
 P. Lungchukiet1,2
P. Lungchukiet1,2
- 1 Department of Physiology, Queen’s University, Kingston, ON, Canada
- 2 Gastrointestinal Diseases Research Unit, Department of Medicine, Kingston General Hospital, Kingston, ON, Canada
- 3 Canada Research Chair in Gastrointestinal Cell Physiology, Queen’s University, Kingston, ON, Canada
Tumor necrosis factor alpha (TNFα) and its receptor TNFR1 play a central role in the development of colitis-associated colon cancer. To understand a role for the extracellular calcium-sensing receptor (CaSR) and its non-canonical Wnt mediators, Wnt5a/Ror2, we used reductionistic systems. We added lipopolysaccharide (LPS) to mouse peritoneal macrophages, RAW264.7 cells, a murine macrophage cell line, and 18Co colonic myofibroblasts, to stimulate TNFα secretion and then activated endogenous CaSR. CaSR activation inhibited TNFα secretion, which in RAW264.7 cells knockdown of CaSR by short-interfering RNA (siRNA) duplex reversed. LPS-stimulated NFκB promoter activity in RAW264.7 cells was inhibited by CaSR activation with Ca2+ or other polyvalent CaSR agonists. Reducing CaSR expression with siRNA duplex prevented this inhibition. Following LPS addition to CaSR–HEK cells or RAW264.7 macrophages, CaSR stimulation deneddylated Cullin1. Wnt5a added to HT-29 cells which overexpressed Ror2 or T84 monolayers treated with 3 mM Ca2+ reduced TNFR1 protein expression ∼70%. TNFα/INFγ addition to high resistance T84 monolayers reduced transepithelial resistance 50% within 4 h. CaSR activation (3 mM Ca2+) together with rhWnt5a (200 ng/ml) prevented this reduction while Wnt3a addition had no effect. LPS-stimulated TNFα secretion from RAW264.7 cells was not effected by rhWnt5a but increased 10-fold by Wnt3a. Together our results suggest that following LPS challenge, CaSR activation will inhibit NFκB activity and reduce TNFα secretion from macrophages and stroma while Wnt5a/Ror2 engagement on intestinal epithelia reduces TNFR1 expression, allowing TNFα signaling to be titrated. Our results also suggest that canonical Wnt signaling may enhance TLR4 stimulation of TNFα secretion from murine macrophages.
Introduction
Increased intake of dairy foods and calcium (Ca2+) has recently been shown to dramatically lower the risk of colorectal cancer in men and women (Lee et al., 2010). The molecular mechanism(s) responsible for this reduction are unknown (Park et al., 2009). Tumor necrosis factor alpha (TNFα), a potent inflammatory cytokine, is a critical requirement for the development of colitis-associated colon cancer (Popivanova et al., 2008). A major target of increased dietary calcium is the extracellular calcium-sensing receptor (CaSR; Brown and MacLeod, 2001), a G-protein coupled receptor, which is present on both colonic epithelia and sub-epithelial myofibroblasts (MacLeod et al., 2007; Peiris et al., 2007; Pacheco and MacLeod, 2008). Our laboratory has demonstrated that a unique paracrine axis exists, in vitro, which may be stimulated by Ca2+ activating the CaSR on colonic epithelia and the subepithelia which inhibits proliferation and stimulates differentiation (MacLeod et al., 2007; Pacheco and MacLeod, 2008). This axis is the production of both Wnt5a from the sub-epithelial myofibroblasts and Ror2, an orphan tyrosine kinase which works as a receptor for Wnt5a, on the epithelia. Screening Wnt5a/Ror2 on intestinal epithelia by microarray, we observed that TNFR1 transcript was substantially reduced while TNFR2 remained unchanged. Crohn’s Disease (CD) and ulcerative colitis (UC) are the two forms of inflammatory bowel diseases (IBD). Although the etiology remains unknown, a large body of work has demonstrated that the involvement of damaged epithelia and activated immune cells in the inflamed mucosa play important role in their pathogenesis (McKay et al., 1996; Targan et al., 1997; Nielson et al., 2000; Bruewer et al., 2003). In addition to the problem of persistent intestinal inflammation in IBD, one of the critical complications in patients with long-standing IBD over ∼10 years after onset is colitis-associated cancer (Rutter et al., 2004).
Tumor necrosis factor alpha has been implicated in the development of colitis (Rutter et al., 2004; Belenkaya et al., 2008) and pharmacological blockade with monoclonal antibodies has demonstrated great efficacy in treatment of IBD patients (Pan et al., 2008). Using mice deficient in type 1 TNF receptor (p55/TNFR1) Popivanova et al. (2008) used the AOM/DSS model of colitis-associated cancer and compared wild type with TNFR1 knockout mice. Abrogation of TNF signaling ameliorated their colitis (as assessed by reduced tissue injury, inflammatory cell infiltrates, and cytokine expression in the mucosa). Loss of TNF signaling also greatly suppressed the colitis-associated cancer (tumor number and size). Further work demonstrated the importance of TNFR1 deletion on the immune cells rather than the intestinal epithelia, consistent with the interpretation that TNFα production in mucosa by resident cells was required for activation of the immune cells, which in turn were important in the subsequent development of the colon cancer-i.e., TNFα was needed to stimulate a tumor promoting immune response. A strong candidate of a mediator of NF-κB activation in the epithelia of IBD patients is TNFα, which is markedly elevated in the inflamed intestinal milieu (Pan et al., 2008). A predominant source of TNFα is from macrophages. A major source of macrophages in the lamina propria is from the peritoneum. The kinetics of the phenotype and function of murine peritoneal macrophages following acute inflammation have been well described (Calorini et al., 2000; Wu et al., 2004).
In this study we show that when LPS stimulates TNFα secretion from peritoneal macrophages, a mouse macrophage cell line, or human colonic myofibroblasts, CaSR stimulation by Ca2+ inhibits both NFκB activity and TNFα secretion. We also report that CaSR-stimulated Wnt5a-Ror2 mediates downregulation of TNFR1 transcript and protein on colonic epithelia. We then show that Ca2+ and Wnt5a added to high resistance T84 monolayers are protected from barrier dissolution triggered by TNFα/INFγ addition. In addition, we report that the CaSR can modulate Wnt protein secretion from macrophages and that canonical Wnt signaling can enhance TNFα production from LPS-stimulated macrophages. CaSR activation together with Wnt5a/Ror2 stimulation may afford a novel mechanism to titrate TNFα signaling.
Materials and Methods
Materials
Wnt3a and Wnt5a antibodies, rhWnt5a, TNFα, and INFγ were obtained from R&D systems (Minneapolis, MN, USA). TNFR1, TNFR2, Cullin-1, and Nedd8 antibodies were obtained from Cell Signaling Technology (Boston, MA, USA). Monoclonal anti-β-actin, Neomycin sulfate and Spermine were from Sigma (Saint Louis, MO, USA). Cell culture media Dulbecco’s Modified Eagles Media, with or without calcium were obtained from GIBCO-BRL (Grand Island, NY, USA). The enhanced chemiluminescence supersignal kit was from Pierce (Rockford, IL, USA). LPS was a gift from Dr. Elaine Petrof (Queen’s University, Kingston, ON, Canada).
Cell Culture
The human colon adenocarcinoma HT-29 cell line, T84 monolayers, colonic myofibroblast cells, and murine macrophages RAW264.7 were purchased from the American Type Culture Collection (ATCC; Rockville, MD, USA). Mouse peritoneal macrophages were isolated as previously described (Quesniaux et al., 2010; Wehner et al., 2010). The protocol was approved by the University Animal Care Committee. CaSR-transfected HEK cells were a gift from Ed Brown (HMS, Boston, MA, USA). HT-29, RAW264.7, CaSR–HEK cells were grown in Dulbecco’s modified eagle media (DMEM) supplemented with 10% fetal bovine serum (FBS), and 1.0% PenStrep (all purchased from Invitrogen, Carlsbad, CA, USA). The colonic myofibroblasts were cultured in Minimum essential medium supplemented with 10% FBS and 1.0% PenStrep. All cells were maintained at 37°C, in 95% air, 5% CO2 atmospheric conditions. Cells were passaged weekly with 0.25% trypsin (except for the RAW264.7 cells which were scraped) and used for experimentation within the first six passages.
