

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 13 January 2012
Sec. Vascular Physiology
Volume 3 - 2012 | https://doi.org/10.3389/fphys.2012.00001
This article is part of the Research Topic Vascular macrophages and dendritic cells View all 8 articles
During atherogenesis, blood monocytes transmigrate into the subendothelial space and differentiate toward macrophages and foam cells. The major driver of monocyte–macrophage differentiation is macrophage colony-stimulating factor (M-CSF). M-CSF-induced macrophages are important promoters of atherogenesis as demonstrated in M-CSF and M-CSF receptor knock out mice. However, M-CSF is not the only relevant promoter of macrophage differentiation. The platelet chemokine CXCL4 also prevents monocyte apoptosis and promotes macrophage differentiation in vitro. It is secreted from activated platelets and has effects on various cell types relevant in atherogenesis. Knocking out the Pf4 gene coding for CXCL4 in Apoe−/− mice leads to reduced atherogenesis. Thus, it seems likely that CXC4-induced macrophages may have specific pro-atherogenic capacities. We have studied CXC4-induced differentiation of human macrophages using gene chips, systems biology, and functional in vitro and ex vivo experiments. Our data indicate that CXCL4-induced macrophages are distinct from both their M-CSF-induced counterparts and other known macrophage polarizations like M1 macrophages (induced by lipopolysaccharide and interferon-gamma) or M2 macrophages (induced by interleukin-4). CXCL4-induced macrophages have distinct phenotypic and functional characteristics, e.g., the complete loss of the hemoglobin–haptoglobin (Hb–Hp) scavenger receptor CD163 which is necessary for effective hemoglobin clearance after plaque hemorrhage. Lack of CD163 is accompanied by the inability to upregulate the atheroprotective enzyme heme oxygenase-1 in response to Hb–Hp complexes. This review covers the current knowledge about CXCL4-induced macrophages. Based on their unique properties, we have suggested to call these macrophages “M4.” CXCL4 may represent an important orchestrator of macrophage heterogeneity within atherosclerotic lesions. Further dissecting its effects on macrophage differentiation may help to identify novel therapeutic targets in atherogenesis.
During the development of atherosclerotic lesions, blood monocytes adhere to the endothelium, transmigrate into the subendothelial space, and differentiate toward macrophages and foam cells (Galkina and Ley, 2009; Moore and Tabas, 2011). This differentiation process is potentially driven by various factors including cell–cell contact, components of the extracellular matrix, pro- and anti-inflammatory cytokines, and chemokines as well as by lipoproteins, most importantly oxidized low density lipoprotein (oxLDL; Moore and Tabas, 2011). Macrophages are very versatile, and depending on the local microenvironment they can assume different phenotypes. In this context, one has to distinguish between the processes of “macrophage differentiation” and “macrophage polarization” (Wolfs et al., 2011).
The term macrophage differentiation describes the changes that occur after monocytes have entered the arterial wall and transform from free floating, spherical cells into adherent cells of irregular shape that take up antigen and migrate within the arterial wall. Macrophage differentiation is accompanied by substantial changes in gene expression as demonstrated in a transcriptome analysis of monocyte–macrophage differentiation induced by macrophage colony-stimulating factor (M-CSF; Martinez et al., 2006). In vitro, most changes occur within the first 3 days, even though the differentiation process is not complete at this stage and some genes may return to their initial expression levels later on (Martinez et al., 2006).
The best studied inducer of monocyte–macrophage differentiation is M-CSF (Stanley et al., 1978). In vitro, M-CSF prevents monocyte apoptosis and drives macrophage differentiation as demonstrated by downregulation of CD14 and upregulation of macrophage markers like CD68 (Martinez et al., 2006). The important role of M-CSF in atherogenesis and atherosclerosis has been demonstrated by knocking out the CSF1 gene coding for M-CSF as well as knocking out the CSF1R gene coding for the M-CSF receptor (Yoshida et al., 1990; de Villiers et al., 1998). Both knock outs resulted in significantly reduced atherogenesis in atherosclerosis-prone Apoe−/− mice. M-CSF has become a widely used growth factor when generating monocyte-derived human macrophages in vitro. Accordingly, monocyte macrophage differentiation induced by M-CSF has been extensively studied at the transcriptional and functional level (Martinez et al., 2006; Cho et al., 2007).
Macrophage colony-stimulating factor is continuously present in the circulation and thereby maintains survival of circulating monocytes as well as monocyte macrophage differentiation (Tushinski et al., 1982; Hanamura et al., 1988). This distinguishes M-CSF from other growth factors that have been associated with macrophage differentiation, among them granulocyte–macrophage colony-stimulating factor (GM-CSF) or the platelet chemokine CXCL4. A role for the latter in monocyte macrophage differentiation has been proposed in 2000 by Scheuerer et al. (2000) and will be discussed in more detail below.
