

- Division of Gastroenterology, Hepatology and Nutrition, Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
The human microbiota presents a highly active metabolic that influences the state of health of our gastrointestinal tracts as well as our susceptibility to disease. Although much of our initial microbiota is adopted from our mothers, its final composition and diversity is determined by environmental factors. Westernization has significantly altered our microbial function. Extensive experimental and clinical evidence indicates that the westernized diet, rich in animal products and low in complex carbohydrates, plus the overuse of antibiotics and underuse of breastfeeding, leads to a heightened inflammatory potential of the microbiota. Chronic inflammation leads to the expression of certain diseases in genetically predisposed individuals. Antibiotics and a “clean” environment, termed the “hygiene hypothesis,” has been linked to the rise in allergy and inflammatory bowel disease, due to impaired beneficial bacterial exposure and education of the gut immune system, which comprises the largest immune organ within the body. The elevated risk of colon cancer is associated with the suppression of microbial fermentation and butyrate production, as butyrate provides fuel for the mucosa and is anti-inflammatory and anti-proliferative. This article will summarize the work to date highlighting the complicated and dynamic relationship between the gut microbiota and immunity, inflammation and carcinogenesis.
Introduction
The distal intestine is populated by a stunning quantity of bacteria, comprised of relatively few phyla that are highly diverse at the species level. This heterogeneous composition presents a vibrant metabolically active life-form, sharing the same habitat as our enteric organs. Realistically, enteric microbiota are a continuum, with members existing somewhere along the spectrum between symbiosis and pathogenicity. As the microbiota co-evolved with humans, they have provided us with genetic and metabolic characteristics that we have not had to develop on our own, such as enabling us to be able to harvest otherwise inaccessible nutrients and synthesizing vitamins (O’Keefe et al., 2009). Resistant starch and non-starch polysaccharides that evade human digestion provide energy for commensal bacteria. In turn, microbes metabolize these undigested nutrients to produce short-chain fatty acids (SCFA), including butyrate, the primary nourishment for colonocytes which possesses potent anti-proliferative and anti-inflammatory properties (Pryde et al., 2002). Alternatively, enteric microbes may enhance disease pathogenesis at the cellular and molecular level via pro-inflammatory mechanisms (Table 1).

Table 1. Cellular and molecular evidence for the role of the microbiota in inflammation and disease.
Evolution of the Microbiota in Atopic Disease
Microbial colonization of the gastrointestinal tract begins at birth, changes rapidly during the first year of life, and thereafter remains fairly constant (Abraham and Cho, 2009). During infancy, symbiotic bacteria have the capacity to promote growth and healing, induce angiogenesis, optimize nutrient absorption, attenuate intestinal inflammation, and prime the innate and the adaptive arms of the immune system (Vaarala, 2003). Aberrations in the biodiversity of enteric microbiota can contribute to individual differences in immunologic behavior during and subsequent to childhood. Western nations, for instance, have experienced consistent increases in the incidence of allergic diseases in the past few decades, which may be due to a lack of microbial exposure during infancy – the “hygiene hypothesis” – or adoption of a Western diet (Strachan, 1989; Wang et al., 2008). A synchronous decrease in the incidence of infectious diseases in developed countries has occurred subsequent to the implementation of antibiotics, vaccination, and improvements in hygiene (Bach, 2002).
Mounting evidence suggests that diet and the microbiota, independently or in conjunction, influence the risk of developing atopic disease. The constitution of enteric microbiota may be a consequence of one’s country of origin. Infants living in developing countries have been shown to be colonized at younger ages with fecal bacteria and have more rapid transfer of enteric microbial strains than infants living in developed countries (Adlerberth et al., 1998). Diversity is considerably greater in rural African than European children (Figure 1), with a predominance of resistant polysaccharide hydrolyzers (Prevotella and Xylanibacter) and relative absence of inflammatory Enterobacteriaceae (De Filippo et al., 2010). Compared to children without atopic illnesses, microbial species of children who manifest atopic sensitization demonstrate a reduced ratio of bifidobacteria to clostridia when they are infants (Kalliomaki et al., 2001a). Important determinants of enteric microbiotic composition in infants appear to be the mode of delivery, maternal diet, type of infant feeding (breast milk or formula), gestational age, infant hospitalization, antibiotic use by the infant, and the presence of siblings (Penders et al., 2006). One study comparing the enteric microbial species constitutions of 7-year-old children noted that those delivered by Cesarean section had significantly lower quantities of clostridia and bifidobacteria (Salminen et al., 2004). Additionally, infants who are treated frequently with antibiotics have a heightened likelihood of developing asthma (Marra et al., 2009).