Preparation of Wnt3a and Wnt5a Conditioned Media
Mouse L-cells, stably transfected with either Wnt3a or Wnt5a plasmid, were seeded on 10 cm plates and supplemented with 10% DMEM, as previously described. After 24 h, cells were then incubated in 0.5 mM Ca2+ DMEM containing 0.1% L-Glutamine and 1.0% PenStrep and supplemented with 0.2% bovine serum albumin. This media was collected 48 and 96 h afterward. Upon collection, media was centrifuged at 175 × g for 5 min and the remaining supernatant was isolated, divided into aliquots, and kept at −30°C for later use.
Short-Interfering RNA Duplex Generation and Transfection
Short-interfering RNA (SiRNA) duplexes against the CaSR were synthesized following the protocol we previously reported (MacLeod et al., 2007; Peiris et al., 2007; Pacheco and MacLeod, 2008). Briefly after BLAST analysis of target sites in the extracellular domain of the CaSR, we synthesized siRNA directed against nucleotides 371–390 utilizing a siRNA Construction Kit (Silencer siRNA Construction Kit, Ambion) according to the manufacturer’s protocol. RAW264.7 cells were transfected with 50 nM of either siRNA against the CaSR or the scrambled sequence using Superfect reagent following manufacturer’s instructions (Qiagen, Valencia, CA, USA).
Transient Transfections and Western Blotting
All DNA plasmid transfections completed for subsequent Western blot analysis were performed using the Superfect transfection reagent, according to the manufacturer’s instructions (Qiagen, Mississauga, ON, Canada).
For the TNFR1 westerns, HT29 cells were transiently transfected with 1 μg of either pcDNA3.1 or WT–Ror2 for 24 h, serum starved in 0.5 mM CaCl2 DMEM for 24 h and treated with Wnt5a conditioned medium (CM) for 18 h. Cells were washed twice with ice-cold phosphate-buffered saline (PBS) containing 1 mM sodium vanadate and 25 mM NaF, and then 100 μl of ice-cold lysis buffer was added (20 mM Tris HCL, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 25 mM NaF, 1% Triton X-100, 10% glycerol, 1 mM dithiothreitol, 1 mM sodium Vanadate, 50 mM glycerophosphate, and a cocktail of proteases inhibitors (10 μg/ml each of aprotinin, leupeptin, calpain, pepstatin, and soybean trypsin inhibitor as well as 100 μg/ml Pefabloc). NaF, sodium vanadate, and Pefabloc were freshly prepared on the same day of the experiments. After sonicating for 5 s, lysates were centrifuged at 10,000 × g for 10 min at 4°C and processed as described (37). TNFR1 was detected using a 1:1000 dilution of primary antibody in TBS containing 0.1% Tween 20 (v/v) and 5% BSA. β-actin was detected with 1:10,000 dilution of primary antibody in TBST with 5% milk.
To measure Cullin-1 denedylation in the presence of calcium, CaSR–HEK cells were incubated in serum-free, 0.5 mM CaCl2 DMEM for 24 h and treated with 10 ng/ml LPS in the presence of either 0.5 mM CaCl2 or 3 mM CaCl2 for 18 h. Lysates were prepared as described above. Western analysis was done using a 1:1000 dilution of both Cullin-1 and Nedd8 antibodies in TBS containing 0.1% Tween 20 (v/v) and 5% BSA.
For the determination of Wnt3a or Wnt5a from RAW264.7 cells, early passage cells were grown on 100 mm dishes. Cells were transiently transfected with 1 μg of either WT–CaSR or DN–CaSR (R185Q) for 24 h, incubated for 24 h in serum-free, 0.5 mM Ca2+ DMEM (as above). This medium was removed and substituted with the same medium supplemented with 0.5 mM Ca2+ or 3 mM Ca2+, or 0.5 mM Ca2+ with LPS (10 ng/ml) for 18 h. At the end of the incubation period (18 h), CM was harvested and concentrated to 16× by Amicon Ultra filter devices (Millipore, Carrigtwohill, Ireland). Cell lysates were prepared as above. Western blot analysis was done as previously described (MacLeod et al., 2007).
For all western analysis, bands were visualized by chemiluminescence as previously described. Quantitation of the bands was done using NIH Image J v1.34 and a Personal Densitometer (Molecular Dynamics).
TNFα Elisa Analysis
Cells were seeded in 24-well plates (Sarstedt, Montreal, QC, Canada), incubated in serum-free, low Ca2+ medium for 18 h followed by 6 h treatments. CM was collected and resuspended in 300 μL. The amount of secreted TNFα was measured using a TNFα Quantikine ELISA kit (R&D, Minneapolis, MN, USA) as per manufacturer’s instructions.
Luciferase Reporter Assay
RAW264.7 cells were seeded in 24-well plates and transiently transfected 24 h later with 2 μg of NF-κB response element-driven firefly luciferase reporter plasmid (Clontech, Palo Alto, CA, USA) and 0.2 μg of thymidine kinase promoter-driven Renilla reporter plasmid (Promega) using PolyFect transfection reagent as per manufacturer’s instructions (Qiagen, Mississauga, ON, Canada). Cells were serum starved in 0.5 mM CaCl2 DMEM for 18 h and treated with 10 ng/ml LPS and either 0.5 mM CaCl2 or 3 mM CaCl2 for 6 h. Neomycin Sulfate and Spermine where required were added with LPS. Cells were harvested and NF-κB luciferase assays were performed by using the Dual Luciferase Assay according to the manufacturer’s instructions (Promega). Luminescence was measured in a Berthold Lumat 9047 Luminometer (Oakridge, TN, USA). Co-transfection with thymidine kinase–Renilla, which displays constitutive low levels of activity, was used as an internal control against which to normalize the NF-κB luciferase data. Experiments were performed in triplicate.
Transepithelial Electrical Resistance Measurements
T-84 epithelial cells were seeded to form confluent monolayers as to develop high electrical resistance on Transwell permeable supports (area of 0.33 cm2) in 24 well plates (Corning Inc.; Corning, NY, USA). The transwells filters were pre-coated in our laboratory with collagen (Sigma, Oakville, ON, Canada). The transepithelial resistance was measured using an epithelial voltmeter (WPI, Inc; Sarasota, FL, USA) and STX2 electrodes. High resistance T84 monolayers were treated overnight with 3 mM CaCl2 and treated with either 2.5 ng/ml INFγ in the basolateral reservoir and 10 ng/ml TNFα to both sides or TNFα alone. Wnt5a CM was added to both treatments for 4 h and the effect on transepithelial electrical resistance (TER) was measured.
Statistics
Data are presented as means ± SE of at least three separate experiments. Data were analyzed by Student’s t-test or ANOVA when appropriate. p < 0.05 was set as the statistically significant difference level.
Results
High ca2+ (3 mM) Inhibits LPS-Stimulated TNFα Secretion from Peritoneal Macrophages, RAW264 Macrophages and 18Co Colonic Myofibroblasts
As CaSR expression has been reported in macrophages (Olszak et al., 2000) and colonic myofibroblasts (Peiris et al., 2007; Pacheco and MacLeod, 2008), we stimulated TNFα secretion from these cells by incubation with LPS (10 ng/ml), then compared the amount of TNFα secretion in medium containing LPS and either low Ca2+ (0. 5 mM) or high Ca2+ (3.0 mM). CaSR activity is stimulated by 3 mM Ca2+ and not activated in 0.5 mM Ca2+. The high Ca2+ addition significantly reduced the LPS-stimulated TNFα secretion from mouse peritoneal macrophages (Figure 1A), from a mouse macrophage cell line, RAW264 (Figure 1B) and from the colonic myofibroblasts (Figure 1C). The most TNFα was secreted from the peritoneal macrophages, while the least was from the myofibroblasts. The amounts of LPS-stimulated TNFα from these two types of cells varied by two orders of magnitude, with the RAW264 macrophage cell line secreting 10-fold less then the peritoneal cells. Addition of high Ca2+ inhibited the LPS-stimulated TNFα secretion comparably in all three cell types.