The term macrophage polarization describes the ability of fully differentiated macrophages to respond to external stimuli by changing their phenotypic and functional characteristics. It was almost two decades ago, that Gordon et al. for the first time described the “alternative” polarization of macrophages induced by interleukin-(IL-)4 (Stein et al., 1992). They identified expression of the mannose receptor as a key feature of these macrophages, which they named “M2 macrophages” (as opposed to “classically” polarized macrophages induced by lipopolysaccharide (LPS) or interferon-gamma (IFN-γ), which currently are called “M1” macrophages). Since then, macrophage polarization has been extensively studied resulting in the extended paradigm of M1/M2a–c macrophage polarization (Gordon and Taylor, 2005; Martinez et al., 2008; Mantovani et al., 2009). Briefly, M1 macrophages can be induced by LPS/IFN-γ and reflect the Th1 response of T cells, i.e., they are considered pro-inflammatory which is reflected by their expression of IL-1β, IL-6, IL-8, or tumor necrosis factor-alpha (TNF-α). M2 macrophages can be induced by IL-4 (M2a), immune complexes (M2b), or IL-13/IL-10 (M2c) and largely reflect the Th2 response of T cells, i.e., they are considered rather anti-inflammatory which is reflected by their expression of IL-10, CD36, scavenger receptor-A, or mannose receptor.
Both macrophage differentiation and polarization may result in similar phenotypes: Thus, GM-CSF may induce M1-like macrophages (Stoger et al., 2010), while M-CSF induces M2-like macrophages (Martinez et al., 2006). An important difference between differentiation and polarization is the fact that, while in many cases polarization may be a reversible process, differentiation seems to be irreversible (Porcheray et al., 2005).
Over the past years, it has become evident that macrophages within human atherosclerotic plaques do not represent a homogeneous cell population, but may consist of several subsets that have distinct phenotypic and functional characteristics. The first report of evidence for the presence M1 and M2 polarized macrophages within human atherosclerotic plaques was published by Bouhlel et al. (2007) who demonstrated expression of both M1 and M2 genes within human atherosclerotic plaques. They furthermore found both the M2 marker mannose receptor and the M1 chemokine macrophage chemotactic protein-1 (MCP-1/CCL2) expressed in plaques. Since then a number of other different macrophage phenotypes have been described in human atherosclerotic lesions.
Waldo et al. (2008) have compared the transcriptomes of M-CSF and GM-CSF-induced macrophages thus comparing two types of macrophage differentiation. They found that the latter were mostly CD68+CD14− and (unless activated with PMA) tended to accumulate less cholesterol in vitro. In human atherosclerotic lesions both CD68+CD14+ M-CSF-induced macrophages and their CD68+CD14− GM-CSF-induced counterparts were Oil red O positive suggesting that both types may actually participate in lipid uptake and become foam cells in vivo. When taking a closer look at the cytokine profile of the GM-CSF-induced macrophages, there seems to be some overlap with the “classically” polarized M1 macrophage (Stoger et al., 2010). This is interesting for two reasons: Firstly, it demonstrates that – as mentioned above – macrophage differentiation and polarization may lead to similar phenotypes. Secondly, it suggests that there may also be some overlap between macrophage and dendritic cells. The latter is based on the finding that differentiation as GM-CSF alone or in combination with IL-4 may promote dendritic cell differentiation from human peripheral blood monocytes (Sallusto and Lanzavecchia, 1994). This idea is supported by the fact that in human macrophages oxLDL induces many genes that have been associated with dendritic cell differentiation (Cho et al., 2007) thus indicating that the strict distinction between macrophages and dendritic cells may be somewhat contrived (Geissmann et al., 2010).
Boyle et al. have studied macrophage polarization induced by hemoglobin as potentially found associated with intra-plaque hemorrhages. It was found that hemoglobin induces a potentially atheroprotective macrophage polarization characterized by expression of IL-10, high levels of the hemoglobin scavenger receptor CD163 and low expression of HLA-DR (Boyle et al., 2011). In a follow-up study, it could be demonstrated that induction of this macrophage polarization type was mediated by the transcription factor Nrf2 and upregulation of the atheroprotective enzyme heme oxygenase-1 (Boyle et al., 2011). This hemorrhage-associated macrophage type (M-HA) seems to be very similar to M2c macrophages induced by IL-10 which is consistent with the fact that hemoglobin-induced upregulation of IL-10 may have auto- and paracrine effects leading to a M2c-like macrophage polarization.
It is extremely likely that our current knowledge on macrophage heterogeneity will significantly expand in the future. We have recently described a novel macrophage differentiation type induced by the platelet chemokine CXCL4 (Gleissner et al., 2010b). This differentiation type will be discussed in further detail below.