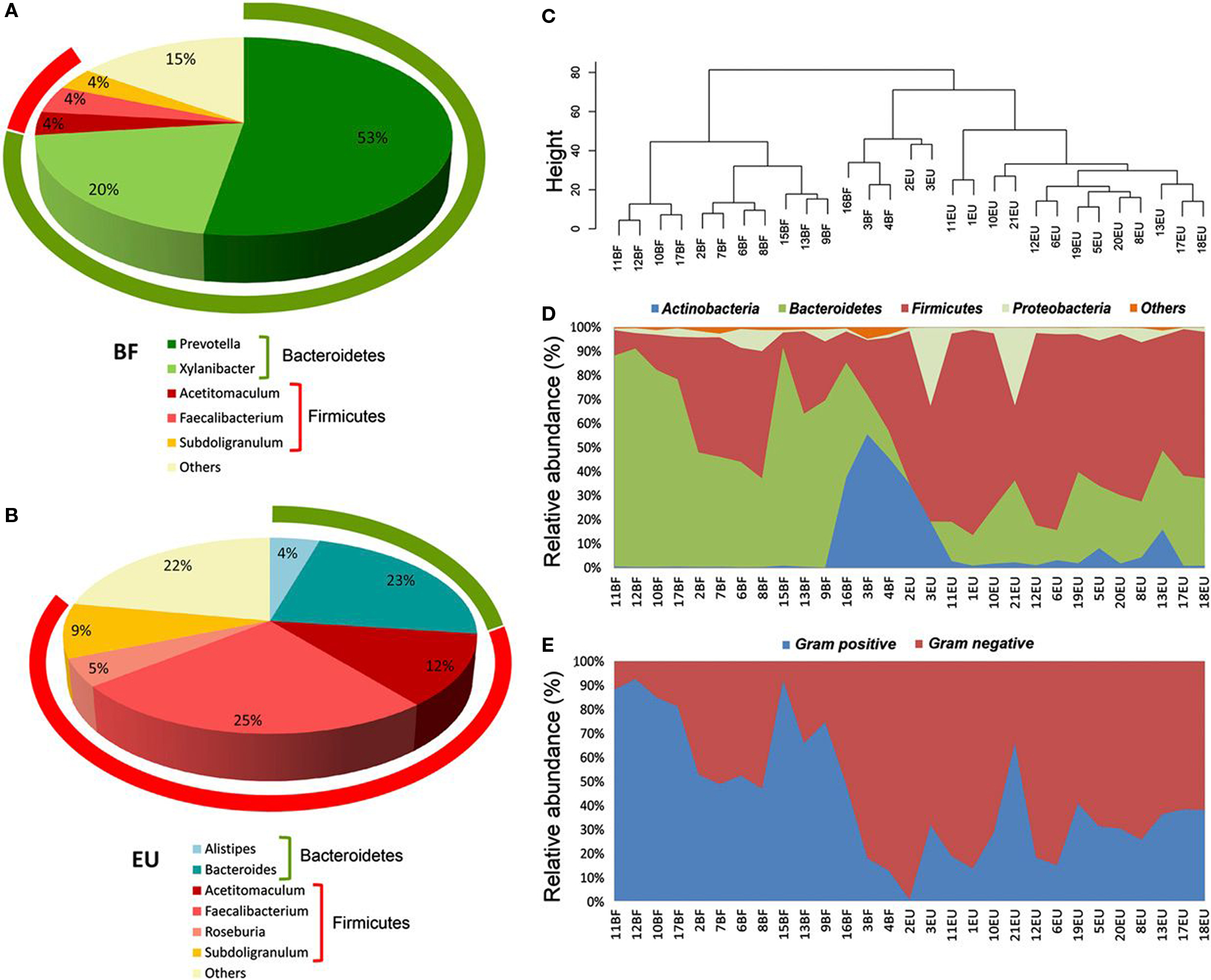
Figure 1. 16S rRNA gene surveys reveal a clear separation of two children populations investigated. (A,B) Pie charts of median values of bacterial genera present in fecal samples of Burkina Faso (BF) and European Union (EU) children (>3%) found by RDP classifier v. 2.1. Rings represent corresponding phylum (Bacteroidetes in green and Firmicutes in red) for each of the most frequently represented genera. (C) Dendrogram obtained with complete linkage hierarchical clustering of the samples from BF and EU populations based on their genera. The subcluster located in the middle of the tree contains samples taken from the three youngest (1–2 years old) children of the BF group (16BF, 3BF, and 4BF) and two 1-year-old children of the EU group (2EU and 3EU). (D) Relative abundances (percentage of sequences) of the four most abundant bacterial phyla in each individual among the BF and EU children. Blue area in middle shows abundance of Actinobacteria, mainly represented by Bifidobacterium genus, in the five youngest EU and BF children. (E) Relative abundance (percentage of sequences) of Gram-negative and Gram-positive bacteria in each individual. Different distributions of Gram-negative and Gram-positive in the BF and EU populations reflect differences in the two most represented phyla, Bacteroidetes and Firmicutes. Adapted from De Filippo et al. (2010).
Breast feeding is associated with a diminished risk of atopic diseases, and alteration of the mother’s diet to avoid n−6 saturated fat and include supplementation with Lactobacillus rhamnosus GG (ATCC 53103), has been shown to reduce risk by 50% (Kalliomaki et al., 2001b). Diet in early life may also affect the likelihood of developing allergic illnesses. For instance, infants fed a strictly organic diet were found to have a significantly lower likelihood of developing eczema (Kummeling et al., 2008). Dietary lipids, particularly n−6 long-chain polyunsaturated fatty acids, have a pro-inflammatory effect through the increased synthesis of prostaglandin E2; their consumption can contribute to the development of allergies and asthma (Black and Sharpe, 1997). N−3 polyunsaturated fatty acids, on the other hand, possess a demonstrated ability to decrease the inflammatory response via their reduction in prostaglandin E2 levels (Kobayashi et al., 2006). Diet may, thus, affect the development of allergic disease both directly by its effect on inflammation and indirectly through its influence on the composition and activity of the microbiota.
