


- Medical Proteomics Laboratory, Institute of Medical Science, University of Tokyo, Minato-ku, Tokyo, Japan
Signal transduction systems coordinate complex cellular information to regulate biological events such as cell proliferation and differentiation. Although the accumulating evidence on widespread association of signaling molecules has revealed essential contribution of phosphorylation-dependent interaction networks to cellular regulation, their dynamic behavior is mostly yet to be analyzed. Recent technological advances regarding mass spectrometry-based quantitative proteomics have enabled us to describe the comprehensive status of phosphorylated molecules in a time-resolved manner. Computational analyses based on the phosphoproteome dynamics accelerate generation of novel methodologies for mathematical analysis of cellular signaling. Phosphoproteomics-based numerical modeling can be used to evaluate regulatory network elements from a statistical point of view. Integration with transcriptome dynamics also uncovers regulatory hubs at the transcriptional level. These omics-based computational methodologies, which have firstly been applied to representative signaling systems such as the epidermal growth factor receptor pathway, have now opened up a gate for systems analysis of signaling networks involved in immune response and cancer.
Introduction
Signal transduction networks are known to regulate complex biological events in orchestration with subsequent transcriptional regulation (Hunter, 2000; Schlessinger, 2000). Previous in-depth analyses on cell signaling under a variety of experimental conditions have revealed many of the key molecules and related events that result in each biological effect. Regarding the intensively studied signaling systems such as the epidermal growth factor (EGF) receptor pathway, the accumulated experimental evidence has clearly demonstrated the complexity of the interaction network involved in the signaling (Oda et al., 2005; Jones et al., 2006). As phosphorylation-dependent protein interaction networks play a major role in transmitting signals, a comprehensive and fine description of their status would contribute substantially toward understanding the regulatory mechanisms at the system level. Recent proteomics technology based on high-resolution mass spectrometry (MS) has enabled us to quantitatively describe the activation dynamics on phosphorylated signaling molecules in a comprehensive and unbiased manner (Blagoev et al., 2004; Zhang et al., 2005; Olsen et al., 2006; Oyama et al., 2009). Computational systems analysis based on the phosphoproteome dynamics data paves the way to theoretical approaches for defining regulatory principles that govern complicated signaling processes. Some statistical methodologies including mathematical modeling (Tasaki et al., 2006, 2010), Bayesian network (Bose et al., 2006; Guha et al., 2008), or partial least square regression (Wolf-Yadlin et al., 2006; Kumar et al., 2007) have already been applied to EGF signaling. An integrated approach based on both phosphoproteomic and transcriptomic data has also revealed a global view of cellular regulation at the transcriptional level (Oyama et al., 2011). In this article, we introduce the recent progress of proteomics-driven computational analyses applied to the signaling behavior of representative biological pathways and the potential impact on the system-level analyses of heterogeneous signaling networks related to immune response and cancer.
Emergence of High-Throughput Phosphoproteomics Technology for Large-Scale Identification and Quantification of Cellular Phosphorylated Molecules
Recent advancement in liquid chromatography–tandem mass spectrometry (LC–MS/MS) measurement technology has greatly improved the throughput and sensitivity of protein measurements. We can now identify thousands of proteins in a single study (Brunner et al., 2007; de Godoy et al., 2008). In order to efficiently describe the status of phosphorylated molecules, a variety of biochemical methodologies have been developed for their enrichment. Immobilized metal affinity chromatography (IMAC; Stensballe et al., 2001; Ficarro et al., 2002), strong cation exchange (SCX) chromatography (Ballif et al., 2004; Beausoleil et al., 2004), metal oxide chromatography (MOC; Pinkse et al., 2004; Larsen et al., 2005) were intensively evaluated as core analytical methodologies in the previous reports. For targeting tyrosine phosphorylation, anti-phosphotyrosine antibodies were applied to efficiently purify the corresponding molecules (Rush et al., 2005). Through these sophisticated enrichment methods, current shotgun proteomics technology based on high-resolution LC–MS/MS has enabled the detection of thousands of phosphorylated molecules from representative cell lines such as human HeLa cells (Figure 1; Olsen et al., 2006, 2010).
