


- Experimental Cardiology Group, Academic Medical Center, Amsterdam, Netherlands
Sodium channel blockers are associated with arrhythmic sudden death, although they are considered antiarrhythmic agents. The mechanism of these opposing effects is unknown. We used a model of induction of ventricular fibrillation (VF) based on selective perfusion of the vascular beds of isolated porcine hearts (n = 8). One bed was perfused with sotalol (220 µM), the adjacent bed with pinacidil (80 µM), leading to repolarization heterogeneity (late repolarization in the sotalol-, early in the pinacidil-area). Premature stimulation from the area with the short action potential was performed. Epicardial activation/repolarization mapping was done. In three of the eight hearts VF was inducible prior to infusion of flecainide. In those hearts the Fibrillation Factor (FF), the interval between the earliest repolarization of the premature beat (S2) in the early repolarizing (pinacidil) domain, and the last S2-activation in the late repolarizing (sotalol) domain, was significantly shorter than in the hearts without VF (33 ± 22 vs 93 ± 11 ms, m ± SEM, p < 0.05). In the three hearts with VF flecainide was infused in the pinacidil domain after defibrillation. This led to shortening of the line of block, local delay of S2 activation and repolarization, an increase in FF and failure to induce VF. In the five hearts without VF, flecainide was subsequently infused in the sotalol domain. This led to a local delay of S2 activation, a shortening of FF (by 47 ± 3 ms) and successful induction of VF in three hearts. In the two remaining hearts FF did not decrease enough (maximally 13 ms) to allow re-entry. Sodium channel blockade applied to myocardium with a short refractory period is antifibrillatory whereas sodium channel blockade of myocardium with a long refractory period is profibrillatory. Our study provides a mechanistic basis for pro- and antiarrhythmic effects of sodium channel blockers in the absence of structural heart disease.
Introduction
Sodium channel blockade was originally considered an antiarrhythmic strategy (Lie et al., 1978 ; Coromilas et al., 1995 ). The CAST-study, however, has demonstrated that it also may provoke arrhythmic death (The Cardiac Arrhythmia Suppression Trial [CAST] Investigators, 1989 ). Also, loss of function mutations in sodium channel are associated with life-threatening arrhythmias (Zimmer and Surber, 2010 ). Slowing of conduction in the presence of an arrhythmogenic substrate (acute myocardial infarction or fibrosis associated with heart failure) is a common explanation for the proarrhythmic effect of sodium channel blockers in the CAST study (Echt et al., 1991 ). It is, however, not clear by which mechanism sodium channel blockade can be proarrhythmic in the absence of structural abnormalities. We addressed this question in an porcine model of ventricular fibrillation (VF) based on heterogeneity in repolarization (Coronel et al., 2009 ).
In a previous study (Coronel et al., 2009 ) we have shown that repolarization heterogeneity may lead to conduction block, but not necessarily to re-entry. In the present study we slowed conduction by regional infusion of the sodium channel blocker flecainide, and hypothesized that conduction slowing by sodium channel blockade can be either pro- or antiarrhythmic depending on the area of administration relative to the repolarization gradient. Figure 1 illustrates our hypothesis. In the presence of a preexisting repolarization gradient a premature stimulus (S2) is delivered to the zone with the short action potential. The difference between the repolarization time of the premature beat in the tissue proximal to the line of block and the activation time of the premature activation front distal to the line of block determines whether or not re-entry occurs (Coronel et al., 2009 ). We have termed this difference the “Fibrillation Factor” (FF). When FF is small there is unidirectional conduction block and re-entry occurs, but when it is large the premature wavefront meets a line of bidirectional block and re-entry is prevented.
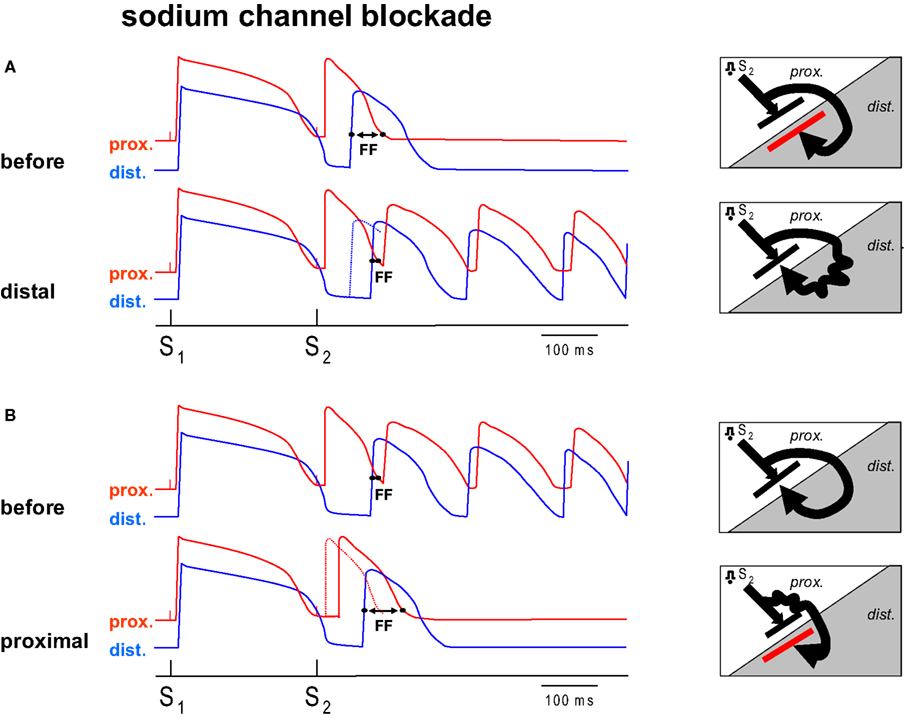
Figure 1. Diagram of the hypothesis. (A,B) Each show two hypothetical action potential tracings from two sides of a region of repolarization dispersion. The distal (dist.) side of this region has a prolonged action potential compared to the proximal side (prox.). Each tracing shows the response of a basic stimulus (S1) and a premature stimulus (S2). The S2 is conducted with a delay towards the distal side because of unidirectional block between the regions. The Fibrillation Factor (FF) is defined by the difference in proximal repolarization time and the distal activation time of the premature beat following S2. If no re-entry can be induced infusion of sodium channel blockade into the distal region (A, distal) shifts the distal activation time of S2 to a later moment (the dotted line represents the action potential before flecainide), decreases FF and results in re-entry. (B) Shows the mirror condition. If VF can be induced, prolongation of the FF can be achieved by sodium channel blockade of the proximal tissue, leading to a delay of proximal S2 repolarization secondary to the delay in activation, and an antifibrillatory effect (B, proximal). The diagrams (right) show the corresponding activation patterns. Distal application of sodium channel blockade leads to the conversion of idirectional block to unidirectional block, proximal application of sodium channel blockade to conversion from unidirectional block to bidirectional block (red line).