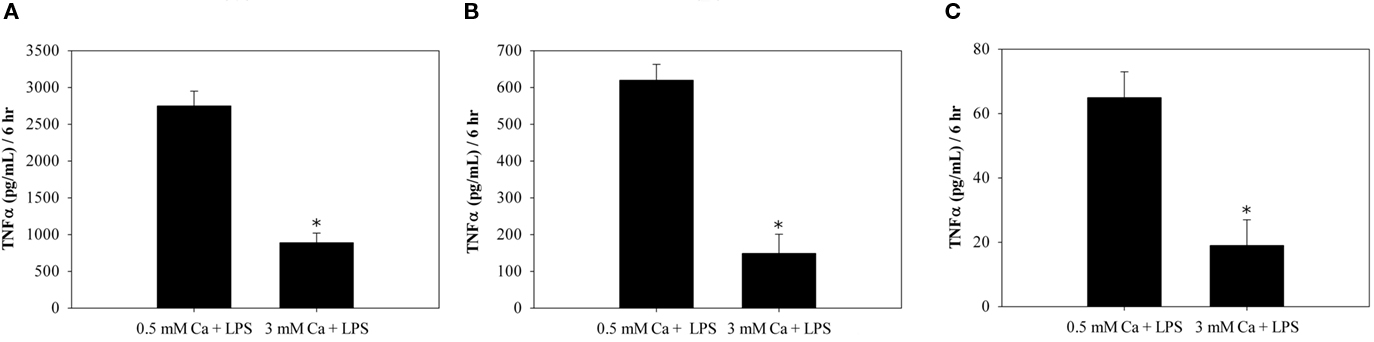
Figure 1. High calcium inhibits LPS-stimulated TNFα secretion from mouse peritoneal macrophages (A), mouse macrophage (RAW264) (B), and colonic myofibroblasts (C). Cells were serum starved for 18 h and treated with LPS in serum-free medium supplemented with either 0.5 mM CaCl2 or 3 mM CaCl2. The amount of TNFα secretion was measured using a TNFα ELISA. *p < 0.05, n = 3.
CaSR Mediates Inhibition of LPS-Stimulated TNFα Secretion and NFκb Promoter Activity
To determine if the CaSR-mediated the effects of high Ca2+ on the LPS-stimulated TNFα secretion, we used RAW264 cells and first compared other well established CaSR agonists added with LPS in low Ca2+ (0.5 mM) medium. We then we knocked down expression of the CaSR by transient transfection with siRNA duplex directed against the CaSR compared with cells transfected by a scrambled siRNA duplex (Figure 2A). Both Neomycin sulfate (350 μM) and spermine (2 mM) added in low Ca2+ medium together with LPS significantly reduced the LPS-stimulated TNFα secretion. Transient transfection with siRNA against the CaSR prevented the high Ca2+ inhibition of the LPS-stimulated TNFα (Figure 2A). The amount of TNFα secreted from the RAW264 cells transfected with a scrambled siRNA duplex and treated with 3 mM Ca2+ was significantly less then from cells comparably treated but transfected with siRNA against the CaSR (pg TNF/ml/6 h: 2048 ± 112 vs 9659 ± 1082, p < 0.05, n = 4). Together, these experiments strongly suggest that CaSR activation was responsible for the inhibition of LPS-stimulated TNFα secretion from the RAW264 macrophages. Because LPS will activate TNFα synthesis through an NFκB pathway, we then asked if CaSR activation altered LPS-stimulated NFκB reporter activity in the RAW264 cells (Figure 2B). High Ca2+ (3 mM) and other CaSR agonists (Neomycin sulfate, 350 μM) or spermine (2 mM) added with LPS had a comparable effect significantly inhibiting the LPS-stimulated activity (Figure 2B). In cells transiently transfected with siRNA duplex against the CaSR and exposed to 3 mM Ca2+ and LPS as in the earlier experiments, there was no inhibition of LPS-stimulated NFκB reporter activity. Cells transfected with scrambled siRNA duplex and exposed to 3 mM Ca2+ and LPS had significantly less activity compared with cells transfected with siRNA duplex against the CaSR (% RLU of 0.5 mM Ca2+ + LPS: 46 ± 8 vs 112 ± 9, p < 0.05, n = 4). These experiments suggested CaSR stimulation was inhibiting NFκB activity, which could lead to a reduction in TNFα secretion.
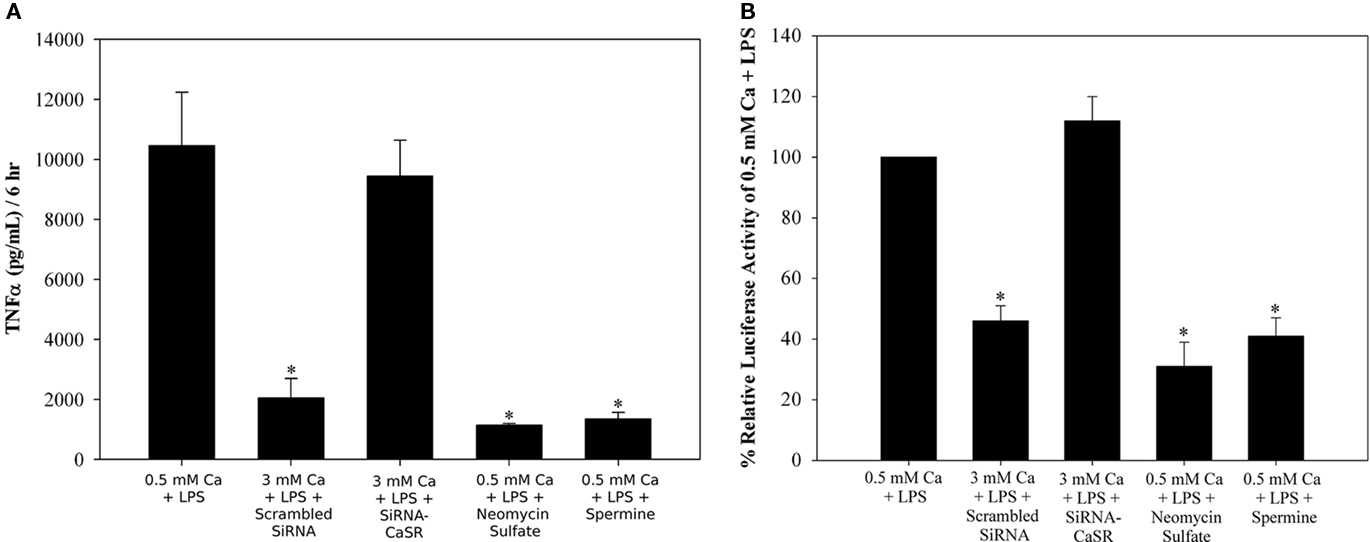
Figure 2. Calcium-sensing receptor mediates high calcium inhibition of LPS-stimulated TNFα secretion and NFκB activity from RAW264.7 macrophages. RAW264 cells were transfected with 50 pmol of either Scrambled SiRNA or SiRNA against CaSR [and a NFκB luciferase reporter in (B)] for 24 h, serum starved in 0.5 mM CaCl2 for 18 h and treated with 10 ng/mL LPS and either 0.5 mM CaCl2 or 3 mM CaCl2 for an additional 6 h. Neomycin sulfate and Spermine were added with LPS in the appropriate samples. (A) Conditioned medium was used in a TNFα ELISA to measure the secretion of TNFα. (B) Lysates were recovered and a dual luciferase assay was done to measure the NFκB reporter activity. *p < 0.05, n = 4.
CaSR Activity Results in Deneddylation of Cullin1 in HEK and RAW264 Macrophages
NFκB activity requires the E-type ubiquitin ligase Cullin1 to ubiquitinate IκB. The ability of Cullin1 to work this way requires it to be covalently modified with Nedd8. To determine whether CaSR influenced the status of Cullin1, we challenged both stably transfected CaSR–HEK cells and RAW264 cells in the presence of LPS with either low Ca2+ (0.5 mM) or high Ca2+ (3 mM) medium and 18 h later prepared lysates and measured by Western blot both Nedd8 and Cullin1 (Figure 3). Cells in the low Ca2+ medium with LPS had the characteristic two molecular weights of Cullin1, representing the covalently modified Cullin1 (as Cullin1–Nedd8) and the lower weight Cullin1. These cells also expressed immunoreactive Nedd8. In contrast, cells treated with 3 mM Ca2+ in the presence of LPS had substantially reduced Cullin1–Nedd8. The ratio of Neddylated Cullin1/Cullin1 in the RAW264.7 cells was significantly reduced compared with cells treated with 0.5 mM Ca2+ and LPS (38 ± 9%, p < 0.05, n = 3). Comparable reduction of the ratio of neddylated Cullin1/Cullin1 was also observed in the CaSR-transfected HEK cells. Treatment of non-transfected HEK cells with low and high Ca2+, in a comparable fashion, demonstrated no differences in the ratio of Neddylated Cullin1/Culllin1 (0.74 ± 0.04 vs 0.76 ± 0.07, n = 3). These results strongly suggest that CaSR activation in both RAW264 macrophages and CaSR-transfected HEK cells stimulates deneddylation of Cullin1.