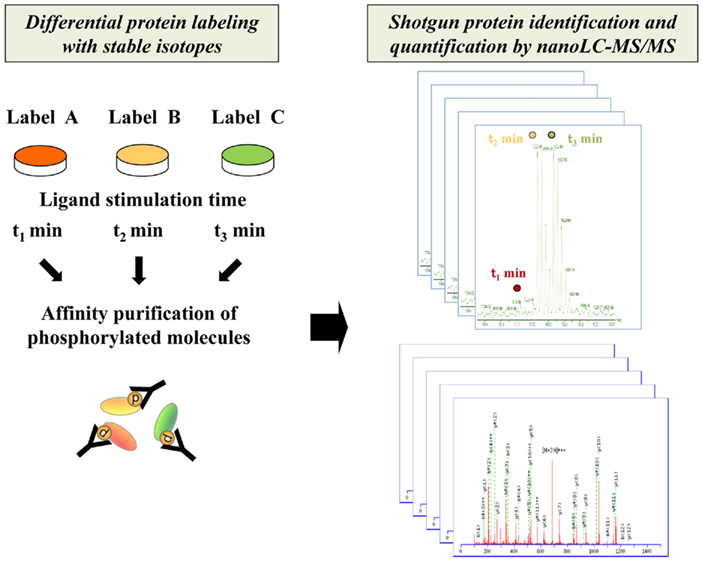
Figure 1. Schematic procedure for comprehensive identification and quantification of phosphorylated proteins by shotgun proteomics technology. The phosphorylated molecules differentially encoded with stable isotopes for each interval of stimulation are enriched through affinity purification and analyzed by high-resolution nanoLC–MS/MS system.
Another important advance in MS-based systems analysis is development of protein/peptide labeling strategies for quantitative proteomics (Figure 1). Several methodologies for in vivo/in vitro labeling have been established for relative quantification of the activation status of signaling molecules. The representative in vivo protein labeling methodology termed stable isotope labeling by amino acids in cell culture (SILAC) can be conducted by incorporating distinguishable stable isotopes into specific amino acid residues such as lysine and arginine during cell culture (Ong et al., 2002, 2003). Another approach to introduce differential labels in vitro is chemical tagging of specific amino acid residues such as cysteine. The isotope-coded affinity tag (ICAT), which consists of a cysteine-directed reactive group, a linker with stable isotope signatures, and a biotin tag, is applied to purify labeled peptides by biotin–avidin affinity (Gygi et al., 1999; Han et al., 2001). As for amine-directed tagging, the isobaric tag for relative and absolute quantitation (iTRAQ) enables comparative quantification of four or eight samples in a single analysis (Ross et al., 2004).
By combining these technologies, time-resolved activation profiles of ligand-induced phosphoproteome were depicted in a quantitative manner (Figure 2). The original approach to describe phosphotyrosine-dependent signaling dynamics led to the identification of 81 effectors in human HeLa cells upon EGF stimulation (Blagoev et al., 2004). The global phosphoserine/threonine/tyrosine-related proteome analysis for the EGF signaling system in the same cell line yielded a network-wide view of the dynamic behavior of 6,600 phosphorylation sites on 2,244 proteins (Olsen et al., 2006).
