


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 03 November 2010
Sec. Mitochondrial Research
Volume 1 - 2010 | https://doi.org/10.3389/fphys.2010.00141
Electro-mechanical and Ca alternans is a beat-to-beat alternation of action potential duration, contraction strength and Ca transient amplitude observed in cardiac myocytes at regular stimulation frequency. Ca alternans is a multifactorial process that is causally linked to cardiac arrhythmias. At the cellular level, conditions that increase fractional release from the sarcoplasmic reticulum or reduce diastolic Ca sequestration favor the occurrence of alternans. Mitochondria play a significant role in cardiac excitation–contraction coupling and Ca signaling by providing the energy for contraction and ATP-dependent processes and possibly by serving as Ca buffering organelles. Here we tested the hypothesis that impairment of mitochondrial function generates conditions that favor the occurrence of Ca alternans. Alternans were elicited by electrical pacing (>1 Hz) in single cat atrial myocytes and intracellular Ca ([Ca]i) was measured with the fluorescent Ca indicator Indo-1. The degree of alternans was quantified as the alternans ratio (AR = 1 − S/L, where S/L is the ratio of the small to the large amplitude of a pair of alternating Ca transients). Dissipation of mitochondrial membrane potential (with FCCP) as well as inhibition of mitochondrial F1/F0-ATP synthase (oligomycin), electron transport chain (rotenone, antimycin, CN−), Ca-dependent dehydrogenases and mitochondrial Ca uptake or extrusion, all enhanced AR and lowered the threshold for the occurrence of Ca alternans. The data indicate that impairment of mitochondrial function adversely affects cardiac Ca cycling leading to proarrhythmic Ca alternans.
Cardiac alternans describes the cyclic, beat-to-beat variations in contraction amplitude (mechanical alternans), action potential duration (electrical or APD alternans), and Ca transient amplitude (Ca alternans) at constant stimulation frequency (e.g., Wohlfart, 1982). Alternans occurs in cardiac failure (e.g., Dumitrescu et al., 2002), during myocardial ischemia, and is believed to be an important factor in the pathogenesis of arrhythmias (Dilly and Lab, 1988; Konta et al., 1990) and the development of re-entry phenomena (Rubenstein and Lipsius, 1995; Berger, 2000; Pastore and Rosenbaum, 2000). Clinically, alternans is manifest as pulsus alternans and specific changes in the ST segment of the electrocardiogram (e.g., Uno, 1991; Surawicz and Fisch, 1992; Lab and Seed, 1993; Pastore et al., 1999). T-wave alternans has been linked to ventricular fibrillation and sudden cardiac death under various conditions including acute myocardial ischemia (Smith et al., 1988; Verrier and Nearing, 1994) and long-QT syndrome (Shimizu and Antzelevitch, 1999). T-wave alternans is now established as one of the strongest marker of susceptibility to sudden cardiac death (Rosenbaum et al., 1994; Walker and Rosenbaum, 2003). Thus, cardiac alternans is thought to be a precursor to life-threatening arrhythmias as both occur in settings of myocardial ischemia, genetic channelopathies, and drug and electrolyte disturbances (Walker and Rosenbaum, 2003). Atrial fibrillation is the most common cardiac arrhythmia that affects a significant segment of the elderly population and is linked to high risk of embolytic complications and stroke (for review see Nattel, 2002). Moreover, atrial fibrillation has been linked to APD alternans (Narayan et al., 2002; Hiromoto et al., 2005). Thus alternans is not only a well established risk factor for ventricular dysrhythmias, but also for atrial arrhythmia.
In intact hearts, cardiac muscle preparations or isolated myocytes, alternans can be induced experimentally by pacing (see review Euler, 1999), acidosis (Orchard et al., 1991; Kapur et al., 2009), low extracellular [Ca] (Badeer et al., 1967), hypothermia (e.g., Spencer et al., 1992), and as we demonstrated, by direct interference with cellular energy metabolism through inhibition of glycolysis (Huser et al., 2000b; Kockskamper and Blatter, 2002; Kockskamper et al., 2005). Furthermore, β-adrenergic stimulation, hypercalcemia, cardiac glycosides, and Ca channel blockers can counteract the likelihood for the occurrence of alternans by increasing the critical pacing threshold leading to alternans (see Euler, 1999). There is strong evidence that alternans is ultimately linked to alterations in myocardial Ca homeostasis and impaired [Ca]i regulation (for review see Blatter et al., 2003). Numerous factors involved in excitation–contraction coupling (ECC) and Ca-induced Ca release (CICR) have been proposed to be involved in Ca alternans, including alternating voltage-dependent L-type Ca current (ICa,L) that triggers CICR (Fox et al., 2002; Li et al., 2009), calmodulin-dependent protein kinase II activity (Livshitz and Rudy, 2007), SERCA activity (Kameyama et al., 2003; Xie et al., 2008; Cutler et al., 2009), redox modulation (Belevych et al., 2009), and status of the sarcoplasmic reticulum (SR) ryanodine receptor (RyR) Ca release channel (Huser et al., 2000b; Diaz et al., 2002), activation of IP3-receptor dependent Ca release (Zima and Blatter, 2004), temporal delay in Ca movement between SR Ca uptake and release sites (Lab and Lee, 1990; Kihara and Morgan, 1991), SR Ca load (Eisner et al., 2000; Diaz et al., 2004; Picht et al., 2006; Li et al., 2009), and SR Ca buffering by calsequestrin (Restrepo et al., 2008). (For details see reviews Euler, 1999; Rosenbaum, 2001; Blatter et al., 2003; Walker and Rosenbaum, 2003; Eisner et al., 2006; Sato et al., 2006; Weiss et al., 2006; Clusin, 2008; Myles et al., 2008). Ca alternans shows regional differences (Cordeiro et al., 2007) and can be subcellularly inhomogeneous (Diaz et al., 2004; Aistrup et al., 2006, 2009; Gaeta et al., 2009). We have demonstrated that these inhomogeneities are particularly pronounced in atrial tissue. Atrial myocytes have a poorly developed or lack entirely a T-tubular system and the mechanism of Ca release during ECC differs significantly from ventricular myocytes (Huser et al., 1996; Sheehan and Blatter, 2003). Due to these unique structural features Ca alternans in atrial cells are spatially inhomogeneous revealing transverse and longitudinal gradients of the degree of Ca alternans and subcellular regions alternating out-of-phase (Kockskamper and Blatter, 2002). These pronounced and complex subcellular inhomogeneities are linked to propagating arrhythmogenic Ca waves (Kockskamper and Blatter, 2002; Xie and Weiss, 2009).
The multitude of experimental conditions and interventions that cause and modulate Ca and electro-mechanical alternans suggest that Ca alternans is a multifactorial process. This is consistent with the fact that a comprehensive mechanism that can explain and predict the occurrence of cardiac alternans, has not been established to date. Nonetheless, invaluable insight has come from computational models (Shiferaw et al., 2003; Sato et al., 2006; Shiferaw and Karma, 2006; Hayashi et al., 2007; Jordan and Christini, 2007; Livshitz and Rudy, 2007; Sato et al., 2007; Mahajan et al., 2008; Rovetti et al., 2010) of cardiac Ca signaling, and ion current activity during ECC and APD alternans (for a comprehensive review see Weiss et al., 2006). From these studies it became evident that two parameters play equally important roles: fractional SR Ca release, and the efficiency of Ca sequestration. Fractional Ca release refers to the non-linear relationship between SR Ca load and amount of Ca (or % of SR Ca content) released by CICR, where at a higher load a larger fraction is liberated upon activation of CICR (Bassani et al., 1995). Ca sequestration is referred to here as a phenomenological parameter that includes Ca reuptake via SERCA, extrusion via NCX and plasmalemmal Ca-ATPase (PMCA), cytosolic buffering, diastolic SR Ca leak, and finally mitochondrial uptake. In general, factors increasing fractional release promote, and factors increasing Ca sequestration efficiency protect against alternans.
