 Sushil K. Sarna1,2*
Sushil K. Sarna1,2*- 1 Enteric Neuromuscular Disorders and Visceral Pain Center, Division of Gastroenterology, Department of Internal Medicine, The University of Texas Medical Branch at Galveston, Galveston, TX, USA
- 2 Department of Neuroscience and Cell Biology, The University of Texas Medical Branch at Galveston, Galveston, TX, USA
The development of IBS symptoms – altered bowel function and abdominal cramping in a subset of adult subjects exposed to severe enteric infections opened up an unprecedented opportunity to understand the etiology of this poorly understood disorder. Perhaps, for the reasons that these symptoms follow a severe enteric infection, and mucosal biopsy tissues are readily available, the focus of most studies thus far has been to show that mild/low-grade mucosal inflammation persisting after the initial infection has subsided causes the IBS symptoms. Parallel studies in non-infectious IBS patients, who did not have prior enteritis, showed similar mild mucosal inflammation. Together, these studies examined the mucosal infiltration of specific immune cells, increase of select inflammatory mediators, mast cell and enterochromaffin cell hyperplasia, and epithelial permeability. In spite of the fact that the data on these topics were not consistent among different studies and clinical trials with prednisone, fluoxetine, and ketotifen failed to provide relief of IBS symptoms, the predominant conclusions were that mild mucosal inflammation is the cause of IBS symptoms. However, the circular smooth muscle cells, and myenteric neurons are the primary regulators of gut motility function, while primary afferent neurons and CNS play essential roles in induction of visceral hypersensitivity – no explanation was provided as to how mild mucosal inflammation causes dysfunction in cells far removed. Accumulating evidence shows that mild mucosal inflammation in IBS patients is in physiological range. It has little deleterious effects on cells within its own environment and therefore it is unlikely to affect cells in the muscularis externa. This review discusses the disconnect between the focus on mild/low-grade mucosal inflammation and the potential mechanisms and molecular dysfunctions in smooth muscle cells, myenteric neurons, and primary afferent neurons that may underlie IBS symptoms.
IBS is a complex disorder – its origins are unknown and the conventional diagnostic approaches are unable to identify an organic cause. The absence of an identifiable abnormality frustrates physicians – they cannot respond satisfactorily to provide relief of symptoms; it frustrates patients – they see no end to morbidity the disorder carries; it frustrates pharmaceutical companies – their attempts to develop therapeutic agents have repeatedly fallen short; it frustrates scientists – they are unable to pinpoint the cellular and system wide mechanisms of organ dysfunction. Is it possible that an open-minded look at post-infectious IBS (PI-IBS) will give a new sense of direction that would move us forward in managing a disorder that causes significant morbidity and carries huge socio-economic burden?
Chaudhary and Truelove (1962) first reported that about a quarter of IBS patients in their study had an episode of severe diarrhea in recent past. This unusual observation did not receive immediate attention it deserved. A publication, almost 32 years later, sparked wider interest in the development of IBS symptoms in about a quarter of subjects exposed to enteric infections in a community setting (McKendrick and Read, 1994). This study noted that the severity of acute illness during enteritis is greater in patients, who developed PI-IBS+ than in those who did not PI-IBS−. Other publications in the following years confirmed these findings (Gwee et al., 1996; Neal et al., 1997; Spiller et al., 2000; Spiller, 2003, 2007; Mearin et al., 2005). Later studies in larger cohorts of patients found that the probability of developing PI-IBS is small if diarrhea lasts less than a week – thereafter it increases proportionally with the duration of symptoms during enteric infection, specifically diarrhea (Neal et al., 1997; Wang et al., 2004). The PI-IBS patients present with the symptoms of altered bowel function – diarrhea (∼63%), constipation (∼24%), and alternating diarrhea/constipation (∼13%), intermittent abdominal cramping and urgency (Dunlop et al., 2003a).
The above findings present an unprecedented opportunity to understand the etiology of at least one type of IBS – PI-IBS – and possibly open up the window to understand it in all types of IBS. In this regard, McKendrick and Read (1994) speculated that several risk factors – “increase in epithelial permeability, elevation of neuropeptides, stimulation of primary afferent nerves by inflammatory mediators, axonal reflexes generated by degranulating mast cells, and contractions of vascular and intestinal smooth muscle” might contribute to the symptoms of PI-IBS. Taking cues from these speculations, several clinical studies have investigated these and other potential mechanisms that might contribute to the symptoms of motility dysfunction, intermittent abdominal cramping, and urgency in PI-IBS patients. The goal of this review is to discuss these findings in light of our current clinical and basic science understandings of regulation of gut colonic motility function, visceral hypersensitivity (VHS), immune responses, and epithelial barrier function. The focus is on understanding the roles of smooth muscle cells, enteric neurons, and primary afferent neurons in causing motility dysfunction and VHS to colorectal mechanical events – rapid and large amplitude compression by colonic contractions or distension.
Altered Motility Function and Persistent Mucosal Inflammation in PI-IBS
Motor Correlates of Diarrhea/Constipation and Urgency
Most publications treat motility dysfunction in IBS as a black box – this might limit our ability to understand the underlying mechanisms that cause it. This section briefly introduces the motor correlates of the symptoms of IBS – diarrhea, constipation, and urgency (Sarna, 2010). The enteric neurons and circular smooth muscle cells are the core regulators of motility function. The inputs from the central nervous system, spinal cord, inflammatory mediators, stress mediators, and hormones modulate the function of these two cell types to influence motility function/dysfunction. Therefore, one must look for the innate cellular mechanisms of motility dysfunction in these two cell types and which of the above factors modulate them.
The colon generates three distinct types of contractions – giant migrating contractions (GMCs), rhythmic phasic contractions (RPCs), and tone (Sarna, 2010). Each type of contraction has a distinct motor function. Two critical requirements for gut contractions to propel luminal contents are that they occlude the lumen partially or completely and propagate in the anal direction (Sarna, 2010). The extent of luminal occlusion relates to the amplitude of the contraction. The GMCs are large amplitude contractions that strongly occlude the limen and propagate over long distances in the colon – thus producing mass movements. The strong compression of a colon segment by a GMC also initiates descending inhibition that facilitates rapid propulsion by accommodating the large bolus being propelled (Sarna, 2007). When GMCs occur in the distal colon, the descending inhibition relaxes the internal anal sphincter to allow passage of feces during defecation. The mass movement by a GMC provides the propulsive force for rapid expulsion of feces (Bampton et al., 2000; Figure 1). The rapid and forceful propulsion against closed anal sphincters distends the intervening segment to induce the sensation of urgency. By contrast, the RPCs in the colon are much smaller in amplitude and they do not propagate or propagate over short distances – causing mainly mixing and turning over of luminal contents (Sarna, 2010). Increase in tone by itself does not cause propulsion or mixing and turning over of luminal contents – however, it potentiates these functions of RPCs. In health, the GMCs occur only a few times a day.
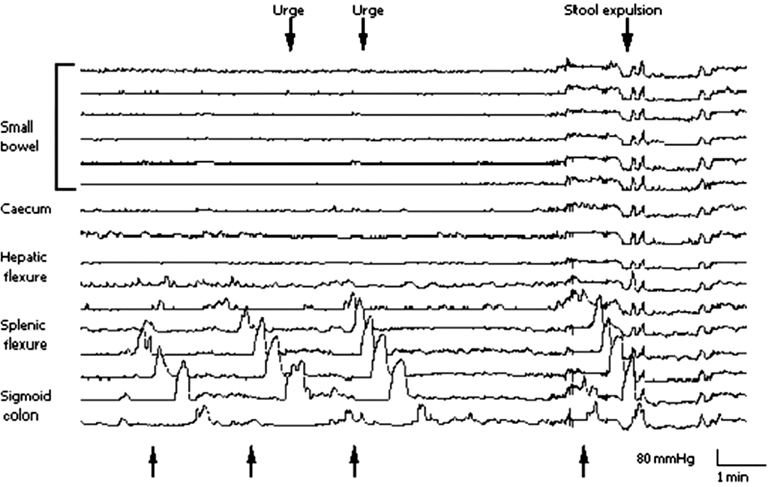
Figure 1. Spontaneous GMCs in the colon associated with urges to defecate and defecation. The first three GMCs (shown by upward arrows) started near the splenic flexure and terminated in the sigmoid colon. The second and third GMCs caused urges to defecate. The fourth GMC propagated to the end of the colon and caused defecation. (Reproduced with permission from Bampton et al., 2000.)
Accumulating evidence from studies of colonic motility in healthy subjects and in patients with diarrhea-predominant IBS (IBS-D), constipation-predominant IBS (IBS-C), and alternating constipation/diarrhea-predominant IBS (IBS-C/D) and in experimental models shows that diarrhea in IBS-D patients results from a significant increase in the frequency of GMCs and constipation occurs due to a decrease in their frequency (Bassotti et al., 1988, 2003; Chey et al., 2001; Rao et al., 2004). The RPCs play a minor role in these motor dysfunctions. Therefore, the mechanisms that regulate the stimulation and propagation of GMCs lie at the root of motor dysfunction in IBS. Note that the smooth muscle cells and myenteric neurons together regulate all three types of colonic contractions (Sarna, 2010).
Mucosal Inflammation in PI-IBS
A few studies noted an increase in the numbers of select types of immune cells and inflammatory mediators – T lymphocytes, macrophages, IL-1β, and IL-2 mRNA in the epithelial cells and lamina propria over those in healthy controls at various times after the initial enteric infection (Gwee et al., 1999, 2003; Spiller et al., 2000; Wang et al., 2004). Other studies noted similar increases in these immune cells and inflammatory mediators in the mucosa of IBS patients, who did not have a recent episode of enteritis (non-infectious/non-inflammatory IBS, NI-IBS. (Chadwick et al., 2002; Lee et al., 2008; Cremon et al., 2009). The suggestion was that this mucosal inflammation might account for motility dysfunction in IBS patients. All studies characterized the immune response in IBS patients as mild/low-grade. However, the unanswered questions are: (1) is this mild immune response sub-clinical or pathogenic? (2) Does it extend to the muscularis externa to cause smooth muscle and myenteric neuronal dysfunctions? The inflammatory mediators have to be in direct contact with their effector cells to impair their function. The biological effects of inflammation depend on its intensity.