Figure 1 A shows a condition prior to the infusion of flecainide, when re-entry does not occur, because the wavefront traveling around the line of block reaches the distal site at a time when the proximal zone has not yet recovered from excitability (FF is large). We hypothesize that by adding flecainide to the distal zone, the wavefront in the distal zone is delayed and now arrives late enough for the proximal tissue to be re-excited (FF has become smaller). If prior to application of sodium channel blockade VF does occur (FF is small, Figure 1 B), administration of flecainide to the proximal tissue causes a delay of local activation (and therefore repolarization) so that the wavefront reaches the distal site too early to allow re-entry (FF has increased). In this scenario, conduction slowing in the distal area is proarrhythmic, whereas conduction slowing in the proximal area is antiarrhythmic. This hypothesis was tested in this study in which we specifically addressed the question whether regional infusion of a sodium channel blocker would cause an increase or decrease in FF and a concomitant corresponding anti- or profibrillatory effect.
Materials and Methods
This study was approved by the local ethical committee on animal experimentation and conforms to the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health (NIH publication No. 85-23, revised 1996).
Preparation
Pigs (n = 8; weight 37.5 ± 1.8 kg (mean ± SEM)) were premedicated with 350 mg ketamine (Nimatek, Animal Health BV, Bladel, The Netherlands), 80 mg azaperone (Stresnil, Janssen, The Netherlands, Janssen-Cilag, High Wycombe, UK), and 0.5 mg atropine (Centrafarm Services, Etten-Leur, The Netherlands) intramuscularly and anesthetized with 20 mg/kg pentobarbital (Nembutal, CevaState Animale, Naaldwijk, The Netherlands) intravenously. The animals were intubated and ventilated with room air and isoflurane (Forene, Abbott, Hoofddorp, The Netherlands). Expiratory CO2 was monitored. Heparin, 5000 international units (LeoPharma, Breda, The Netherlands) was injected intravenously. Blood was collected, and the heart was isolated via a midsternal thoracotomy.
The heart was perfused in a Langendorff setup using a mixture of blood and Tyrode’s solution (pH = 7.35–7.45). Atrioventricular block was created by crushing the AV nodal area. The left anterior descending coronary artery (LAD) was freed over a distance of 5 mm, above the first diagonal branch. A ligature was passed underneath the LAD, and a cannula was introduced via a small incision into the LAD. The cannula was fixed by tying the ligature and was connected to a separate perfusion system via a miniature heat exchanger. The temperature of both perfusion areas was controlled by separate heat exchangers in each perfusion limb. Infusion pumps were connected to the side branch of the LAD cannula and to the aortic cannula for the administration of sotalol and/or flecainide. Pinacidil was added to the perfusate by mixing it in the main container to reach a final concentration of 80 µM.
Infusion pump rates were adjusted to obtain calculated concentrations of 220 µM sotalol and 6 µM flecainide. The LAD perfused area had a temperature of 37.6 ± 0.30°C, the remainder of the heart was at 37.4 ± 0.23°C. Flow in the LAD was set at 30 ml/min, flow in the remainder of the heart at 70 ml/min. The absence of ST-T segment changes indicated absence of local ischemia (recording methods see below).
Flecainide was selectively infused in either vascular bed, depending on the preexisting inducibility of VF.
Electrophysiology
A rectangular grid of 11 × 11 electrodes (gold, 5 mm interelectrode distance) was sutured over the border between the myocardium perfused by the LAD and the rest of the heart. The cyanotic border was identified prior to application of the electrode by a 30-s occlusion of the LAD. Correct positioning of the electrode was verified by creating a 5-min occlusion of the LAD and analyzing the border between the region with and without electrophysiological signs of ischemia. After restoration of blood flow the heart was allowed to recover for at least 60 min before measurements were begun. Full recovery was defined by the return of ST-segment elevation to the isoelectric line and a stable value of refractoriness in the LAD-area.