Fractional Ca release and Ca sequestration critically depend on two factors: (i) availability of ATP to fuel Ca pumps and to serve as substrate for phosphorylation processes, and (ii) organelles capable of storing Ca. For both tasks mitochondria play a central role. Mitochondria affect ECC and Ca signaling in cardiac cells at different levels. As a key source of ATP, mitochondria provide the fuel for the activation of the contractile machinery, and for ATP consuming Ca pumps (SERCA and PMCA). ATP is further required for phosphorylation of Ca handling proteins (SERCA, RyR, NCX) and also acts as a direct modulator (through MgATP) of RyR activity. Mitochondria are capable of storing significant amounts of Ca through uptake of Ca mainly via the electrogenic mitochondrial Ca uniporter (MCU) driven by the electrochemical gradient across the inner mitochondrial membrane (IMM), a rapid mode of Ca uptake (RaM) (Sparagna et al., 1995; Gunter et al., 2000; Buntinas et al., 2001), and possibly via mitochondrial RyR (Beutner et al., 2001, 2005; Altschafl et al., 2007) localized in the IMM. In cardiac cells Ca extrusion from mitochondria occurs predominantly via the mitochondrial NCX, whereas a participation of the mitochondrial permeability transition pore has remained controversial. A highly debated and still unresolved issue is whether kinetics and affinities of Ca transporters in cardiac mitochondria allow for Ca uptake rapid enough to allow [Ca]mito to follow beat-to-beat changes of cytosolic [Ca]i (for review see Huser et al., 2000a; Dedkova and Blatter, 2008; O’Rourke and Blatter, 2009). There is experimental support for two different, and to some extent mutually exclusive models of how [Ca]mito responds to oscillatory (beat-to-beat) changes of [Ca]i. This may occur either as a slow integration of cytosolic [Ca]i transients (Miyata et al., 1991; Moravec and Bond, 1991; Sedova et al., 2006), or as beat-to-beat oscillations of [Ca]mito (Isenberg et al., 1993; Robert et al., 2001; Maack et al., 2006).
Mitochondrial [Ca] levels directly modulate mitochondrial functions. Elevated [Ca]mito levels play an important role in the initiation of cell death pathways (Crompton, 1999) and activate several key enzymes in the mitochondrial matrix to enhance ATP production. Oxidation of metabolic substrates in the tricarboxylic acid cycle (TCA) by mitochondrial dehydrogenases is coupled with mitochondrial ATP synthesis through the electron transport chain (ETC). The TCA cycle contains four Ca-dependent mitochondrial dehydrogenases (glycerol 3-phosphate dehydrogenase, pyruvate dehydrogenase, NAD-linked isocitrate dehydrogenase, and 2-oxoglutarate dehydrogenase). Activation of mitochondrial dehydrogenases accelerates the production of NADH, which promotes the generation of the electrochemical proton gradient (ΔΨm) that in turn provides the driving force for the F1/F0-ATP synthase, thus maintaining ATP production. Thus, Ca homeostasis and mitochondrial energy metabolism are intimately interconnected. Nonetheless, the direct effects of mitochondrial function on Ca alternans has not been studied in depth.
Therefore, in the present study we tested the hypothesis that impairment of mitochondrial function, either by reducing Ca buffering or inhibition of energy metabolism, enhances the susceptibility for pacing-induced Ca alternans in atrial tissue. Part of this work has been presented in abstract form (Florea and Blatter, 2005).
Cat atrial myocytes were isolated according to the method described previously (Wu et al., 1991). All protocols and procedures were approved by the Institutional Animal Care and Use Committee. Adult cats of either sex were anesthetized with sodium pentobarbital (50 mg kg−1) and hearts were excised, mounted on a Langendorff apparatus and retrogradely perfused through the aorta with oxygenated collagenase-containing solution (37°C). Dissociated cells were placed on glass coverslips and allowed to attach for 15 min. All experiments were performed at room temperature (22–25°C) where the threshold frequency for pacing-induced Ca alternans is generally lower than at body temperature (Huser et al., 2000b). During experiments, cells were continuously superfused with standard Tyrode solution containing (mM): 140 NaCl, 5 KCl, 1 MgCl2, 10 glucose, 10 HEPES, 2 CaCl2, and pH 7.3 (adjusted with NaOH). All inhibitors, agonists, and antagonists were added to normal Tyrode solution from stock solutions.
For [Ca]i measurements, atrial myocytes were loaded with the membrane-permeable form of the fluorescent Ca indicator indo-1/AM (5 μM; Invitrogen/Molecular Probes, Carlsbad, CA, USA) in standard Tyrode solution for 15–20 min at room temperature, followed by a >20 min de-esterification interval in dye-free media. Indo-1 loaded cells were excited at 360 nm and emission signals were collected simultaneously at 410 nm (F410) and 485 nm (F485) using photomultiplier tubes. Fluorescence signals were background subtracted and [Ca]i changes were expressed as changes in ratio R = F410/F485. Ca transients and Ca alternans were elicited by electrical field stimulation (voltage set ∼50% above threshold) using a pair of platinum electrodes. Pacing-induced stable Ca alternans was elicited by increasing the stimulation frequency from 0.5 to >1 Hz. The degree of Ca alternans was quantified as the alternans ratio (AR). The AR was defined as 1 − S/L, where S/L is the ratio of the small (S) to the large (L) amplitude Ca transient during a pair of alternating Ca transients (Wu and Clusin, 1997; Kockskamper and Blatter, 2002). Thus, the AR had values between 1 and 0, with AR = 0 indicating no alternans and AR = 1 indicating the highest possible degree of alternans, with only every other stimulation resulting in a measurable Ca transient.
Oligomycin, antimycin, sodium cyanide (CN−), α-keto-β-methylvaleric acid sodium salt (KMVA), ruthenium red (RuR), and carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Ruthenium 360 (Ru360) and cyclosporin A (CsA) were obtained from Calbiochem (San Diego, CA, USA). CGP-37157 was obtained from Tocris (Ellisville, MO, USA). Cells were paced continuously during application of inhibitors.
Results are presented as mean ± standard error of the mean for the indicated number (n) of cells. Statistical differences were determined using Student’s t-test for paired or unpaired data. Differences were considered statistically significant at p < 0.05.
The electrochemical potential (ΔΨm) across the IMM is sustained by the mitochondrial respiratory chain (or electron transport chain, ETC) and drives mitochondrial ATP synthesis and provides the driving force for mitochondrial Ca uptake. To test whether inhibition of mitochondrial function affects the propensity for pacing-induced Ca alternans in atrial myocytes we exposed cells to the protonophore FCCP (0.1 μM). FCCP dissipates ΔΨm by uncoupling electron transport from ATP synthesis. As shown in Figure 1A, FCCP enhanced pacing-induced Ca alternans. FCCP increased the AR by 34% from 0.50 ± 0.04 (control) to 0.67 ± 0.06 (FCCP; n = 16; p < 0.05). Furthermore, we showed previously that cardiac mitochondrial Ca uptake is blocked in the presence of FCCP (Sedova et al., 2006).