Inflammatory bowel disease (IBD; comprised of ulcerative colitis and Crohn’s disease) and microscopic colitis (lymphocytic colitis and collagenous colitis) are two major inflammatory conditions in the colon. These and other inflammatory conditions show a very clear gradient in the intensity of immune response (Cremon et al., 2009; Figure 2). The inflammation in IBD is transmural and most severe of all when the infiltration/activation of immune cells and generation of oxidative/nitrosative stresses are considered (Pravda, 2005; Shi et al., 2011). The increase of these inflammatory mediators in the muscularis externa impairs smooth muscle and myenteric neuronal functions in vivo and in vitro by their presence in the microenvironments of these cells. The intensity of inflammation in IBD is severely pathogenic – it disrupts the mucosal layer and impairs the functions of almost all cell types in the colon wall. During remission, the intensity of the mucosal immune response decreases closer to that in microscopic colitis (Figure 2). Endoscopic examination shows apparently normal macroscopic appearance in patients with microscopic colitis. However, this level of inflammation compromises the epithelial transport function, resulting in secretory diarrhea in microscopic colitis (Protic et al., 2005). There is no evidence that the mildly pathogenic mucosal inflammation in microscopic colitis spreads to the muscularis externa to impair smooth muscle and enteric neuronal functions.
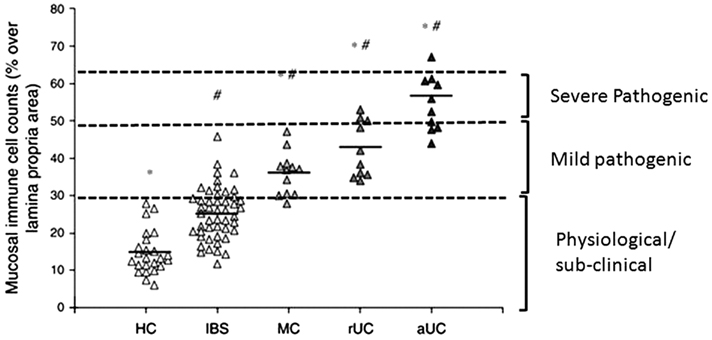
Figure 2. Relative counts of total immune cells in colonic lamina propria of healthy controls (HC), and patients with irritable bowel syndrome (IBS), microscopic colitis (MC), ulcerative colitis in remission (rUC), and active ulcerative colitis (aUC). The total cell counts increase almost linearly with the severity of inflammation and its pathogenic effects. Up to a certain level, the total cell counts are physiological/sub-clinical. Thereafter, the intensity of pathogenic effects increases with increases in total immune cell counts. (Reproduced with permission and modified from Cremon et al., 2009.)
The luminal pathogens continually challenge the epithelial barrier in healthy subjects. However, the mucosal immune system keeps small challenges from these pathogens in check by maintaining a low-level immune response. This level of inflammation is physiological – it does not affect integrity of the epithelial barrier, epithelial transport function, and it prevents the inflammatory response from spreading to the outer layers – submucosa and muscularis externa. The intensity of mucosal immune activation in IBS patients is significantly less than that in lymphocytic colitis patients, but the infiltrates in both types of patients are significantly greater than those in healthy controls subjects (Chadwick et al., 2002; Cremon et al., 2009; Figure 2). However, the intraepithelial lymphocytes (IELs) in IBS patients are well within the normal range of less than 20 per 100 epithelial cells in healthy subjects. There is no evidence that the intensity of immune response in IBS patients significantly impairs the epithelial transport function. Therefore, even though there is a statistical increase in the infiltration/activation of a few types of immune cells in IBS patients, the intensity of mucosal inflammation remains within the physiological/sub-clinical range (Figure 2). It is noteworthy that only about 50% of IBS patients show a significant infiltration of immunocytes – they are undetectable in the remaining 50% of the patients, even though they have the same symptoms as those with mild mucosal inflammation (Cremon et al., 2009).
Other findings support the gradient in mucosal inflammation in inflammatory diseases of the colon. Myeloperoxidase (MPO), a marker of neutrophil activation, measured by mucosal patch technique, increases significantly in active ulcerative colitis (300-fold), inactive colitis (fivefold), collagen colitis (fivefold), celiac disease (sixfold), and IBS (fourfold; Kristjansson et al., 2004). The corresponding fold-increases for eosinophil cationic protein (ECP), a marker of eosinophil activation are 11 in ulcerative colitis, 1 in inactive colitis, 2.5 in collagen colitis, 1 in celiac disease, and 1 in IBS – only the 11-fold increase in ulcerative colitis patients is significant. The eosinophil granulocyte is a potent proinflammatory cell that releases several highly toxic proteins, including ECP and major basic protein (MBP; Venge and Peterson, 1989). The fecal leukocyte markers in IBS patients are not different from those in healthy controls – they are modestly elevated in collagenous colitis and several-fold in IBD patients (Silberer et al., 2005; Lettesjo et al., 2006).
The PI-IBS or IBS patients do not have secretory diarrhea. They have motor diarrhea – frequent unformed stools (Read, 1986). Motor diarrhea results from frequent mass movements due to increase in the frequency of GMCs, which significantly decreases the duration of fecal contact with the mucosa. Of course, a sub-clinical impairment of epithelial function might contribute marginally to unformed stools. Smooth muscle cells and myenteric neurons are the final regulators of GMCs as well as the other two types of gut contractions – RPCs and tone. Only the inflammatory mediators, stress mediators, hormones, and neurotransmitters that directly make contacts with the smooth muscle cells/myenteric neurons can alter their function. There is no evidence that a sub-clinical mucosal immune activation in IBS patients spreads to the muscularis externa. One report of low-grade infiltration of lymphocytes in the jejunal myenteric plexi of a heterogeneous group of IBS patients with severe symptoms is hard to interpret or relate to the symptoms of IBS (Tornblom et al., 2002), which are primarily due to colonic motor dysfunction.
Several other lines of evidence speak against a role of mild mucosal inflammation in motility dysfunction in PI-IBS/NI-IBS patients.
1. Prednisone treatment in a reasonably large cohort of PI-IBS patients decreases the lamina propria T lymphocyte counts and probably other inflammatory cells without a concurrent improvement in the symptoms of abdominal pain, diarrhea, or urgency substantiates the hypothesis that the physiological/sub-clinical inflammation is not the underlying factor in IBS symptoms (Dunlop et al., 2003b).
2. The increases in T lymphocytes in lamina propria and IELs in PI-IBS and NI-IBS are not consistent among different studies – some reporting significant increases, while others reporting no change (Gwee et al., 1999; Spiller et al., 2000; Dunlop et al., 2003a; Lee et al., 2008). More importantly, one study found that the lamina propria T lymphocytes remained significantly elevated in patient controls (those who have enteritis, but do not develop PI-IBS) over healthy controls (Dunlop et al., 2003a). The surface IELs were not different among PI-IBS, patient controls, and healthy controls. The crypt IELs numbers were lower in PI-IBS and patient controls vs. healthy controls. The symptoms of IBS had subsided in patient controls at this time, which questions the role of elevated lymphocytes in causing them.
3. The intensities of infiltration of immunocytes in IBS-D and IBS-C patients do not differ (Cremon et al., 2009). The same stimulus could not explain the diametrically opposite effects on colonic motor function in IBS-D and IBS-C patients.
4. Rectal hypersensitivity, and colon transit are similar in PI-IBS+ and PI-IBS− patients following enteric infection, even though the PI-IBS+ have elevated mucosal inflammatory cell counts over those who did not develop IBS symptoms (PI-IBS−; Gwee et al., 1999).
Lessons Learnt
1. The inflammatory response is a continuum (Figure 1). A basal level of immune activation is necessary to protect the gut wall from the hostile environment in the gut lumen – physiological inflammation. The next is the sub-clinical level, where the inflammation increases significantly over the physiological level; however, it is not enough to impair the function of cells in its environment (PI-IBS and NI-IBS). The third level is pathogenic, which impairs the function of cells in its environment accompanied with mild structural damage (Microscopic colitis). The fourth is severe pathogenic, which impairs the function of cells in its environment accompanied with severe structural damage and/or change in phenotype (IBD).
2. The symptoms of IBS relate to disturbances in the frequency of GMCs. The monitoring of these contractions is central to identifying the causative factors of motility dysfunction in IBS.
3. The biopsy tissues are available from IBS patients and healthy subjects – however, the mild/inconsistent inflammatory response detected in mucosal biopsies is unable to predict smooth muscle/enteric neuronal dysfunction in the muscularis externa that directly regulate normal and abnormal frequency of GMCs and hence motility dysfunction in PI-IBS and NI-IBS patients.
4. Even though the symptoms of IBD and IBS patients are similar, they do not have the same etiologies, such as inflammation.
5. The similarity of the immunological infiltrate in PI-IBS and NI-IBS patients suggests that the prior enteric infection is unlikely to be the cause of mild mucosal inflammation in these patients.
Enterochromaffin (EC) Cell Hyperplasia and Motility Dysfunction in PI-IBS
The findings of EC cell hyperplasia in the mucosa of PI-IBS/NI-IBS patients are divergent – some found a significant, but modest, increase (Spiller et al., 2000; Dunlop et al., 2003a) – others did not (Coates et al., 2004; Park et al., 2006). The hyperplasia of a cell type does not necessarily mean that it will release more of its contents, which depends on complex cell signaling and microenvironment – this is one of the reasons that immunohistochemical staining alone is insufficient to establish a cause and effect relationship. EC cell hyperplasia, if it is present, does not translate into an increase in the release of 5-HT in the mucosa of IBS patients (Coates et al., 2004). Paradoxically, the mucosal serotonin decreases in IBS patients even though the plasma serotonin increases (Dunlop et al., 2005). The source of increase plasma serotonin is unknown and there is no evidence that it has a significant effect on colonic motility or VHS. The serotonin transporter (SERT) mRNA expression also does not change in IBS patients (Camilleri et al., 2007). Recent studies show that the mucosal release of 5-HT is not required to stimulate GMCs in response to distention in the guinea pig and mouse colons (Keating and Spencer, 2010; Spencer et al., 2011). Overall, the available evidence weighs heavily against a potential role of EC cell hyperplasia in causing motility dysfunction or VHS in PI-IBS or NI-IBS patients.