Platelets do not only represent an important mediator of hemostasis, but do also play an important role in inflammation and immunity. Platelets represent an important reservoir of chemokines (Gleissner et al., 2008; Gleissner, 2012). CXCL4 (formerly known as platelet factor 4, PF4) is one of the most abundant platelet chemokines and is released from platelet α-granules upon platelet activation into the blood in micromolar concentrations as determined in serum (Brandt et al., 2000). CXCL4 has been described to have effects on various cell types relevant for atherogenesis, among them endothelial cells, T cells, monocytes, macrophages, and dendritic cells (Gleissner et al., 2008; Gleissner, 2012). Accordingly, there is considerable evidence suggesting that CXCL4 plays an important role in atherosclerosis. The presence of CXCL4 within human carotid atherosclerotic plaques could be demonstrated to correlate with clinical parameters like lesion grade or the presence of symptoms (Pitsilos et al., 2003). Also, knocking out the Pf4 gene coding for CXCL4 in Apoe−/− deficient mice led to reduced atherosclerotic lesion formation suggesting a pro-atherogenic role of CXCL4 (Sachais et al., 2007).
It is unknown whether CXCL4 affects monocytes in the blood stream. In vitro, monocytes treated with CXCL4 have been shown to become cytotoxic for EC but not epithelial cells (Scheuerer et al., 2000). This effect was mediated β2-integrin ICAM-1 interaction and required generation of reactive oxygen species in monocytes. The earliest known time point for interaction between CXCL4 and blood monocytes is during monocyte adhesion to the endothelium. Activated platelets have been demonstrated to deposit CXCL4 on the endothelium of Apoe−/− deficient mice (Huo et al., 2003). Also, CXCL4 has been demonstrated to promote monocyte adhesion to the endothelium in conjunction with CCL5 (formerly known as RANTES) to a larger extent than each of the chemokines alone (von Hundelshausen et al., 2005). The fact that interrupting this interaction using a CCL5 receptor antagonist (Met-RANTES) reduces lesion size in Apoe−/− mice supports the pathophysiological relevance of CXCL4 in this context (Koenen et al., 2009).
Based on the presence of CXCL4 in atherosclerotic lesions (Pitsilos et al., 2003), it is possible that it may affect monocyte macrophage differentiation during atherogenesis. To better understand the potential role of CXCL4-induced macrophages in atherosclerosis, we have recently performed a comprehensive analysis of the transcriptome of human CXCL4-induced macrophages and compared it to that of M-CSF-induced (M0) macrophages as well as M1 and M2 polarized macrophages (Gleissner et al., 2010b). As expected, we found that both M-CSF- and CXCL4-induced macrophages share strong phenotypic similarities. They express similar mRNA and protein levels of leukocyte and myeloid markers like CD45, CD14, and CD68. Furthermore, their transcriptomes show a very high level of correlation (r = 0.934, P < 0.0001). By contrast, we also found significant differences between both macrophage types. Three hundred seventy-five genes showed significantly differential expression, 206 of them being overexpressed in CXCL4-induced macrophages. When comparing the gene expression data with those of M1 and M2 macrophages published earlier by Martinez et al. (2006), we found that CXCL4 neither induced M1-, nor M2 polarization in macrophages. This was evident when looking at the expression levels of typical M1 or M2 markers, but could also be confirmed by statistical methods like gene set enrichment analysis (Subramanian et al., 2005), modified principal components analysis (PCA), and hierarchical clustering (Gleissner et al., 2010b).
These findings suggest that CXCL4 induces macrophage differentiation resulting in specific phenotypic and functional characteristics, which will be discussed in more detail below. Based on these results, we have suggested calling these macrophages M4, a nomenclature that we believe reflects both the fact that these cells are distinct from M1 and M2 macrophages and that they can be induced by CXCL4. Accordingly, in the following paragraphs CXCL4-induced macrophages will be referred to as M4 macrophages. Figure 1 summarizes how M4 macrophages may fit in the monocyte macrophage network present in human atherosclerosis. The following sections will discuss some specific characteristics of the M4 macrophage with potential relevance to atherosclerosis (Figure 2).
