



94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 19 December 2011
Sec. Redox Physiology
volume 2 - 2011 | https://doi.org/10.3389/fphys.2011.00104
Hydrogen sulfide (H2S) and nitric oxide (NO) are both gasotransmitters that can elicit synergistic vasodilatory responses in the in the cardiovascular system, but the mechanisms behind this synergy are unclear. In the current study we investigated the molecular mechanisms through which H2S regulates endothelial NO production. Initial studies were performed to establish the temporal and dose-dependent effects of H2S on NO generation using EPR spin trapping techniques. H2S stimulated a twofold increase in NO production from endothelial nitric oxide synthase (eNOS), which was maximal 30 min after exposure to 25–150 μM H2S. Following 30 min H2S exposure, eNOS phosphorylation at Ser 1177 was significantly increased compared to control, consistent with eNOS activation. Pharmacological inhibition of Akt, the kinase responsible for Ser 1177 phosphorylation, attenuated the stimulatory effect of H2S on NO production. Taken together, these data demonstrate that H2S up-regulates NO production from eNOS through an Akt-dependent mechanism. These results implicate H2S in the regulation of NO production in endothelial cells, and suggest that deficiencies in H2S signaling can directly impact processes regulated by NO.
Hydrogen sulfide (H2S) and nitric oxide (NO) are both gasotransmitters (Hosoki et al., 1997; Wang, 2003) that function in the cardiovascular system. Recent reports indicate that the NO and H2S signaling pathways interact on a variety of levels, both in vitro and in vivo (Geng et al., 2007; Kubo et al., 2007a,b; Yong et al., 2008). Exogenous NaHS, a chemical source of H2S, enhances NO-mediated relaxation up to 13-fold in isolated rat aorta (Hosoki et al., 1997). Treatment of Langendorff-perfused Sprague-Dawley rat hearts with NaHS immediately following ischemia confers cardioprotection through NOS activation (Yong et al., 2008). In a study on the pro-angiogenic effects of NaHS in cultured endothelial cells, Akt phosphorylation was induced after 30 min when the cells were exposed to 10–200 μM NaHS (Cai et al., 2007). However, this study measured NO metabolites (nitrite) instead of NO directly, and reported that there was no increase in NO metabolites with NaHS treatment (Cai et al., 2007). Therefore, it is not clear whether this phosphorylation resulted in an increase in NO bioavailability. In contrast, other in vitro studies indicate that incubation with NaHS or H2S gas-bubbled buffer decreases eNOS activity in aortic rings (Geng et al., 2007; Kubo et al., 2007b), cell culture (Geng et al., 2007), and recombinant eNOS (Kubo et al., 2007a), as well as the NO metabolites nitrite and nitrate (Geng et al., 2007). However, in these studies NaHS incubation occurred 1–6 h before measurement of eNOS activity or NO metabolites. Since H2S is volatile and oxidizes rapidly in the presence of oxygen and free divalent metals (Tapley et al., 1999), key signaling events mediated by H2S may have occurred before the activity measurement was performed. There also exists direct cross-talk between NO and H2S, and much work has been done investigating their interaction (Whiteman and Moore, 2009). There is speculation that an inert, nitrosothiol-like intermediate forms from the reaction of the two gases, which may serve as a biological sink or storage source of NO (Whiteman et al., 2006), and there is also evidence that the interaction of the two gases may lead to formation of nitroxyl (HNO), at least in the heart (Yong et al., 2010).
In the present study we investigated the ability of H2S, administered as the chemical source Na2S, to acutely modulate NO bioavailability in a cultured endothelial cell system and direct measurement of NO, with a specific focus on the potential mechanism of action through Akt.
Endothelial cell growth supplement was purchased from Upstate (Temecula, CA, USA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and were of the highest quality available, unless otherwise noted.