Unipolar cathodal stimulation was performed through one of the electrodes in the electrode grid overlying the circumflex area. One to three stimulus positions were tested sequentially. The anode was placed at the aortic root. Premature beats were introduced after each train of eight beats with coupling intervals ranging from the basic cycle length of 600 ms down to the refractory period. Control recordings were made of a basic beat and a premature beat prior to the interventions. Local unipolar electrograms were recorded against a reference electrode at the aortic root using a data acquisition system [Biosemi, Amsterdam, The Netherlands, sampling rate 2000 Hz, filtering DC 1 kHz (3 dB)].
Analysis of the electrograms was performed offline using a custom-made analysis program (Potse et al., 2002 ). Local activation times were measured at the moment of the minimum dV/dt of the initial deflection, and local repolarization times at the moment of the maximum dV/dt of the T wave (Coronel et al., 2006 ). When determination of activation times was difficult because of fractionation of the signals, Laplacian electrograms were constructed to aid in the detection of local activation (Coronel et al., 2000 ). Regional conduction delay induced by flecainide was quantified by averaging activation times within the respective perfusion domains, before and after administration of flecainide. In the distal tissue, the local activation times were corrected for the time of arrival of the activation wave in the distal domain.
Contrary to our previous study where pinacidil was infused in the LAD area and sotalol in the remainder of the heart, we now infused sotalol in the LAD and pinacidil in the rest of the heart. To facilitate comparison with the activation maps in that study we rotated the activation maps 180°. The FF is defined as the difference between the activation time of the premature activation front distal to the line of block and the repolarization time of the tissue proximal to the line of block (Coronel et al., 2009 ).
Protocol
At time zero (t = 0), the LAD was perfused with the normal blood-Tyrode’s mixture, and pinacidil was administered to the rest of the heart (the proximal zone) to shorten the repolarization time. At t = 10 min, sotalol was added to the LAD area (distal zone) to prolong local repolarization time. Before one recirculation period expired, recirculation was stopped and the venous blood was not reentered into the perfusion apparatus. From this moment onward a stable condition could be maintained for about 30 min until we ran out of perfusate.
We attempted to induce VF by a premature stimulus applied to the proximal zone from t = 15 to 18 min. If VF occurred within this period, the heart was defibrillated immediately and flecainide was added to the proximal zone. If VF did not occur, flecainide was added to the distal area while the combination of the perfusate was otherwise maintained. Subsequent attempts to induce VF were made between 22 and 30 min. As many recordings as possible were made of the premature beats and any ensuing arrhythmias from the time at which repolarization heterogeneity was created by selective perfusion with the drugs. When VF occurred, the heart was immediately defibrillated by a DC shock, stimulation was resumed and a period of 1 min was allowed before induction of VF was re-attempted. The selection of the relevant successful and unsuccessful attempts to induce VF was made on the following grounds. First, pacing was performed with the same coupling interval and from the same stimulus site before and after flecainide. Second, VF induction immediately before administration of flecainide was compared to VF-induction at the moment on which the maximum conduction slowing induced by flecainide occurred. As a result of this selection we were able to compare 8 pairs of total of 68 VF-inductions.
Drugs
Sotalol (Sotacor vials 10 mg/ml) was obtained from Bristol–Myers–Squibb (Woerden, The Netherlands). Pinacidil (pinacidil monohydrate P154) was obtained from Sigma Aldrich (Zwijndrecht, The Netherlands), Tambocor (flecainide acetate) from 3 M Nederland (Zoeterwoude, The Netherlands).
Presentation of Data and Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM). Comparisons were tested for differences with the use of the paired t-test or the Mann–Whitney Rank Sum test, when appropriate p < 0.05 was accepted as the level of statistical significance.
Results
The magnitude of differences in repolarization time obtained by infusion of sotalol and pinacidil was on average 96 ± 11 ms (n = 8). In three of the eight hearts VF was inducible prior to infusion of flecainide. As expected, the FF was significantly shorter in the hearts with VF than in the hearts without VF (33 ± 22 vs 93 ± 11 ms, m ± SEM, p < 0.05).
Figure 2 shows an example of the proarrhythmic effect of the application of flecainide to the myocardium with late repolarization. Maps are shown of activation times following application of a premature stimulus to the proximal zone (p, pinacidil) with a coupling interval of 260 ms. Prior to flecainide infusion (top panels) the premature wavefront arrives after 340 ms at the tissue distal to the line of block, too early to re-excite the proximal tissue which has a repolarization time following S2 of 401 ms (not shown). Hence no VF ensues. Selected electrograms from either regions show that the T-wave is positive in the area with the pinacidil (proximal, p) and predominantly negative in the area with sotalol (distal, d). The premature activation of the distal area is early (dotted line) relative to the proximal repolarization (dot).