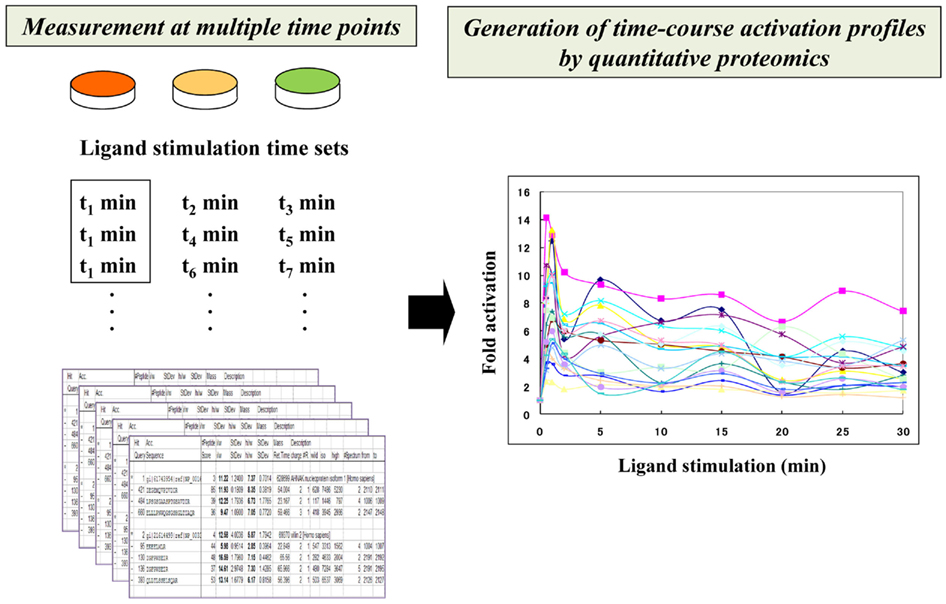
Figure 2. Time-resolved description of signaling networks by quantitative proteomics. Time-course activation profiles of phosphorylated molecules are generated through integration of a series of fold activation data that were measured at different time points.
In a recent study, a highly time-resolved description of EGF/EGFR signaling was measured in human epithelial A431 cells (Oyama et al., 2009). The quantitative activation data on the EGF-regulated tyrosine-phosphoproteome were measured at 10 time points after EGF stimulation (0, 0.5, 1, 2, 5, 10, 15, 20, 25, and 30 min), generating a detailed view of their multi-phase network dynamics. In this study, temporal perturbation of the signaling dynamics was also conducted with a kinase inhibitor to clearly distinguish between sensitive and robust pathways to this treatment. This approach showed that phosphoproteomics-based time-resolved description of the network dynamics functioned as an analytical basis for evaluating temporal perturbation effects in relation to specific signaling interactions, leading us to obtain a system-level view of the regulatory relationships in signaling dynamics.
Computational Modeling of Signal Transduction Networks Based on Quantitative Phosphoproteome Data
Although phosphoproteomics-based temporal description of signaling networks provides system-level information on dynamic regulation of signal transduction via phosphorylation/dephosphorylation, the most important challenge for elucidating the mechanistic aspects of signal transduction is the establishment of statistical methodologies for performing computational modeling with increasing species, states, and reactions over the signaling network. In a recent study, some computational frameworks have been developed for analyzing flux-based signaling information on quantitative phosphoproteomics data (Figure 3). In the initial approach, self-organizing maps were applied to identify EGF signaling modules based on time-resolved description of 78 tyrosine phosphorylation sites on 58 proteins in human mammary epithelial 184A1 cells (Zhang et al., 2005). The cells with varying human ErbB2 (HER2) expression levels were further analyzed to characterize HER2-mediated signaling effects on cell behavior (Wolf-Yadlin et al., 2006). Partial least squares regression (PLSR) was applied to estimate the phosphotyrosine clusters exhibiting self-similar temporal activation profiles, leading to identification of the signals that were strongly correlated with cell migration and proliferation and could function as a “network gage” of cell fate control (Wolf-Yadlin et al., 2006; Kumar et al., 2007).