The release of serotonin from EC cells in response to villus stimulation by the ingested meal activates the intrinsic sensory neurons (Bulbring and Crema, 1959), which in turn stimulate the release of excitatory and inhibitory neurotransmitters from the myenteric motor neurons via interneurons (Grider and Jin, 1994; Gershon, 2005). Thereafter, the myenteric neuronal circuits and smooth muscle excitation–contraction coupling together determine whether the resulting contractions will be GMCs, RPCs, or TCs (Sarna, 2006, 2010). As noted earlier, each type of contraction has a different effect on motility function. It is too simplistic to assume that an increase or decrease in the availability of serotonin in the mucosa alters postprandial transit in intact organisms. This is one of the reasons that partial or full 5-HT4 agonists are marginally effective or effective only in a subset of IBS patients in improving the global symptoms of IBS (Brandt et al., 2002).
Additional evidence that speaks against a potential role of EC cell hyperplasia as a causative factor for motility dysfunction or VHS in IBS patients is:
1. A double blind, randomized, placebo-controlled clinical trial with serotonin reuptake inhibitor fluoxetine found no significant effect on rectal hypersensitivity and the symptoms of abdominal pain, bloating, flatulence, incomplete evacuation and urgency in a mixed group of IBS-C, IBS-D and IBS-C/D patients (Kuiken et al., 2003) – suggesting that none of their symptoms were due to a decrease in the availability of serotonin in the mucosa or elsewhere.
2. The mucosal 5-HT, tryptophan hydroxylase1, SERT are all significantly reduced in the mucosal biopsies of ulcerative colitis, IBS-D and IBS-C patients (Coates et al., 2004). A similar decrease in mucosal 5-HT could not explain faster colonic transit in ulcerative colitis and IBS-D patients and slower transit in IBS-C patients. Consequently, a small increase in EC cell numbers noted by some studies is unlikely to be the cause of IBS symptoms.
Lesson Learnt
1. There is little evidence that a decrease or increase in serotonin release/availability in the mucosa decreases or increases the frequency of GMCs. Note that the GMCs occur only a few times (2–5) per day, while serotonin release is stimulated constantly due to continuous presence of fecal material in the colon.
Mast Cell Hyperplasia and Visceral Hypersensitivity in PI-IBS/NI-IBS
Factors Contributing to Abdominal Pain
The sensation of pain in response to colorectal distension (CRD) by a balloon is a standard test to evaluate hypersensitivity of the afferent neurons or impaired processing of supraspinal signals that might contribute to the symptom of intermittent abdominal pain/cramping in IBS patients. The sensation in this test lasts for the duration of balloon distension, which shows that hypersensitivity, by itself, does not cause the sensation of intermittent cramping – a mechanical event, such as colon wall distension, is required. The patients feel similar cramping episodes in their routine lives without balloon distensions in their colons. Obviously, in routine life, the colon contractions, instead of balloon distension, distort afferent nerve endings to generate afferent signals.
As discussed earlier in this review, the colon generates three distinct types of contractions, RPCs, GMCs, and TCs. The GMCs have the largest amplitude and they can propagate uninterruptedly over long distances in the colon to produce mass movements. They also generate descending inhibition of ongoing RPCs and tone, especially of the anal internal anal sphincter to relax it in preparation for defecation (Bassotti et al., 1999). The intensity of afferent signals generated by mechanical stimuli in the gut is proportional to the amplitude of contraction or distension, which makes GMCs the prime candidates to generate nociceptive signals. Note that the RPCs are present somewhere in the colon all the time. However, their amplitudes are invariably sub nociceptive threshold – if that were not the case, we will feel abdominal pain all the time. The complimentary roles of VHS and GMCs in producing the symptom of abdominal cramping are discussed in depth elsewhere (Sarna, 2010). The following provides a brief summary as background for discussion of the role of mast cell hyperplasia/activation in generating the symptom of intermittent abdominal cramping in IBS patients.
1. In health, the maximum amplitude of colonic GMCs is sub nociceptive threshold. Therefore, their infrequent presence in the colon is not painful (Figure 3A).
2. The amplitudes of GMCs nearly double to above nociceptive threshold in the colons of IBS-D patients with moderate to severe diarrhea (Chey et al., 2001). In this condition, VHS is not required for the sensation of abdominal cramping, when a GMC occurs in the colon (Figure 3B). However, concurrent VHS would worsen the sensation. It is noteworthy that, on average, only about 50% of IBS patients demonstrate VHS, although more that 90% of them complain of abdominal cramping (Prior et al., 1990; Lembo et al., 1994; Kuiken et al., 2005; Posserud et al., 2007; Camilleri et al., 2008).
3. The second scenario is that a significant increase in VHS lowers the nociceptive threshold to a level such that the afferent signals generated by normal amplitude GMCs (about 110 mm Hg) generate nociceptive signals to cause the sensation of abdominal cramping (Figure 3C).
4. Of the three types of colonic contractions, only the GMCs contract colon segments strongly enough to produce descending inhibition of RPCs and relaxation of internal anal sphincter as the GMC approaches it (Sarna, 2010). The purpose of descending inhibition is to prevent the distal receiving segment from contracting, and at the same time allow it to relax to accommodate the large contents of mass movement without generating afferent signals. However, if the descending inhibition were impaired, the receiving segment would distend under tension to generate afferent signals of its own, which add up to those produced by the GMC itself (Figure 3D). Similarly, if the internal sphincter does not relax due to impaired descending inhibition, the approaching GMS will push fecal contents against a closed anal sphincter to distend the intervening segment and send afferent signals to add to those being generated by the GMC. The combined afferent signals might exceed the nociceptive threshold to induce abdominal cramping (Figure 3D). In this case, also, VHS is not required, but, if present, it would worsen the sensation.
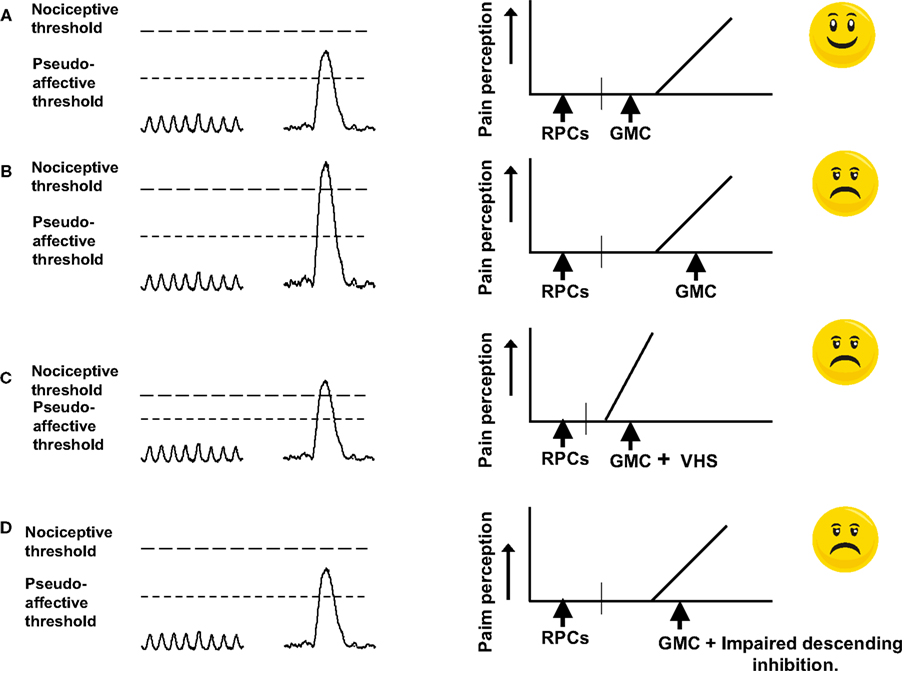
Figure 3. Cartoon showing the conditions under which GMCs generate afferent signals that exceed the nociceptive threshold. (A) In normal state, the afferent signal generated by a GMC is above the pseudoaffective threshold, but below the nociceptive threshold. The descending inhibition prevents the receiving segment from generating tone and hence afferent signals. No sensation of pain develops. (B) Due to enteric neuromuscular dysfunction, the amplitude of GMCs increases so that the afferent signals generated by them exceed the nociceptive threshold and painful sensation results. (C) Visceral hypersensitivity of the afferent neurons or impaired processing of signals in the CNS effectively lowers the nociceptive threshold so that the afferent signal generated by a GMC of normal amplitude exceeds the threshold for perception of pain. (D) Due to impairment of descending inhibition, the receiving segment generates tone and sends afferent signals. These signals add to those generated by the GMC. The combined signal exceeds the nociceptive threshold to induce the sensation of pain (Reproduced with permission from Sarna, 2010).