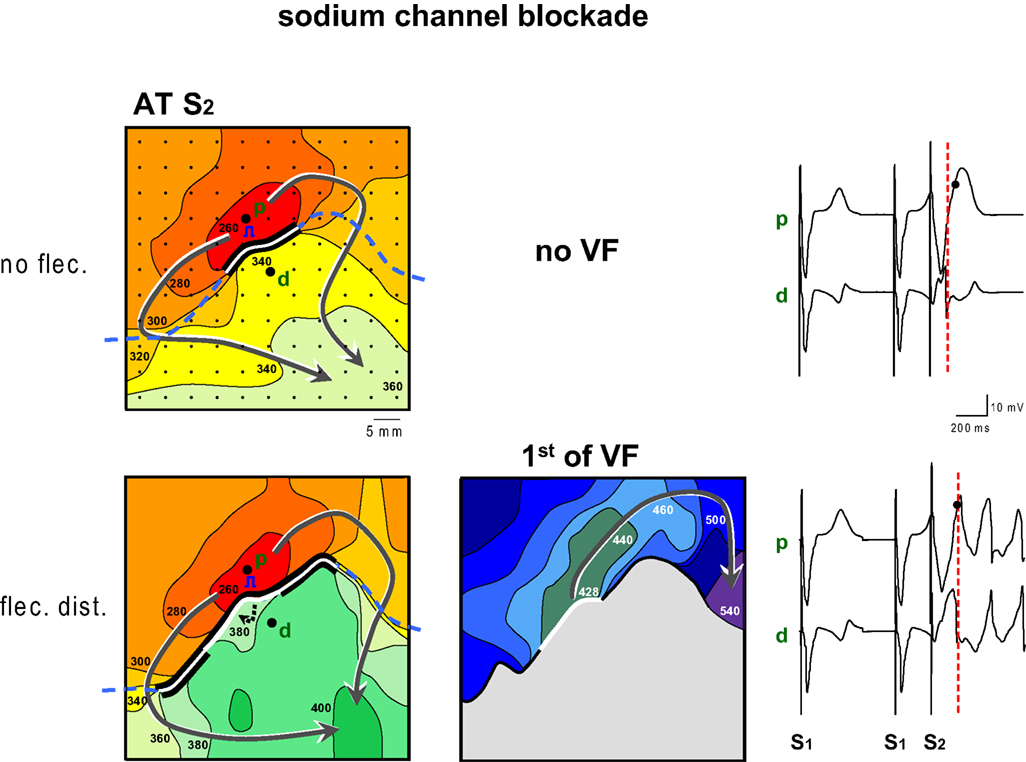
Figure 2. Activation maps of the premature beat (following S2) before and after application of flecainide (flec) to the region with prolonged repolarization (distal). Prior to infusion of flecainide no VF could be induced, because local distal activation preceded local proximal repolarization. Flecainide infusion the distal domain caused a decrease of the FF and induction of VF. The dashed line indicates the epicardial interface between the perfusion beds. Small dots indicate electrode positions. Bold dots indicate positions from which electrograms (right panels) were recorded (p, proximal; d distal). Lines are 20 ms isochrones. Red dotted vertical line in the electrograms indicate the moment of distal activation.
The lower panels of Figure 2 show activation maps after the addition of flecainide to the distal zone. The activation pattern of the proximal area is unaltered compared to the top panel. The wavefront reaches the tissue distal to the line of block after 380 ms, much later than before infusion of flecainide, and now is able to re-excite the proximal tissue after 428 ms, leading to VF. The repolarization time of the proximal tissue is 403 ms (not shown). The pattern of the isochrones suggests that the tissue distal to the line of block is activated from deeper layers. The local electrograms, recorded from the same sites as in the top panel now demonstrate that the time difference between local distal S2 activation (dotted line) and proximal S2 repolarization (dot) has decreased.
Figure 3 shows an example of two electrograms simultaneously recorded from the proximal (red) and distal (blue) perfusion beds. With longer distal exposure to flecainide local S2 activation (dotted line) is progressively delayed in the distal domain, until it encroaches on the proximal S2 repolarization (circle) and VF ensues (bottom).
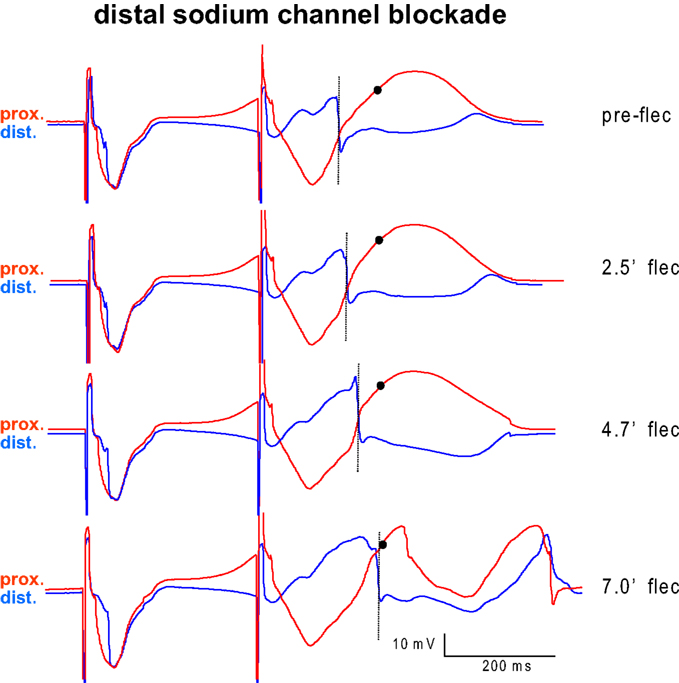
Figure 3. Example of two electrograms simultaneously recorded from the myocardium proximal (red) and distal (blue) from the interface between the perfusion beds. Proximal tissue was perfused with pinacidil, distal tissue was perfused with sotalol. The last basic beat and the first premature beat are shown before (top) and several minutes after infusion of flecainide into the distal (blue) domain. With longer exposure to flecainide local activation (dotted line) is progressively delayed in the distal domain, until it encroaches on the proximal S2 repolarization (circle) and VF ensues (bottom).
Regional infusion of flecainide into the proximal region led to an activation time delay of both the basic and premature beats (S1 and S2) of about 7 ms (averaged activation time differences in the proximal perfusion domain) (Figure 4 A). As expected (flecainide was not infused in this domain) propagation in the distal perfusion domain was not affected (not shown).