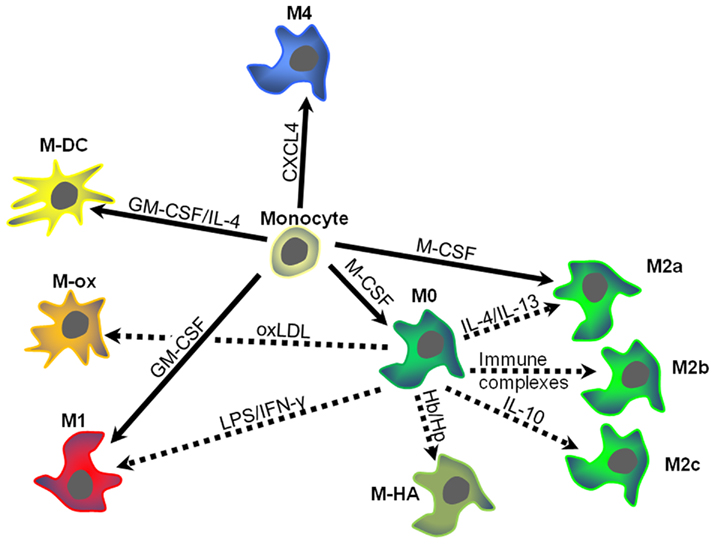
Figure 1. Schematic overview of macrophage heterogeneity within atherosclerotic plaques. Monocytes entering the vascular wall may develop differentially depending on the predominant micromilieu. Different growth factors, chemokines, or lipoproteins may induce specific types of macrophage polarization. Dotted arrows indicate monocyte–macrophage differentiation, while solid arrows signify macrophage polarization (for details see text). Arrows are labeled with the key drivers of differentiation or polarization. Colors and distances indicate similarities and differences between the different macrophage types. M-DC, dendritic cells; M-ox, foam cells induced by oxidized low density lipoprotein (oxLDL); M-HA, hemorrhage-associated macrophages. CXCL4, platelet factor 4; GM-CSG, granulocyte–macrophage colony-stimulating factor; Hb/Hp hemoglobin–haptoglobin complexes; IFN-γ, interferon-gamma; IL, interleukin; LPS, lipopolysaccharide.
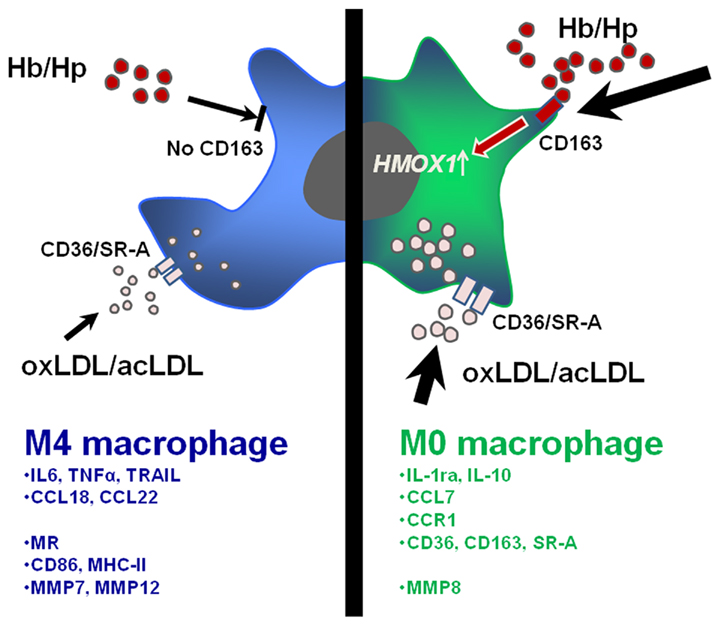
Figure 2. Specific functional and phenotypic features of CXCL4-induced M4 macrophages (blue/left) and M0 macrophages (green/right). M4 macrophages lose the ability to take up hemoglobin–haptoglobin (Hb–Hp) complexes via the receptor CD163 and subsequently upregulate the atheroprotective enzyme heme oxzgenase-1 as seen in M-CSF-induced macrophages. Furthermore, M4 macrophages express lover levels of scavenger receptors CD36/SR-A leading to fewer uptake of oxidized or acetylated low density lipoprotein (oxLDL/acLDL) as compared to M0 macrophages. The lower part of the figure indicates a selection of relevant cytokines, chemokines, chemokine receptors, surface receptors, molecule involved in T cell activation, and matrix metalloproteinases overexpressed in M4 or M0 macrophages (mRNA).
Matrix metalloproteinases (MMPs) play an important role in atherosclerosis as they may degrade the extracellular matrix and thereby promote plaque destabilization and adverse events caused by plaque rupture including myocardial infarction and stroke (Newby, 2008). Accordingly, inhibition of MMP expression and activity may be considered atheroprotective as it may lead to stable plaques with low likelihood of plaque rupture and subsequent atherothrombosis.
When comparing MMP gene expression in M4 macrophages with that in M-CSF-induced macrophages, we found a very heterogeneous profile with some MMPs being significantly higher expressed in M4 (e.g., MMP7 and MMP12) while others showing significantly higher expression in M0 macrophages (e.g., MMP8; Gleissner et al., 2010b). One has to keep in mind that MMPs are tightly regulated including regulation of gene and protein expression as well as modulation of activity by cathepsins, which are needed to activate some MMPs and which partly are also differentially expressed in M4 and M0 macrophages (cathepsin B and K significantly higher in M4 macrophages; Newby, 2008). Accordingly, based on the currently available gene expression data, it is not possible to definitively assess the functional effects of CXCL4 on MMP expression. Further functional experiments are needed to show whether M4 macrophages really contribute to plaque destabilization to a larger extent then their M-CSF-induced counterparts.