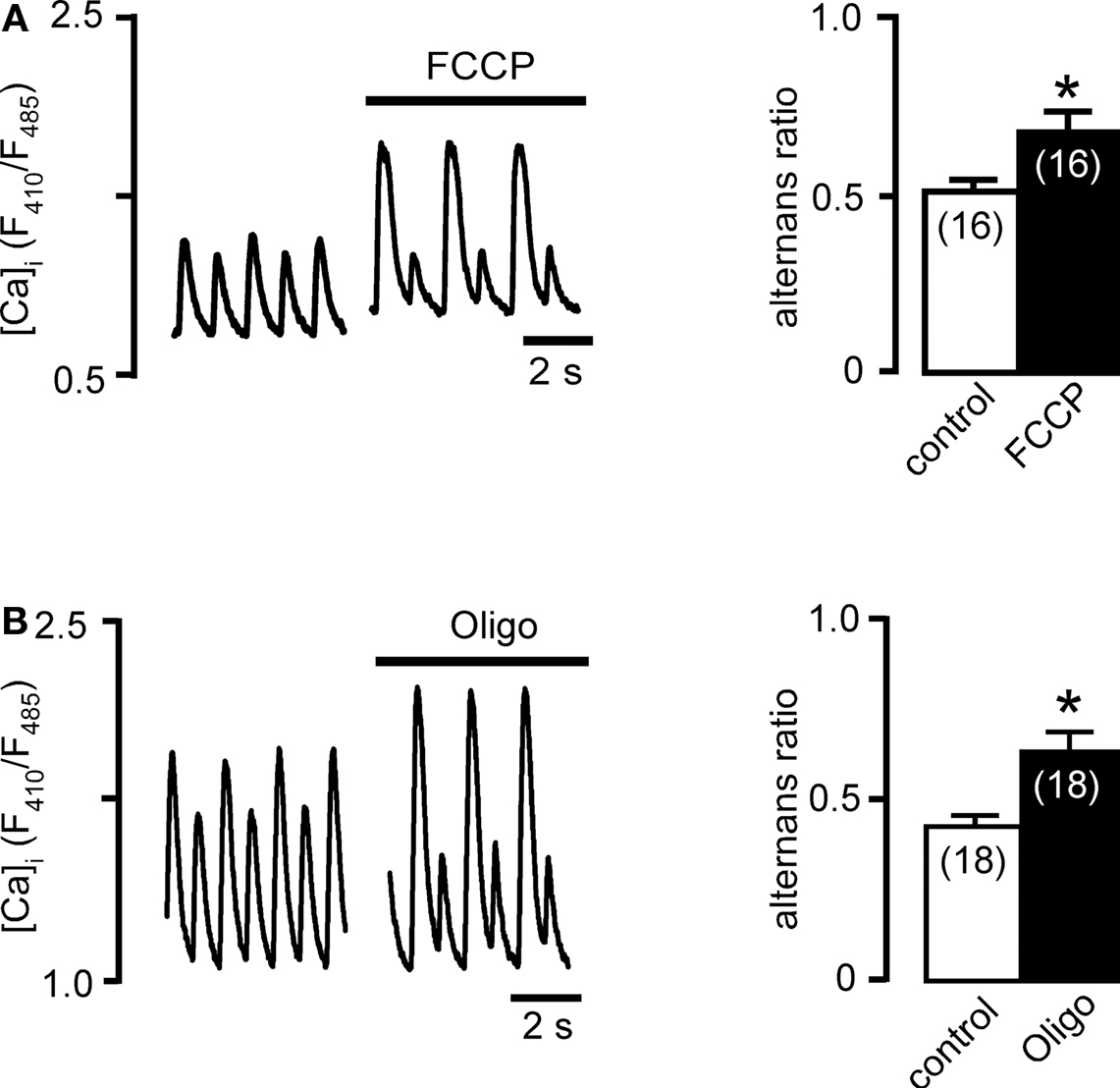
Figure 1. Effects of mitochondrial membrane potential and ATP production on Ca alternans. (A) Left: Ca transients recorded before and after treatment with FCCP (0.1 μM) to collapse the mitochondrial membrane potential (ΔΨm). Right: average AR in control conditions and in the presence of FCCP. (B) Effect of inhibition of mitochondrial F1/F0-ATP synthase with oligomycin (Oligo, 1 μg/ml) on Ca alternans. Numbers in parentheses indicate the number of individual cells tested. *significantly different from control at P < 0.05.
Direct inhibition of mitochondrial ATP synthesis by exposure to oligomycin, an inhibitor of the mitochondrial F1/F0-ATP synthase, also enhanced Ca alternans (Figure 1B). In atrial myocytes revealing pacing-induced Ca alternans under control stimulation frequencies, oligomycin increased AR by 50%, from 0.42 ± 0.03 (control) to 0.63 ± 0.04 (n = 18; p < 0.05).
Inhibition of ATP synthesis and dissipation of ΔΨm (which profoundly affects mitochondrial energy metabolism and ATP synthesis as well as mitochondrial Ca homeostasis) enhanced Ca alternans. The following experiments were designed to dissect further how these key mitochondrial functions interfere with the susceptibility to Ca alternans.
During increased workload of the heart, accumulation of ADP and elevated [Ca]i stimulate mitochondrial respiration, and enhance F1/F0-ATP synthase and TCA dehydrogenase activity in order to match increased metabolic demands by accelerating mitochondrial ATP production. Increasing pacing rate results in both, increase in [Ca]i and ATP demand but also in Ca alternans. We therefore tested whether inhibition of mitochondrial respiration or blockade of the TCA cycle generates conditions favorable to the occurrence of Ca alternans. As shown in Figures 2A–C inhibition of mitochondrial ETC complex I (rotenone, 5 μM), complex III (antimycin, 5 μg/ml), and complex IV (CN−, 1 mM) led to an enhancement of Ca alternans to various, but always significant degrees (Figure 2E) between 31% (rotenone) and 82% (CN−). The combined application of antimycin and oligomycin increased AR further (115% increase compared to control).
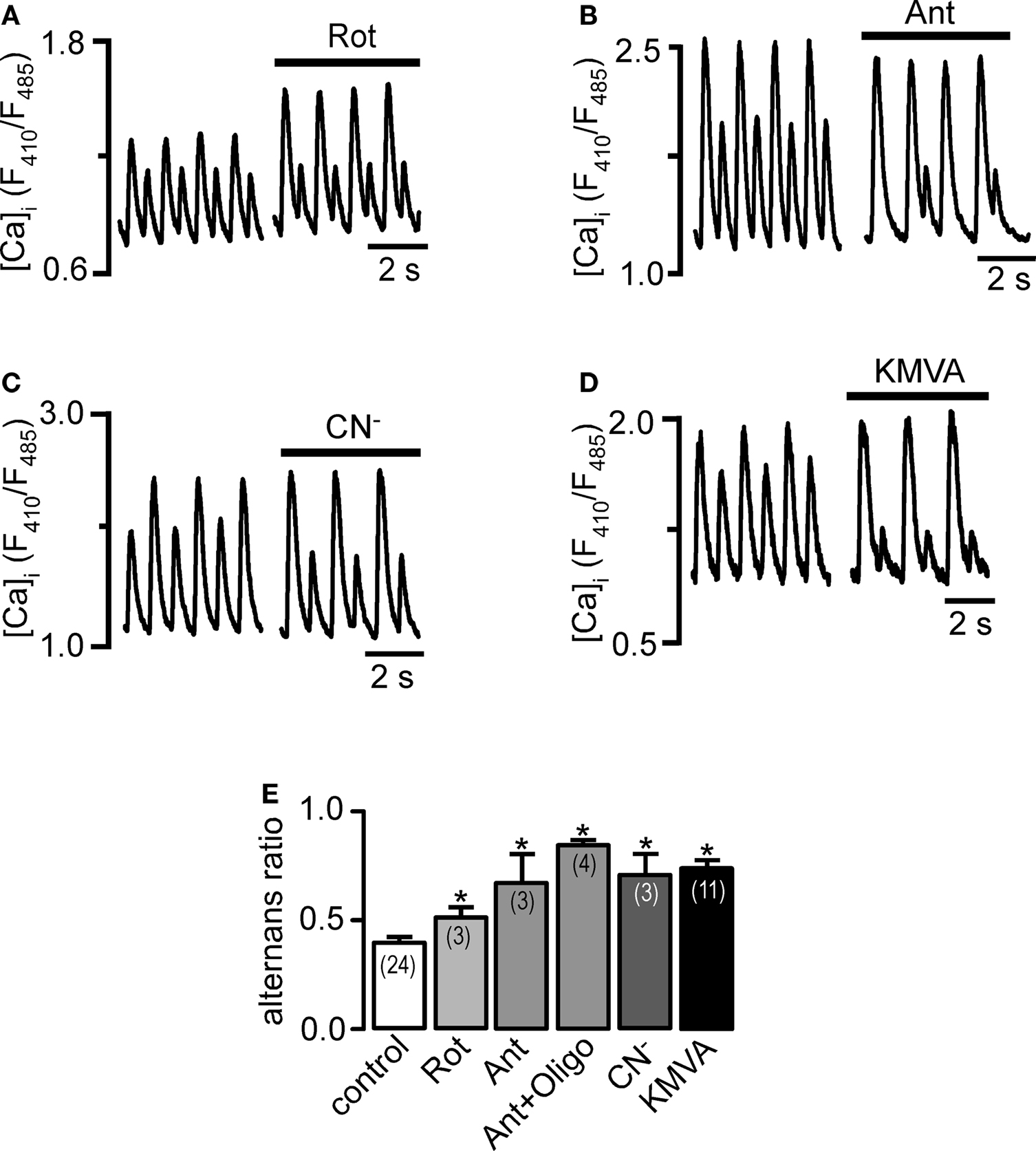
Figure 2. Effects of inhibition of mitochondrial respiration and metabolism on Ca alternans. (A) Rotenone (Rot, 5 μM), (B) antimycin (Ant, 5 μg/ml), (C) cyanide (CN−, 1 mM), and (D) KMVA (5 mM) enhanced pacing-induced Ca alternans. (E) Average AR during mitochondrial inhibition. Numbers in parentheses indicate the number of individual cells tested. *Significantly different from control at P < 0.05.