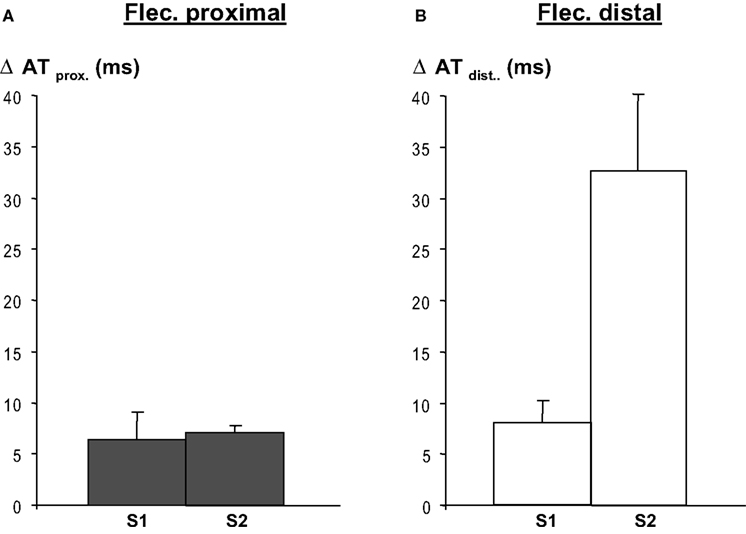
Figure 4. Regional flecainide (flec) infusion causes activation delay in the respective regions following a basic beat (S1) and a premature beat (S2), and both during proximal (panel A)and distal (panel B) application of flecainide. After S2 conduction delays are more pronounced when flecainide is infused in the distal region than in the proximal regional, because the activation pathway is longer than in the latter. Distal activation times were calculated relative to the earliest arrival at the border between the perfusion beds. Activation delays in the domain not perfused with flecainide, no change in activation time occurred (not shown).
When flecainide was infused in the distal area (as in Figure 2 ) the local-distal-activation delays of S1 were of similar magnitude as after infusion in the proximal area. However, activation delays following S2 were larger related to the activation path that was longer after S2 than after S1. Distal flecainide infusion did not affect proximal activation times (not shown), as expected. The activation delay induced by flecainide was statistically significant both for S1 and S2 (p < 0.001, n = 8 each, Mann–Whitney Rank sum test).
Figure 5 shows the antiarrhythmic effect of flecainide when administered to the proximal zone. Before the infusion of flecainide, a premature impulse given after 240 ms causes re-entry and VF. The premature wavefront arrives at the distal side of the line of block after 394 ms while the repolarization time of the proximal tissue is 382 ms (not shown), hence FF is small. The conduction slowing of the S2 beat induced by flecainide has a twofold effect: (1) the later arrival of the activation wave at the interface between the perfusion beds causes a shortening of the line of block and therefore an earlier arrival of the activation wave at the distal side of the line of block (after 371 ms, compared to 394 ms before flecainide) (2) the proximal S2 repolarization time increased from 382 to 401 ms. These two combined effects of flecainide cause the impulse to arrive too early at the distal side of the block relative to proximal S2 repolarization. This causes an increase in FF and failure to induce VF.
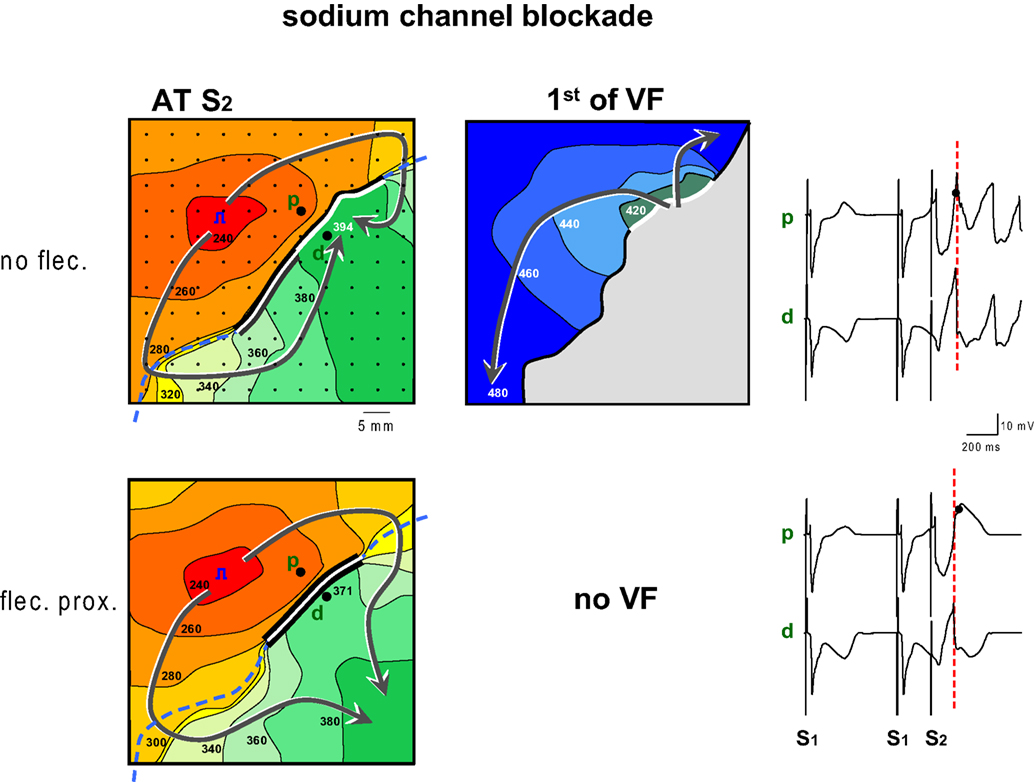
Figure 5. Activation maps of the premature beat (following S2) before and after application of flecainide to the region with shortened repolarization (proximal). Prior to infusion of flecainide (flec) VF could be induced, because local distal activation coincided with local proximal repolarization. Flecainide infusion into the proximal domain caused an increase of the FF and failure of induction of VF. The increase of FF was reached by two mechanisms: first the proximal line of block was shortened because the activation front reached the line of block later. This also led to an earlier activation of the tissue distal to the line of block. Second because proximal S2 repolarization was delayed. Symbols as in Figure 2 .