CXCL4-induced macrophages were expected to be more prone to foam cell formation induced by modified (i.e., acetylated or oxidized) low density lipoprotein (LDL) as this could represent one mechanism by which CXCL4 may promote atherosclerosis. Notably, CXCL4 has been shown to inhibit binding and uptake of LDL through its receptor which might enhance oxidation of LDL and has been shown to be related to about 10-fold increase in the amount of esterified oxLDL in macrophages (Sachais et al., 2002; Nassar et al., 2003). In these cell culture experiments, CXCL4 and native or oxLDL were present at the same time and CXCL4 could be shown to mediate binding and uptake of LDL suggesting that CXCL4 may thereby promote foam cell formation in atherosclerotic lesions. By contrast, by comparing gene expression of receptors for native and modified LDL in M4 and M0 macrophages, we found that CXCL4-induced macrophages expressed similar levels of the LDLR gene coding for the LDL receptor, but significantly lower levels of the CD36 and MSR1 genes coding for CD36 and scavenger receptor-A, which both account for the majority of modified LDL uptake (Kunjathoor et al., 2002; Gleissner et al., 2010b). In functional in vitro experiments, this resulted in reduced uptake of DiI-labeled acetylated or oxLDL compared to M-CSF-induced macrophages. It is likely that these discrepancies reflect different experimental settings. Thus, Sachais et al. (2002) and Nassar et al. (2003) studied the interactions of CXCL4 with native and modified LDL, which were simultaneously present in the cell culture. Also, while Nassar et al. (2003) looked at CXCL4 effects on murine macrophages, our focus was on human macrophage differentiation and CXCL4 was present during the entire process of monocyte macrophage differentiation. It has been recently demonstrated that murine and human monocytes differ in expression of numerous genes including those coding for receptors that may be important for LDL metabolism including CD36 (Ingersoll et al., 2010). Accordingly, species specific differences may also play an important role regarding the effects of CXCL4 on macrophage differentiation.
One of the genes significantly downregulated by CXCL4 during monocyte–macrophage differentiation is CD163 which codes for the hemoglobin–haptoglobin scavenger receptor (Gleissner et al., 2010a). Hemoglobin–haptoglobin complexes as well as free hemoglobin are bound by CD163 resulting in upregulation of heme oxygenase-1 (Schaer et al., 2006). Heme oxygenase-1 in myeloid cells has been shown to be atheroprotective as demonstrated in several mouse models (Juan et al., 2001; Yet et al., 2003; Orozco et al., 2007). Thus, the inability to appropriately respond to hemoglobin, e.g., as a consequence of intra-plaque hemorrhage may be unfavorable in patients with atherosclerotic plaques. This is also supported by data from Boyle et al. (2009, 2011) who have demonstrated that intra-plaque hemorrhage induces a CD163+ (M2c-like) macrophage type that seems to be atheroprotective.
While M-CSF induces upregulation of CD163 gene und protein expression during monocyte macrophage differentiation, CXCL4 treatment resulted in complete loss of CD163 expression on the mRNA and protein level within hours (Gleissner et al., 2010a). This effect was not caused by shedding as is seen after treatment with LPS (Buechler et al., 2000), as culture supernatants did not reveal increased levels of sCD163. Functionally, downregulation of CD163 resulted in the loss of heme oxygenase-1 upregulation upon exposure to hemoglobin–haptoglobin complexes confirming the functional relevance of the in vitro findings. It is noteworthy that once CD163 had been downregulated after exposure to CXCL4, cells were unable to upregulate the receptor after CXCL4 had been removed. Even strong inducers of CD163 like M-CSF or IL-10 were unable to rescue CD163 expression suggesting that CXCL4 induces a regulatory program that irreversible.
Immunohistochemistry of human post mortem coronary arteries revealed the presence of CD68+CD163+ as well as CD68+CD163− macrophages, furthermore there was an inverse correlation between message for CD163 and PF4 (the latter coding for CXCL4; Gleissner et al., 2010a). Considering that PF4 is exclusively expressed in megakaryocytes and platelets and CD163 expression is restricted to myeloid cells, we concluded that the presence of large amounts of CXCL4 is associated with low levels of CD163 supporting the in vivo relevance of CXCL4-induced downregulation of CD163.