Colon Cancer
Robust experimental and clinical evidence demonstrates that chronic inflammation increases the risk of neoplastic transformation (Ekbom et al., 1990; Baron and Sandler, 2000; Coussens and Werb, 2002; Garcea et al., 2005). Not surprisingly, studies have linked colonic carcinogenesis to chronic inflammation generated by “pathogenic” bacteria. Although specific bacterial species have yet to be proven to be the cause of colon cancer, the minor gut colonizer Streptococcus bovis has been implicated in some clinical studies. Experimental investigations into its carcinogenic potential showed that bacterial proteins upregulated COX-2 production and promoted cellular proliferation by triggering mitogen-activated protein kinases which can increase the incidence of cell transformation as well as the rate of acquiring genetic mutations (Baron and Sandler, 2000; Biarc et al., 2004). Pro-inflammatory cytokines act as tumor promoters by regulating cytokines, chemokines, adhesion molecules, and angiogenesis (Coussens and Werb, 2002). A recent study of multiple intestinal neoplasia mice showed that infection with enterotoxin-producing Bacteroides fragilis organisms that produce inflammatory diarrhea in humans triggered colitis and strongly induced colonic tumors (Wu et al., 2009). Clinical support for the importance of chronic inflammation is provided by the observation that colon cancer risk is increased five-fold in patients with chronic ulcerative colitis (Ekbom et al., 1990). Furthermore, the risk of cancer in inflammatory bowel disease (IBD) is considerably higher if the disease affects the colon, rather than the small intestine, indicating that the carcinogenic effects of bacteria and inflammation may be additive. Finally, it has been clearly established that the use of anti-inflammatory medications suppresses colon cancer risk (Baron and Sandler, 2000).
Substantial epidemiological evidence also supports a primary role for diet in the genesis of colorectal carcinoma (Doll and Peto, 1981). Comparative studies from high and low risk populations indicate that elevated intake of red meat and animal fat increase colon cancer risk, while greater consumption of fiber reduces risk. The effect of diet on colonic carcinogenesis may be mediated by the microbiota, which can either maintain colonic health or promote chronic inflammation (O’Keefe et al., 2009). Fermentation of indigestible carbohydrate, such as fiber and resistant starch, maintains colonic health by producing SCFA such as acetate, propionate, and butyrate. Unlike most other cells in the body, colonocytes use butyrate as their preferred energy source. Furthermore, butyrate appears to be fundamental in the maintenance of cellular homeostasis and a normal colonocyte phenotype (Topping and Clifton, 2001). SCFAs have anti-inflammatory effects, either directly by regulating the release of prostaglandin E2, cytokines, and chemokines from human immune cells (Cox et al., 2009), or indirectly by their ability to support the growth of probiotic species, such as bifidobacteria and lactobacilli (Wang et al., 2002; Delcenserie et al., 2008). SCFAs also possess immune-modulating and anti-inflammatory actions by binding certain G-coupled receptors which may help to stimulate the normal resolution of inflammatory responses in colonocytes (Maslowski et al., 2009). Colonic blood flow as well as fluid and electrolyte uptake are also enhanced by SCFA production (Topping and Clifton, 2001).
Butyrate, in particular, can modulate inflammation and affect tumorigenesis. Evidence shows that risk reduction may be related to the effects of butyrate on activating the apoptosis cascade and inducing growth arrest of tumors by histone hyperacetylation (Avivi-Green et al., 2000). In vitro, butyrate demonstrates numerous additional anti-inflammatory properties, including decreasing the effects of pro-inflammatory cytokines and their expression by inhibiting NF kappa B activation and abolishing lipopolysaccharide (LPS)-induced expression of cytokines by peripheral blood mononuclear cells (Segain et al., 2000). Butyrate also inhibits the in vitro preneoplastic hyperproliferation-induced tumor promoters and arrests the growth of neoplastic colonocytes (Pryde et al., 2002). A fascinating recent study provides experimental evidence that butyrate maintains colonic motility, and therefore “housekeeping,” by modifying histone acetylation in the colonic myenteric plexus, leading to an increased proportion of cholinergic neurons and accelerated colonic transit (Soret et al., 2010).
The negative side of fermentation is the production of hydrogen. Excess hydrogen can damage living cells by impairing NAD regeneration and inhibiting respiration (Gibson et al., 1993). This would not only injure the colonic mucosa (in the extreme case causing pneumatosis intestinalis), but would suppress the bacterial population size, thereby inhibiting its potential to feed itself and the colonic mucosa. The microbiota have evolved to circumvent this problem by expanding their diversity to contain organisms – archaeal hydrogenotrophic methanogens (e.g., Methanobrevibacter smithii) – specialized to consume hydrogen to produce the non-cytotoxic end-product methane (Wolin, 1976). The net effect is an enormous increase in the generation capacity of SCFAs from complex carbohydrates which ensures the health of both the microbiota and the colonic epithelium. As evidence, we observed that methanogenesis was considerably higher in low cancer risk rural Africans consuming high complex carbohydrate diets compared to high risk Americans who eat little fiber (O’Keefe, 2001).
Industrialized man’s diet has changed from containing mainly unprocessed vegetables and grains to favoring meat, animal products, and refined cereals. This evolution reduces the quantity of complex carbohydrates or fiber that reaches the colon and alters the composition of the residue to contain more sulfur. The alteration in colonic milieu affects the diversity of the microbiota as sulfate stimulates the alternative growth of sulfur-reducing bacteria (SRBs, e.g., Desulfovibrio vulgaris), another form of hydrogenotrophic bacteria, which outcompete methanogens for hydrogen under these conditions (Gibson et al., 1988). Unfortunately, the metabolic end-product of SRBs, hydrogen sulfide (H2S), differs from methane, being inflammatory and genotoxic. H2S impairs cytochrome oxidase, suppresses butyrate utilization, inhibits the synthesis of mucus and the methylation of DNA (Gibson et al., 1993), and is genotoxic via the generation of free radicals (Attene-Ramos et al., 2007). Christl et al. (1992) demonstrated that 70% of British fecal samples contained SRBs and the remainder produced methane, whereas the reverse was the case with rural black South Africans (Gibson et al., 1988). The link to diet was illustrated by further studies showing that a high meat diet increased fecal sulfide (Magee et al., 2000), and increasing colonic sulfide suppressed methanogenesis (Christl et al., 1992). The clinical significance of this finding is exemplified by observing that colonic overgrowth with SRBs exists, together with suppression in butyrate production in patients with ulcerative colitis (Roediger, 1993), another condition that increases colon cancer risk. Interestingly, ulcerative colitis is also rare in native Africans (O’Keefe et al., 1983). While SRBs play an essential role in the external environment by suppressing the release of methane gas – one of the “greenhouse” gases – from sewers and marshes into the atmosphere (Guisasola et al., 2008), it seems that humans, unlike the carnivores, have had insufficient exposure time to a westernized diet to evolve mechanisms for preventing H2S injury.