Overall, a re-entrant activation pattern was mapped in three of the six hearts that fibrillated. In the other hearts a complete re-entrant pathway was not demonstrable, but the FF could be calculated. In Figure 6 the FF before and after administration of flecainide is plotted for the three hearts in which flecainide applied to the distal zone converted a bidirectional line of block (without re-entry) into a unidirectional block and to VF. Proarrhythmia was associated with a decrease of the FF induced by flecainide. In the two hearts in which flecainide was given distally, the shortening of the FF was not enough to cause re-entry. In three hearts that fibrillated before administration of flecainide, the drug infused in the proximal zone prolonged the FF, and VF could no longer be induced.
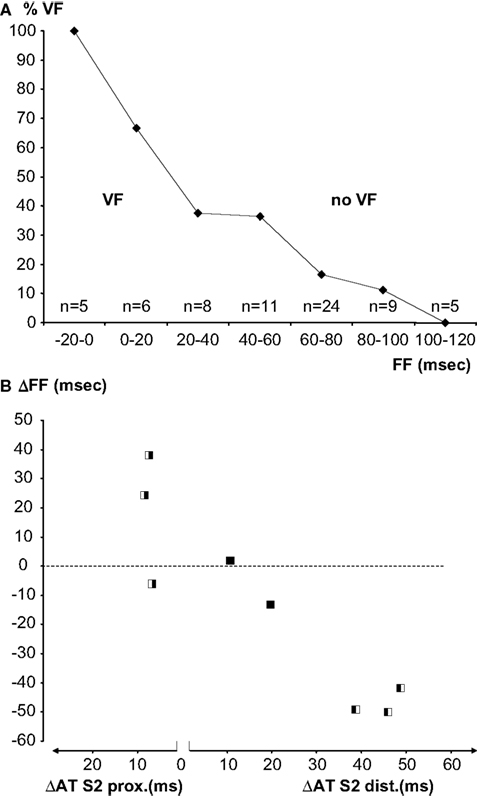
Figure 6. (A) Percentage of successful inductions of VF in bins of 20 ms FF. With increasing FF the induction success decreases. Numbers represent all induction attempts in eight experiments. (B) Individual paired observations (see Materials and Methods) of the relation between the S2-activation delay induced by flecainide and the FF. The symbols indicate the inducibility of VF before and after application of flecainide (white VF, black no VF, left before, right after flecainie). The left panel represents increase in activation delay in the proximal (pinacidil) domain, the right panel activation delay in the distal (sotalol) domain. Slowing of activation in the proximal domain is antiarrhythmic, slowing in the distal domain is proarrhythmic.
Figure 6 A shows a summary of all VF-induction attempts as a function of FF. Observations were binned in FF classes of 20 ms. With decreasing FF, the proportion of VF induction was more successful. Figure 6 B shows the change of FF of the eight paired observations of VF induction as a function of the activation delay induced by flecainide in each of the perfusion beds. Note that the left panel of the figure shows increased activation delay caused by proximal and the right panel activation delay caused by distal infusion of flecainide. The symbols indicate whether a change in inducibility of VF occurred (white VF, black no VF). The experiments in which no change in VF inducibility occurred were characterized by a small change in FF and a small change in activation time.
Discussion
In this study we have demonstrated that sodium channel blockade can be either proarrhythmic or antiarrhythmic depending on its site of application relative to the repolarization gradient. We chose a model of induction of VF based on heterogeneity in repolarization (Coronel et al., 2009 ) in which VF can be reproducibly induced after premature stimulation from the area with the shorter repolarization time(Coronel et al., 2009 ). The differential effect of sodium channel blockade depends on the timing of the arrival of the premature activation at the distal side of the line of activation block relative to repolarization of the proximal side (the Fibrillation Factor, FF), which is critical for the induction of re-entry and VF. A reduction of FF is associated with a profibrillatory effect and can be achieved by application of sodium channel blockade to the site with the longer action potential duration. Conversely, an antifibrillatory effect can be produced by sodium channel blockade of the tissue with the short action potential. Our study shows that a small delay of activation (see Figure 4 , about 7 ms.) caused by infusion of flecainide makes the difference between successful induction of VF or not.
Activation delay induced by sodium channel blockade has two effects relevant for the magnitude of the FF: In the case of conduction slowing in the area with short action potentials it delays the arrival of the premature wavefront to the tissue with prolonged action potential duration and thereby shortens the area of (proximal) activation block, causing earlier activation of the tissue distal to the line of block. Furthermore, it secondarily delays repolarization of the premature wave and thereby increases the FF (antiarrhythmic). Conversely, conduction slowing of the basic beat in the tissue with prolonged repolarization delays repolarization further, causing a lengthening of the line of proximal activation block. Also, the premature wavefront is delayed causing a reduction of the FF.