Bovine arterial endothelial cells (BAECs) were cultured in DMEM (1.0 g L glucose) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and endothelial cell growth supplement (5 mg L). Culture flasks were maintained in a 37°C incubator at 5.0% CO2. Adherent endothelial cells were grown in six-well plates for EPR measurements and in 100 mm dishes for protein expression measurements.
Sodium sulfide (Na2S), an H2S donor, was made into a saturated stock solution in distilled water and maintained at 4°C. At this temperature, the concentration of a saturated solution of Na2S is 1.72 M. From this stock, Na2S dilutions were made in Krebs buffer, of which 1.0 mL was added per well of a six-well plate, and 3.0 mL was added per 100 mm Petri dish. In aqueous solution, hydrogen sulfide exists in equilibrium as H2S, HS−, and S2−. Therefore, the term “H2S” used throughout this manuscript refers these chemical species in aqueous solution from the addition of Na2S.
The Akt inhibitor Triciribine was used to prevent the phosphorylation of eNOS (Dieterle et al., 2009). Triciribine (5.0 μmol L) was added in Krebs buffer 30 min before experiments. Cells were washed with phosphate-buffered saline (PBS) before and after addition of Triciribine.
Spin-trapping measurements of NO were performed using a Bruker E-scan spectrometer (BrukerBioSpin Corporation, Billerica, MA, USA) with the iron spin trapping complex N-methyl-D-glucamine dithiocarbamate (Fe–MGD) (Cardounel and Zweier, 2002; Cardounel et al., 2007). For measurements of NO produced by BAECs, cells were cultured as described above and spin trapping was performed on cells grown in six-well plates (1 × 106 cells well). In these studies, cells attached to the substratum were utilized since scraping or enzymatic removal leads to injury and membrane damage with impaired NO generation. The medium from each well was removed and the cells were washed with PBS (without CaCl2 or MgCl2). Cells in six-well plates were treated individually so that all six wells received the same treatment (i.e., addition of Krebs buffer or Na2S in Krebs buffer) for the various experiments. Next, 0.15 ml of Krebs buffer containing the NO spin trap FE–MGD (0.5 mmol L Fe2+, 5.0 mmol L MGD), and calcium ionophore (A23187, 1 μmol L) was added to each well and the plates were incubated at 37°C under a humidified environment containing 5% CO2/95% O2 for 20 min (Cardounel and Zweier, 2002; Cardounel et al., 2007). Following incubation, the medium from two wells was removed and pooled as one 0.3 ml sample, frozen in liquid nitrogen and stored at −80°C. This yielded three samples per plate. The frozen NO spin-trap samples are stable, and were later individually thawed, after which trapped NO in the supernatants was quantified using EPR. Spectra were obtained using the following parameters: 20 mW microwave power, 3.16 G modulation amplitude, and 100 kHz modulation frequency.
Six 100 mm dishes were set up with two control treatments (no Na2S addition), and the cells in four plates were exposed to 150 μM Na2S. Two of these plates were sampled 15 min later, and two were sampled 30 min later. To sample each plate, BAECs from the 100 mm dishes were scraped and suspended in 300 μl radioimmunoprecipitation assay (RIPA) buffer with Halt protease inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL, USA), placed on ice, and sonicated to lyse the cells and suspend the protein. The suspension was centrifuged at 12000 × g for 20 min at 4°C and the supernatant removed, frozen in liquid nitrogen, and stored at −80°C. Western blotting was performed using commercially available polyclonal antibodies for eNOS and Ser 1177 eNOS (BD Biosciences, San Jose, CA, USA), monoclonal β-actin (Cell Signaling Technology, Danvers, MA, USA), and secondary antibody conjugated to alkaline phosphatase (Sigma-Aldrich St. Louis, MO, USA). Protein was separated using SDS-PAGE and transferred onto PVDF membrane (Immobilon P, Millipore, Billerica, MA, USA). Using the Snap-ID system (Millipore, Billerica, MA, USA) membranes were blocked in 0.005% non-fat milk in phosphate-buffered saline with 0.05% Tween-20 (PBST). Primary antibodies were diluted 1:333 in PBST and secondary antibody diluted 1:3,333 in blocking solution. Chemiluminescent substrate (DuoLux, Vector Laboratories, Burlingame, CA, USA) was used to generate a chemiluminescent signal, captured with a digital imager (GeneSnap, Syngene, Frederick, MD, USA). Protein from each 100 mm dish of cells (N = 2 dishes per treatment) was run on three separate blots, for a total of six independent measurements per treatment group. Blot images were analyzed using commercial software (Quantity One, BioRad, Hercules, CA, USA).