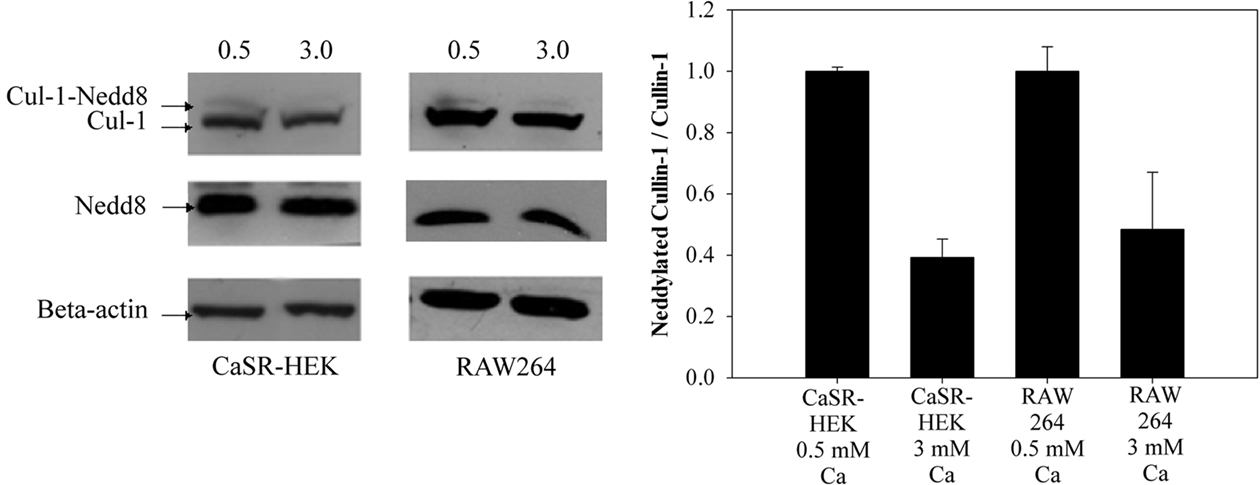
Figure 3. Calcium-sensing receptor activation stimulates denedylation of Cullin-1. CaSR–HEK and RAW264 cells were serum starved for 18 h in 0.5 mM CaCl2 and treated with 10 ng/ml LPS in the presence of either 0.5 mM CaCl2 or 3 mM CaCl2.
Wnt5a/Ror2 Reduces Expression of TNFR1 (p55) on HT-29 and T84 Monolayers
Tumor necrosis factor alpha will signal through two receptors, p55 and p75 (TNFR1 and 2 respectively). Because it had been shown earlier that CaSR activation would increase expression of Ror2 on intestinal epithelia, and that CaSR activation of colonic myofibroblasts would stimulate secretion of Wnt5a (Pacheco and MacLeod, 2008), we asked if Wnt5a/Ror2 engagement influenced expression of TNFR1. Ror2 was overexpressed in HT-29 cells and rhWnt5 added to the cells 48 h later and allowed to incubate for 18 h. Semi-quantitative RT-PCR analysis of cDNA from these cells demonstrated Wnt5a/Ror2 reduced amplicons of TNFR1, while not effecting expression of TNFR2. Western blot of HT-29 cells comparably treated demonstrated that Wnt5a/Ror2 decreased TNFR1 protein ∼70% (Figure 4) compared with cells transfected with empty plasmid. T84 cells grown on Transwells for 12 days to generate a high transepithelial resistance (TER = 3000 Ω/cm2) were first incubated treated overnight with 3 mM Ca2+ (to increase Ror2), then rhWnt5 (200 ng/ml) was added for a further 12 h, Western blot analysis demonstrated a comparable reduction in TNFR1 protein (83 ± 4%, n = 4, p < 0.05) compared with monolayers which did not receive rhWnt5a addition.
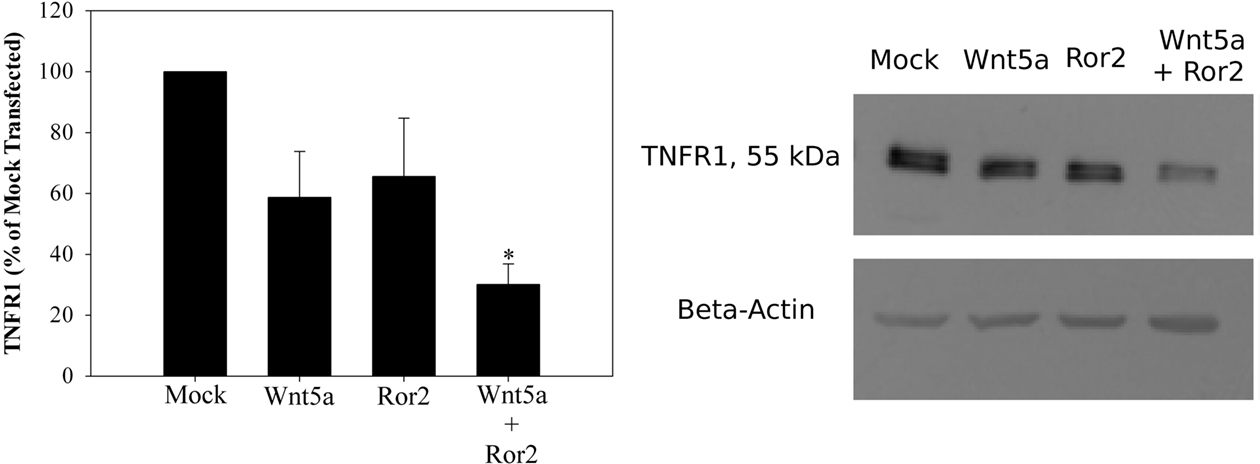
Figure 4. Wnt5a/Ror2 decrease TNFR1 protein expression. HT29 cells were transiently transfected with 1 μg WT–Ror2 (or 1 μg pcDNA3.1 as a control) for 24 h, serum starved for 24 h and treated with Wnt5a conditioned medium for 18 h. Lysates were recovered and western blot analysis demonstrated a 70% reduction in TNFR1 protein expression in Wnt5a treated, Ror2 overexpressed HT29 cells. *p < 0.05, n = 4.
Wnt5a/Ror2/CaSR Prevents Decline in TER Stimulated by TNFα in T84 Monolayers
Using high resistance T84 monolayers, we first treated these cells overnight with 3 mM Ca2+, to increase expression of Ror2. We then either “primed” the monolayer by adding INFγ (2.5 ng/ml) to the basolateral reservoir and added TNFα (10 ng/ml) to both sides in the continued presence of 3 mM Ca2+, or just added TNFα alone without priming, and compared the change in TER after 4 h following addition of various Wnt proteins to the apical chamber. Without Wnt protein addition, but in the presence of 3 mM Ca2+ the TER declined significantly (Ω/cm2: 3105 ± 139 vs 1513 ± 37, p < 0.05, n = 6, Figure 5). In the presence of a 1:1 dilution of CM from Wnt5a-secreting L-cells, this decline was prevented (Ω/cm2: 2750 ± 247 vs 2500 ± 180, ND, n = 6), irrespective of the prior priming with INFγ (Ω/cm2: 2930 ± 170 vs 2758 ± 69 Ω/cm2, ND, n = 6). Addition of rhWnt5a (200 ng/ml) to TNFα/INFγ-primed monolayers also inhibited the decline at 4 h (Ω/cm2: 2845 ± 172 vs 1513 ± 37, p < 0.05, n = 6). Addition of a 1:1 dilution of CM from Wnt3a-secreting L-cells did not prevent the TNFα/INFγ reduction in TER at 4 h (Ω/cm2: 2981 ± 155 vs 1468 ± 148, p < 0.05 n = 6). These results strongly suggest that Wnt5a, but not Wnt3a, can prevent early TNFα induced reduction in TER in model epithelial membranes.