Han and Moe (1964) formulated in 1964 the concept that heterogeneity in refractory periods was proarrhythmic. Boineau et al. (1980) published activation maps of “figure of 8” re-entry in the canine atrium, induced by a premature stimulus applied to the site with the shortest refractory period, in an atrium with large inhomogeneities in refractory periods (ranging from 150 to 210 ms). In the same year, re-entry leading to VF in response to a spontaneous premature beat was described in acutely ischemic myocardium (Janse et al., 1980 ). Again, this was partly due to large inhomogeneities in refractory periods, those at the normal side of the ischemic border, where the premature beat originated, being short, those in the center of the ischemic zone being long. The differences in refractory period amounted to more than 100 ms (Capucci et al., 1985 ). Kuo et al. (1983) created differences in ventricular action potential duration by local warming during global hypothermia. They found that in the presence of regional differences in action potential duration of 111 ± 16 ms, a premature stimulus delivered to the region with the shortest action potential induced arrhythmias. In our previous study (Coronel et al., 2009 ) we showed that although a repolarization gradient is a prerequisite for the occurrence of conduction block, the restitution properties of the tissue with the shorter repolarization time determines whether or not re-entry occurs. If the “FF” is small, re-entry occurs, if it is large there is bidirectional conduction block.
Earlier studies have shown that drugs that flatten the restitution curve, such as calcium channel blockers, the electro-mechanical uncoupler diacetyl monoxime, and bretylium, did indeed prevent the induction of VF and converted existing fibrillation into a regular rhythm (Riccio et al., 1999 ; Garfinkel et al., 2000 ; Gelzer et al., 2008 ). We expect that flattening of the restitution curve will prolong the FF and will be antifibrillatory, independent of the maximum slope of the restitution curve.
In all cases of flecainide infusion, a statistically significant increase in activation time following S1 and S2 occurred within the relevant domain (Figure 4 ). It is remarkable that a small difference in activation time can have a large effect on VF inducibility. The activation maps presented in this and a previous study make clear that the critical arrival time of the activation wave at the distal side of the border relative to proximal repolarization is a matter of milliseconds. In this regards the FF resembles the all or none effect of cardiac activation following refractoriness.
It is conceivable that the beta-blocking effects of sotalol may have contributed to the observed effects. However, we did pace the heart at a regular heart rate and the antiarrhythmic bradycardic effects of beta-blockade can be excluded. Secondly, the pro- and antifibrillatory effects of flecainide were tested under similar conditions, both in the presence of sotalol and pinacidil. Also, flecainide has been described to block potassium currents and therefore may have influenced the repolarization gradients. We did observe an increase in repolarization gradient after application of flecainide in some experiments. However, conduction slowing in itself also contributes to this effect. Indeed, after proximal application of flecainide the increase in conduction delay was, on average of the same magnitude as the increase in repolarization. Therefore, the potassium blocking affects of flecainide probably have played a minor role in our results. A possible explanation for this is that sodium channel blockade also reduces the contribution of the sodium current to the action potential plateau and thereby reduces action potential duration.
The model of VF inducibility based on large and steep gradients in repolarization that we have used is artificial and likely does not directly reflect a pathophysiological condition. However, the model resembles that of acute myocardial ischemia (Janse et al., 1980 ), where conduction slowing coincides with repolarization heterogeneity and where a premature beat originates from the area with the shorter repolarization. These conditions combine into the ideal small FF for the induction of VF. Furthermore, it is difficult to extrapolate our finding to the clinical condition where sodium channel blockers are systemically administered. Whether sodium channel blockers will be pro- or antiarrhythmic depends on which of the two opposing effects predominate.
It is remarkable that large repolarization gradients are required for the induction of VF, and that VF does not occur spontaneously, even in these extreme conditions. The line of activation block occurring upon premature stimulation from the area with the shorter repolarization depends on the coupling interval and the site of stimulation. If the premature beat originates closely from the border immediately after recovery from inactivation, the line of block is longest and is more likely to lead to re-entry and VF.
The model is limited by the restricted time frame in which measurements can be made. Therefore long term effects of the drugs cannot be studied. Secondly, only epicardial mapping was performed and no information is present about the intramural gradients of repolarization. On the other hand, the experiments address the question whether flecainide is able to prevent of facilitate the induction of VF. This is independent of the location of the mapping electrode.
Interventions aimed at increasing conduction velocity are therefore expected to be antiarrhythmic. Indeed, in the 1-week old canine myocardial infarction model this has been demonstrated (Lau et al., 2009 ). Alternatively, local RNA interference by siRNA to suppress local sodium channel function could be applied to the area of the earliest repolarization time in order to prevent reentrant arrhythmias. Because a positive T-wave indicates tissue with early repolarization and a negative T-wave a region with late repolarization, this may open the way for gene therapy directed by the morphology of the T-wave of the local electrogram.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors are grateful for fruitful discussions with Dr. T. Opthof and to Charly Belterman and Wim ter Smitte for excellent technical support.
References
Boineau, J. P., Schuessler, R. B., Mooney, C. R., Miller, C. B., Wylds, A. C., Hudson, R. D., Borremans, J. M., and Brockus, C. W. (1980). Natural and evoked atrial flutter due to circus movement in dogs. Rule of abnormal atrial pathways, slow conduction, nonuniform refractory period distribution and premature beats. Am. J. Cardiol. 45, 1167–1181.
Capucci, A., Coronel, R., Fabius, M. A. W., and Janse, M. J. (1985). Electrophysiologic mechanisms of ventricular arrhythmias in acute ischemia: further observations. New Trends Arrhythmias 1, 41–56.