Mast Cell Hyperplasia in PI-IBS
Some studies found mast cell hyperplasia and an increase in the release of their products (histamine, tryptase) in the mucosal biopsies from the rectum of PI-IBS and NI-IBS patients (O’Sullivan et al., 2000; Park et al., 2003; Barbara et al., 2004, 2007; Cenac et al., 2007; Cremon et al., 2009). Others found a positive correlation between the mast cell numbers in close proximity (less than 5 μm) to nerve endings and IBS symptoms. Based on these findings, they proposed that the products of the mast cells in close proximity to enteric nerve endings induce VHS (Barbara et al., 2004, 2007). There are several limitations of this hypothesis. (1) These studies did not identify that the mucosal nerves closely associated with mast cells were indeed extrinsic afferent nerves. Instead, they prepared supernatants from the mucosal biopsies and added them to the colons of mice to show that they activate afferent neurons. However, they did not demonstrate that the concentrations of mast cell products in the supernatants were similar to those in the intact mucosa. The method of preparation of supernatants could easily concentrate the mast cell products enough to show a pharmacological effect in the mouse colon. The mast cell products are stable in supernatants – they degrade rapidly in situ in the intact mucosa. In addition, the spontaneous release of mast cell mediators is an inappropriate biomarker for the role of mast cells in evaluating their role in disease (Klooker et al., 2010). (2) Ulcerative colitis patients show significant mast cell hyperplasia (Middel et al., 2001) and yet most studies show that these patients do not develop VHS (Chang et al., 2000). (3) Other studies found no significant increase in rectosigmoid mast cells or their products in the mucosal biopsies of PI-IBS and/or NI-IBS patients (Spiller et al., 2000; Chadwick et al., 2002; Dunlop et al., 2003c; Wang et al., 2004; Cenac et al., 2007; Klooker et al., 2010). Mast cell hyperplasia occurs in diarrhea-predominant PI-IBS patients, but not in constipation-predominant or alternating diarrhea/constipation patients – yet patients in all three types of IBS feel abdominal cramping (Lee et al., 2008). (4) Statistical correlation between two biological variables does not establish a cause and effect relationship between them. A third independent variable might regulate teach of them independently. Morphological and immunohistochemical observations, by themselves, are insufficient to establish cause and effect relationships. Close association does not automatically prove causation.
Several additional lines of evidence speak against a role of mast cell hyperplasia close to mucosal nerve endings as the underlying cause of the symptom of abdominal cramping in IBS patients.
1. Clinical trials with ketotifen, a mast cell stabilizer (Klooker et al., 2010), and mesalazine (Corinaldesi et al., 2009) showed no effect on abdominal pain, bloating, or bowel habits. Mesalazine reduced mucosal immune cells, including mast cells. Ketotifen increased the threshold for discomfort and improved IBS symptoms without a change in mucosal mast cell numbers or release of their products, histamine, and tryptase, discounting a role of immune cells, including mast cells in generating the symptoms of IBS. This study found that the mucosal mast cell numbers and spontaneous release of tryptase were lower in IBS patients than in healthy volunteers.
2. A well-designed and rigorously executed study found that patients with low-grade chronic inflammation in inactive/mild ulcerative colitis had a higher threshold to recto-sigmoidal distension than healthy controls (Chang et al., 2000). By contrast, IBS patients with similar chronic inflammatory conditions in the mucosa have lower sensory thresholds to recto-sigmoidal distension. The IBS, ulcerative colitis patients, and healthy controls did not differ in psychological symptom scores and rectal compliance. Chronic mild inflammation of the esophagus and stomach also do not associate with visceral mechanical hyperalgesia (Fass et al., 1998; Mertz et al., 1998).
3. Accumulating evidence from human (Lembo et al., 1997) and animal (Lynn and Blackshaw, 1999; Brierley et al., 2004) studies show that the splanchnic nerves with mechanoreceptors in the muscularis externa/serosa mediate afferent signaling in response to rapid balloon distensions – GMCs cause rapid gut wall compressions. In vitro, the mucosal afferents do not respond to circumferential stretch – but, they respond to mild mucosal stoking by von Frey hairs that simulates the flow of digesta (Lynn and Blackshaw, 1999; Brierley et al., 2004). In vivo, the mucosal afferents respond to slow distensions that mimic slow lumen filling, but not to rapid balloon distension (Lembo et al., 1997). The desensitization of mucosal mechanoreceptors by lidocane does not block pain perception to rapid balloon distension in IBS patients – it blocks the sensation to slow distension (Lembo et al., 1994). The inflammation in the lamina propria/epithelium is not in the microenvironment of the mechanoreceptors of splanchnic afferents in the muscularis externa/serosa to modulate their function.
Taken together, the above findings show that chronic mild inflammation of the mucosa in IBS patients does not relate to VHS. This review does not discuss the roles of mast cells in experimental models of IBD or IBS.
Lessons Learnt
1. Visceral hypersensitivity and generation of GMCs are both critical players in generating the symptom of intermittent abdominal cramping in IBS patients. VHS, by itself, does not produce this sensation – a mechanical event, such as, a strong colonic contraction or colon wall distension or both are essential. The sensation of cramping lasts as long as a GMC is present anywhere in the colon (∼1–2 min).
2. Spatial association, by itself, does not establish cause and effect relationship.
Epithelial Permeability and Post-Infectious IBS
The interphase between the gut and the lumen is a sophisticated barrier that keeps the luminal microbes, incompletely digested proteins, and pathogens at bay, while allowing the nutrients to pass through. However, this barrier is vulnerable to adverse stimuli from the lumen as well as from biological/pathological events within the gut wall. An increase in luminal pathogenic load impairs the barrier. The subsequent access of the microbes or their products to the sterile structures in the gut wall initiates an immune response, whose intensity, and profile depend upon the volume and phenotypes of pathogens. To make things worse, the inflammatory mediators might further erode the barrier (Chavez et al., 1999), setting up a vicious cycle until the anti-inflammatory mechanisms achieve a dynamic equilibrium. Biological events, such as psychological stress, operate from within the gut wall. These events activate resident immunocytes, such as mast cells, whose products impair the barrier to cause influx of luminal pathogens, which might enhance barrier dysfunction until a dynamic equilibrium is achieved. Fortunately, the adverse effects of mucosal inflammatory response are sub-clinical within a wide range of this dynamic equilibrium.
In context of the above, diarrhea-predominant IBS patients show increase in colonic permeability (Spiller et al., 2000; Dunlop et al., 2006) – constipation-predominant patients do not (Dunlop et al., 2006). Other studies found no differences in small intestinal permeability between patients, who developed PI-IBS and those who did not, 2 years following acute gastroenteritis (Marshall et al., 2004) and in NI-IBS patients vs. healthy subjects (Dainese et al., 1999; Tibble et al., 2002). A subset of diarrhea-predominant PI-IBS patients showed a small, but statistically significant, increase in small intestinal permeability above the conventional cut off limit of 0.03 in lactulose/mannitol ratio (Spiller et al., 2000). In another group of diarrhea-predominant IBS patients, the majority (61%) had small intestinal permeability within the normal range – yet they presented with symptoms similar to those with permeability above the normal range (Zhou et al., 2009). The patients with increase in small intestinal permeability had higher functional bowel disorder severity index. It is likely that chronic stress caused increase in small intestinal permeability, rather that small intestinal permeability causing VHS to CRD. Note that the small intestinal permeability in collagenous colitis patients with secretory diarrhea also does not differ from that in controls (Wildt et al., 2006). This is hardly surprising because collagen colitis should correlate with changes in colonic permeability, rather than to changes in small intestinal permeability. Small intestinal or colonic permeability does not correlate with colonic transit or bowel frequency/week (Dunlop et al., 2006). It is hard to assign a significance to increase in small intestinal permeability to the motility dysfunction or abdominal cramping in IBS patients, which relate mostly to the abnormalities in the colon. Overall, small intestinal permeability is an unlikely biomarker of IBS.
Intestinal inflammation and a systemic response to antigens can develop in the absence of disruption to intestinal permeability (Sydora et al., 2006a,b).
Lesson Learnt
1. Measurements of small intestinal permeability cannot predict similar changes in the colon and they do not relate to the primary symptoms of IBS – diarrhea/constipation and VHS.
2. The GMCs are ultra-strong contractions and their frequency increases in IBS-D patients. Frequent strong compressions of the colon wall by GMCs could damage the epithelium to increase its permeability. The frequency of GMCs decreases in IBS-C patients – they show no change in colonic permeability. Note that the concurrent increase in the frequency of GMCs and mucosal friability in ulcerative colitis patients results in bleeding and mucus discharge (see Sarna, 2010 for details). Bleeding or mucus discharge does not occur in IBS-D patients because of intact mucosal structure.
Genetics, Epigenetics, and Post-Infectious IBS
Genetic and Epigenetic Basis of Disease
Organs develop from a single zygote under the guidance of genomic and epigenetic information inherited from both parents. The genome has the code (DNA sequences) for the correct synthesis of all proteins required by an organism – the epigenome has the means to set the transcription rates of genes encoding these proteins. The expression of a unique group of proteins at the correct levels imparts phenotype to each cell type in an organ. Epigenetic mechanisms regulate gene expression without a change in DNA sequence. DNA replication mechanisms during meiotic and mitotic cell divisions maintain its integrity except under extreme environmental conditions, such as toxins and ionizing radiation. By contrast, the epigenetic mechanisms are highly sensitive to cellular environment throughout life, especially during fetal and neonatal stages of development.
A mutation in gene sequence, inherited from the parents, causes a Mendelian or simple disease – e.g., sickle cell anemia, hemophilia, and cystic fibrosis, if it is at a locus in the coding region critical for the synthesis of correct amino acid sequence of the protein, or it is in 5′ flanking promoter region that is essential to initiate transcription. These diseases have early onset in life and worsen progressively (Childs, 1995). By contrast, complex diseases arise from a combination of changes in one or more cell phenotypes that accumulate over a period. The search for gene mutations underlying complex diseases –diabetes, most cancers, and asthma has largely been unsuccessful. Complex diseases exhibit an inheritable component, but do not follow Mendel’s laws. For example, discordance of monozygotic twins reaches 30–50% in diabetes, 70% in multiple sclerosis and rheumatoid arthritis, and 80% in breast cancer (King et al., 1992). Differential exposures to environments usually explain these discordances. Complex diseases tend to appear later in life, show gender differences, parent-of-origin effects, remissions, and relapses and more than one peak (Howard et al., 2000). Functional Bowel Disorders fall into the category of complex diseases. These disorders are likely to relate to epigenetic dysregulation.