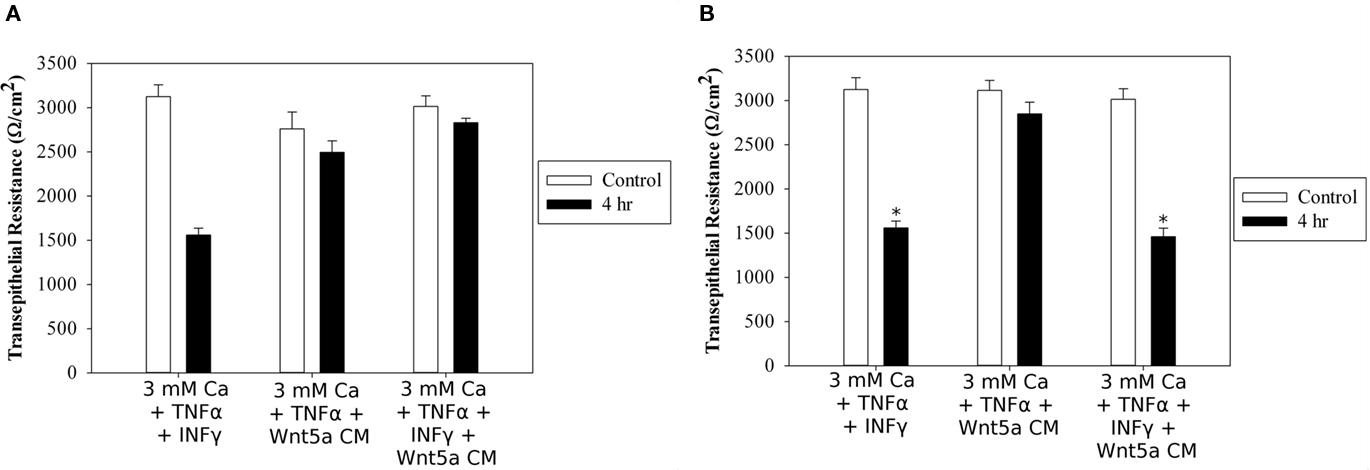
Figure 5. Wnt5a prevents reduction in TER stimulated by TNFα in T84 monolayers but Wnt3a does not. (A) High resistance T84 monolayers were treated overnight with 3 mM CaCl2 and treated with either 2.5 ng/ml INFγ in the basolateral reservoir and 10 ng/ml TNFα to both sides or TNFα alone. Wnt5a conditioned medium was added to both treatments for 4 h and the effect on TER was measured. (B) Monolayers were treated as above with both INFγ and TNFα. Either 200 ng/ml rhWnt5a or Wnt3a conditioned medium was added to the apical chamber for 4 h and the change in TER was measured. * p < 0.05, n = 6.
Wnt5a Status of RAW264.7 Cells and Wnt Protein Effects on TNFα Secretion
In a final set of experiments, we asked if CaSR activation altered Wnt protein secretion from the macrophages and if Wnt protein influenced TNFα secretion. We measured Wnt5a in the CM of RAW264.7 cells that had been transiently transfected with either wild type CaSR or a natural dominant negative CaSR R185Q, and compared the Wnt protein in CM with amount in the lysate by Western blot (Figure 6A). Addition of 3 mM Ca2+ for 18 h reduced the Wnt5a protein secreted from RAW264.7 cells; overexpression of R187Q reduced the inhibition of Wnt5a secretion caused by high Ca2+ (Figure 6A). Addition of CM from L-Wnt5a-secreting cells had no effect on LPS-stimulated TNFα secretion. Addition of rhWnt5a (200 ng/ml) also did not change the amount of TNFα secretion from these cells. However, CM from L-Wnt3a-secreting cells substantially increased the amount of LPS-stimulated TNFα secreted (Figure 6B). Wnt3a CM increased the LPS-stimulated TNFα (pg/mg/6 h: 51,000 ± 640 vs 582 ± 26, p < 0.001, n = 4). These results suggest the CaSR can mediate Wnt protein secretion from macrophages and that the canonical Wnt protein, Wnt3a, can increase TNFα secretion from macrophages.
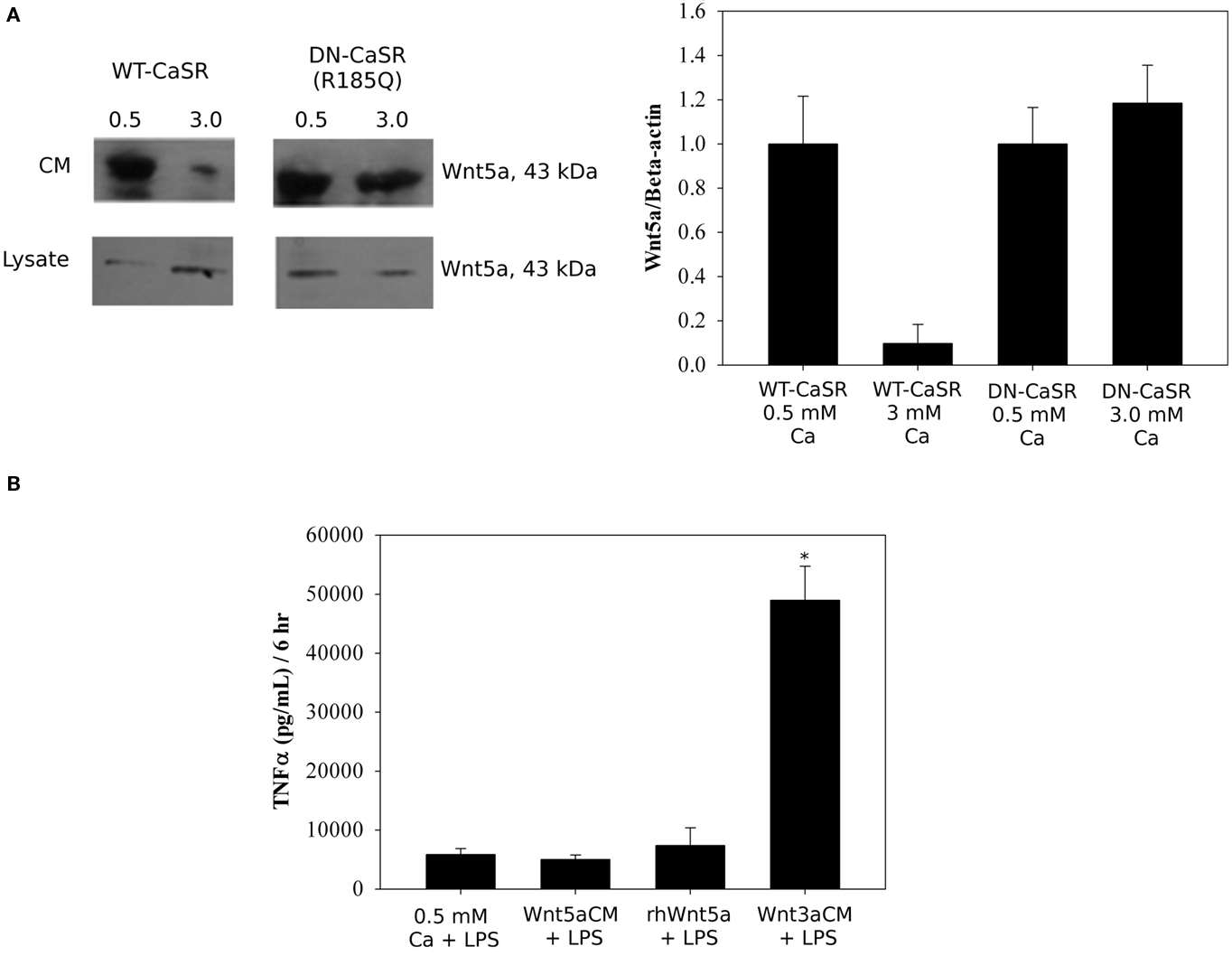
Figure 6. Calcium-sensing receptor stimulates the secretion of Wnt5a and Wnt3a increases LPS-stimulated secretion of TNFα. (A) RAW264.7 cells were transiently transfected with 1 μg of either WT–CaSR or DN–CaSR for 24 h. Cells were then serum starved in 0.5 mM CaCl2 for 24 h followed by treatment with either 0.5 mM CaCl2 or 3 mM CaCl2 for an additional 18 h. Both the conditioned medium and lysates were analyzed by Western blot. RAW264.7 cells were also treated with LPS (10 ng/ml) and the conditioned medium probed with Wnt5a. (B) RAW264.7 cells were serum starved for 24 h in 0.5 mM CaCl2 and treated with 10 ng/ml LPS and either 200 ng/ml rhWnt5a or Wnt3a conditioned medium. Conditioned medium was collected and TNFα secretion was measured using a TNFα ELISA kit.*p < 0.0011, n = 4.