We tested the involvement of the TCA cycle by inhibiting α-ketoglutarate dehydrogenase (αKGDH) with KMVA (5 mM). As shown in Figure 2D inhibition of one of the key Ca-dependent dehydrogenases of the TCA cycle caused a significant increase of the AR from 0.39 ± 0.06 to 0.74 ± 0.06 (+90%; n = 11; p < 0.05).
In summary, inhibition of the mitochondrial ETC and TCA cycle significantly increased AR indicating that impaired mitochondrial energy metabolism generates proarrhythmic conditions in the form of Ca alternans.
We tested the hypothesis whether inhibition of mitochondrial Ca uptake via MCU or disruption of the mitochondrial Ca extrusion pathway via NCXmito had effects on pacing-induced Ca alternans. As shown previously ruthenium red (RuR) abolished Ca uptake by cardiac mitochondria (Sedova et al., 2006). Inhibition of MCU with RuR (10 μM) increased AR on average from 0.40 ± 0.02 (control; n = 52) to 0.60 ± 0.07 (n = 12; p < 0.05) or by 50%. The more specific MCU inhibitor Ru360 (Matlib et al., 1998) had a more profound effect and increased AR by 78% (AR in the presence of 5 μM Ru360: 0.71 ± 0.15; n = 5; p < 0.05).
Next, we selectively inhibited mitochondrial Ca extrusion via NCXmito. As shown previously, in cat cardiac mitochondria physiological Ca extrusion occurs solely via NCXmito (Sedova et al., 2006). NCXmito was blocked with the benzothiazepine compound CGP-37157 (Cox et al., 1993). CGP-3715 at 2.5 μM increased AR by 98% to 0.79 ± 0.15 (n = 16; p < 0.05), whereas a higher CGP-37157 concentration of 10 μM lead to nearly maximal alternans (AR = 0.94 ± 0.04; n = 12; p < 0.05).
Among the inhibitors of mitochondrial Ca cycling used, CGP-37157 had the most profound effect on the AR. As shown previously, with each Ca transient mitochondria accumulate Ca which is normally counterbalanced by extrusion via NCXmito. However during inhibition of NCXmito, Ca entering mitochondria is not returned to the cytosol, resulting in mitochondrial Ca overload. Elevated matrix [Ca] is a known inducer of the mitochondrial permeability transition pore, thus mitochondrial Ca overload my lead to PTP opening (Dedkova and Blatter, 2008), dissipation of ΔΨm (Huser et al., 1998), and ultimately loss of mitochondrial function. We used the PTP inhibitor cyclosporin A (CsA, 10 μM) to test whether blockade of PTP could counteract the effects of CGP-37157 on Ca alternans. Figure 3D shows an example where CGP-37157 (2.5 μM) caused a maximum degree of alternans where every other stimulation failed to evoke a Ca transient (AR = ∼1). Subsequent exposure to CsA (10 μM) partially rescued alternans by improving AR on average to 0.58 ± 0.07 (n = 6). While CsA was not capable of normalizing Ca alternans completely, the results indicate that mitochondrial Ca overload followed by Ca-induced PTP opening affects Ca alternans negatively and contributes to proarrhythmic conditions.
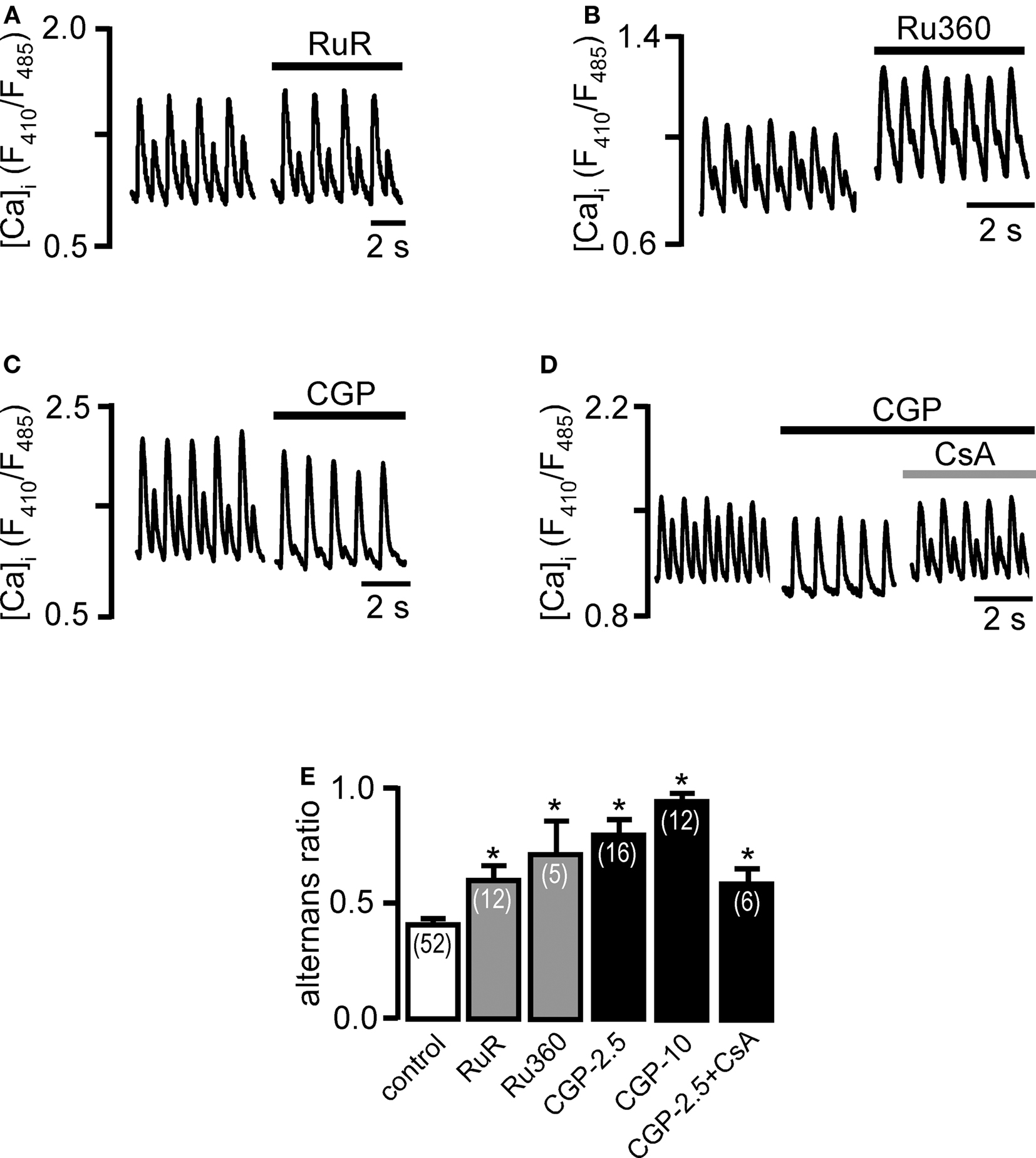
Figure 3. Effects of inhibition of mitochondrial Ca cycling on Ca alternans. Ca alternans recordings before and during treatment with (A) ruthenium red (RuR, 10 μM), (B) Ru360 (5 μM), and (C) CGP-37157 (CGP, 2.5 μM). (D) Cyclosporin A (CsA, 10 μM) partially reversed the effect of CGP-37157 (2.5 μM). (E) Average AR during inhibition of mitochondrial Ca cycling. Numbers in parentheses indicate the number of individual cells tested. *Significantly different from control at P < 0.05. CGP-2.5: 2.5 μM CGP-37157. CGP-10: 10 μM CGP-37157.