A high meat diet is usually associated with elevated fat intake, and fat itself has been shown to influence cancer risk (Ferguson, 2010). Fat stimulates the synthesis and enterohepatic circulation of primary bile acids, such as cholic acid and chenodeoxycholic acid, which are mostly reabsorbed, although a fraction reaches the colon – and this quantity increases on a high fat diet (Reddy, 1981). If the colonic microbiota contains 7α-dehydroxylating bacteria (chiefly clostridial species), the primary bile acids are converted to the secondary bile acids, namely deoxycholic acid (DCA) and lithocholic acid, which are well recognized co-carcinogens with lithogenic properties (Nagengast et al., 1995; Ridlon et al., 2006). Higher fecal and plasma levels of DCA have been observed in patients with colon cancer (Hori et al., 1998), and our studies have shown that levels were considerably higher in healthy, high risk African Americans compared to low risk healthy rural Africans (Ou et al., 2011). Our recent studies have confirmed that 7α-dehydroxylating bacteria are less common in developing countries, where gallstones and colon cancer are rare (Wells et al., 2000; O’Keefe et al., 2007). Figure A1 displays the relationship between food, the enteric microbiota, and the intestinal mucosa in regards to macronutrient metabolism and inflammation.
Finally, an abundant supply of colonic carbohydrate residues can stimulate the synthesis of vitamins by the resident microbiota. In our studies in rural Africans, we measured constituents of colonic evacuants following polyethylene glycol (PEG) consumption (O’Keefe et al., 2007). As expected, total colonic SCFA and butyrate were significantly higher in rural Africans than in Caucasians and African Americans. More surprising was that colonic folate and biotin content was similar in all three groups despite considerably lower dietary intake in Africans. Considering that content was higher than normal dietary intake, this indicated avid net synthesis by the microbiota. Folate is synthesized by some strains of specific microbes, e.g., bifidobacteria, from glycolysis and tricarboxylic acid cycle intermediates (Watson, 2006). Studies have shown that increasing colonic carbohydrate, in the form of oligosaccharides or resistant starch, enhances the production rate of folate by microbiota (Pompei et al., 2007). The importance of luminal folate and biotin is evident in recent studies demonstrating that active colonic mucosal transport mechanisms exist for these vitamins and that both vitamins play key roles in DNA synthesis and repair (O’Keefe et al., 2009).
Inflammatory Bowel Disease
Inflammatory bowel disease comprises two distinct, but overlapping, entities – Crohn’s disease and ulcerative colitis. There are approximately 1.4 million people living in the United States with IBD and most cases are diagnosed in individuals in their teens or twenties (Loftus Jr. et al., 2002). The etiology of IBD has been described as on-going intestinal mucosal inflammation that hinges on microbial or environmental factors and immune deficiencies in genetically predisposed individuals (Abraham and Cho, 2009). Etiologically, immune system dysfunction, an abnormally permeable mucosal barrier, or imbalances in beneficial and harmful species of enteric bacteria are hypothesized as contributing to IBD risk. Clinical and epidemiologic data demonstrate that a network of inflammatory components interacts to incite IBD (Abt and Artis, 2009). Examples of the influence of microbially perpetuated inflammation include the association of IL-10 mutations with ulcerative colitis (Franke et al., 2008) and elevated levels of mucosal IgG directed against the normal microbiota in IBD patients (Cummings et al., 2003). Additionally, mutations in genes involved in bacterial peptidoglycan recognition (NOD2/CARD15) are known risk factors for Crohn’s disease (Abraham and Cho, 2009) and recent studies have shown that the microbiota can mediate the effects of these genetic defects (Rogler, 2010).
Rates of IBD in modernized nations are rising. Between the 1950s and 1990s, the incidence of Crohn’s disease nearly tripled in Northern Europe (Farrokhyar et al., 2001). The “hygiene hypothesis” has again been indicted (Strachan, 1989). This hypothesis posits that the growing reliance on antibiotics and the sometimes obsessive focus on maintaining an anti-bacterial environment for children may be triggers for IBD on a population level. Studies of migrants, for instance, have shown that relocating from a developing to a developed nation increases the risk of IBD and risk is greatest if relocation occurs during infancy or early childhood (Bernstein and Shanahan, 2008). Regional differences in rates of IBD indicate that its etiology is exceptionally complex, with rates in the southern United States being lower than other US regions, but those of Olmstead county (Minnesota) being more similar to rates in Wales (UK) rather than neighboring Canadian provinces (Bernstein et al., 2006; Gunesh et al., 2008). The incidence of IBD in African and Asian countries, such as Korea, is only a fraction of what it is in the United States and the United Kingdom (Yang et al., 2008).
Clinical observations of IBD patients and animal models highlight the role of bacteria, viruses, and mycobacteria in intestinal inflammation, although no specific microbe or profile of microbes have to date been proven to be causative and it must be remembered that the disease itself and its treatment may also disturb the microbiota. Murine models of colitis require bacteria for inflammation to occur, while other animal models have shown that antibiotics that target anaerobes and Gram-positive organisms such as enterococci are protective against ulcerative colitis – particularly when they are administered prior to the induction of inflammation (Cummings et al., 2003). Antibiotics can also be effective treatments for some IBD patients (Elson et al., 2005). IL-2 deficient, germ-free rats develop colitis due to a non-pathogenic E. coli mpk strain but not with exposure to B. vulgatus (Waidmann et al., 2003).