Polymorphisms and PI-IBS/NI-IBS
The DNA sequence of approximately 3.2 billion bases is roughly 99.9% similar between any two individuals – the remaining 0.1% (approx. 7 million bases) makes individuals unique. These variations in sequences among individuals, called mutations, polymorphism, insertions, and deletions are inheritable. Variations may be in one base (single nucleotide polymorphism, SNP) or in a sequence of bases of variable length. A variation in one base in less than 1% (arbitrary number) of the population is a mutation – greater than this number is polymorphism. A vast majority of these variations have no effects on the attributes of humans because only about 3% of the DNA sequence contains the coding and regulatory (promoters) sequences – the rest of the DNA sequence is mostly non-coding and non-regulatory – however, it might contain some enhancer and repressor elements that can influence transcription of distant genes. In essence, these variations have the potential to affect the attributes of individuals or make them susceptible to diseases, if they are in the coding or the regulatory region. In the coding region, they would have a detrimental effect if the altered amino acid changes the protein coded by the gene. In the regulatory region, these variations could reset the rate of transcription of the gene, if they are within the recognition sequences (or close to it) of the transcription factors that regulate the constitutive or evoked expression of the affected gene. Even though most polymorphisms are harmless, they might alter the response of therapeutic agents or environmental toxins because they might activate transcription factors not generally utilized in health.
A recent thorough and rigorous study identified SNPs in the coding and regulatory regions of genes regulating IL-6, CDH1, and Toll-like receptor-9 (TLR-9) in PI-IBS patients (Villani et al., 2010). The investigators chose these candidate genes on the assumption that a mild immune cell infiltration and increase in epithelial permeability are the causes of altered motility and visceral sensory functions in IBS patients. However, as per discussions in previous sections, there is little evidence to support this assumption. In addition, the frequencies of alleles in PI-IBS patients were of the same order of magnitude as in controls, which beg the question why did they not cause symptoms in controls. It is noteworthy, that a change in phenotype due to polymorphism is permanent – the IBS symptoms are relapsing and recurring. Polymorphism studies in NI-IBS patients have focused on genes encoding the SERT, TNFα, and IL-10 – all of which lack compelling evidence to show that they play a role in motility dysfunction in IBS patients (see earlier sections). These studies yielded conflicting or negative findings regarding polymorphisms in these genes (Pata et al., 2002; Kim et al., 2004; Lee et al., 2004; Yeo et al., 2004; van der Veek et al., 2005) – meta analysis found no correlation between polymorphism in SERT gene and IBS symptoms (Van Kerkhoven et al., 2007). Taken together, there is little compelling evidence for a genetic background for IBS. Several reviews have discussed additional serious limitations of polymorphism data in explaining IBS (Park and Camilleri, 2005; Saito and Talley, 2008; Vermeire et al., 2010).
The studies on polymorphisms are descriptive and inconclusive unless followed up by further studies to establish that the SNP affects the binding of one or more transcription factors to the regulatory region to affect the transcription rate of the gene, or to show that the SNP in the coding region affects the synthesis of the protein encoded by the gene. A more effective approach is to identify first a protein that alters smooth muscle, myenteric neuronal, or afferent neuronal function and then investigate whether its gene has a mutation in its coding or regulatory region that explains altered protein expression in cohorts of IBS patients. Note that aberrant protein synthesis is not a feature of complex diseases –biological effects of polymorphisms do not fade away and recur. While it is hard to envisage a role of polymorphisms in a complex disease, such as IBS, polymorphisms might explain varying intensities of enteritis in subjects exposed to the same infectious agent. Polymorphisms might also explain differential sensitivities of IBS patients to therapeutic agents (Camilleri et al., 2002).
Epigenetic Dysregulation and PI-IBS/NI-IBS
Early life psychological trauma/stress is a known risk factor for the development of IBS in adulthood (Chitkara et al., 2008). Gut inflammation is another major early life stressful condition (Lowman et al., 1987; Chitkara et al., 2008). The annual episodes of diarrhea in U.S. children less than 5-year-old ranges from 20 to 35 million (Pont et al., 2009). These diarrheal episodes lead to about 22,000 hospitalizations per year, indicating the severity of enteritis. The epigenetic programming of genes begins at the start of fetal development and continues in early life. During these periods, epigenetic mechanisms program the genes for normal health in adulthood. However, in the face of stressful conditions, they reprogram the genes for immediate survival. Unfortunately, the reprogramming of genes might persist in adulthood, resulting in aberrant rates of gene expressions to cause organ dysfunction.
Four mechanisms constitute epigenetic regulation of gene transcription – transcription factors, DNA methylation, histone codes (acetylation, methylation, phosphorylation, ubiquitination, and sumoylation of lysine residues on the N-terminal tails of histone proteins), and micro RNAs (Bhaumik et al., 2007). The post-translational modification of histone proteins alters the interaction between DNA and chromatin, which regulates the access of transcription factors to their recognition sequences to initiate gene transcription. Epigenetic programming/reprogramming is gene, stimulus and cell-type specific. Unlike mutations/polymorphisms, the epigenetic programming is dynamic – it responds to environmental stimuli throughout life, especially during vulnerable stages of fetal and neonatal developments.
Animal Models of IBS
The symptoms of IBS arise from dysfunctions in smooth muscle cells, myenteric neurons, afferent neurons, and possibly the central nervous system. The access to these tissues for scientific investigations in humans is severely limited. Biopsy tissues are available, but experiences with PI/IBS and NI-IBS patients show that they might have little use in predicting dysfunctions of the above cell types associated directly with the symptoms of IBS. The alternate approach is to develop animal models of sub-types of IBS with the full understanding that animal models do not mimic full human diseases. However, these models can provide valuable information on specific aspects of human disease. Testing of these findings in human patients for diagnostic and therapeutic purposes will advance the field.
Numerous animal models support the hypothesis that neonatal inflammatory or psychological insults result in two cardinal IBS-like dysfunctions in adulthood – altered motility function and VHS. A neonatal inflammatory insult (trinitrobenzene sulfonic acid, 130 mg/kg) enhances smooth muscle reactivity to ACh, accelerates colon transit, increases fecal pellet output per 24-h and increases fecal water content in adulthood (Choudhury et al., 2009). These effects result from alterations in the expressions of key genes that regulate smooth muscle excitation–contraction coupling and sensitivity of primary afferent neurons to cause IBS-like symptoms (Choudhury et al., 2009). A similar inflammatory insult in adult rats does not cause diarrhea-like conditions, 6–8 weeks later. Inflammatory insult with mustard oil, colon irritation, or maternal deprivation in neonates results in VHS to CRD in adulthood (D:Al-Chaer:2000]; Coutinho et al., 2002; Barreau et al., 2004).
It is noteworthy that the inflammatory/psychological stress in neonates has to be intense and/or chronic to cause dysfunctions in adulthood (D:Al-Chaer:2000]; Coutinho et al., 2002; Barreau et al., 2004; Choudhury et al., 2009). For example, a smaller dose of TNBS or duration of maternal deprivation in neonates does not cause colonic motor or sensory dysfunction in adulthood (Choudhury et al., 2009). The question arises why these stressors induce persistent VHS and colonic motor dysfunction in neonates, but not in adults. The answer is epigenetic dysregulation during the vulnerable stages of fetal and neonatal developments. As the organisms develop from a single zygote, the DNA replicates at each asymmetric or symmetric cell division. In addition, at each cell division, the epigenetic mechanisms program the expression of each gene to impart cell phenotype. Inherited epigenetic information programs genes for normal health in adulthood. However, in the face of intense or chronic psychological/chemical stress, the epigenetic mechanisms reprogram the genes to protect vital organs in neonates, such as the heart, to ensure survival. This aberrant epigenetic regulation also affects the programming of genes in other organs that are vulnerable at the time of the insult. Unfortunately, this reprogramming persists in adulthood to cause organ dysfunctions, resulting in complex diseases (Nuyt, 2008).
Neonatal inflammatory insult increases the acetylation and trimethylation of lysine residue nine on histone H3 to enhance the association of RNA polymerase II with the core promoter region of the Cacna1c. This gene encodes the pore-forming α1C subunit of Cav1.2b (L-type) calcium channels in smooth muscle cells (Saada et al., 2003). This epigenetic modulation increases the transcription of this gene (Li and Sarna, 2011). The resulting increase of calcium influx through these channels enhances the amplitude of colonic contractions, accelerates colonic transit, and fecal output (Shi et al., 2007; Choudhury et al., 2009). It is noteworthy that a similar insult in adulthood does not produce these effects emphasizing the vulnerability of genes during development. However, an inflammatory insult in adult rats causes DNA damage that recovers partially after the insult – the persistent damage associates with suppression of smooth muscle reactivity to ACh (Choi et al., 2011a).
Clinical studies show a strong link between early life infection and development of IBS and dyspepsia in adulthood (Saps et al., 2008). Eighty-seven percentage of children in the age range of 3–19 years developed IBS following acute gastroenteritis [Salmonella (54%), Campylobacter (32%), and Shigella (14%)] and 24% develop dyspepsia (Saps et al., 2008). It is noteworthy that in animal model, neonatal inflammatory insult concurrently causes gastric hypersensitivity to gastric distension (Winston and Sarna, 2011) and moderate delay in gastric emptying (Choi et al., 2011b) – the two cardinal symptoms of functional dyspepsia. The concurrent induction of IBS-like symptoms in the colon and functional dyspepsia-like symptoms in the stomach following neonatal inflammatory insult highlights overlap seen in subsets of patients with IBS and functional dyspepsia (Geeraerts and Tack, 2008).
Lessons Learnt
1. Epigenetic regulation modifies the expression of selective genes in cells following changes in their microenvironment.
2. If the changes in microenvironment occur during the vulnerable stages of fetal and neonatal development, the changes in expressions of selective genes may persist into adulthood to cause complex diseases, such as IBS.