All data were analyzed using one-way ANOVA with Dunnett’s post hoc test for significant differences from a control, with alpha ≤0.05 considered significant (Prism 5.0, Graph Pad Software, La Jolla, CA, USA).
Initial experiments were conducted to establish the time course of H2S effects on endothelial NO production. BAECs were exposed to a chemical source of H2S, Na2S (100 μM) for 5, 30, 60, 90, 120 and 240 min (Figure 1A). At each time point, endothelial-derived NO production was measured using EPR. Results demonstrated an 87% increase in mean NO production at 30 min post H2S treatment (ANOVA, p < 0.001). This effect was not observed at later time points, suggesting a transient activation of eNOS. Post hoc statistical testing also revealed that levels of NO were significantly lower than control at the 90 and 240 min time points, albeit a small difference in magnitude compared to the significant increase observed at 30 min. Subsequent experiments, in which NO production was measured 30 min after the addition of 5–600 μM Na2S, were performed to establish the dose–response for the H2S effects (Figure 1B). Results demonstrated a 39–62% increase in mean NO production at Na2S concentrations between 40–150 μM (ANOVA, p < 0.001).
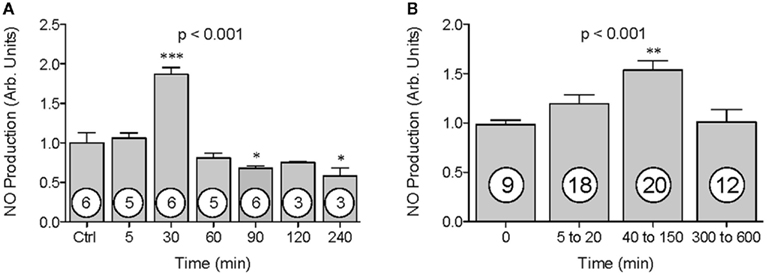
Figure 1. The effect of H2S on NO production by BAECs. NO production was measured using EPR spectroscopy. NO values (y-axis) are presented in arbitrary units and have been normalized to control. Time (A) or Na2S treatment concentration range (B) are shown on the x-axis. Data are shown as mean ± SE. Statistical analysis was performed using a one-way ANOVA (ANOVA p-value is in center of figure) with Dunnett’s post hoc test. Numbers inside circles denote the N for each column. An asterisk denotes values significantly different from control by post hoc test. *p < 0.05, **p < 0.01, ***p < 0.001.
The transient nature of the H2S effects on endothelial NO production suggested a change in eNOS phosphorylation status. Therefore, western blotting was used to determine the phosphorylation state of eNOS after addition of H2S at 15 and 30 min. Total eNOS expression was unchanged for all treatments (Figure 2A, ANOVA, p = 0.831), but after 30 min of incubation in the presence of 150 μM Na2S, eNOS phosphorylation at Ser 1177 increased by 100% compared to control (Figure 2B, ANOVA p < 0.001). Furthermore, the ratio of phosphorylated Ser 1177 eNOS to total eNOS increased by 139% after 30 min compared to control (Figure 2C–D, ANOVA p = 0.0033). To determine whether increased Ser-1177 phosphorylation was responsible for the augmented NO production, BAECs were pretreated for 30 min with the Akt inhibitor Triciribine (5 μM), after which the cells were exposed to 150 μM Na2S. Triciribine prevented both the increase in eNOS ser1177 phosphorylation as well as the augmentation of endothelial NO production observed with H2S treatment (Figure 3A,B, ANOVA p < 0.01). These results clearly indicate that H2S released from Na2S increases endothelial NO production through Akt activation and subsequent increased phosphorylation of eNOS at Ser 1177.