Dysbiosis may also play a role in the etiology and severity of IBD, as some patients demonstrate lower enteric bacterial diversity with reduced proportions of butyrate-producers (Frank et al., 2007) and “protective” bacteria such as lactobacilli and bifidobacteria (Eckburg and Relman, 2007). This suggests that the mucosa is “at risk,” exemplified by the heightened incidence and severity of Clostridium difficile colitis among IBD patients both in hospitalized and outpatient settings (Ananthakrishnan et al., 2009; Clayton et al., 2009). C. difficile thrives in a dysbiotic, “permissive,” environment and is suppressed by metabolically active butyrate-producers. However, butyrate deficiency may not provide the whole explanation as there is evidence for decreased butyrate utilization in ulcerative colitis and restoration of butyrate-producers may not reverse the inflammation (Thibault et al., 2010). Other microbial products may better explain the ability of butyrate-producers contained within Clostridium cluster IV and XIVa to decrease intestinal inflammation and necrosis in the mouse model (Sokol et al., 2008). Sokol et al. (2008) showed that the supernatant of a culture of a member of cluster IV, Faecalibacterium prausnitzii, reduced colitis and mortality in the mouse model, even if administered orally or intraperitoneally.
Dietary factors may also influence risk of IBD. A Japanese study found a strong correlation between increased intake of animal protein and n−6 polyunsaturated fatty acids, and decreased intake of n−3 polyunsaturated fatty acids over the course of 20 years with Crohn’s disease (Shoda et al., 1996). IBD is also more likely to occur following a bout of acute gastroenteritis. Researchers in Spain studied 43,013 patients aged 20–74 years who had experienced an episode of acute infectious gastroenteritis and found that the estimated incidence rate of IBD was 68.4 per 100,000 person-years after an episode of gastroenteritis but 29.7 per 100,000 person-years in an age-, sex-, and calendar-matched controls (Garcia Rodriguez et al., 2006). There was an especially greater risk of developing Crohn’s disease during the first year after the infective episode (hazard ratio, 6.6; 95% CI, 1.9–22.4) although confounding of Crohn’s symptoms with a bout of gastroenteritis might explain this observation. IBD studies also note that patients have greater risk of developing gastrointestinal infections; an IBD twin study noted greater rates of self-reported enteric infections among patients than their non-affected twin (Halfvarson et al., 2006). Nonetheless, environmental triggers could be as important as genetic ones in the etiology of IBD. Studies of monozygotic twins in which only one twin is affected with IBD note that the unaffected twin may have altered mucosal glycosylation, indicative of “preinflammatory” NFkappaB activation which would be acquired rather than inherited (Bodger et al., 2006).
Conclusion
Enteric organisms share a common environment with the mucosa and have evolved in tandem to ensure survival of what has been described as the “superorganism.” In health, the microbiota prevents overgrowth with pathogens and simultaneously educates the gut immune system in immunotolerance and defense. Disturbance of the microbiota by diet, hygiene, antibiotics, or disease leads to “dysbiosis,” increasing the potential for mucosal injury and inflammation which, with time, may trigger a variety of mucosal and systemic diseases depending upon the host’s genetic susceptibility. The final result of the soluble protein products of inflammatory cells is tissue damage, which can manifest in a variety of disease states, including eczema, colonic neoplasia, or IBD. Each of these entities requires a number of interactive factors to evolve and a prerequisite genetic susceptibility. Reproductive and lifestyle factors in developed nations, including advancements in public healthy hygiene measures, could help foster an environment that diminishes the protective effects of the enteric microbiota. Nonetheless, we can make a concerted effort to write fewer antibiotic prescriptions for infants, support breastfeeding for new mothers and discourage elective C-sections unless conditions necessitate them. Additionally, education about protective dietary constituents should be embraced by clinicians. For instance, encouraging patients to include ample fiber or complex carbohydrates in their diet, supplementing with probiotics, choosing “healthy” n−3 fats over pro-inflammatory n−6 fats, and limiting meat consumption, may optimize health and longevity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abt, M. C., and Artis, D. (2009). The intestinal microbiota in health and disease: the influence of microbial products on immune cell homeostasis. Curr. Opin. Gastroenterol. 25, 496–502.
Adlerberth, I., Jalil, F., Carlsson, B., Mellander, L, Hanson, L. A., Larsson, P., Khalil, K., and Wold, A. E. (1998). High turnover rate of Escherichia coli strains in the intestinal flora of infants in Pakistan. Epidemiol. Infect. 121, 587–598.
Ananthakrishnan, A. N., Issa, M., and Binion, D. G. (2009). Clostridium difficile and inflammatory bowel disease. Gastroenterol. Clin. North Am. 38, 711–728.
Angulo, S., Llopis, M., Antolin, M., Gironella, M., Sans, M., Malagelada, J. R., Pique, J. M., Guarner, F., and Panes, J. (2006). Lactobacillus casei prevents the upregulation of ICAM-1 expression and leukocyte recruitment in experimental colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G1155–G1162.
Attene-Ramos, M. S., Wagner, E. D., Gaskins, H. R., and Plewa, M. J. (2007). Hydrogen sulfide induces direct radical-associated DNA damage. Mol. Cancer Res. 5, 455–459.
Avivi-Green, C., Polak-Charcon, S., Madar, Z., and Schwartz, B. (2000). Apoptosis cascade proteins are regulated in vivo by high intracolonic butyrate concentration: correlation with colon cancer inhibition. Oncol. Res. 12, 83–95.
Bach, J. F. (2002). The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 347, 911–920.
Baron, J. A., and Sandler, R. S. (2000). Nonsteroidal anti-inflammatory drugs and cancer prevention. Annu. Rev. Med. 51, 511–523.