3. The relapsing/recurring changes in symptoms of IBS do not make them candidates for genetic mutations/polymorphisms.
Conclusion
Mounting evidence suggests that post-translational modifications of histone proteins by environmental stimuli at various stages of development and maturity are major factors in a variety of diseases, including cancer, inflammatory disorders, and autoimmune diseases (Egger et al., 2004). Based on these advances, novel histone deacetylase inhibitors are under development to treating complex diseases – some types of cancers and diabetes mellitus (Butler et al., 2002; Kumagai et al., 2007; Lawless et al. 2009; Christensen et al. 2011). The investigations of epigenetic dysregulation in functional bowel disorders are beginning to emerge. Overall, a paradigm shift from technology-driven approach (ready availability of mucosal biopsies and blood samples, immunofluorescence and assay methodologies) to hypothesis-driven approach (investigations of smooth muscle cells, enteric neurons, visceral afferent neurons, and CNS) will advance our understanding of functional bowel disorders and help in development of novel therapies.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported in part by NIDDK Grants DK 32346, DK 72414, DK 079952, and DK088796.
References
Al-Chaer, E. D., Kawasaki, M., and Pasricha, P. J. (2000). A new model of chronic visceral hypersensitivity in adult rats induced by colon irritation during postnatal development. Gastroenterology 119, 1276–1285.
Bampton, P. A., Dinning, P. G., Kennedy, M. L., Lubowski, D. Z., deCarle, D., and Cook, I. J. (2000). Spatial and temporal organization of pressure patterns throughout the unprepared colon during spontaneous defecation. Am. J. Gastroenterol. 95, 1027–1035.
Barbara, G., Stanghellini, V., De Giorgio, R., Cremon, C., Cottrell, G. S., Santini, D., Pasquinelli, G., Morselli-Labate, A. M., Grady, E. F., Bunnett, N. W., Collins, S. M., and Corinaldesi, R. (2004). Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology 126, 693–702.
Barbara, G., Wang, B., Stanghellini, V., de Giorgio, R., Cremon, C., Di Nardo, G., Trevisani, M., Campi, B., Geppetti, P., Tonini, M., Bunnett, N. W., Grundy, D., and Corinaldesi, R. (2007). Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology 132, 26–37.
Barreau, F., Cartier, C., Ferrier, L., Fioramonti, J., and Bueno, L. (2004). Nerve growth factor mediates alterations of colonic sensitivity and mucosal barrier induced by neonatal stress in rats. Gastroenterology 127, 524–534.
Bassotti, G., Chistolini, F., Marinozzi, G., and Morelli, A. (2003). Abnormal colonic propagated activity in patients with slow transit constipation and constipation-predominant irritable bowel syndrome. Digestion 68, 178–183.
Bassotti, G., Gaburri, M., Imbimbo, B. P., Rossi, L., Farroni, F., Pelli, M. A., and Morelli, A. (1988). Colonic mass movements in idiopathic chronic constipation. Gut 29, 1173–1179.
Bassotti, G., Iantorno, G., Fiorella, S., Bustos-Fernandez, L., and Bilder, C. R. (1999). Colonic motility in man: features in normal subjects and in patients with chronic idiopathic constipation. Am. J. Gastroenterol. 94, 1760–1770.
Bhaumik, S. R., Smith, E., and Shilatifard, A. (2007). Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 14, 1008–1016.
Brandt, L. J., Bjorkman, D., Fennerty, M. B., Locke, G. R., Olden, K., Peterson, W., Quigley, E., Schoenfeld, P., Schuster, M., and Talley, N. (2002). Systematic review on the management of irritable bowel syndrome in North America. Am. J. Gastroenterol. 97(11 Suppl.), S7–S26.
Brierley, S. M., Jones, R. C. III, Gebhart, G. F., and Blackshaw, L. A. (2004). Splanchnic and pelvic mechanosensory afferents signal different qualities of colonic stimuli in mice. Gastroenterology 127, 166–178.
Bulbring, E., and Crema, A. (1959). The release of 5-hydroxytryptamine in relation to pressure exerted on the intestinal mucosa. J. Physiol. (Lond.) 146, 18–28.
Butler, L. M., Zhou, X., Xu, W. S., Scher, H. I., Rifkind, R. A., Marks, P. A., and Richon, V. M. (2002). The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 99, 11700–11705.
Camilleri, M., Andrews, C. N., Bharucha, A. E., Carlson, P. J., Ferber, I., Stephens, D., Smyrk, T. C., Urrutia, R., Aerssens, J., Thielemans, L., Göhlmann, H., van den Wyngaert, I., and Coulie, B. (2007). Alterations in expression of p11 and SERT in mucosal biopsy specimens of patients with irritable bowel syndrome. Gastroenterology 132, 17–25.
Camilleri, M., Atanasova, E., Carlson, P. J., Ahmad, U., Kim, H. J., Viramontes, B. E., McKinzie, S., and Urrutia, R. (2002). Serotonin-transporter polymorphism pharmacogenetics in diarrhea-predominant irritable bowel syndrome. Gastroenterology 123, 425–432.
Camilleri, M., McKinzie, S., Busciglio, I., Low, P. A., Sweetser, S., Burton, D., Baxter, K., Ryks, M., and Zinsmeister, A. R. (2008). Prospective study of motor, sensory, psychologic, and autonomic functions in patients with irritable bowel syndrome. Clin. Gastroenterol. Hepatol. 6, 772–781.
Cenac, N., Andrews, C. N., Holzhausen, M., Chapman, K., Cottrell, G., Andrade-Gordon, P., Steinhoff, M., Barbara, G., Beck, P., Bunnett, N. W., Sharkey, K. A., Ferraz, J. G., Shaffer, E., and Vergnolle, N. (2007). Role for protease activity in visceral pain in irritable bowel syndrome. J. Clin. Invest. 117, 636–647.
Chadwick, V. S., Chen, W., Shu, D., Paulus, B., Bethwaite, P., Tie, A., and Wilson, I. (2002). Activation of the mucosal immune system in irritable bowel syndrome. Gastroenterology 122, 1778–1783.
Chang, L., Munakata, J., Mayer, E. A., Schmulson, M. J., Johnson, T. D., Bernstein, C. N., Saba, L., Naliboff, B., Anton, P. A., and Matin, K. (2000). Perceptual responses in patients with inflammatory and functional bowel disease. Gut 47, 497–505.
Chaudhary, N. A., and Truelove, S. C. (1962). The irritable colon syndrome. A study of the clinical features, predisposing causes, and prognosis in 130 cases. Q. J. Med. 31, 307–322.
Chavez, A. M., Menconi, M. J., Hodin, R. A., and Fink, M. P. (1999). Cytokine-induced intestinal epithelial hyperpermeability: role of nitric oxide. Crit. Care Med. 27, 2246–2251.
Chey, W. Y., Jin, H. O., Lee, M. H., Sun, S. W., and Lee, K. Y. (2001). Colonic motility abnormality in patients with irritable bowel syndrome exhibiting abdominal pain and diarrhea. Am. J. Gastroenterol. 96, 1499–1506.
Childs, B. (1995). “A logic of disease,” in The Metabolic and Molecular Bases of Inherited Disease, 7th Edn, eds C. R. Scriver, W. S. Sly, B. Childs, A. L. Beaudet, D. Valle, K. W. Kinzler, and B. Vogelstein (New York, NY: Mcgraw-Hill), 229–257.
Chitkara, D. K., van Tilburg, M. A., Blois-Martin, N., and Whitehead, W. E. (2008). Early life risk factors that contribute to irritable bowel syndrome in adults: a systematic review. Am. J. Gastroenterol. 103, 765–774; quiz 775.
Choi, K., Chen, J., Mitra, S., and Sarna, S. K. (2011a). Impaired integrity of DNA after recovery from inflammation causes persistent dysfunction of colonic smooth muscle. Gastroenterology. PMID: 21745450. [Epub ahead of print].
Choi, K., Shi, X. Z. P., and Sarna, S. K. (2011b). Epigenetic modulation of Mlck1 gene contributes to delayed gastric emptying in a rat model of functional dyspepsia. Gastroenterology 140, S70–S71.
Choudhury, B. K., Shi, X. Z., and Sarna, S. K. (2009). Gene plasticity in colonic circular smooth muscle cells underlies motility dysfunction in a model of postinfective IBS. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G632–G642.
Christensen, D. P., Dahllöf, M., Lundh, M., Rasmussen, D. N., Nielsen, M. D., Billestrup, N., Grunnet, L. G., and Mandrup-Poulsen, T. (2011). Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. 17, 378–390.
Coates, M. D., Mahoney, C. R., Linden, D. R., Sampson, J. E., Chen, J., Blaszyk, H., Crowell, M. D., Sharkey, K. A., Gershon, M. D., Mawe, G. M., and Moses, P. L. (2004). Molecular defects in mucosal serotonin content and decreased serotonin reuptake transporter in ulcerative colitis and irritable bowel syndrome. Gastroenterology 126, 1657–1664.
Corinaldesi, R., Stanghellini, V., Cremon, C., Gargano, L., Cogliandro, R. F., De Giorgio, R., Bartesaghi, G., Canovi, B., and Barbara, G. (2009). Effect of mesalazine on mucosal immune biomarkers in irritable bowel syndrome: a randomized controlled proof-of-concept study. Aliment. Pharmacol. Ther. 30, 245–252.
Coutinho, S. V., Plotsky, P. M., Sablad, M., Miller, J. C., Zhou, H., Bayati, A. I., McRoberts, J. A., and Mayer, E. A. (2002). Neonatal maternal separation alters stress-induced responses to viscerosomatic nociceptive stimuli in rat. Am. J. Physiol. Gastrointest. Liver Physiol. 282, G307–G316.
Cremon, C., Gargano, L., Morselli-Labate, A. M., Santini, D., Cogliandro, R. F., De Giorgio, R., Stanghellini, V., Corinaldesi, R., and Barbara, G. (2009). Mucosal immune activation in irritable bowel syndrome: gender-dependence and association with digestive symptoms. Am. J. Gastroenterol. 104, 392–400.