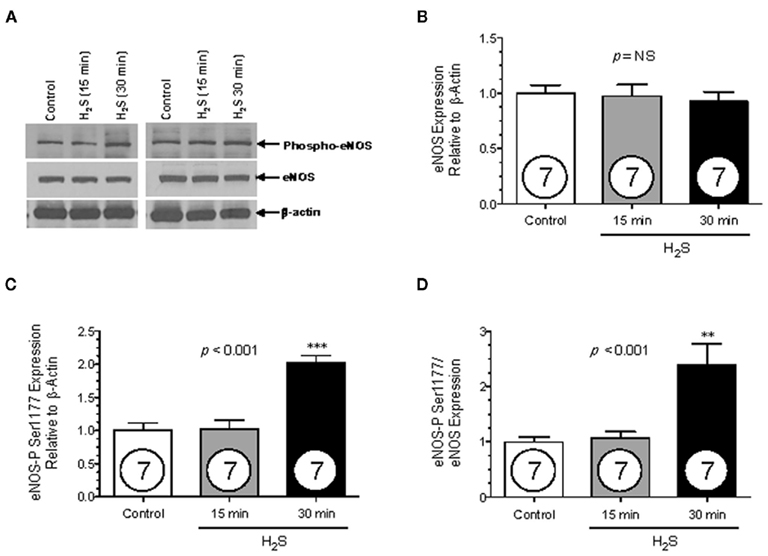
Figure 2. The effect of H2S on eNOS phosphorylation. Protein expression (y-axis, normalized to β-actin) was measured from BAECs untreated (Ctrl), or exposed to 15 or 30 min of 150 μM Na2S (x-axis). Densiometric analysis values have been normalized to control. (A) Representative Western blots. (B) Total eNOS expression (C) eNOS phosphorylated at Ser 1177 expression. (D) The ratio eNOS phosphorylated at Ser 1177 expression over total eNOS expression. Statistical analysis was performed using a one-way ANOVA (ANOVA p-value is in center of figure) with Dunnett’s post hoc test. Numbers inside circles denote the N for each column. An asterisk denotes values significantly different from control by post hoc test. **p < 0.01, ***p < 0.001.
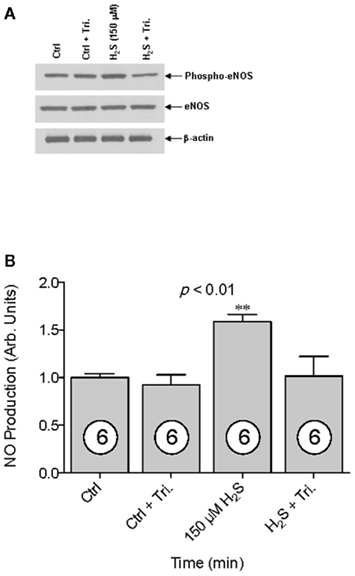
Figure 3. The effect of Akt inhibition on H2S-stimulated NO production. (A) Western blot analysis demonstrating total, ser1177-eNOS (phospho-eNOS), and actin in the presence and absence of the AKT inhibitor Triciribine (5 μM). (B) NO production was measured using EPR spectroscopy. NO values (y-axis) are presented in arbitrary units and have been normalized to control. NO was measured from untreated (Ctrl), Triciribine treated (Ctrl + Tri.), 150 μM Na2S treated (150 μM H2S), and 150 μM Na2S treated with Triciribine (H2S + Tri). Statistical analysis was performed using a one-way ANOVA (ANOVA p-value is in center of figure) with Dunnett’s post hoc test. Numbers inside circles denote the N for each column. An asterisk denotes values significantly different from control by post hoc test, **p < 0.01.