Coromilas, J., Saltman, A. E., Waldecker, B., Dillon, S. M., and Wit, A. L. (1995). Electrophysiological effects of flecainide on anisotropic conduction and reentry in infarcted canine hearts. Circulation 91, 2245–2263.
Coronel, R., De Bakker, J. M. T., Wilms-Schopman, F. J. G., Opthof, T., Linnenbank, A. C., Belterman, C. N., and Janse, M. J. (2006). Monophasic action potentials and activation recovery intervals as measures of ventricular action potential duration: experimental evidence to resolve some controversies. Heart Rhythm 3, 1043–1050.
Coronel, R., Wilms-Schopman, F. J. G., de Groot, J. R., Janse, M. J., Van Capelle, F. J. L., and De Bakker, J. M. T. (2000). Laplacian electrograms and the interpretation of complex ventricular activation patterns during ventricular fibrillation. J. Cardiovasc. Electrophysiol. 1119–1128.
Coronel, R., Wilms-Schopman, F. J. G., Opthof, T., and Janse, M. J. (2009). Dispersion of repolarization and arrhythmogenesis. Heart Rhythm 6, 537–543.
Echt, D. S., Liebson, P. R., Mitchell, L. B., Peters, R. W., Obias-Manno, D., Barker, A. H., Arensberg, D., Baker, A., Friedman, L., Greene, H. L., Huther, M. L., Richardson, D. W., and CAST investigators. (1991). Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 324, 781–788.
Garfinkel, A., Kim, Y. H., Voroshilovsky, O., Qu, Z., Kil, J. R., Lee, M. H., Karagueuzian, H. S., Weiss, J. N., and Chen, P. S. (2000). Preventing ventricular fibrillation by flattening cardiac restitution. Proc. Natl. Acad. Sci. U.S.A. 97, 6061–6066.
Gelzer, A. R. M., Koller, M. L., Otani, N. F., Fox, J. J., Enyeart, M. W., Hooker, G. J., Riccio, M. L., Bartoli, C. R., and Gilmour, R. F., Jr. (2008). Dynamic mechanism for initiation of ventricular fibrillation in vivo. Circulation 118, 1123–1129.
Han, J., and Moe, G. K. (1964). Nonuniform recovery of excitability in ventricular muscle. Circ. Res. 14, 44–61.
Janse, M. J., Van Capelle, F. J. L., Morsink, H., Kléber, A. G., Wilms-Schopman, F., Cardinal, R., Naumann d’Alnoncourt, C., and Durrer, D. (1980). Flow of “injury” current and patterns of excitation during early ventricular arrhythmias in acute regional myocardial ischemia in isolated porcine and canine hearts. Evidence for two different arrhythmogenic mechanisms. Circ. Res. 47, 151–165.
Kuo, C. S., Munakata, K., Reddy, C. P., and Surawics, B. (1983). Characteristics and possible mechanisms of ventricular arrhythmia dependent on the dispersion of action potential durations. Circulation 67, 1356–1367.
Lau, D. H., Clausen, C., Sosunov, E. A., Shlapakova, I. N., Anyukhovsky, E. P., Danilo, P. Jr., Rosen, T. S., Kelly, C., Duffy, H. S., Szabolcs, M. J., Chen, M., Robinson, R. B., Lu, J., Kumari, S., Cohen, I. S., and Rosen, M. R. (2009). Epicardial border zone overexpression of skeletal muscle sodium channel SkM1 normalizes activation, preserves conduction, and suppresses ventricular arrhythmia: an in silico, in vivo, in vitro study. Circulation 119, 19–27.
Lie, K. I., Liem, K. L., Willebrands, A. F., Lourdidtz, W. J., Janse, M. J., and Durrer, D. (1978). The effectiveness of intramuscular lidocaine in the prevention of ventricular fibrillation within one hour after injection. Am. J. Cardiol. 42, 486–488.
Potse, M., Linnenbank, A. C., and Grimbergen, C. A. (2002). Software design for analysis of multichannel intracardial and body surface electrocardiograms. Comput. Methods Programs Biomed. 69, 225–236.
Riccio, M. L., Koller, M. L., and Gilmour, R. F. Jr. (1999). Electrical restitution and spatiotemporal organization during ventricular fibrilation. Circ. Res. 84, 955–963.
The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. (1989). Preliminary report: effect of encainide, and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N. Engl. J. Med. 321, 406–412.
Keywords: sodium channel blockers, ventricular fibrillation, antiarrhythmic effects, proarrhythmic effects, repolarization heterogeneity, re-entry
Citation: Coronel R, Wilms-Schopman FJG and Janse MJ (2010) Anti- or profibrillatory effects of Na+ channel blockade depend on the site of application relative to gradients in repolarization. Front. Physio. 1:10. doi: 10.3389/fphys.2010.00010
This article was submitted to Frontiers in Cardiac Electrophysiology, a specialty of Frontiers in Physiology.
Received: 07 April 2010;
Paper pending published: 20 April 2010;
Accepted: 06 May 2010;
Published online: 07 June 2010
Edited by:
Marcel v. d. Heyden, University Medical Center, NetherlandsReviewed by:
Gudrun Antoons, Katholieke Unversiteit Leuven, BelgiumMilan Stengl, Charles University, Czech Republic
Morten B. Thomsen, University of Copenhagen, Denmark
Copyright: © 2010 Coronel, Wilms-Schopman and Janse. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Ruben Coronel, Experimental Cardiology Group, Academic Medical Center, Room K2-112, Meibergdreef 9, 1105 AZ Amsterdam, Netherlands. e-mail:ci5jb3JvbmVsQGFtYy51dmEubmw=