M. P., Schulze-Osthoff, K., and Bantel, H. (2007). Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology 46, 1498–1508.
Wullaert, A., van Loo, G., Heynick, K., and Beyaert, R. (2007). Hepatic tumor necrosis factor signaling and nclear factor-kappaB: effects on liver homeostasis and beyond. Endocr. Rev. 28, 365–386.
Yoon, J. H., and Gores, G. J. (2002). Death receptor-mediated apoptosis and the liver. J. Hepatol. 37, 400–410.
Zender, L., Hutker, S., Liedtke, C., Tillmann, H. L., Zender, S., Mundt, B., Waltemathe, M., Gosling, T., Flemming, P., Malek, N. P., Trautwein, C., Manns, M. P., Kuhnel, F., and Kubicka, S. (2003). Caspase 8 small interfering RNA prevents acute liver failure in mice. Proc. Natl. Acad. Sci. U.S.A. 105, 9053–9058.
Keywords: acute liver failure, apoptosis, ATP, caspases, death receptors, necrosis, oxidative stress, tumor necrosis factor
Citation: Bantel H and Schulze-Osthoff K (2012) Mechanisms of cell death in acute liver failure. Front. Physio. 3:79. doi: 10.3389/fphys.2012.00079
Received: 03 February 2012; Accepted: 16 March 2012;
Published online: 02 April 2012.
Edited by:
Ali Canbay, University Hospital Essen, GermanyReviewed by:
Eileen F. Grady, University of California at San Francisco, USACopyright: © 2012 Bantel and Schulze-Osthoff. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Heike Bantel, Department of Gastroenterology, Hepatology and Endocrinology, Hannover Medical School, Carl-Neuberg-Strasse 1, D-30625 Hannover, Germany. e-mail:QmFudGVsLkhlaWtlQG1oLWhhbm5vdmVyLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.