Bernstein, C. N., and Shanahan, F. (2008). Disorders of a modern lifestyle: reconciling the epidemiology of inflammatory bowel diseases. Gut 57, 1185–1191.
Bernstein, C. N., Wajda, A., Svenson, L. W., MacKenzie, A., Koehoorn, M., Jackson, M., Fedorak, R., Israel, D., and Blanchard, J. F. (2006). The epidemiology of inflammatory bowel disease in Canada: a population-based study. Am. J. Gastroenterol. 101, 1559–1568. [Erratum appears in Am. J. Gastroenterol. 2006 Aug;101(8):1945].
Biarc, J., Nguyen, I. S., Pini, A., Gosse, F., Richert, S., Thierse, D., Van Dorsselaer, A., Leize-Wagner, E., Raul, F., Klein, J. P., and Scholler-Guinard, M. (2004). Carcinogenic properties of proteins with pro-inflammatory activity from Streptococcus infantarius (formerly S. bovis). Carcinogenesis 25, 1477–1484.
Black, P. N., and Sharpe, S. (1997). Dietary fat and asthma: is there a connection? Eur. Respir. J. 10, 6–12.
Bodger, K., Halfvarson, J., Dodson, A. R., Campbell, F., Wilson, S., Lee, R., Lindberg, E., Jarnerot, G., Tysk, C., and Rhodes, J. M. (2006). Altered colonic glycoprotein expression in unaffected monozygotic twins of inflammatory bowel disease patients. Gut 55, 973–977.
Christl, S. U., Gibson, G. R., and Cummings, J. H. (1992). Role of dietary sulphate in the regulation of methanogenesis in the human large intestine. Gut 33, 1234–1238.
Clayton, E. M., Rea, M. C., Shanahan, F., Quigley, E. M., Kiely, B., Hill, C., and Ross, R. P. (2009). The vexed relationship between Clostridium difficile and inflammatory bowel disease: an assessment of carriage in an outpatient setting among patients in remission. Am. J. Gastroenterol. 104, 1162–1169.
Cox, M. A., Jackson, J., Stanton, M., Rojas-Triana, A., Bober, L., Laverty, M., Yang, X., Zhu, F., Liu, J., Wang, S., Monsma, F., Vassileva, G., Maguire, M., Gustafson, E., Bayne, M., Chou, C. C., Lundell, D., and Jenh, C. H. (2009). Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E(2) and cytokines. World J. Gastroenterol. 15, 5549–5557.
Cummings, J. H., Macfarlane, G. T., and Macfarlane, S. (2003). Intestinal bacteria and ulcerative colitis. Curr. Issues Intest. Microbiol. 4, 9–20.
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., Collini, S., Pieraccini, G., and Lionetti, P. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696.
Delcenserie, V., Martel, D., Lamoureux, M., Amiot, J., Boutin, Y., and Roy, D. (2008). Immunomodulatory effects of probiotics in the intestinal tract. Curr. Issues Mol. Biol. 10, 37–54.
Doll, R., and Peto, R. (1981). The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 66, 1191–1308.
Eckburg, P. B., and Relman, D. A. (2007). The role of microbes in Crohn’s disease. Clin. Infect. Dis. 44, 256–262.
Ekbom, A., Helmick, C., Zack, M., and Adami, H. O. (1990). Ulcerative colitis and colorectal cancer. A population-based study. N. Engl. J. Med. 323, 1228–1233.
Elson, C. O., Cong, Y., McCracken, V. J., Dimmitt, R. A., Lorenz, R. G., and Weaver, C. T. (2005). Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol. Rev. 206, 260–276.
Farrokhyar, F., Swarbrick, E. T., and Irvine, E. J. (2001). A critical review of epidemiological studies in inflammatory bowel disease. Scand. J. Gastroenterol. 36, 2–15.
Frank, D. N., St Amand, A. L., Feldman, R. A., Boedeker, E. C., Harpaz, N., and Pace, N. R. (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. U.S.A. 104, 13780–13785.
Franke, A., Balschun, T., Karlsen, T. H., Sventoraityte, J., Nikolaus, S., Mayr, G., Domingues, F. S., Albrecht, M., Nothnagel, M., Ellinghaus, D., Sina, C., Onnie, C. M., Weersma, R. K., Stokkers, P. C., Wijmenga, C., Gazouli, M., Strachan, D., McArdle, W. L., Vermeire, S., Rutgeerts, P., Rosenstiel, P., Krawczak, M., Vatn, M. H., Group, I. S., Mathew, C. G., and Schreiber, S.(2008). Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat. Genet. 40, 1319–1323.
Garcea, G., Dennison, A. R., Steward, W. P., and Berry, D. P. (2005). Role of inflammation in pancreatic carcinogenesis and the implications for future therapy. Pancreatology 5, 514–529.
Garcia Rodriguez, L. A., Ruigomez, A., and Panes, J. (2006). Acute gastroenteritis is followed by an increased risk of inflammatory bowel disease. Gastroenterology 130, 1588–1594.
Gibson, G. R., Cummings, J. H., and Macfarlane, G. T. (1988). Competition for hydrogen between sulphate-reducing bacteria and methanogenic bacteria from the human large intestine. J. Appl. Bacteriol. 65, 241–247.
Gibson, G. R., Macfarlane, G. T., and Cummings, J. H. (1993). Sulphate reducing bacteria and hydrogen metabolism in the human large intestine. Gut 34, 437–439.
Guisasola, A., de Haas, D., Keller, J., and Yuan, Z. (2008). Methane formation in sewer systems. Water Res. 42, 1421–1430.