While there is a lot of evidence that CXCL4 induces a specific macrophage type with phenotypic and functional characteristics that may be of importance in vascular disease, the mechanisms by which CXCL4 exerts its effects on monocytes and macrophages have not been clarified in detail yet. Other than all other known CXC-chemokines, CXCL4 lacks the ELR amino acid sequence at its C terminus that mediates binding to the chemokines receptors CXCR1 and CXCR2 (Gear and Camerini, 2003). Thus, while in microvascular endothelial cells CXCL4 seems to bind exclusively to the CXCR3B splice variant of the chemokine receptor CXCR3 (Lasagni et al., 2003), in T cells both splice variants seem to serve as receptors for CXCL4 (Mueller et al., 2008). By contrast, we could not confirm substantial CXCR3 expression on human monocytes by flow cytometry, nor did blocking CXCR3 prevent CXCL4-induced monocyte macrophage differentiation (Gleissner et al., 2010a). By contrast, treatment of cells with chlorate (which prevents synthesis of glycosaminoglycans on the cell surface, Greve et al., 1988) significantly attenuated the effects of CXCL4 on CD163 expression suggesting a chondroitin sulfate proteoglycan as potential receptor as previously demonstrated in neutrophils (Brandt et al., 2000). Most likely, these proteoglycans are linked to a thus far unknown core protein.
CXCL4-induced signal transduction has recently been reviewed by Kasper and Petersen (2011). There is good evidence that CXCL4 signals via differential pathways in different cell types. It seems that while CXCR3B-dependent signaling involves Gs proteins, signaling via proteoglycans seems to differ between cell types. As mentioned above, CXCL4 signaling in monocyte is most likely mediated by chondroitin sulfate proteoglycans (Gleissner et al., 2010). Kasper et al. (2007) have shown that Src-kinases and GTPase Ras are essential for CXCL4 signaling in monocytes. Furthermore, it could be demonstrated that CXCL4 induces increased activity of JNK, Ras, Syk tyrosine kinase, PI3K, and in addition phosphorylation of p38 MAP kinase and Erk (Kasper et al., 2007). Most of these events occur within minutes after exposure of monocytes to CXCL4 and are associated with acute respiratory burst. Interestingly, prevention of monocyte apoptosis is related to delayed activation of Erk after 6 h suggesting that this delayed pathway may be responsible for CXCL4-induced monocyte–macrophage differentiation (Kasper et al., 2007). It is noteworthy that while M1 and M2 polarization represent reversible processes (Porcheray et al., 2005), as mentioned above CXCL4-induced macrophage differentiation seems to be irreversible (Gleissner et al., 2010).
There is no doubt that CXCL4 is important in atherogenesis. It has been convincingly demonstrated that the pro-atherogenic effects of CXCL4 are most likely multifactorial, including effects on leukocyte recruitment to the arterial wall as well as direct effects on endothelial cells and various types of leukocytes. While monocyte–macrophage differentiation induced by CXCL4 was described more than 10 years ago, it was only recently that the specific characteristics of these CXCL4-induced macrophages have been studied in more detail. Based on transcriptomic and experimental data in vitro and ex vivo in human atherosclerotic lesions, we suggest that monocyte-derived macrophages that have differentiated under the influence of CXCL4 may represent a unique macrophage set with relevance to atherosclerotic disease. We have therefore suggested calling this macrophage type “M4.” Further experiments are necessary to identify the receptor, the exact signaling pathways involved, and the functional consequences of CXCL4-induced macrophage differentiation. Future research will be needed to establish whether M4 macrophages represent a promising therapeutic target in human atherosclerosis.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Bouhlel, M. A., Derudas, B., Rigamonti, E., Dievart, R., Brozek, J., Haulon, S., Zawadzki, C., Jude, B., Torpier, G., Marx, N., Staels, B., and Chinetti-Gbaguidi, G. (2007). PPARgamma activation primes human monocytes into alternative m2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143.
Boyle, J. J., Harrington, H. A., Piper, E., Elderfield, K., Stark, J., Landis, R. C., and Haskard, D. O. (2009). Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 174, 1097–1108.
Boyle, J. J., Johns, M., Lo, J., Chiodini, A., Ambrose, N., Evans, P. C., Mason, J. C., and Haskard, D. O. (2011). Heme induces heme oxygenase 1 via nrf2: role in the homeostatic macrophage response to intraplaque hemorrhage. Arterioscler. Thromb. Vasc. Biol. 31, 2685–2691.
Brandt, E., Petersen, F., Ludwig, A., Ehlert, J. E., Bock, L., and Flad, H. D. (2000). The beta-thromboglobulins and platelet factor 4: blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J. Leukoc. Biol. 67, 471–478.
Buechler, C., Ritter, M., Orso, E., Langmann, T., Klucken, J., and Schmitz, G. (2000). Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J. Leukoc. Biol. 67, 97–103.
Cho, H. J., Shashkin, P., Gleissner, C. A., Dunson, D., Jain, N., Lee, J. K., Miller, Y., and Ley, K. (2007). Induction of dendritic cell-like phenotype in macrophages during foam cell formation. Physiol. Genomics 29, 149–160.
de Villiers, W. J., Smith, J. D., Miyata, M., Dansky, H. M., Darley, E., and Gordon, S. (1998). Macrophage phenotype in mice deficient in both macrophage-colony-stimulating factor (op) and apolipoprotein e. Arterioscler. Thromb. Vasc. Biol. 18, 631–640.
Galkina, E. V., and Ley, K. (2009). Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 27, 165–197.