Refilling of the SR Ca store after each AP-induced Ca release occurs via SERCA under the consumption of ATP. We tested the hypothesis whether inhibition of mitochondrial metabolism could lower cytosolic ATP levels enough to affect SERCA function and subsequently refilling of the SR. Under conditions of declined cytosolic ATP levels beat-to-beat alternations in the degree of refilling of the SR could ultimately lead to Ca alternans. To test this hypothesis we challenged individual myocytes in the absence and presence of mitochondrial inhibitors twice during pacing-induced Ca alternans, and we compared the amplitude of the caffeine-evoked Ca release after the large (L) and the small (S) Ca transient. Figure 4A (left) shows that the amplitude of the caffeine induced Ca transients were the same irrespective whether they were evoked after a large or a small transient during alternans. This is consistent with our earlier observation that alternating diastolic SR content was not a requirement for the occurrence of Ca alternans (Huser et al., 2000b; Picht et al., 2006). Treatment with oligomycin (1 μg/ml) lead to an enhancement of Ca alternans (increase in AR; Figure 4A right), however SR Ca content was not affected and did not differ whether caffeine (10 mM) was applied after a large or small Ca transient. Similar results were obtained for treatments with CGP-37157 and KMVA (Figure 4B). Thus, despite presumed suppression of mitochondrial ATP production, metabolism and Ca extrusion in the presence of oligomycin, KMVA and CGP-37157, SR Ca replenishment does not appear to be affected. This suggests that cytosolic ATP levels are still sufficiently high to allow for normal SERCA activity and/or ATP for SERCA is supplied from a different source. This is consistent with the observation that ATP derived from glycolysis is utilized preferentially by membrane ion transport mechanisms, including the SERCA pump (Xu et al., 1995). Furthermore, we have shown that inhibition of glycolysis lowers the threshold for the occurrence of electro-mechanical and Ca alternans (Huser et al., 2000b; Kockskamper and Blatter, 2002). This may occur via a mechanism involving reduced ATP supply for SERCA, but based on our observations we also suggested that periodic alterations in the gain of ECC, i.e., the efficacy of a given trigger signal to release Ca from the SR, are controlled locally in the microenvironment of the SR Ca release sites by mechanisms utilizing ATP, produced by glycolytic enzymes closely associated with the release channel. Alternatively, cat atrial cells have low levels of phospholamban expression (Luss et al., 1999), therefore SERCA pumps are more efficient in pumping Ca into the SR, which might contribute to an unchanged SR load.
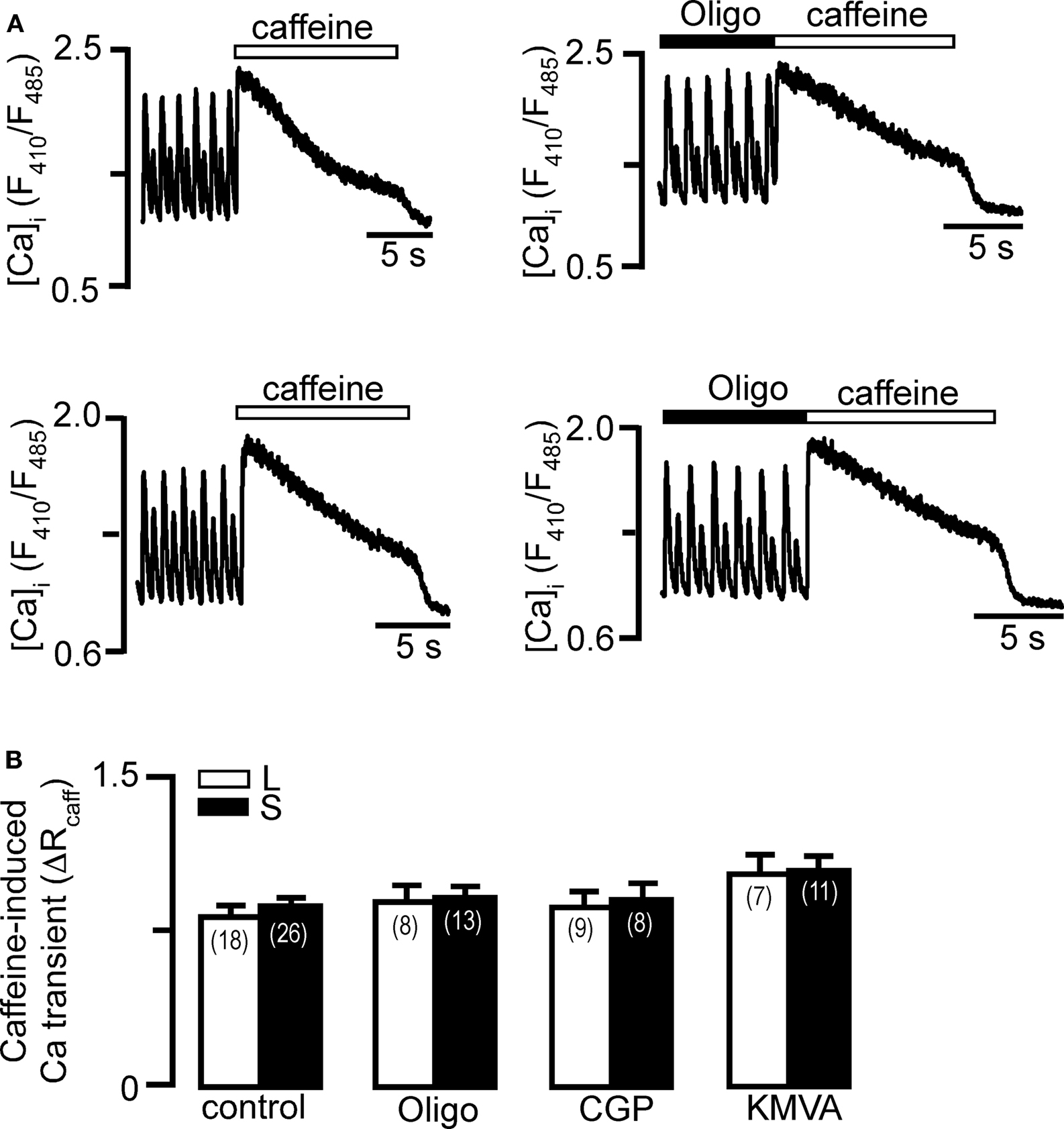
Figure 4. Effects of mitochondrial inhibition on SR Ca load. Caffeine (10 mM) was applied to myocytes revealing stable pacing-induced Ca alternans to measure SR load. Caffeine was applied immediately following the large (L) and small (S) amplitude Ca transient, in the absence (control) and presence of mitochondrial inhibitors. (A) Effect of oligomycin (Oligo, 1 μg/ml). (B) Average SR load during Ca alternans after large and small amplitude Ca transient in control, and in the presence of oligomycin, CGP-37157 (CGP, 2.5 μM) and KMVA (5 mM). SR Ca load is quantified as the amplitude of the caffeine induced Ca transient where Δ R = Rpeak − Rdiastole. Numbers in parentheses indicate the number of individual cells tested.
Our study shows that any intervention that interferes with mitochondrial functions, either by impairing mitochondrial ATP production or mitochondrial Ca buffering, enhanced Ca alternans. All pathways tested here, from direct inhibition of the mitochondrial F1/F0-ATP synthase, over collapsing ΔΨm, inhibition of ETC and Ca-dependent mitochondrial dehydrogenases to blocking mitochondrial Ca uptake and extrusion, directly or indirectly affect diastolic Ca sequestration. As outlined earlier, factors diminishing Ca sequestration generate favorable conditions for Ca alternans. Diastolic Ca sequestration typically occurs against electrical and/or chemical gradients for Ca ions, thus involve energy consuming processes and the availability of ATP. Therefore, one would expect that reduced mitochondrial ATP production is paralleled by impaired Ca removal from the cytosol. It remains to be determined whether this occurs via reduced reuptake of Ca into the SR, diminished Ca extrusion across the plasma membrane or even reduced mitochondrial Ca uptake. Inhibition of the mitochondrial uniporter indeed enhanced Ca alternans. We have shown previously that mitochondria accumulate Ca when exposed to higher frequencies of Ca transients, however elevated matrix [Ca] will eventually reduce the electrochemical gradient for mitochondrial Ca uptake. It remains a matter of debate (recently discussed in O’Rourke and Blatter, 2009) whether beat-to-beat cytosolic Ca transients translate into oscillatory changes of [Ca]mito, or whether changes in beating frequency are merely reflected by changes in steady-state [Ca]mito. Based on our previous investigations it is unlikely that [Ca]mito alternans would be observed at pacing frequencies used in this study (Sedova et al., 2006). Nevertheless, several studies have shown that under conditions of increased workload, as it may occur during β-adrenergic stimulation or high frequency pacing, mitochondrial Ca transients could be discerned (reviewed e.g., in Huser et al., 2000a; Dedkova and Blatter, 2008; O’Rourke and Blatter, 2009). Therefore, it is conceivable that during high frequency stimulation leading to Ca alternans, mitochondrial Ca alternans could be observed, however to date there is no experimental evidence that cytosolic Ca alternans are paralleled by alternans of [Ca]mito.