Discussion
Our current findings are illustrated in the accompanying schematic (Figure 7). In the current experiments we demonstrate that following LPS challenge, mouse peritoneal macrophages, a mouse macrophage cell line, RAW264.7, and a human colonic myofibroblast cell line, 18Co, responded to increased extracellular calcium with a reduction in TNFα secretion. The mediator of inhibition was the extracellular CaSR, as knockdown of the CaSR reversed both the high Ca2+-mediated inhibition of TNFα secretion and LPS-stimulated NFκB reporter activity. CaSR stimulation was found to deneddylate Cullin1, consistent with a mechanism which could inhibit NFκB activity. Because CaSR stimulation will increase Wnt5a secretion from colonic myofibroblasts while on the overlying epithelia CaSR stimulation increases amounts of the Wnt5a receptor, Ror2, we assessed whether TNFα induced damage to a model epithelial barrier was influenced by Wnt5a. Addition of Wnt5a to intestinal epithelia that were overexpressing Ror2 substantially reduced protein expression of TNFR1. Exogenous Wnt5a prevented TNFα-stimulated decreases in transepithelial resistance in epithelial monolayers, while addition of the canonical signaling Wnt3a family member did not influence TNFα-stimulated barrier reduction. Addition of exogenous Wnt5a did not effect LPS-stimulated TNFα secretion from mouse macrophages, while Wnt3a substantially increased LPS-stimulated TNFα secretion from these cells. Together our results suggest to us that CaSR stimulation, together with the sub-epithelial and epithelial effectors of CaSR stimulation, manifested as Wnt5a/Ror2 engagement, can titrate TNFα signaling through the combined reduction of TNFα and its receptor TNFR1.
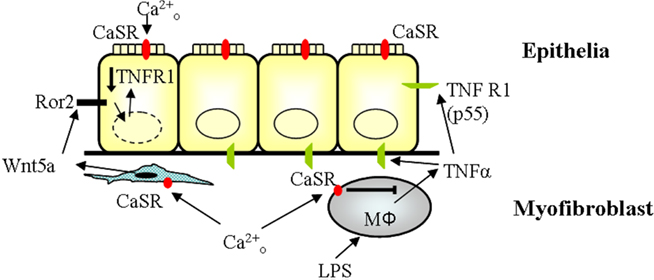
Figure 7. Multi-cell role of CaSR on TNF signaling. TNFR1 signaling is inhibited by CaSR through two mechanisms. CaSR increases the secretion of wnt5a from the myofibroblasts, which interacts with Ror2 and leads to a decrease in TNFR1 expression. CaSR also inhibits the secretion of TNFα from macrophages, thereby interrupting TNFR1 signaling.
High Ca2+ (3 mM) addition to stimulate the CaSR of macrophages and myofibroblasts previously challenged with LPS, reduced TNFα secretion. The presence of the CaSR has previously been shown on monocytes/macrophages and macrophage cell lines (Olszak et al., 2000). RAW264.7 is a well defined monocyte/macrophage cell line which has been extensively characterized for its macrophage-like responses (Boutdot et al., 2010). Reducing CaSR expression using previously characterized siRNA duplex transient transfection (MacLeod et al., 2007; Peiris et al., 2007; Pacheco and MacLeod, 2008), prevented the reduction caused by high Ca2+ in RAW264.7 cells, consistent with CaSR-mediating the inhibition of TNFα secretion. Other CaSR polyvalent agonists, such as neomycin sulfate and spermine (Brown and MacLeod, 2001), had comparable effects as high Ca2+ inhibiting LPS-stimulated NFκB reporter activity in the macrophage cell line, consistent with CaSR-mediating inhibition of NFκB activity. Because siRNA duplex against the CaSR prevented the high Ca2+-inhibition of NFκB reporter activity in these experiments, we conclude that it was activation of the CaSR which mediated the inhibition of NFκB activity to subsequently reduce LPS-stimulated TNFα secretion from the macrophages. CaSR activation has previously been shown to increase TNFα production in mTAL cells, through pertussis toxin-sensitive and PKC requiring pathway(s) (Wang et al., 2003; Abdullah et al., 2008). Indeed, in mature rabbit osteoclasts, calcium-sensing receptor activation has been shown to stimulate nuclear translocation of NFκB (Mentaverri et al., 2006). These studies differ from our current studies as they did not use LPS to stimulate NFκB prior to CaSR activation. Our current studies suggest that CaSR activation after NFκB has been stimulated (at least through TLR4 activation) inhibits NFκB activity, leading to inhibition of LPS-stimulated TNFα secretion. We speculate that CaSR stimulation generates a negative feedback inhibition of Toll-like receptor mediated NFκB activation in macrophages and stromal cells.
Insight into how CaSR activation inhibits NFκB activity came from our experiments measuring the amount of neddylated Cullin1 in macrophages and CaSR-transfected HEK cells. Inhibitors of NFκB (IκBs) hold the NFκB transcription factors in the cytoplasm (Cardozo and Pagano, 2004). Proteosomal degradation of IκB proteins that inhibit NFκB is a highly regulated process. The E3 SCF ubiquitin ligase specific to IκB family members comprising SKP1, Cullin1, and the F-box domain of β-TrCP, is responsible for the polyubiquitination of IκB (Read et al., 2000; Pan et al., 2004; Khoury et al., 2007). The activity of the E3 ligases requires the regulatory subunit of Cullin1 to be posttranslationally modified by the ubiquitin-like protein Nedd8. Loss of Nedd8 modification results in loss of SCF activity. Deneddylated Cullin1 is incapable of ubiquitination of IκB. Lack of ubiquitination of IκB generates inactivation of NFκB. Adenosine activating the A2b receptor inhibits TNFα secretion from macrophages by stimulating deneddylation of Cullin1 (Khoury et al., 2007). Glutamine will also stimulate deneddylation of Cullin1, reducing NFκB signaling and TNFα release in a post-sepsis model, but the status of TNFR1 was not addressed in these studies (Singleton and Wishmeyer, 2008). Because CaSR activation will reduce the ratio of neddylated Cullin1 to Cullin1, our results suggest to us that CaSR stimulation inhibits NFκB activity at the level of proteosomal degradation of IκB proteins. Further studies are required to identify the signalosome responsible for the loss of Nedd8 modification after CaSR stimulation.
Wnt5a added to epithelial cells where Ror2 was either overexpressed or increased by CaSR activation, reduced expression of TNFR1, and prevented TNFα-induced changes in transepithelial resistance of a model epithelial barrier. Wnt5a/Ror2 engagement has been shown to increase CDX2 expression and sucrase–isomaltase expression in model epithelia (Pacheco and MacLeod, 2008). The signaling cascades responsible for the former increases are JNK, Casein kinase, and Src kinase activation, while Wnt5a/Ror2 production of villin requires only pERK activation. Wnt5a/Ror2 signaling has recently been reviewed (Minami et al., 2010; Nishita et al., 2010; Sato et al., 2010) and appears both cell and tissue specific, highlighting that in many cell types, Ror2 will also work as a co-receptor allowing canonical Wnt signaling to occur. Nevertheless Wnt5a/Ror2 activation reduced TNFR1 protein expression substantially in model intestinal epithelia. Histone deacetylase inhibitors added to non-small lung cell cancer cells down-regulate TNFR1 transcript and protein, as well as reduce surface exposure of TNFR1, leading to substantially reduced NFκB signaling in response to exogenous TNFα addition (Singleton and Wishmeyer, 2008). It is not known if Wnt5a/Ror2 alter histone deacetylase activity in enterocytes.
Wnt5a/Ror2 activation in intestinal epithelia leading to a reduction in TNFR1 will be addressed in a separate study. Nevertheless, Wnt5a prevented the barrier damaging effects of exogenous TNFα addition to a model epithelial barrier in the current study. Addition of Wnt3a, a prototypical canonical Wnt signaling family member, did not prevent the reduction in transepithelial resistance we observed after TNFα addition. Consistent with other studies (Ye et al., 2006) we interpret the reduction of transepithelial resistance after TNFα addition to increases in paracellular permeability because of tight junction disruption. While it is possible that Wnt5a/Ror2 engagement prevents tight junction disruption by altering myosin light chain kinase (MLCK) activity or expression, the simplest explanation of our results is due to the reduction in TNFR1 protein. We included INFγ in the T84 experiments because others had shown that INFγ could prime Caco-2 monolayers to respond to TNFα addition by increasing expression of TNFR2 (Wang et al., 2005, 2006). These studies suggested that the deleterious effect of TNFα on epithelial barriers was mediated predominantly through TNFR2. Our current studies did not use Caco-2 monolayers. We found that with INFγ or without INFγ, the effect of exogenous TNFα addition was comparable on T84 monolayer resistance, and that the presence of INFγ did not effect Wnt5a prevention of the TNFα-induced reduction of transepithelial resistance. We also found that Wnt5a/Ror2 engagement did not alter TNFR2 expression in HT-29 cells and T84 monolayers (data not shown). The absence of a “priming” effect of INFγ on the T84 monolayers may reflect either expression levels of TNFR2 in these cells, or an effect of CaSR activation on TNFR2. Our current results are best described as CaSR-mediating an increase in Ror2 on the epithelia (Pacheco and MacLeod, 2008) allowing CaSR-mediated Wnt5a (Pacheco and MacLeod, 2008) from the sub-epithelial myofibroblasts to engage Ror2 leading to a reduction in TNFR1. The reduced TNFR1 prevents TNFα signaling that would otherwise lead to tight junction disruption and epithelial barrier loss.