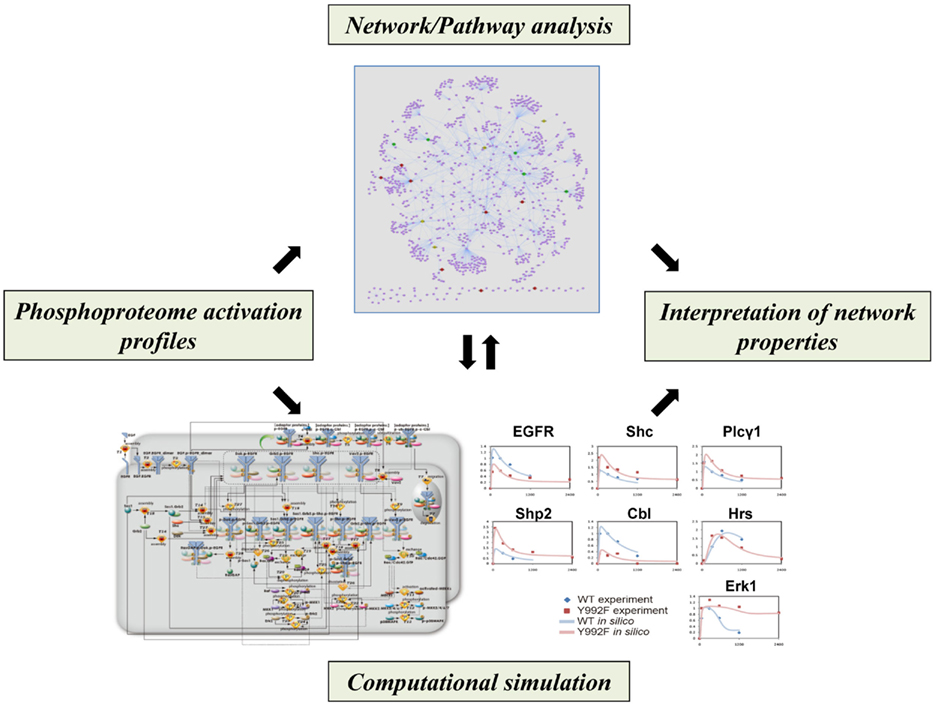
Figure 3. Computational approaches for analyzing network properties of phosphorylation-dependent signaling behavior. Phosphoproteomics-based network models, in combination with literature-based network/pathway information, can be sophisticated to interpret regulatory aspects of signaling dynamics.
Bayesian network modeling based on multiple sets of quantitative phosphoproteome data could generate probabilistic networks that represented core aspects of the models with a directed graph of influence on protein phosphorylation. In combination with the literature-based protein–protein interaction data on the EGFR/ErbB signaling, this statistical approach not only recapitulated known portions of the signaling pathways but also inferred novel relationships between the related molecules (Bose et al., 2006; Guha et al., 2008). In a recent study, a computational framework based on data assimilation was also developed for analyzing mutated EGFR signaling through phosphoproteomics-driven numerical modeling (Tasaki et al., 2010). The hybrid functional petri net with extension (HFPNe) is a computational modeling architecture which can deal with discrete biological events as well as continuous ones and enables us to analyze temporal data on biological entities such as phosphorylated signaling molecules within the data assimilation framework. The HFPNe-based computational modeling of aberrant EGFR signaling led to reduction of the factors responsible for mutational effect to several alterations in the reaction parameters and provided a mechanistic description of the disorders of their cell signaling networks at the system level.
Phosphoproteome dynamics data can be integrated with the transcriptome dynamics to analyze the regulatory mechanisms more systematically. In a very recent study, time-resolved phosphoproteome and transcriptome data on 17β-estradiol (E2) and heregulin (HRG)-induced signal-transcription programs were quantitatively analyzed to elucidate regulatory pathways in breast cancer signaling (Oyama et al., 2011). Reconstruction of protein interaction networks based on the phosphoproteome data shed light on the activated signaling molecules over the network, while statistical evaluation of transcription factor-binding site motif significance for the entire gene expression data led us to focus on the core transcriptional regulators. Functional association of these factors using pathway databases revealed ligand-dependent signal-transcription regulatory programs in both of wild type and drug-resistant breast cancer MCF-7 cells, leading us to extract the pathways activated in drug-resistant cells.
Future Prospects
Recent advances in proteomics technology have presented us with a system-wide view of phosphorylation-dependent signaling network dynamics in a quantitative manner. Mathematical analysis of phosphoproteomics-based networks will lead to a better understanding of the critical factors controlling network behavior and provide a computational platform to explore potential drug targets for specific disease conditions and theoretically estimate the effect of the corresponding drugs on a network-wide scale prior to clinical application. As signaling network structures depend on cellular context (Morandell et al., 2008), cell-specific signaling network architectures need to be described independently using phosphoproteomics to characterize the behavior of each signaling system. Although this emerging technology has been applied to only a limited fraction of signaling networks including the EGFR pathway, further accumulation and integration of phosphoproteome data on heterogeneous immune and cancer signaling networks should accelerate elucidation of general and condition-specific principles that govern signaling network behavior and pave the way to understanding complex cellular responses from a systems perspective.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge our colleagues at the Institute of Medical Science, University of Tokyo for helpful discussions and comments. This work was supported by Grants-in-Aid for Scientific Research (C) and Scientific Research on Innovative Areas from Japan Society for the Promotion of Science (JSPS) and The Ministry of Education, Culture, Sports, Science and Technology (MEXT).