Certain pathological conditions can be even more favorable for Ca alternans. For example, in heart failure where SERCA expression is reduced and Ca release from the SR (Ca leak) is increased, or during acute cardiac ischemia (where SR Ca load is initially unaffected, but SERCA activity is diminished due to reduced ATP levels), the heart is pushed into instability due to diminished Ca sequestration, all conditions that enhance the propensity of Ca alternans. On the other hand, under β-adrenergic stimulation SERCA activity and consequently SR Ca uptake and load are increased, leading to enhanced fractional release that tends to promote alternans. Increased SERCA activity, however, also increases the efficiency of Ca sequestration, resulting in protection against alternans. Whether β-adrenergic stimulation favors (de Diego et al., 2008) or protects (Huser et al., 2000b) against alternans and alternans-related arrhythmias depends on which β-adrenergic effects predominate.
Another key controversial question in the alternans field is whether beat-to-beat alterations in SR Ca content are either causative (Diaz et al., 2004) or not associated with Ca alternans (Huser et al., 2000b). It has been suggested that diminished Ca reuptake and instability in the beat-to-beat feedback control of SR content leads to Ca alternans (Eisner et al., 2005). We addressed this question with a refined method of direct dynamic measurements of [Ca]SR during pacing-induced alternans (Picht et al., 2006). We found that diastolic [Ca]SR fluctuations can occur during alternans, where a large amplitude Ca release is preceded by a higher diastolic Ca content and vice versa. Interestingly, however, was our observation that Ca alternans can also readily occur without significant diastolic [Ca]SR alternations. These results suggest that factors other than SR Ca load, such as refractoriness (Kornyeyev et al., 2010; Rovetti et al., 2010) and restitution properties of ion currents and CICR from the SR via RyR, aberrant RyR openings (Armoundas, 2009) or, as shown here, metabolic activity and Ca cycling properties of mitochondria, are centrally involved in the mechanism underlying frequency-induced cardiac alternans.
Mitochondrial Ca signaling and energy metabolism are highly intertwined, thus the experimental dissection of the exact mechanism by which impaired mitochondrial function causes Ca alternans remains a difficult task. Our study relied exclusively on the pharmacological challenge of various mitochondrial targets ranging from mitochondrial ATP synthase, membrane potential, electron transport chain, Ca-dependent dehydrogenases to Ca uptake and extrusion pathways. While many of the pharmacological tools employed here are considered rather specific for the respective target, this approach still faces the difficulty that any interference with the metabolic function (ATP production) of mitochondria also affects mitochondrial and cellular Ca homeostasis, and vice versa any disturbance of mitochondrial Ca cycling interferes with mitochondrial energy metabolism (e.g., via Ca-dependent dehydrogenases). The oligomycin experiments (Figure 1B) clearly show that inhibition of mitochondrial ATP production enhances Ca alternans, thus sufficient ATP supply is protective against proarrhythmic Ca alternans. This finding is in line with our earlier studies where we showed that inhibition of glycolysis (and glycolytic ATP production) generates conditions that favor Ca alternans (Huser et al., 2000b; Kockskamper and Blatter, 2002; Kockskamper et al., 2005). Clearly, experimental interventions that impair mitochondrial Ca uptake and extrusion, and therefore mitochondrial Ca buffering, also enhance the propensity of Ca alternans (Figure 3). Whether this effect on Ca alternans occurs through a modification of cytosolic Ca transients or via alterations of Ca-dependent steps of mitochondrial energy metabolism remains to be determined. However, our experiments indicate that both are equally important. In line with the multifactorial nature of Ca and electro-mechanical alternans we can conclude from this study that sufficient mitochondrial energy (ATP) production and Ca buffering are both important and necessary safeguards against the occurrence of proarrhythmic Ca alternans.
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
This work was supported by grants from the National Institutes of Health (HL62231 and HL80101 to Lothar A. Blatter), and the American Heart Association, Midwest Affiliate (to Stela M. Florea).
Aistrup, G. L., Kelly, J. E., Kapur, S., Kowalczyk, M., Sysman-Wolpin, I., Kadish, A. H., and Wasserstrom, J. A. (2006). Pacing-induced heterogeneities in intracellular Ca2+ signaling, cardiac alternans, and ventricular arrhythmias in intact rat heart. Circ. Res. 99, e65–e73.
Aistrup, G. L., Shiferaw, Y., Kapur, S., Kadish, A. H., and Wasserstrom, J. A. (2009). Mechanisms underlying the formation and dynamics of subcellular calcium alternans in the intact rat heart. Circ. Res. 104, 639–649.
Altschafl, B. A., Beutner, G., Sharma, V. K., Sheu, S. S., and Valdivia, H. H. (2007). The mitochondrial ryanodine receptor in rat heart: a pharmaco-kinetic profile. Biochim. Biophys. Acta 1768, 1784–1795.
Armoundas, A. A. (2009). Mechanism of abnormal sarcoplasmic reticulum calcium release in canine left-ventricular myocytes results in cellular alternans. IEEE Trans. Biomed. Eng. 56, 220–228.
Badeer, H. S., Ryo, U. Y., Gassner, W. F., Kass, E. J., Cavaluzzi, J., Gilbert, J. L., and Brooks, C. M. (1967). Factors affecting pulsus alternans in the rapidly driven heart and papillary muscle. Am. J. Physiol. 213, 1095–1101.
Bassani, J. W., Yuan, W., and Bers, D. M. (1995). Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 268, C1313–1319.
Belevych, A. E., Terentyev, D., Viatchenko-Karpinski, S., Terentyeva, R, Sridhar, A., Nishijima, Y., Wilson LD, Cardounel, A. J., Laurita, K. R., Carnes, C. A., Billman, G. E., and Gyorke, S. (2009). Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc. Res. 84, 387–395.
Berger, R. D. (2000). Repolarization alternans: toward a unifying theory of reentrant arrhythmia induction. Circ. Res. 87, 1083–1084.
Beutner, G., Sharma, V. K., Giovannucci, D. R., Yule, D. I., and Sheu, S. S. (2001). Identification of a ryanodine receptor in rat heart mitochondria. J. Biol. Chem. 276, 21482–21488.
Beutner, G., Sharma, V. K., Lin, L., Ryu, S. Y., Dirksen, R. T., and Sheu, S. S. (2005). Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochim. Biophys. Acta. 1717, 1–10.
Blatter, L. A., Kockskamper, J., Sheehan, K. A., Zima, A. V., Huser, J., and Lipsius, S. L. (2003). Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J. Physiol. 546, 19–31.
Buntinas, L., Gunter, K. K., Sparagna, G. C., and Gunter, T. E. (2001). The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim. Biophys. Acta 1504, 248–261.
Clusin, W. T. (2008). Mechanisms of calcium transient and action potential alternans in cardiac cells and tissues. Am. J. Physiol. Heart Circ. Physiol. 294, H1–H10.
Cordeiro, J. M., Malone, J. E., Di Diego, J. M., Scornik, F. S., Aistrup, G. L., Antzelevitch, C., and Wasserstrom, J. A. (2007). Cellular and subcellular alternans in the canine left ventricle. Am. J. Physiol. Heart Circ. Physiol. 293, H3506–H3516.
Cox, D. A., Conforti, L., Sperelakis, N., and Matlib, M. A. (1993). Selectivity of inhibition of Na( + )-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J. Cardiovasc. Pharmacol. 21, 595–599.
Crompton, M. (1999). The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341 (Pt 2), 233–249.