Gunesh, S., Thomas, G. A., Williams, G. T., Roberts, A., Hawthorne, A. B., and Thomas, G. A. O. (2008). The incidence of Crohn’s disease in Cardiff over the last 75 years: an update for 1996-2005. Aliment. Pharmacol. Ther. 27, 211–219.
Halfvarson, J., Jess, T., Magnuson, A., Montgomery, S. M., Orholm, M., Tysk, C., Binder, V., and Jarnerot, G. (2006). Environmental factors in inflammatory bowel disease: a co-twin control study of a Swedish–Danish twin population. Inflamm. Bowel Dis. 12, 925–933.
Hori, T., Matsumoto, K., Sakaitani, Y., Sato, M., and Morotomi, M. (1998). Effect of dietary deoxycholic acid and cholesterol on fecal steroid concentration and its impact on the colonic crypt cell proliferation in azoxymethane-treated rats. Cancer Lett. 124, 79–84.
Ivanov, II, Frutos Rde, L., Manel, N., Yoshinaga, K., Rifkin, D. B., Sartor, R. B., Finlay, B. B., and Littman, D. R.(2008). Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349.
Kalliomaki, M., Kirjavainen, P., Eerola, E., Kero, P., Salminen, S., and Isolauri, E. (2001a). Distinct patterns of neonatal gut microflora in infants in whom atopy was and was not developing. J. Allergy Clin. Immunol. 107, 129–134.
Kalliomaki, M., Salminen, S., Arvilommi, H., Kero, P., Koskinen, P., and Isolauri, E. (2001b). Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet 357, 1076–1079.
Kobayashi, N., Barnard, R. J., Henning, S. M., Elashoff, D., Reddy, S, T., Cohen, P., Leung, P., Hong-Gonzalez, J., Freedland, S. J., Said, J., Gui, D., Seeram, N. P., Popoviciu, L. M., Bagga, D., Heber, D., Glaspy, J. A., and Aronson, W. J. (2006). Effect of altering dietary omega-6/omega-3 fatty acid ratios on prostate cancer membrane composition, cyclooxygenase-2, and prostaglandin E2. Clin. Cancer Res. 12, 4662–4670.
Kummeling, I., Thijs, C., Huber, M., Van De Vijver, L. P., Snijders, B. E., Penders, J., Stelma, F., Van Ree, R., Van Den Brandt, P. A., and Dagnelie, P. C. (2008). Consumption of organic foods and risk of atopic disease during the first 2 years of life in the Netherlands. Br. J. Nutr. 99, 598–605.
Loftus, E. V. Jr., and Sandborn, W. J. (2002). Epidemiology of inflammatory bowel disease. Gastroenterol. Clin. North Am. 31, 1–20.
Magee, E. A., Richardson, C. J., Hughes, R., and Cummings, J. H. (2000). Contribution of dietary protein to sulfide production in the large intestine: an in vitro and a controlled feeding study in humans. Am. J. Clin. Nutr. 72, 1488–1494.
Marra, F., Marra, C. A., Richardson, K., Lynd, L. D., Kozyrskyj, A., Patrick, D. M., Bowie, W. R., and Fitzgerald, J. M. (2009). Antibiotic use in children is associated with increased risk of asthma. Pediatrics 123, 1003–1010.
Maslowski, K. M., Vieira, A. T., Ng, A., Kranich, J., Sierro, F., Yu, D., Schilter, H., C., Rolph, M. S., Mackay, F., Artis, D., Xavier, R. J., Teixeira, M. M., and Mackay, C. R. (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286.
Mazmanian, S. K., Liu, C. H., Tzianabos, A. O., and Kasper, D. L. (2005). An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118.
Mazmanian, S. K., Round, J. L., and Kasper, D. L. (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625.
Nagengast, F. M., Grubben, M. J., and van Munster, I. P. (1995). Role of bile acids in colorectal carcinogenesis. Eur. J. Cancer 31A, 1067–1070.
O’Keefe, S. J. (2001). The African way of life and colon cancer risk. Am. J. Gastroenterol. 96, 3220–3221.
O’Keefe, S. J., Chung, D., Mahmoud, N., Sepulveda, A. R., Manafe, M., Arch, J., Adada, H., and Van Der Merwe, T. (2007). Why do African Americans get more colon cancer than Native Africans? J. Nutr. 137, 175S–182S.
O’Keefe, S. J., Ou, J., Aufreiter, S., O’Connor, D., Sharma, S., Sepulveda, J., Fukuwatari, T., Shibata, K., and Mawhinney, T. (2009). Products of the colonic microbiota mediate the effects of diet on colon cancer risk. J. Nutr. 139, 2044–2048.
O’Keefe, S. J., Thusi, D., and Epstein, S. (1983). The fat and the thin – a survey of nutritional status and disease patterns among urbanized Black South Africans. S. Afr. Med. J. 63, 679–683.
Ou, J., DeLany, J. P., Zhang, M., Sharma, S., and O’Keefe, S. J. (2011). Association between low colonic short chain fatty acids and high bile acids in high colon cancer risk populations. Nutr. Cancer (in press).
Penders, J., Thijs, C., Vink, C., Stelma, F. F., Snijders, B., Kummeling, I., Van Den Brandt, P. A., and Stobberingh, E. E. (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521.
Pompei, A., Cordisco, L., Amaretti, A., Zanoni, S., Matteuzzi, D., and Rossi, M. (2007). Folate production by bifidobacteria as a potential probiotic property. Appl. Environ. Microbiol. 73, 179–185.
Pryde, S. E., Duncan, S. H., Hold, G. L., Stewart, C. S., and Flint, H. J. (2002). The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 217, 133–139.
Ridlon, J. M., Kang, D. J., and Hylemon, P. B. (2006). Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 47, 241–259.
Rogler, G. (2010). The importance of gut microbiota in mediating the effect of NOD2 defects in inflammatory bowel disease. Gut 59, 153–154.