References
Ballif, B. A., Villén, J., Beausoleil, S. A., Schwartz, D., and Gygi, S. P. (2004). Phosphoproteomic analysis of the developing mouse brain. Mol. Cell. Proteomics 3, 1093–1101.
Beausoleil, S. A., Jedrychowski, M., Schwartz, D., Elias, J. E., Villén, J., Li, J., Cohn, M. A., Cantley, L. C., and Gygi, S. P. (2004). Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12130–12135.
Blagoev, B., Ong, S. E., Kratchmarova, I., and Mann, M. (2004). Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat. Biotechnol. 22, 1139–1145.
Bose, R., Molina, H., Patterson, A. S., Bitok, J. K., Periaswamy, B., Bader, J. S., Pandey, A., and Cole, P. A. (2006). Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc. Natl. Acad. Sci. U.S.A. 103, 9773–9778.
Brunner, E., Ahrens, C. H., Mohanty, S., Baetschmann, H., Loevenich, S., Potthast, F., Deutsch, E. W., Panse, C., de Lichtenberg, U., Rinner, O., Lee, H., Pedrioli, P. G., Malmstrom, J., Koehler, K., Schrimpf, S., Krijgsveld, J., Kregenow, F., Heck, A. J., Hafen, E., Schlapbach, R., and Aebersold, R. (2007). A high-quality catalog of the Drosophila melanogaster proteome. Nat. Biotechnol. 25, 576–583.
de Godoy, L. M., Olsen, J. V., Cox, J., Nielsen, M. L., Hubner, N. C., Fröhlich, F., Walther, T. C., and Mann, M. (2008). Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254.
Ficarro, S. B., McCleland, M. L., Stukenberg, P. T., Burke, D. J., Ross, M. M., Shabanowitz, J., Hunt, D. F., and White, F. M. (2002). Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20, 301–305.
Guha, U., Chaerkady, R., Marimuthu, A., Patterson, A. S., Kashyap, M. K., Harsha, H. C., Sato, M., Bader, J. S., Lash, A. E., Minna, J. D., Pandey, A., and Varmus, H. E. (2008). Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proc. Natl. Acad. Sci. U.S.A. 105, 14112–14117.
Gygi, S. P., Rist, B., Gerber, S. A., Turecek, F., Gelb, M. H., and Aebersold, R. (1999). Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999.
Han, D. K., Eng, J., Zhou, H., and Aebersold, R. (2001). Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat. Biotechnol. 19, 946–951.
Jones, R. B., Gordus, A., Krall, J. A., and Macbeath, G. (2006). A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature 439, 168–174.
Kumar, N., Wolf-Yadlin, A., White, F. M., and Lauffenburger, D. A. (2007). Modeling HER2 effects on cell behavior from mass spectrometry phosphotyrosine data. PLoS Comput. Biol. 3, e4. doi:10.1371/journal.pcbi.0030004
Larsen, M. R., Thingholm, T. E., Jensen, O. N., Roepstorff, P., and Jorgensen, T. J. (2005). Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 4, 873–886.
Morandell, S., Stasyk, T., Skvortsov, S., Ascher, S., and Huber, L. A. (2008). Quantitative proteomics and phosphoproteomics reveal novel insights into complexity and dynamics of the EGFR signaling network. Proteomics 8, 4383–4401.
Oda, K., Matsuoka, Y., Funahashi, A., and Kitano, H. (2005). A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 1, 0010.
Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., and Mann, M. (2006). Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648.
Olsen, J. V., Vermeulen, M., Santamaria, A., Kumar, C., Miller, M. L., Jensen, L. J., Gnad, F., Cox, J., Jensen, T. S., Nigg, E. A., Brunak, S., and Mann, M. (2010). Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3.