Cutler, M. J., Wan, X., Laurita, K. R., Hajjar, R. J., and Rosenbaum, D. S. (2009). Targeted SERCA2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circ. Arrhythm. Electrophysiol. 2, 686–694.
de Diego, C., Chen, F., Xie, L. H., Dave, A. S., Thu, M., Rongey, C., Weiss, J. N., and Valderrabano, M. (2008). Cardiac alternans in embryonic mouse ventricles. Am. J. Physiol. Heart Circ. Physiol. 294, H433–H440.
Dedkova, E. N., and Blatter, L. A. (2008). Mitochondrial Ca(2+) and the heart. Cell Calcium 44, 77–91.
Diaz, M. E., Eisner, D. A., and O’Neill, S. C. (2002). Depressed ryanodine receptor activity increases variability and duration of the systolic Ca2+ transient in rat ventricular myocytes. Circ. Res. 91, 585–593.
Diaz, M. E., O’Neill, S. C., and Eisner, D. A. (2004). Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ. Res. 94, 650–656.
Dilly, S. G., and Lab, M. J. (1988). Electrophysiological alternans and restitution during acute regional ischaemia in myocardium of anaesthetized pig. J. Physiol. 402, 315–333.
Dumitrescu, C., Narayan, P., Efimov, I. R., Cheng, Y., Radin, M. J., McCune, S. A., and Altschuld, R. A. (2002). Mechanical alternans and restitution in failing SHHF rat left ventricles. Am. J. Physiol. Heart Circ. Physiol. 282, H1320–H1326.
Eisner, D. A., Choi, H. S., Diaz, M. E., O’Neill, S. C., and Trafford, A. W. (2000). Integrative analysis of calcium cycling in cardiac muscle. Circ Res. 87, 1087–1094.
Eisner, D. A., Diaz, M. E., Li, Y., O’Neill, S. C., and Trafford, A. W. (2005). Stability and instability of regulation of intracellular calcium. Exp. Physiol. 90, 3–12.
Eisner, D. A., Li, Y., and O’Neill, S. C. (2006). Alternans of intracellular calcium: mechanism and significance. Heart Rhythm 3, 743–745.
Euler, D. E. (1999). Cardiac alternans: mechanisms and pathophysiological significance. Cardiovasc. Res. 42, 583–590.
Florea, S. M., and Blatter, L. A. (2005). The role of β-adrenergic signaling and mitochondria for Ca2+ alternans modulation in atrial myocytes. Biophys. J. 88, 135a.
Fox, J. J., McHarg, J. L., and Gilmour, R. F. Jr. (2002). Ionic mechanism of electrical alternans. Am. J. Physiol. Heart Circ. Physiol. 282, H516–H530.
Gaeta, S. A., Bub, G., Abbott, G. W., and Christini, D. J. (2009). Dynamical mechanism for subcellular alternans in cardiac myocytes. Circ. Res. 105, 335–342.
Gunter, T. E., Buntinas, L., Sparagna, G., Eliseev, R., and Gunter, K. (2000). Mitochondrial calcium transport: mechanisms and functions. Cell Calcium 28, 285–296.
Hayashi, H., Shiferaw, Y., Sato, D., Nihei, M., Lin, S. F., Chen, P. S., Garfinkel, A., Weiss, J. N., and Qu, Z. (2007). Dynamic origin of spatially discordant alternans in cardiac tissue. Biophys. J. 92, 448–460.
Hiromoto, K., Shimizu, H., Furukawa, Y., Kanemori, T., Mine, T., Masuyama, T., and Ohyanagi, M. (2005). Discordant repolarization alternans-induced atrial fibrillation is suppressed by verapamil. Circ. J. 69, 1368–1373.
Huser, J., Blatter, L. A., and Sheu. S. S. (2000a). Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? J. Bioenerg. Biomembr. 32, 27–33.
Huser, J., Wang, Y. G., Sheehan, K. A., Cifuentes, F., Lipsius, S. L., and Blatter, L. A. (2000b). Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J. Physiol. 524 (Pt 3), 795–806.
Huser, J., Lipsius, S. L., and Blatter, L. A. (1996). Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J. Physiol. 494 (Pt 3), 641–651.
Huser, J., Rechenmacher, C. E., and Blatter, L. A. (1998). Imaging the permeability pore transition in single mitochondria. Biophys. J. 74, 2129–2137.
Isenberg, G., Han, S., Schiefer, A., and Wendt-Gallitelli, M. F. (1993). Changes in mitochondrial calcium concentration during the cardiac contraction cycle. Cardiovasc. Res. 27, 1800–1809.
Jordan, P. N., and Christini, D. J. (2007). Characterizing the contribution of voltage- and calcium-dependent coupling to action potential stability: implications for repolarization alternans. Am. J. Physiol. Heart Circ. Physiol. 293, H2109–H2118.
Kameyama, M., Hirayama, Y., Saitoh, H., Maruyama, M., Atarashi, H., and Takano, T. (2003). Possible contribution of the sarcoplasmic reticulum Ca(2+) pump function to electrical and mechanical alternans. J. Electrocardiol. 36, 125–135.
Kapur, S., Wasserstrom, J. A., Kelly, J. E., Kadish, A. H., and Aistrup, G. L. (2009). Acidosis and ischemia increase cellular Ca2+ transient alternans and repolarization alternans susceptibility in the intact rat heart. Am. J. Physiol. Heart Circ. Physiol. 296, H1491–H1512.
Kihara, Y., and Morgan, J. P. (1991). Abnormal Cai2+ handling is the primary cause of mechanical alternans: study in ferret ventricular muscles. Am. J. Physiol. 261, H1746–H1755.
Kockskamper, J., and Blatter, L. A. (2002). Subcellular Ca2+ alternans represents a novel mechanism for the generation of arrhythmogenic Ca2+ waves in cat atrial myocytes. J. Physiol. 545, 65–79.
Kockskamper, J., Zima, A. V., and Blatter, L. A. (2005). Modulation of sarcoplasmic reticulum Ca2+ release by glycolysis in cat atrial myocytes. J. Physiol. 564, 697–714.
Konta, T., Ikeda, K., Yamaki, M., Nakamura, K., Honma, K., Kubota, I., and Yasui, S. (1990). Significance of discordant ST alternans in ventricular fibrillation. Circulation 82, 2185–2189.
Kornyeyev, D., Reyes, M., and Escobar, A. L. (2010). Luminal Ca(2+) content regulates intracellular Ca(2+) release in subepicardial myocytes of intact beating mouse hearts: effect of exogenous buffers. Am. J. Physiol. Heart Circ. Physiol. 298, H2138–H2153.
Lab, M. J., and Lee, J. A. (1990). Changes in intracellular calcium during mechanical alternans in isolated ferret ventricular muscle. Circ. Res. 66, 585–595.
Li, Y., Diaz, M. E., Eisner, D. A., and O’Neill, S. (2009). The effects of membrane potential, SR Ca2+ content and RyR responsiveness on systolic Ca2+ alternans in rat ventricular myocytes. J. Physiol. 587, 1283–1292.
Livshitz, L. M., and Rudy, Y. (2007). Regulation of Ca2+ and electrical alternans in cardiac myocytes: role of CAMKII and repolarizing currents. Am. J. Physiol. Heart Circ. Physiol. 292, H2854–H2866.
Luss, I., Boknik, P., Jones, L. R., Kirchhefer, U., Knapp, J., Linck, B., Luss, H., Meissner, A., Muller, F. U., Schmitz, W., Vahlensieck, U., and Neumann, J. (1999). Expression of cardiac calcium regulatory proteins in atrium v ventricle in different species. J. Mol. Cell Cardiol. 31, 1299–1314.
Maack, C., Cortassa, S., Aon, M. A., Ganesan, A. N., Liu, T., and O’Rourke, B. (2006). Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 99, 172–182.
Mahajan, A., Shiferaw, Y., Sato, D., Baher, A., Olcese, R., Xie, L. H., Yang, M. J., Chen, P. S., Restrepo, J. G., Karma, A., Grafinkel, A., Qu, Z., and Weiss, J. (2008). A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys. J. 94, 392–410.