Salminen, S., Gibson, G. R., McCartney, A. L., and Isolauri, E. (2004). Influence of mode of delivery on gut microbiota composition in seven year old children. Gut 53, 1388–1389.
Segain, J. P., Raingeard de la Bletiere, D., Bourreille, A., Leray, V., Gervois, N., Rosales, C., Ferrier, L., Bonnet, C., Blottiere, H. M., and Galmiche, J. P. (2000). Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut 47, 397–403.
Shoda, R., Matsueda, K., Yamato, S., and Umeda, N. (1996). Epidemiologic analysis of Crohn disease in Japan: increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan. Am. J. Clin. Nutr. 63, 741–745.
Sokol, H., Pigneur, B., Watterlot, L., Lakhdari, O., Bermudez-Humaran, L, G., Gratadoux, J, J., Blugeon, S., Bridonneau, C., Furet, J. P., Corthier, G., Grangette, C., Vasquez, N., Pochart, P., Trugnan, G., Thomas, G., Blottiere, H. M., Dore, J., Marteau, P., Seksik, P., and Langella, P. (2008). Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U.S.A. 105, 16731–16736.
Soret, R., Chevalier, J., De Coppet, P., Poupeau, G., Derkinderen, P., Segain, J. P., and Neunlist, M. (2010). Short-chain fatty acids regulate the enteric neurons and control gastrointestinal motility in rats. Gastroenterology 138, 1772–1782.e4.
Thibault, R., Blachier, F., Darcy-Vrillon, B., De Coppet, P., Bourreille, A., and Segain, J. P. (2010). Butyrate utilization by the colonic mucosa in inflammatory bowel diseases: a transport deficiency. Inflamm. Bowel Dis. 16, 684–695.
Topping, D. L., and Clifton, P. M. (2001). Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 81, 1031–1064.
Vaarala, O. (2003). Immunological effects of probiotics with special reference to lactobacilli. Clin. Exp. Allergy 33, 1634–1640.
Waidmann, M., Bechtold, O., Frick, J. S., Lehr, H. A., Schubert, S., Dobrindt, U., Loeffler, J,. and Bohn, E. (2003). Bacteroides vulgatus protects against Escherichia coli-induced colitis in gnotobiotic interleukin-2-deficient mice. Gastroenterology 125, 162–177.
Wang, M., Karlsson, C., Olsson, C., Adlerberth, I., Wold, A, E., Strachan, D. P., Martricardi, P. M., Aberg, N., Perkin, M. R., Tripodi, S., Coates, A. R., Hesselmar, B., Saalman, R., Molin, G., and Ahrne, S. (2008). Reduced diversity in the early fecal microbiota of infants with atopic eczema. J. Allergy Clin. Immunol. 121, 129–134.
Wang, X., Brown, I. L., Khaled, D., Mahoney, M. C., Evans, A. J., and Conway, P. L. (2002). Manipulation of colonic bacteria and volatile fatty acid production by dietary high amylose maize (amylomaize) starch granules. J. Appl. Microbiol. 93, 390–397.
Watson, A. J. (2006). An overview of apoptosis and the prevention of colorectal cancer. Crit. Rev. Oncol. Hematol. 57, 107–121.
Wells, J. E., Berr, F., Thomas, L. A., Dowling, R. H., and Hylemon, P. B. (2000). Isolation and characterization of cholic acid 7alpha-dehydroxylating fecal bacteria from cholesterol gallstone patients. J. Hepatol. 32, 4–10.
Wolin, M. J. (1976). “Interactions between H2-producing and methane producing species,” in Microbial Formation and Utilization of Gases, eds H. G. Schlegel, G. Gottschalk, and N. Pfennig (Gottingen: Goltze Press), 141–150.
Wu, S., Rhee, K. J., Albesiano, E., Rabizadeh, S., Wu, X., Yen, H. R., Huso, D. L., Brancati, F. L., Wick, E., McAllister, F., Housseau, F., Pardoll, D. M., and Sears, C. L. (2009). A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022.
Yan, F., Cao, H., Cover, T. L., Whitehead, R., Washington, M. K., and Polk, D. B. (2007). Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology 132, 562–575.
Yang, S. K., Yun, S., Kim, J. H., Park, J. Y., Kim, H. Y., Kim, Y. H., Chang, D. K., Kim, J. S., Song, I. S., Park, J. B., Park, E. R., Kim, K. J., Moon, G., and Yang, S. H. (2008). Epidemiology of inflammatory bowel disease in the Songpa-Kangdong district, Seoul, Korea, 1986–2005: a KASID study. Inflamm. Bowel Dis. 14, 542–549.
Keywords: microbiota, colon cancer, allergy, inflammatory bowel disease, diet
Citation: Greer JB and O’Keefe SJ (2011) Microbial induction of immunity, inflammation, and cancer. Front. Physio. 1:168. doi: 10.3389/fphys.2010.00168
Received: 29 October 2010;
Paper pending published: 29 November 2010;
Accepted: 23 December 2010;
Published online: 26 January 2011.
Edited by:
Hong Xiang Hui, Southern Medical University, ChinaReviewed by:
Xiaoning Zhao, Cedars-Sinai Medical Center at Los Angeles, USAShuping Vincent Wu, VA Greater Los Angeles Healthcare System, USA
George Tang, Cedars-Sinai Medical Center, USA
Hongxiang Hui, Southern Medical University, China
Copyright: © 2011 Greer and O’Keefe. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Stephen John O’Keefe, Division of Gastroenterology, Hepatology and Nutrition, Department of Medicine, University of Pittsburgh School of Medicine, 570 Scaife Hall, 3550 Terrace Street, Pittsburgh, PA 15213, USA. e-mail:c2pva2VlZmVAcGl0dC5lZHU=