Although an early study showed a synergistic effect of H2S on NO-induced relaxation of blood vessel rings (Hosoki et al., 1997), later studies showed that H2S inhibited eNOS activity in aortic rings and cell culture, as well as in recombinant proteins (Geng et al., 2007; Kubo et al., 2007a,b). However, these later studies measured eNOS activity 1–6 h after H2S (NaHS) addition and did not directly measure the NO produced after addition of H2S, as we have in this study. Since H2S is volatile and rapidly oxidizes in the presence of oxygen and free divalent metals (Tapley et al., 1999), we hypothesized that H2S acts within minutes of its application, not hours, and therefore that an hour or longer delay between H2S application and measurement of eNOS activity or, in our case, NO production could fail to detect an effect. Here we demonstrate that H2S from Na2S increases NO production from endothelial cells within 30 min.
The comparatively rapid action of Na2S addition lead us to suspect that H2S was stimulating phosphorylation of eNOS at Ser 1177. We investigated by measuring total eNOS and phosphorylated eNOS protein expression. While total eNOS remained constant for all treatment groups, there was a significant increase in both phosphorylated eNOS and the ratio of phosphorylated eNOS to total eNOS after 30 min of 150 μM Na2S exposure. To confirm that H2S-induced NO production was dependent on eNOS phosphorylation, pharmacological inhibition of Akt was used to prevent phosphorylation of eNOS at Ser 1177. Inhibition of Akt prevented the increase in NO production in cells exposed to Na2S, but did not significantly affect NO production in control cells. Nonetheless, it should be noted that Akt alone is not the sole mechanism of phosphorylation at Ser 1177. AMPK, PKA, and CaMKII can also contribute to phosphorylation of Ser 1177 (Fleming, 2010). Their contribution to the increase in NO by H2S cannot be ruled out by this study, and therefore they remain targets of further research.
While we assume that H2S gas is causing this action, hydrogen sulfide exists as H2S, HS−, and S2− in solution. With the extracellular ratio of H2S/HS− being between 1:3 and 1:5 and the intracellular ratio being approximately equal (Olson and Donald, 2009), HS− may also be causing the up-regulation. To our knowledge, there is as yet no definitive demonstration that only H2S and not HS− is causing the observed effects of hydrogen sulfide.
The concentrations of Na2S used in this study may be considered supraphysiological, given the recent finding that the circulating concentration of hydrogen sulfide is probably in the nanomolar range and not in the 10 up to 300 μM range of previous reports, many of which have used methods of measurement that artificially inflated the amount of free H2S detected by the assay by releasing bound sulfur as well as detecting free H2S (Whitfield et al., 2008; Whiteman and Moore, 2009). However, the intracellular concentrations of H2S have yet to be quantified, and are currently not known.
The data in the present study suggest a novel mechanism of endogenous H2S signaling: up-regulation of NO production via Akt-dependent phosphorylation of eNOS at Ser1177, although the mechanism by which H2S activates Akt is unknown. While it remains to be tested in vivo, upstream regulation of NO production by H2S could represent a novel and potentially important regulatory mechanism in NO signaling, and could further implicate a dysfunction in endogenous H2S signaling in cardiovascular disease and other pathologies.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Arthur Pope, Scott Forbes, and Kanchana Karrupiah for their help with cell culture. This work was supported by a T32 HL083810 to Benjamin L. Predmore and NIH HL081734 to Arturo J. Cardounel.
Cai, W. J., Wang, M. J., Moore, P. K., Jin, H. M., Yao, T., and Zhu, Y. C. (2007). The novel proangiogenic effect of hydrogen sulfide is dependent on akt phosphorylation. Cardiovasc. Res. 76, 29–40.