Gear, A. R., and Camerini, D. (2003). Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation 10, 335–350.
Geissmann, F., Manz, M. G., Jung, S., Sieweke, M. H., Merad, M., and Ley, K. (2010). Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661.
Gleissner, C. A. (2012). Platelet-derived chemokines in atherogenesis: what’s new? Curr. Vasc. Pharmacol. (in press).
Gleissner, C. A., Shaked, I., Erbel, C., Bockler, D., Katus, H. A., and Ley, K. (2010a). CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ. Res. 106, 203–211.
Gleissner, C. A., Shaked, I., Little, K. M., and Ley, K. (2010b). CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J. Immunol. 184, 4810–4818.
Gleissner, C. A., von Hundelshausen, P., and Ley, K. (2008). Platelet chemokines in vascular disease. Arterioscler. Thromb. Vasc. Biol. 28, 1920–1927.
Gordon, S., and Taylor, P. R. (2005). Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964.
Greve, H., Cully, Z., Blumberg, P., and Kresse, H. (1988). Influence of chlorate on proteoglycan biosynthesis by cultured human fibroblasts. J. Biol. Chem. 263, 12886–12892.
Hanamura, T., Motoyoshi, K., Yoshida, K., Saito, M., Miura, Y., Kawashima, T., Nishida, M., and Takaku, F. (1988). Quantitation and identification of human monocytic colony-stimulating factor in human serum by enzyme-linked immunosorbent assay. Blood 72, 886–892.
Huo, Y., Schober, A., Forlow, S. B., Smith, D. F., Hyman, M. C., Jung, S., Littman, D. R., Weber, C., and Ley, K. (2003). Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein e. Nat. Med. 9, 61–67.
Ingersoll, M. A., Spanbroek, R., Lottaz, C., Gautier, E. L., Frankenberger, M., Hoffmann, R., Lang, R., Haniffa, M., Collin, M., Tacke, F., Habenicht, A. J., Ziegler-Heitbrock, L., and Randolph, G. J. (2010). Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 115, e10–e19.
Juan, S.-H., Lee, T.-S., Tseng, K.-W., Liou, J.-Y., Shyue, S.-K., Wu, K. K., and Chau, L.-Y. (2001). Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein e-deficient mice. Circulation 104, 1519–1525.
Kasper, B., Brandt, E., Brandau, S., and Petersen, F. (2007). Platelet factor 4 (CXC chemokine ligand 4) differentially regulates respiratory burst, survival, and cytokine expression of human monocytes by using distinct signaling pathways. J. Immunol. 179, 2584–2591.
Kasper, B., and Petersen, F. (2011). Molecular pathways of platelet factor 4/CXCL4 signaling. Eur. J. Cell Biol. 90, 521–526.
Koenen, R. R., von Hundelshausen, P., Nesmelova, I. V., Zernecke, A., Liehn, E. A., Sarabi, A., Kramp, B. K., Piccinini, A. M., Paludan, S. R., Kowalska, M. A., Kungl, A. J., Hackeng, T. M., Mayo, K. H., and Weber, C. (2009). Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 15, 97–103.
Kunjathoor, V. V., Febbraio, M., Podrez, E. A., Moore, K. J., Andersson, L., Koehn, S., Rhee, J. S., Silverstein, R., Hoff, H. F., and Freeman, M. W. (2002). Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 277, 49982–49988.
Lasagni, L., Francalanci, M., Annunziato, F., Lazzeri, E., Giannini, S., Cosmi, L., Sagrinati, C., Mazzinghi, B., Orlando, C., Maggi, E., Marra, F., Romagnani, S., Serio, M., and Romagnani, P. (2003). An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by ip-10, mig, and I-tac, and acts as functional receptor for platelet factor 4. J. Exp. Med. 197, 1537–1549.
Mantovani, A., Garlanda, C., and Locati, M. (2009). Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler. Thromb. Vasc. Biol. 29, 1419–1423.
Martinez, F. O., Gordon, S., Locati, M., and Mantovani, A. (2006). Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177, 7303–7311.
Martinez, F. O., Sica, A., Mantovani, A., and Locati, M. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461.
Moore, K. J., and Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355.
Mueller, A., Meiser, A., McDonagh, E. M., Fox, J. M., Petit, S. J., Xanthou, G., Williams, T. J., and Pease, J. E. (2008). CXCL4-induced migration of activated t lymphocytes is mediated by the chemokine receptor CXCR3. J. Leukoc. Biol. 83, 875–882.
Nassar, T., Sachais, B. S., Akkawi, S., Kowalska, M. A., Bdeir, K., Leitersdorf, E., Hiss, E., Ziporen, L., Aviram, M., Cines, D., Poncz, M., and Higazi, A. A. (2003). Platelet factor 4 enhances the binding of oxidized low-density lipoprotein to vascular wall cells. J. Biol. Chem. 278, 6187–6193.