Wnt5a added to RAW264.7 macrophages which were stimulated with LPS did not further increase TNFα secretion from these cells. However, addition of Wnt3a dramatically stimulated TNFα secretion from these LPS-treated cells. When Wnt secretion into CM was assessed, we found that CaSR activation inhibited Wnt5a secretion. We conclude that CaSR-mediated this effect, since transient transfection with a dominant negative CaSR, R185Q (Peiris et al., 2007) restored Wnt5a secretion from the macrophages. While the determinants of Wnt protein secretion are currently being elucidated, little is known about how a GPCR like the CaSR can mediate the secretion of one Wnt family member over another (Bartscherer et al., 2006; Belenkaya et al., 2008; Angers and Moon, 2009; Jin et al., 2010; Port and Basler, 2010). Our current results suggest that there may be plasticity in regulating the type of Wnt family member secreted from native macrophages compared with activated cells in response to GPCR activation. In this regard, recent studies have demonstrated distinct effects of Wnt5a and Wnt3a in human macrophages. TLR2 and TLR4 activation by Mycobacterium tuberculosis increased expression of Wnt5a and Frizzled5 (Fzd5; Blumenthal et al., 2006). LPS/INFγ treatment increased Wnt5a which appeared to work in an autocrine fashion, using Fzd5 to stimulate CAMKII (Pereira et al., 2008). M. tuberculosis infection of mice has been shown to up-regulate Frizzled1 on macrophages. Frizzled1 is a Frizzled family member which may be activated by Wnt3a. This study found that exogenous Wnt3a to the macrophages inhibited the release of TNFα (Neumann et al., 2010). Our current results differ from these findings through the use of LPS to solely activate TLR4 on the macrophages. We speculate that signaling distal to TLR4 stimulation allows canonical Wnt signaling to cross-talk to increase TNFα secretion.
Our current in vitro findings are interesting in the context of the extensive epidemiological evidence showing increased dietary calcium (and dairy) intake diminishes the onset of colon cancer (Shaukat et al., 2005; Park et al., 2009). It is not understood in several studies (Yang et al., 2008; Newmark et al., 2009) whether it is calcium or vitamin D which is the protective factor. For example, APCmin mice fed increased calcium still develop tumors, while the largest tumor reduction occurred with increased calcium and 1,25-Vitamin D3 (Huerta et al., 2003). As the CaSR promoter contains several VDR (Canaff and Hendy, 2002), and the expression of the colonic CaSR in several mouse strains is quite low (MacLeod and kelly, data not shown), we hypothesize that increased dietary 1,25-Vitamin D increases colonic CaSR expression, allowing calcium activation of the CaSR to stimulate Wnt5a/Ror2 signaling. How does increased dietary calcium diminish colon cancer onset? The amounts of ionized calcium in the colonic lumen after increased dietary calcium have not been measured. Nevertheless, if 80–90% of calcium absorption occurs in the duodenum, a fraction of the non-absorbed calcium may reach the colon. With increased dietary calcium the non-absorbed fraction may be proportionately increased, to activate the CaSR to stimulate Wnt5a/Ror2 interaction and signaling. Further work using in vivo models is required to understand agonist stimulation of the CaSR in the settings of the colon.
In summary we report that following LPS-stimulation of peritoneal macrophages, a murine macrophage cell line and colonic myofibroblasts, activation of the extracellular CaSR, reduced NFκB promoter activity and TNFα secretion. This reduction may be due to CaSR stimulation causing deneddylation of Cullin1. In model epithelial cells and monolayers, the previously documented non-canonical Wnt signaling cascade mediated by the CaSR–Wnt5a interacting with Ror2, substantially reduced TNFR1 protein. Exogenous Wnt5a, but not Wnt3a, when added to epithelial barriers which had been treated with high Ca2+ to activate the CaSR (to produce more Ror2) were protected from barrier disruption caused by TNFα/INFγ addition. Activation of the CaSR on the macrophage cell line altered Wnt protein secretion, and in macrophages treated with LPS, Wnt3a substantially enhanced TNFα secretion. We conclude that our results suggest that after LPS-stimulation, CaSR activation of macrophages, epithelia, and subepithelia working together can titrate the effects of increased TNFα on the intestinal barrier to generate a novel anti-inflammatory effect.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge funding from the Canadian Crohn’s and Colitis Foundation, NSERC, and The Dairy Farmers of Canada.
References
Abdullah, H. I., Pedraza, P. L., McGiff, J. C., and Ferreri, N. R. (2008). CaR activation increases TNF production by mTAL cells via a Gi-dependent mechanism. Am. J. Physiol. Renal Physiol. 294, F345–F354.
Angers, S., and Moon, R. T. (2009). Proximal events in Wnt signal transduction. Nat. Rev. Mol. Cell Biol. 10, 468–477.
Bartscherer, K., Pelte, N., Ingelfinger, D., and Boutros, M. (2006). Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell 125, 523–533.
Belenkaya, T. Y., Wu, Y., Tang, X., Zhou, B., Cheng, L., Sharma, Y. V., Yan, D., Selva, E. M., and Lin, X. (2008). The retromer complex influences Wnt secretion by recycling wntless from endosomes to the trans-Golgi network. Dev. Cell 14, 120–131.
Blumenthal, A., Ehlers, S., Lauber, J., Buer, J., Lange, C., Goldmann, T., Heine, H., Brandt, E., and Reiling, N. (2006). The wntless homolog Wnt5a and its receptor Frizzled 5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood 108, 965–973.
Boutdot, C., Saidak, Z., Boulanouar, A., Petit, L., Gouilleux, F., Massy, Z., Brazier, M., Metaverri, R., and Kamel, S. (2010). Implication of the calcium sensing receptor and P13K/Akt pathway in the extracellular calcium-mediated migration of RAW264.7 osteoclast precursor cells. Bone 46, 1416–1423.
Brown, E. M., and MacLeod, R. J. (2001). Extracellular calcium-sensing and extracellular calcium signaling. Physiol. Rev. 81, 239–297.
Bruewer, M., Lugering, A., Kucharzik, T., Parkos, C. A., Madara, J. L., Hopkins, A. M., and Nusrat, A. (2003). Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J. Immunol. 171, 6164–6172.
Calorini, L., Mannini, A., Bianchini, F., Mugnai, G., and Ruggieri, S. (2000). The change in leukotrienes and lipoxins in activated mouse peritoneal macrophages. Biochim. Biophys. Acta 1484, 87–92.
Canaff, L., and Hendy, G. N. (2002). Human calcium-sensing receptor gene. Vitamin D response elements in promoters P1 and P2 confer transcriptional responsiveness to 1,25-dihydroxyvitamin D. J. Biol. Chem. 277, 30337–30350.
Cardozo, T., and Pagano, M. (2004). The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 5, 739–751.
Huerta, S., Irwin, R. W., Heber, D., Go, V. L., Moatamed, F., Huerta, S., Ou, C., and Harris, D. M. (2003). Intestinal polyp formation in the Apcmin mouse: effects of levels of dietary calcium and altered vitamin D homeostasis. Dig. Dis. Sci. 48, 870–876.
Jin, J., Kittanakom, S., Wong, V., Reyes, B. A., Van Bockstaele, E. J., Stagljar, I., Berrettini, W., and Levenson, R. (2010). Interaction of the mu-opioid receptor with GPR177 (Wntless) inhibits Wnt secretion: potential implications for opioid dependence. BMC Neurosci. 11, 33. doi: 10.1186/1471-2202-11-33
Khoury, J., Ibla, J. C., Neish, A. S., and Cogan, S. P. (2007). Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J. Clin. Investig. 117, 703–711.
Lee, G., Goretsky, T., Managlia, E., Dirisina, R., Singh, A. P., Brown, J. B., May, R., Yang, G. Y., Ragheb, J. W., Evers, B. M., Weber, C. R., Turner, J. R., He, X. C., Katzmann, R. B., Li, L., and Barret, T. A. (2010). Phosphoinositide 3-kinase signaling mediates beta-catenin activation in intestinal epithelial stem and progenitor cells in colitis. Gastroenterology 139, 869–881.
MacLeod, R. J., Hayes, M., and Pacheco, I. (2007). Wnt5a secretion stimulated by the extracellular calcium-sensing receptor inhibits defective Wnt signaling in colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G403–G411.