Cardounel, A. J., Cui, H., Samouilov, A., Johnson, W., Kearns, P., Tsai, A. L., Berka, V., and Zweier, J. L. (2007). Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial no production and vascular function. J. Biol. Chem. 282, 879–887.
Cardounel, A. J., and Zweier, J. L. (2002). Endogenous methylarginines regulate neuronal nitric-oxide synthase and prevent excitotoxic injury. J. Biol. Chem. 277, 33995–34002.
Dieterle, A., Orth, R., Daubrawa, M., Grotemeier, A., Alers, S., Ullrich, S., Lammers, R., Wesselborg, S., and Stork, B. (2009). The akt inhibitor triciribine sensitizes prostate carcinoma cells to trail-induced apoptosis. Int. J. Cancer 125, 932–941.
Fleming, I. (2010). Molecular mechanisms underlying the activation of enos. Pflugers Arch. 459, 793–806.
Geng, B., Cui, Y., Zhao, J., Yu, F., Zhu, Y., Xu, G., Zhang, Z., Tang, C., and Du, J. (2007). Hydrogen sulfide downregulates the aortic l-arginine/nitric oxide pathway in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1608–1618.
Hosoki, R., Matsuki, N., and Kimura, H. (1997). The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 237, 527–531.
Kubo, S., Kurokawa, Y., Doe, I., Masuko, T., Sekiguchi, F., and Kawabata, A. (2007a). Hydrogen sulfide inhibits activity of three isoforms of recombinant nitric oxide synthase. Toxicology 241, 92–97.
Kubo, S., Doe, I., Kurokawa, Y., Nishikawa, H., and Kawabata, A. (2007b). Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide: contribution to dual modulation of vascular tension. Toxicology 232, 138–146.
Olson, K. R., and Donald, J. A. (2009). Nervous control of circulation – the role of gasotransmitters, no, co, and H(2)S. Acta Histochem. 111, 244–256.
Tapley, D. W., Beuttner, G. R., and Shick, J. M. (1999). Free radicals and chemiluminescence as products of the spontaneous oxidation of sulfide in seawater, and their biological implications. Biol. Bull. 196, 52–56.
Whiteman, M., Li, L., Kostetski, I., Chu, S. H., Siau, J. L., Bhatia, M., and Moore, P. K. (2006). Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem. Biophys. Res. Commun. 343, 303–310.
Whiteman, M., and Moore, P. K. (2009). Hydrogen sulfide and the vasculature: a novel vasculoprotective entity and regulator of nitric oxide bioavailability? J. Cell. Mol. Med. 13, 488–507.
Whitfield, N. L., Kreimier, E. L., Verdial, F. C., Skovgaard, N., and Olson, K. R. (2008). A reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, 1930–1937.
Yong, Q. C., Hu, L. F., Wang, S., Huang, D., and Bian, J. S. (2010). Hydrogen sulfide interacts with nitric oxide in the heart: possible involvement of nitroxyl. Cardiovasc. Res. 88, 482–491.
Keywords: hydrogen sulfide, nitric oxide, eNOS, Akt, endothelial cells
Citation: Predmore BL, Julian D and Cardounel AJ (2011) Hydrogen sulfide increases nitric oxide production from endothelial cells by an Akt-dependent mechanism. Front. Physio. 2:104. doi: 10.3389/fphys.2011.00104
Received: 20 January 2011; Paper pending published: 15 February 2011;
Accepted: 01 December 2011; Published online: 19 December 2011.
Edited by:
Heraldo DeSouza, Universidade de São Paulo, BrazilReviewed by:
Matt Whiteman, University of Exeter, UKCopyright: © 2011 Predmore, Julian and Cardounel. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Arturo J. Cardounel, Department of Anesthesiology, Ohio State University, 410 W. 10th Avenue, N411 Doan Hall, Columbus, OH 43210, USA. e-mail:Y2FyZG91bmVsLjFAdWZsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.