Dainese, R., Galliani, E. A., De Lazzari, F., Di Leo, V., and Naccarato, R. (1999). Discrepancies between reported food intolerance and sensitization test findings in irritable bowel syndrome patients. Am. J. Gastroenterol. 94, 1892–1897.
Dunlop, S. P., Coleman, N. S., Blackshaw, E., Perkins, A. C., Singh, G., Marsden, C. A., and Spiller, R. C. (2005). Abnormalities of 5-hydroxytryptamine metabolism in irritable bowel syndrome. Clin. Gastroenterol. Hepatol. 3, 349–357.
Dunlop, S. P., Hebden, J., Campbell, E., Naesdal, J., Olbe, L., Perkins, A. C., and Spiller, R. C. (2006). Abnormal intestinal permeability in subgroups of diarrhea-predominant irritable bowel syndromes. Am. J. Gastroenterol. 101, 1288–1294.
Dunlop, S. P., Jenkins, D., Neal, K. R., and Spiller, R. C. (2003a). Relative importance of enterochromaffin cell hyperplasia, anxiety, and depression in postinfectious IBS. Gastroenterology 125, 1651–1659.
Dunlop, S. P., Jenkins, D., Neal, K. R., Naesdal, J., Borgaonker, M., Collins, S. M., and Spiller, R. C. (2003b). Randomized, double-blind, placebo-controlled trial of prednisolone in post-infectious irritable bowel syndrome. Aliment. Pharmacol. Ther. 18, 77–84.
Dunlop, S. P., Jenkins, D., and Spiller, R. C. (2003c). Distinctive clinical, psychological, and histological features of postinfective irritable bowel syndrome. Am. J. Gastroenterol. 98, 1578–1583.
Egger, G., Liang, G., Aparicio, A., and Jones, P. A. (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463.
Fass, R., Naliboff, B., Higa, L., Johnson, C., Kodner, A., Munakata, J., Ngo, J., and Mayer, E. A. (1998). Differential effect of long-term esophageal acid exposure on mechanosensitivity and chemosensitivity in humans. Gastroenterology 115, 1363–1373.
Geeraerts, B., and Tack, J. (2008). Functional dyspepsia: past, present, and future. J. Gastroenterol. 43, 251–255.
Gershon, M. D. (2005). Nerves, reflexes, and the enteric nervous system: pathogenesis of the irritable bowel syndrome. J. Clin. Gastroenterol. 39(5 Suppl. 3), S184–S193.
Grider, J. R., and Jin, J. G. (1994). Distinct populations of sensory neurons mediate the peristaltic reflex elicited by muscle stretch and mucosal stimulation. J. Neurosci. 14(5 Pt 1), 2854–2860.
Gwee, K. A., Collins, S. M., Read, N. W., Rajnakova, A., Deng, Y., Graham, J. C., McKendrick, M. W., and Moochhala, S. M. (2003). Increased rectal mucosal expression of interleukin 1beta in recently acquired post-infectious irritable bowel syndrome. Gut 52, 523–526.
Gwee, K. A., Graham, J. C., McKendrick, M. W., Collins, S. M., Marshall, J. S., Walters, S. J., and Read, N. W. (1996). Psychometric scores and persistence of irritable bowel after infectious diarrhoea. Lancet 347, 150–153.
Gwee, K. A., Leong, Y. L., Graham, C., McKendrick, M. W., Collins, S. M., Walters, S. J., Underwood, J. E., and Read, N. W. (1999). The role of psychological and biological factors in postinfective gut dysfunction. Gut 44, 400–406.
Howard, R., Rabins, P. V., Seeman, M. V., and Jeste, D. V. (2000). Late-onset schizophrenia and very-late-onset schizophrenia-like psychosis: an international consensus. The International Late-Onset Schizophrenia Group. Am. J. Psychiatry 157, 172–178.
Keating, D. J., and Spencer, N. J. (2010). Release of 5-hydroxytryptamine from the mucosa is not required for the generation or propagation of colonic migrating motor complexes. Gastroenterology 138, 659–70 670 e1–2.
Kim, H. J., Camilleri, M., Carlson, P. J., Cremonini, F., Ferber, I., Stephens, D., McKinzie, S., Zinsmeister, A. R., and Urrutia, R. (2004). Association of distinct alpha(2) adrenoceptor and serotonin transporter polymorphisms with constipation and somatic symptoms in functional gastrointestinal disorders. Gut 53, 829–837.
King, R. A., Rotter, J. I., and Motulsky, A. G. (1992). The Genetic Bases of Common Diseases. Oxford: Oxford University Press.
Klooker, T. K., Braak, B., Koopman, K. E., Welting, O., Wouters, M. M., van der Heide, S., Schemann, M., Bischoff, S. C., van den Wijngaard, R. M., and Boeckxstaens, G. E. (2010). The mast cell stabiliser ketotifen decreases visceral hypersensitivity and improves intestinal symptoms in patients with irritable bowel syndrome. Gut 59, 1213–1221.
Kristjansson, G., Venge, P., Wanders, A., Lööf, L., and Hällgren, R. (2004). Clinical and subclinical intestinal inflammation assessed by the mucosal patch technique: studies of mucosal neutrophil and eosinophil activation in inflammatory bowel diseases and irritable bowel syndrome. Gut 53, 1806–1812.
Kuiken, S. D., Lindeboom, R., Tytgat, G. N., and Boeckxstaens, G. E. (2005). Relationship between symptoms and hypersensitivity to rectal distension in patients with irritable bowel syndrome. Aliment. Pharmacol. Ther. 22, 157–164.
Kuiken, S. D., Tytgat, G. N., and Boeckxstaens, G. E. (2003). The selective serotonin reuptake inhibitor fluoxetine does not change rectal sensitivity and symptoms in patients with irritable bowel syndrome: a double blind, randomized, placebo-controlled study. Clinical Gastroenterol. Hepatol. 1, 219–228.
Kumagai, T., Wakimoto, N., Yin, D., Gery, S., Kawamata, N., Takai, N., Komatsu, N., Chumakov, A., Imai, Y., and Koeffler, H. P. (2007). Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int. J. Cancer 121, 656–665.
Lawless, M. W., Norris, S., O’Byrne, K. J., and Gray, S. G. (2009). Targeting histone deacetylases for the treatment of disease. J. Cell. Mol. Med. 13, 826–852.
Lee, D. Y., Park, H., Kim, W. H., Lee, S. I., Seo, Y. J., and Choi, Y. C. (2004). Serotonin transporter gene polymorphism in healthy adults and patients with irritable bowel syndrome. Korean J. Gastroenterol. 43, 18–22.
Lee, K. J., Kim, Y. B., Kim, J. H., Kwon, H. C., Kim, D. K., and Cho, S. W. (2008). The alteration of enterochromaffin cell, mast cell, and lamina propria T lymphocyte numbers in irritable bowel syndrome and its relationship with psychological factors. J. Gastroenterol. Hepatol. 23, 1689–1694.
Lembo, T., Munakata, J., Mertz, H., Niazi, N., Kodner, A., Nikas, V., and Mayer, E. A. (1994). Evidence for the hypersensitivity of lumbar splanchnic afferents in irritable bowel syndrome. Gastroenterology 107, 1686–1696.
Lembo, T., Munakata, J., Naliboff, B., Fullerton, S., and Mayer, E. A. (1997). Sigmoid afferent mechanisms in patients with irritable bowel syndrome. Dig. Dis. Sci. 42, 1112–1120.
Lettesjo, H., Hansson, T., Peterson, C., Ung, K. A., Ringström, G., Abrahamsson, H., and Simrén, M. (2006). Detection of inflammatory markers in stools from patients with irritable bowel syndrome and collagenous colitis. Scand. J. Gastroenterol. 41, 54–59.
Li, Q. J., and Sarna, S. K. (2011). Developmental origins of irritable bowel syndrome (IBS)-like symptoms: epigenetic dysregulation. Gastroenterology 140, S121.
Lowman, B. C., Drossman, D. A., Cramer, E. M., and McKee, D. C. (1987). Recollection of childhood events in adults with irritable bowel syndrome. J. Clin. Gastroenterol. 9, 324–330.
Lynn, P. A., and Blackshaw, L. A. (1999). In vitro recordings of afferent fibres with receptive fields in the serosa, muscle and mucosa of rat colon. J. Physiol. 518(Pt 1), 271–82.
Marshall, J. K., Thabane, M., Garg, A. X., Clark, W., Meddings, J., Collins, S. M., and WEL Investigators. (2004). Intestinal permeability in patients with irritable bowel syndrome after a waterborne outbreak of acute gastroenteritis in Walkerton, Ontario. Aliment. Pharmacol. Ther. 20, 1317–1322.
McKendrick, M. W., and Read, N. W. (1994). Irritable bowel syndrome – post salmonella infection. J. Infect. 29, 1–3.
Mearin, F., Pérez-Oliveras, M., Perelló, A., Vinyet, J., Ibañez, A., Coderch, J., and Perona, M. (2005). Dyspepsia and irritable bowel syndrome after a Salmonella gastroenteritis outbreak: one-year follow-up cohort study. Gastroenterology 129, 98–104.
Mertz, H., Fullerton, S., Naliboff, B., and Mayer, E. A. (1998). Symptoms and visceral perception in severe functional and organic dyspepsia. Gut 42, 814–822.
Middel, P., Reich, K., Polzien, F., Blaschke, V., Hemmerlein, B., Herms, J., Korabiowska, M., and Radzun, H. J. (2001). Interleukin 16 expression and phenotype of interleukin 16 producing cells in Crohn’s disease. Gut 49, 795–803.
Neal, K. R., Hebden, J., and Spiller, R. (1997). Prevalence of gastrointestinal symptoms six months after bacterial gastroenteritis and risk factors for development of the irritable bowel syndrome: postal survey of patients. BMJ 314, 779–782.
Nuyt, A. M. (2008). Mechanisms underlying developmental programming of elevated blood pressure and vascular dysfunction: evidence from human studies and experimental animal models. Clin. Sci. 114, 1–17.