McKay, D. M., Croitoru, K., and Perdue, M. H. (1996). T cell monocyte interactions regulate epithelial physiology in a coculture model of inflammation. Am. J. Physiol. 270, C418–:C428.
Mentaverri, R., Yano, S., Chattopadhyay, N., Petit, L., Kifor, O., Kamel, S., Terwilliger, E. F., Brazier, M., and Brown, E. M. (2006). The calcium-sensing receptor is directly involved in both osteoclast differentiation and apoptosis. FASEB J. 20, E1945–E1954.
Minami, Y., Oishi, I., Endo, M., and Nishita, M. (2010). Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: their implications in developmental morphogenesis and human diseases. Dev. Dyn. 239, 1–15.
Neumann, J., Schaale, K., Farhat, K., Endermann, T., Ulmer, A. J., Ehlers, S., and Reiling, N. (2010). Frizzled 1 is a marker of inflammatory macrophages, and its ligand Wnt3a is involved in reporgramming Mycobacterium tuberculosis-infected macrophages. FASEB J. 24, 4599–4612.
Newmark, H. L., Yang, K., Kurihara, N., Fan, K., Augenlicht, L. H., and Lipkin, M. (2009). Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis 30, 88–92.
Nielson, O. H., Vainer, B., Madsen, S. M., Seidelin, J. B., and Heegaard, N. H. (2000). Established and emerging biological activity markers of inflammatory bowel disease. Am. J. Gastroenterol. 95, 359–367.
Nishita, M., Enomoto, M., Yamagata, K., and Minami, Y. (2010). Cell/tissue-tropic functions of Wnt5a signaling in normal and cancer cells. Trends Cell Biol. 20, 346–354.
Olszak, I. T., Poznansky, M. C., Evans, R. H., Olson, D., Kos, C., Pollak, M. R., Brown, E. M., and Scadden, D. T. (2000). Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. J. Clin. Investig. 105, 1299–1305.
Pacheco, I. I., and MacLeod, R. J. (2008). Extracellular calcium-sensing receptor (CaSR) stimulates secretion of Wnt5a from colonic myofibroblasts to stimulate CDX2 and sucrase isomaltase using Ror2 on intestinal epithelia. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G748–G759.
Pan, C. L., Baum, P. D., Gu, M., Jorgensen, E. M., Clark, S. G., and Garriga, G. (2008). C. elegans AP-2 and retromer control Wnt signaling by regulating mig-14/Wntless. Dev. Cell 14, 132–139.
Pan, Z. Q., Kentsis, A., Dias, D. C., Yomah, K., and Wu, K. (2004). Nedd8 on cullin: building an expressway to protein destruction. Oncogene 23, 1985–1997.
Park, Y., Leitzmann, M. F., Subar, A. F., Hollenbeck, A., and Schatzkin, A. (2009). Dairy food, calcium and the risk of cancer in the NIH-AARP diet and health study. Arch. Intern. Med. 169, 391–401.
Peiris, D., Pacheco, I., Spencer, C., and MacLeod, R. J. (2007). The extracellular calcium-sensing receptor reciprocally regulates the secretion of BMP-2 and the BMP antagonist noggin in colonic myofibroblasts. Am. J. Physiol. 292, G753–G766.
Pereira, C., Schaer, D. J., Bachli, E. B., Kurrer, M. O., and Schoedon, G. (2008). Wnt5a/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the anti-inflammatory action of activated protein C and interleukin 10. Arterioscler. Thromb. Vasc. Biol. 28, 504–510.
Popivanova, B. K., Kitamura, K., Wu, Y., Konodo, T., Kagaya, T., Kaneko, S., Oshima, M., Fujii, C., and Mukaida, N. (2008). Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 118, 560–570.
Port, F., and Basler, K. (2010). Wnt trafficking: new insights into Wnt maturation, secretion and spreading. Traffic 11, 1265–1271.
Quesniaux, V., Erard, F., and Ryffel, B. (2010). Adjuvant activity on murine and human macrophages. Methods Mol. Biol. 626, 117–130.
Read, M., Brownell, J. E., Gladsheve, T. B., Hottelet, M., Parent, L. A., Coggins, M. B., Pierce, J. W., Podust, V. N., Lou, R., Chau, V., and Palombella, V. J. (2000). Nedd8 modification of Cul-1 activates SCFbetaTrCP-dependent ubiquitination of IkappaBalpha. Mol. Cell Biol. 20, 2326–2333.
Rutter, M., Saunders, B., Wilkinson, K., Rumbles, S., Schfield, G., Kamm, M., Williams, C., Price, A., Talbot, I., and Forbes, A. (2004). Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 126, 451–459.
Sato, A., Yamamoto, H., Sakane, H., Koyama, H., and Kikuchi, A. (2010). Wnt5a regulates distinct signaling pathways by binding to Frizzled 2. EMBO J. 29, 41–54.
Shaukat, A., Arain, T. M., Shahid, A., Irfan, S., and Farrukh, S. (2005). Estrogen replacement therapy for depression in perimenopausal women. J. Coll. Physicians Surg. Pak. 15, 597–600.
Singleton, K. D., and Wishmeyer, P. E. (2008). Glutamine attenuates inflammation and NFkappaB activation via Cullin-1 deneddylation. Biochem. Biophys. Res. Commun. 373, 445–449.
Targan, S. R., Hanauer, S. B., van Deventer, S. J., Mayer, L., Present, D. H., Braakman, T., DeWoody, K. L., Schaible, T. F., and Rutgeerts, P. J. (1997). A short-term study of chimeric monoclonal antibodycA2 to TNF-alpha for Crohn’s disease. N. Engl. J. Med. 337, 1029–1035.
Wang, D., Pedraza, P. L., Abdullah, H. I., McGiff, J. C., and Ferreri, N. R. (2003). Calcium-sensing receptor-mediated TNF production in medullary thick ascending limb cells. Am. J. Physiol. Renal Physiol. 263, F963–F970.
Wang, F., Graham, W. V., Wang, Y., Witkowski, E. D., Schwarz, B. T., and Turner, J. T. (2005). Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 166, 409–419.
Wang, F., Schwarz, B. T., Graham, W. V., Wang, Y., Su, L., Clayburgh, D. R., Abraham, C., and Turner, J. R. (2006). INF-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology 131, 1152–1163.
Wehner, S., Buchholz, B. M., Schuchtrup, S., Rocke, A., Schaefer, N., Lysson, M., Hirner, A., and Kalff, J. C. (2010). Mechanical strain and TLR4 synergistically induce cell-specific inflammatory gene expression in intestinal smooth muscle cells and peritoneal macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G1187–G1197.
Wu, Q., Feng, Y., Yang, Y., Jingliu, Zhou, W., He, P., Zhou, R., Li, X., and Zou, J. (2004). Kinetics of the phenotype and function of murine peritoneal macrophages following acute inflammation. Cell. Mol. Immunol. 1, 57–62.
Yang, K., Lamprecht, S. A., Shinozaki, H., Fan, K., Yang, W., Newmark, H. L., Kopelovich, L., Edelmann, W., Jin, B., Gravaghi, C., Augenlicht, L., Kucherlapati, R., and Lipkin, M. (2008). Dietary calcium and cholecalciferol modulate cyclin D1 expression, apoptosis, and tumorigenesis in intestine of adenomatous polyposis coli1638N/+ mice. J. Nutr. 138, 1658–1663.
Keywords: extracellular calcium-sensing receptor, Wnt5a, Wnt3a, Ror2, non-canonical Wnt signaling, TNFα secretion
Citation: Kelly JC, Lungchukiet P and MacLeod RJ (2011) Extracellular calcium-sensing receptor inhibition of intestinal epithelialTNF signaling requires CaSR-mediated Wnt5a/Ror2 interaction. Front. Physio. 2:17. doi: 10.3389/fphys.2011.00017
Received: 04 November 2010; Paper pending published: 27 November 2010;
Accepted: 31 March 2011; Published online: 05 May 2011.
Edited by:
Peter Hegyi, University of Szeged, HungaryReviewed by:
Diego Alvarez de la Rosa, University of La Laguna, SpainOsvaldo Rey, University of California Los Angeles, USA
Copyright: © 2011 Kelly, Lungchukiet and MacLeod. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: R. John MacLeod, Gastrointestinal Diseases Research Unit, Department of Medicine, Kingston General Hospital, 76 Stuart Street, Rm 3-003, GIDRU Wing, Kingston, ON K7L 2V7, Canada.e-mail:cmptNUBxdWVlbnN1LmNh