Ong, S. E., Blagoev, B., Kratchmarova, I., Kristensen, D. B., Steen, H., Pandey, A., and Mann, M. (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386.
Ong, S. E., Kratchmarova, I., and Mann, M. (2003). Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC). J. Proteome Res. 2, 173–181.
Oyama, M., Kozuka-Hata, H., Tasaki, S., Semba, K., Hattori, S., Sugano, S., Inoue, J., and Yamamoto, T. (2009). Temporal perturbation of tyrosine phosphoproteome dynamics reveals the system-wide regulatory networks. Mol. Cell. Proteomics 8, 226–231.
Oyama, M., Nagashima, T., Suzuki, T., Kozuka-Hata, H., Yumoto, N., Shiraishi, Y., Ikeda, K., Kuroki, Y., Gotoh, N., Ishida, T., Inoue, S., Kitano, H., and Okada-Hatakeyama, M. (2011). Integrated quantitative analysis of the phosphoproteome and transcriptome in tamoxifen-resistant breast cancer. J. Biol. Chem. 286, 818–829.
Pinkse, M. W., Uitto, P. M., Hilhorst, M. J., Ooms, B., and Heck, A. J. (2004). Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal. Chem. 76, 3935–3943.
Ross, P. L., Huang, Y. N., Marchese, J. N., Williamson, B., Parker, K., Hattan, S., Khainovski, N., Pillai, S., Dey, S., Daniels, S., Purkayastha, S., Juhasz, P., Martin, S., Bartlet-Jones, M., He, F., Jacobson, A., and Pappin, D. J. (2004). Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169.
Rush, J., Moritz, A., Lee, K. A., Guo, A., Goss, V. L., Spek, E. J., Zhang, H., Zha, X. M., Polakiewicz, R. D., and Comb, M. J. (2005). Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101.
Stensballe, A., Andersen, S., and Jensen, O. N. (2001). Characterization of phosphoproteins from electrophoretic gels by nanoscale Fe(III) affinity chromatography with off-line mass spectrometry analysis. Proteomics 1, 207–222.
Tasaki, S., Nagasaki, M., Kozuka-Hata, H., Semba, K., Gotoh, N., Hattori, S., Inoue, J., Yamamoto, T., Miyano, S., Sugano, S., and Oyama, M. (2010). Phosphoproteomics-based modeling defines the regulatory mechanism underlying aberrant EGFR signaling. PLoS ONE 5, e13926. doi:10.1371/journal.pone.0013926
Tasaki, S., Nagasaki, M., Oyama, M., Hata, H., Ueno, K., Yoshida, R., Higuchi, T., Sugano, S., and Miyano, S. (2006). Modeling and estimation of dynamic EGFR pathway by data assimilation approach using time series proteomic data. Genome Inform. 17, 226–238.
Wolf-Yadlin, A., Kumar, N., Zhang, Y., Hautaniemi, S., Zaman, M., Kim, H. D., Grantcharova, V., Lauffenburger, D. A., and White, F. M. (2006). Effects of HER2 overexpression on cell signaling networks governing proliferation and migration. Mol. Syst. Biol. 2, 54.
Keywords: signal transduction, phosphoproteomics, quantitative proteomics, computational modeling, systems biology
Citation: Kozuka-Hata H, Tasaki S and Oyama M (2012) Phosphoproteomics-based systems analysis of signal transduction networks. Front. Physio. 2:113. doi: 10.3389/fphys.2011.00113
Received: 20 August 2011;
Accepted: 13 December 2011;
Published online: 03 January 2012.
Edited by:
Kumar Selvarajoo, Keio University, JapanReviewed by:
Bhawana Agarwal, Medical College of Wisconsin, USAJeffrey Varner, Cornell University, USA
Copyright: © 2012 Kozuka-Hata, Tasaki and Oyama. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Masaaki Oyama, Medical Proteomics Laboratory, Institute of Medical Science, University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo 108-8639, Japan. e-mail:bW95YW1hQGltcy51LXRva3lvLmFjLmpw