O’Sullivan, M., Clayton, N., Breslin, N. P., Harman, I., Bountra, C., McLaren, A., and O’Morain, C. A. (2000). Increased mast cells in the irritable bowel syndrome. Neurogastroenterol. Motil. 12, 449–457.
Park, C. H., Joo, Y. E., Choi, S. K., Rew, J. S., Kim, S. J., and Lee, M. C. (2003). Activated mast cells infiltrate in close proximity to enteric nerves in diarrhea-predominant irritable bowel syndrome. J. Korean Med. Sci. 18, 204–210.
Park, J. H., Rhee, P. L., Kim, G., Lee, J. H., Kim, Y. H., Kim, J. J., Rhee, J. C., and Song, S. Y. (2006). Enteroendocrine cell counts correlate with visceral hypersensitivity in patients with diarrhoea-predominant irritable bowel syndrome. Neurogastroenterol. Motil. 18, 539–546.
Park, M. I., and Camilleri, M. (2005). Genetics and genotypes in irritable bowel syndrome: implications for diagnosis and treatment. Gastroenterol. Clin. North Am. 34, 305–317.
Pata, C., Erdal, M. E., Derici, E., Yazar, A., Kanik, A., and Ulu, O. (2002). Serotonin transporter gene polymorphism in irritable bowel syndrome. Am. J. Gastroenterol. 97, 1780–1784.
Pont, S. J., Grijalva, C. G., Griffin, M. R., Scott, T. A., and Cooper, W. O. (2009). National rates of diarrhea-associated ambulatory visits in children. J. Pediatr. 155, 56–61.
Posserud, I., Syrous, A., Lindström, L., Tack, J., Abrahamsson, H., and Simrén, M. (2007). Altered rectal perception in irritable bowel syndrome is associated with symptom severity. Gastroenterology 133, 1113–1123.
Pravda, J. (2005). Radical induction theory of ulcerative colitis. World J. Gastroenterol. 11, 2371–2384.
Prior, A., Maxton, D. G., and Whorwell, P. J. (1990). Anorectal manometry in irritable bowel syndrome: differences between diarrhoea and constipation predominant subjects. Gut 31, 458–462.
Protic, M., Jojic, N., Bojic, D., Milutinovic, S., Necic, D., Bojic, B., Svorcan, P., Krstic, M., and Popovic, O. (2005). Mechanism of diarrhea in microscopic colitis. World J. Gastroenterol. 11, 5535–5539.
Rao, S. S., Sadeghi, P., Beaty, J., and Kavlock, R. (2004). Ambulatory 24-hour colonic manometry in slow-transit constipation. Am. J. Gastroenterol. 99, 2405–2416.
Saada, N., Dai, B., Echetebu, C., Sarna, S. K., and Palade, P. (2003). Smooth muscle uses another promoter to express primarily a form of human Ca(V)1.2 L-type calcium channel different from the principal heart form. Biochem. Biophys. Res. Commun. 302, 23–28.
Saito, Y. A., and Talley, N. J. (2008). Genetics of irritable bowel syndrome. Am. J. Gastroenterol. 103, 2100–2104; quiz 2105.
Saps, M., Pensabene, L., Di Martino, L., Staiano, A., Wechsler, J., Zheng, X., and Di Lorenzo, C. (2008). Post-infectious functional gastrointestinal disorders in children. J. Pediatr. 152, 812–816, 816.e1.
Sarna, S. K. (2006). Molecular, functional and pharmacological targets for the development of gut promotility drugs. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G545–G555.
Sarna, S. K. (2007). Enteric descending and afferent neural signaling stimulated by giant migrating contractions: essential contributing factors to visceral pain. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G572–G581.
Sarna, S. K. (2010). Colonic Motility: From Bench Side to Bedside, in Colonic Motility. San Rafael, CA: Morgan and Claypool Life Sciences, 1–140.
Shi, X. Z., Choudhury, B. K., Pasricha, P. J., and Sarna, S. K. (2007). A novel role of VIP in colonic motility function: induction of excitation-transcription coupling in smooth muscle cells. Gastroenterology 132, 1388–1400.
Shi, X. Z., Winston, J. H., and Sarna, S. K. (2011). Differential immune and genetic responses in rat models of Crohn’s colitis and ulcerative colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G41–G51.
Silberer, H., Küppers, B., Mickisch, O., Baniewicz, W., Drescher, M., Traber, L., Kempf, A., and Schmidt-Gayk, H. (2005). Fecal leukocyte proteins in inflammatory bowel disease and irritable bowel syndrome. Clin. Lab. 51, 117–126.
Spencer, N. J., Nicholas, S. J., Robinson, L., Kyloh, M., Flack, N., Brookes, S. J., Zagorodnyuk, V. P., and Keating, D. J. (2011). Mechanisms underlying distension-evoked peristalsis in guinea-pig distal colon: is there a role for enterochromaffin (EC) cells? Am. J. Physiol. Gastrointest. Liver Physiol. PMID: 21700904. [Epub ahead of print].
Spiller, R. C. (2007). Role of infection in irritable bowel syndrome. J. Gastroenterol. 42(Suppl. 17), 41–47.
Spiller, R. C., Jenkins, D., Thornley, J. P., Hebden, J. M., Wright, T., Skinner, M., and Neal, K. R. (2000). Increased rectal mucosal enteroendocrine cells, T lymphocytes, and increased gut permeability following acute Campylobacter enteritis and in post-dysenteric irritable bowel syndrome. Gut 47, 804–811.
Sydora, B. C., Tavernini, M. M., Doyle, J., and Fedorak, R. N. (2006a). A defect in epithelial barrier integrity is not required for a systemic response to bacterial antigens or intestinal injury in T cell receptor-alpha gene-deficient mice. Inflamm. Bowel Dis. 12, 750–757.
Sydora, B. C., Martin, S. M., Lupicki, M., Dieleman, L. A., Doyle, J., Walker, J. W., and Fedorak, R. N. (2006b). Bacterial antigens alone can influence intestinal barrier integrity, but live bacteria are required for initiation of intestinal inflammation and injury. Inflamm. Bowel Dis. 12, 429–436.
Tibble, J. A., Sigthorsson, G., Foster, R., Forgacs, I., and Bjarnason, I. (2002). Use of surrogate markers of inflammation and Rome criteria to distinguish organic from nonorganic intestinal disease. Gastroenterology 123, 450–460.
Tornblom, H., Lindberg, G., Nyberg, B., and Veress, B. (2002). Full-thickness biopsy of the jejunum reveals inflammation and enteric neuropathy in irritable bowel syndrome. Gastroenterology 123, 1972–1979.
van der Veek, P. P., van den Berg, M., de Kroon, Y. E., Verspaget, H. W., and Masclee, A. A. (2005). Role of tumor necrosis factor-alpha and interleukin-10 gene polymorphisms in irritable bowel syndrome. Am. J. Gastroenterol. 100, 2510–2516.
Van Kerkhoven, L. A., Laheij, R. J., and Jansen, J. B. (2007). Meta-analysis: a functional polymorphism in the gene encoding for activity of the serotonin transporter protein is not associated with the irritable bowel syndrome. Aliment. Pharmacol. Ther. 26, 979–986.
Venge, P., and Peterson, C. (1989). “Eosinophil biochemistry and killing mechanisms,” in Eosinophils in Asthma, eds J. Morley, and I. Colditz (New York: Academic Press).
Vermeire, S., Van Assche, G., and Rutgeerts, P. (2010). Postinfectious irritable bowel syndrome: a genetic link identified? Gastroenterology 138, 1246–1249.
Villani, A. C., Lemire, M., Thabane, M., Belisle, A., Geneau, G., Garg, A. X., Clark, W. F., Moayyedi, P., Collins, S. M., Franchimont, D., and Marshall, J. K. (2010). Genetic risk factors for post-infectious irritable bowel syndrome following a waterborne outbreak of gastroenteritis. Gastroenterology 138, 1502–1513.
Wang, L. H., Fang, X. C., and Pan, G. Z. (2004). Bacillary dysentery as a causative factor of irritable bowel syndrome and its pathogenesis. Gut 53, 1096–1101.
Wildt, S., Madsen, J. L., and Rumessen, J. J. (2006). Small-bowel permeability in collagenous colitis. Scand. J. Gastroenterol. 41, 1044–1049.
Winston, J. H., and Sarna, S. K. (2011). Neonatal inflammatory insult to the colon results in functional dyspepsia-like gastric hypersensitivity in adulthood by epigenetic dysregulation. Gastroenterology 140, S23–S24.
Yeo, A., Boyd, P., Lumsden, S., Saunders, T., Handley, A., Stubbins, M., Knaggs, A., Asquith, S., Taylor, I., Bahari, B., Crocker, N., Rallan, R., Varsani, S., Montgomery, D., Alpers, D. H., Dukes, G. E., Purvis, I., and Hicks, G. A. (2004). Association between a functional polymorphism in the serotonin transporter gene and diarrhoea predominant irritable bowel syndrome in women. Gut 53, 1452–1458.
Keywords: functional bowel disorders, visceral pain, inflammatory bowel disease, enteric neurons, smooth muscle, immune cells, epithelial permeability, gut inflammation
Citation: Sarna SK (2011) Lessons learnt from post-infectious IBS. Front. Physio. 2:49. doi: 10.3389/fphys.2011.00049
Received: 14 July 2011; Accepted: 02 August 2011;
Published online: 23 August 2011.
Edited by:
D. N. Granger, Louisiana State University Health Sciences Center, USAReviewed by:
Julian Panes, Hospital Clinic, University of Barcelona, SpainThorsten Vowinkel, University Hospital Muenster, Germany
Copyright: © 2011 Sarna. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Sushil K. Sarna, Division of Gastroenterology, Department of Internal Medicine, The University of Texas Medical Branch at Galveston, 8.104 Medical Research Building, Galveston, TX 77555-1083, USA. e-mail: sksarna@utmb.edu