Newby, A. C. (2008). Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler. Thromb. Vasc. Biol. 28, 2108–2114.
Orozco, L. D., Kapturczak, M. H., Barajas, B., Wang, X., Weinstein, M. M., Wong, J., Deshane, J., Bolisetty, S., Shaposhnik, Z., Shih, D. M., Agarwal, A., Lusis, A. J., and Araujo, J. A. (2007). Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ. Res. 100, 1703–1711.
Pitsilos, S., Hunt, J., Mohler, E. R., Prabhakar, A. M., Poncz, M., Dawicki, J., Khalapyan, T. Z., Wolfe, M. L., Fairman, R., Mitchell, M., Carpenter, J., Golden, M. A., Cines, D. B., and Sachais, B. S. (2003). Platelet factor 4 localization in carotid atherosclerotic plaques: correlation with clinical parameters. Thromb. Haemost. 90, 1112–1120.
Porcheray, F., Viaud, S., Rimaniol, A. C., Leone, C., Samah, B., Dereuddre-Bosquet, N., Dormont, D., and Gras, G. (2005). Macrophage activation switching: an asset for the resolution of inflammation. Clin. Exp. Immunol. 142, 481–489.
Sachais, B. S., Kuo, A., Nassar, T., Morgan, J., Kariko, K., Williams, K. J., Feldman, M., Aviram, M., Shah, N., Jarett, L., Poncz, M., Cines, D. B., and Higazi, A. A. (2002). Platelet factor 4 binds to low-density lipoprotein receptors and disrupts the endocytic machinery, resulting in retention of low-density lipoprotein on the cell surface. Blood 99, 3613–3622.
Sachais, B. S., Turrentine, T., Dawicki McKenna, J. M., Rux, A. H., Rader, D., and Kowalska, M. A. (2007). Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in c57bl/6 and apoe-/- mice. Thromb. Haemost. 98, 1108–1113.
Sallusto, F., and Lanzavecchia, A. (1994). Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179, 1109–1118.
Schaer, C. A., Schoedon, G., Imhof, A., Kurrer, M. O., and Schaer, D. J. (2006). Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ. Res. 99, 943–950.
Scheuerer, B., Ernst, M., Durrbaum-Landmann, I., Fleischer, J., Grage-Griebenow, E., Brandt, E., Flad, H. D., and Petersen, F. (2000). The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood 95, 1158–1166.
Stanley, E. R., Chen, D. M., and Lin, H. S. (1978). Induction of macrophage production and proliferation by a purified colony stimulating factor. Nature 274, 168–170.
Stein, M., Keshav, S., Harris, N., and Gordon, S. (1992). Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J. Exp. Med. 176, 287–292.
Stoger, J. L., Goossens, P., and de Winther, M. P. (2010). Macrophage heterogeneity: relevance and functional implications in atherosclerosis. Curr. Vasc. Pharmacol. 8, 233–248.
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., Paulovich, A., Pomeroy, S. L., Golub, T. R., Lander, E. S., and Mesirov, J. P. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550.
Tushinski, R. J., Oliver, I. T., Guilbert, L. J., Tynan, P. W., Warner, J. R., and Stanley, E. R. (1982). Survival of mononuclear phagocytes depends on a lineage-specific growth factor that the differentiated cells selectively destroy. Cell 28, 71–81.
von Hundelshausen, P., Koenen, R. R., Sack, M., Mause, S. F., Adriaens, W., Proudfoot, A. E., Hackeng, T. M., and Weber, C. (2005). Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 105, 924–930.
Waldo, S. W., Li, Y., Buono, C., Zhao, B., Billings, E. M., Chang, J., and Kruth, H. S. (2008). Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am. J. Pathol. 172, 1112–1126.
Wolfs, I. M., Donners, M. M., and de Winther, M. P. (2011). Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb. Haemost. 106, 763–771.
Yet, S. F., Layne, M. D., Liu, X., Chen, Y. H., Ith, B., Sibinga, N. E., and Perrella, M. A. (2003). Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 17, 1759–1761.
Keywords: atherosclerosis, macrophage, differentiation, polarization, CXCL4, M4
Citation: Gleissner CA (2012) Macrophage phenotype modulation by CXCL4 in atherosclerosis. Front. Physio. 3:1. doi: 10.3389/fphys.2012.00001
Received: 04 December 2011; Paper pending published: 26 December 2011;
Accepted: 01 January 2012; Published online: 13 January 2012.
Edited by:
Klaus Ley, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Jonathan Steven Alexander, LSU Health Sciences Center in Shreveport, USACopyright: © 2012 Gleissner. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Christian A. Gleissner, Department of Cardiology, University of Heidelberg, Im Neuenheimer Feld 410, D-69120 Heidelberg, Germany. e-mail:Y2hyaXN0aWFuLmdsZWlzc25lckBtZWQudW5pLWhlaWRlbGJlcmcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.