Matlib, M. A., Zhou, Z., Knight, S., Ahmed, S., Choi, K. M., Krause-Bauer, J., Phillips, R., Altschuld, R., Katsube, Y., Sperelakis, N., and Bers, D. M. (1998). Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J. Biol. Chem. 273, 10223–10231.
Miyata, H., Silverman, H. S., Sollott, S. J., Lakatta, E. G., Stern, M. D., and Hansford, R. G. (1991). Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am. J. Physiol. 261, H1123–H1134.
Moravec, C. S., and Bond, M. (1991). Calcium is released from the junctional sarcoplasmic reticulum during cardiac muscle contraction. Am. J. Physiol. 260, H989–H997.
Myles, R. C., Burton, F. L., Cobbe, S. M., and Smith, G. L. (2008). The link between repolarisation alternans and ventricular arrhythmia: does the cellular phenomenon extend to the clinical problem? J. Mol. Cell Cardiol. 45, 1–10.
Narayan, S. M., Bode, F., Karasik, P. L., and Franz, M. R. (2002). Alternans of atrial action potentials during atrial flutter as a precursor to atrial fibrillation. Circulation 106, 1968–1973.
O’Rourke, B., and Blatter, L. A. (2009). Mitochondrial Ca2+ uptake: tortoise or hare? J. Mol. Cell Cardiol. 46, 767–774.
Orchard, C. H., McCall, E., Kirby, M. S., and Boyett, M .R. (1991). Mechanical alternans during acidosis in ferret heart muscle. Circ. Res. 68, 69–76.
Pastore, J. M., Girouard, S. D., Laurita, K. R., Akar, F. G., and Rosenbaum, D.S. (1999). Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 99, 1385–1394.
Pastore, J. M., and Rosenbaum, D. S. (2000). Role of structural barriers in the mechanism of alternans-induced reentry. Circ. Res. 87, 1157–1163.
Picht, E., DeSantiago, J., Blatter, L. A., and Bers, D. M. (2006). Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ. Res. 99, 740–748.
Restrepo, J. G., Weiss, J. N., and Karma, A. (2008). Calsequestrin-mediated mechanism for cellular calcium transient alternans. Biophys. J. 95, 3767–3789.
Robert, V., Gurlini, P., Tosello, V., Nagai, T., Miyawaki, A., Di Lisa, F., and Pozzan, T. (2001). Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 20, 4998–5007.
Rosenbaum, D. S. (2001). T wave alternans: a mechanism of arrhythmogenesis comes of age after 100 years. J. Cardiovasc. Electrophysiol. 12, 207–209.
Rosenbaum, D. S., Jackson, L. E., Smith, J. M., Garan, H., Ruskin, J. N., and Cohen, R. J. (1994). Electrical alternans and vulnerability to ventricular arrhythmias. N. Engl. J. Med. 330, 235–241.
Rovetti, R., Cui, X., Garfinkel, A., Weiss, J. N., and Qu, Z. (2010). Spark-induced sparks as a mechanism of intracellular calcium alternans in cardiac myocytes. Circ. Res. 106, 1582–1591.
Rubenstein, D. S., and Lipsius, S. L. (1995). Premature beats elicit a phase reversal of mechanoelectrical alternans in cat ventricular myocytes. A possible mechanism for reentrant arrhythmias. Circulation 91, 201–214.
Sato, D., Shiferaw, Y., Garfinkel, A., Weiss, J. N., Qu, Z., and Karma, A. (2006). Spatially discordant alternans in cardiac tissue: role of calcium cycling. Circ. Res. 99, 520–527.
Sato, D., Shiferaw, Y., Qu, Z., Garfinkel, A., Weiss, J. N., and Karma, A. (2007). Inferring the cellular origin of voltage and calcium alternans from the spatial scales of phase reversal during discordant alternans. Biophys. J. 92, L33–L35.
Sedova, M., Dedkova, E. N., and Blatter, L. A. (2006). Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am. J. Physiol. Cell Physiol. 291, C840–C850.
Sheehan, K. A., and Blatter, L. A. (2003). Regulation of junctional and non-junctional sarcoplasmic reticulum calcium release in excitation-contraction coupling in cat atrial myocytes. J. Physiol. 546, 119–135.
Shiferaw, Y., and Karma, A. (2006). Turing instability mediated by voltage and calcium diffusion in paced cardiac cells. Proc. Natl. Acad. Sci. U.S.A. 103, 5670–5675.
Shiferaw, Y., Watanabe, M. A., Garfinkel, A., Weiss, J. N., and Karma, A. (2003). Model of intracellular calcium cycling in ventricular myocytes. Biophys. J. 85, 3666–3686.
Shimizu, W., and Antzelevitch, C. (1999). Cellular and ionic basis for T-wave alternans under long-QT conditions. Circulation 99, 1499–1507.
Smith, J. M., Clancy, E. A., Valeri, C. R., Ruskin, J. N., and Cohen, R. J. (1988). Electrical alternans and cardiac electrical instability. Circulation 77, 110–121.
Sparagna, G. C., Gunter, K. K., Sheu, S. S., and Gunter, T. E. (1995). Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J. Biol. Chem. 270, 27510–27515.
Spencer, C. I., Lab, M. J., and Seed, W. A. (1992). Mechanical restitution during alternans in guinea pig papillary muscles. Cardiovasc. Res. 26, 779–782.
Surawicz, B., and Fisch, C. (1992). Cardiac alternans: diverse mechanisms and clinical manifestations. J. Am. Coll. Cardiol. 20, 483–499.
Uno, K. (1991). Mechanisms of pulsus alternans: its relation to alternation of regional contraction and elevated ST segment. Am. Heart J. 122, 1694–1700.
Verrier, R. L., and Nearing, B. D. (1994). Electrophysiologic basis for T wave alternans as an index of vulnerability to ventricular fibrillation. J. Cardiovasc. Electrophysiol. 5, 445–461.
Walker, M. L., and Rosenbaum, D. S. (2003). Repolarization alternans: implications for the mechanism and prevention of sudden cardiac death. Cardiovasc. Res. 57, 599–614.
Weiss, J. N., Karma, A., Shiferaw, Y., Chen, P. S., Garfinkel, A., and Qu, Z. (2006). From pulsus to pulseless: the saga of cardiac alternans. Circ. Res. 98, 1244–1253.
Wohlfart, B. (1982). Analysis of mechanical alternans in rabbit papillary muscle. Acta. Physiol. Scand. 115, 405–414.
Wu, J. Y., Vereecke, J., Carmeliet, E., and Lipsius, S. L. (1991). Ionic currents activated during hyperpolarization of single right atrial myocytes from cat heart. Circ. Res. 68, 1059–1069.
Wu, Y., and Clusin, W. T. (1997). Calcium transient alternans in blood-perfused ischemic hearts: observations with fluorescent indicator fura red. Am. J. Physiol. 273, H2161–H2169.
Xie, L. H., Sato, D., Garfinkel, A., Qu, Z., and Weiss, J. N. (2008). Intracellular Ca alternans: coordinated regulation by sarcoplasmic reticulum release, uptake, and leak. Biophys. J. 95, 3100–3110.
Xie, L. H., and Weiss, J. N. (2009). Arrhythmogenic consequences of intracellular calcium waves. Am. J. Physiol. Heart Circ. Physiol. 297, H997–H1002.
Xu, K. Y., Zweier, J. L., and Becker, L. C. (1995). Functional coupling between glycolysis and sarcoplasmic reticulum Ca2+ transport. Circ. Res. 77, 88–97.
Keywords: mitochondria, intracellular [Ca], Ca alternans, electron transport chain, permeability transition pore, mitochondrial Ca cycling, TCA cycle
Citation: Florea SM and Blatter LA (2010) The role of mitochondria for the regulation of cardiac alternans. Front. Physio. 1:141. doi: 10.3389/fphys.2010.00141
Received: 28 May 2010;
Accepted: 07 October 2010;
Published online: 03 November 2010.
Edited by:
Miguel A. Aon, Johns Hopkins University School of Medicine, USAReviewed by:
Fadi G. Akar, Mount Sinai School of Medicine, USACopyright: © 2010 Florea and Blatter. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Lothar A. Blatter, Department of Molecular Biophysics and Physiology, Rush University Medical Center, 1750 W. Harrison St., Chicago, IL 60612, USA. e-mail:bG90aGFyX2JsYXR0ZXJAcnVzaC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.