 Joseph M. Chalovich
Joseph M. Chalovich
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Phys., 25 January 2024
Sec. Biophysics
Volume 12 - 2024 | https://doi.org/10.3389/fphy.2024.1298482
This article is part of the Research TopicBiophysics of Muscle ContractionView all 6 articles
At low free Ca2+, the actin binding proteins tropomyosin, troponin I, troponin T and troponin C inhibit contraction in striated muscles. Ca2+ activation alters the position of tropomyosin on actin to uncover binding sites for high affinity forms of myosin (i.e., myosin-ADP). Inhibition of contraction is commonly thought to result from steric blocking of myosin binding to actin by tropomyosin. However, myosin-ADP binding to actin is energetically more favorable than localization of tropomyosin in the blocking position. Tropomyosin is an effective inhibitor of binding only at low levels of myosin-ADP. At low free Ca2+, troponin-tropomyosin also inhibits the rate of a step associated with Pi release to about 1% of the maximum rate. This results in accumulation of myosin with bound ATP and ADP-Pi. Such myosin binds weakly to actin. Ca2+ activation increases the rate of Pi release, but not to the maximum value, and increases the population of myosin-ADP. The high affinity binding of myosin-ADP to actin can displace tropomyosin into the fully active position in relation to the amount of myosin-ADP bound. It seems likely that an important outcome of the steric clash between myosin-ADP and tropomyosin is the dual activation by Ca2+ and myosin-ADP. The C-terminal region of troponin T (TnT) contributes to the incomplete activation by Ca2+ alone. Because this region of TnT is highly conserved, the ability of myosin-ADP to move tropomyosin may be more important than any restriction that tropomyosin may place on myosin binding.
I often ask medical and graduate students to explain how striated muscle contraction is regulated. The common response is that tropomyosin sterically blocks myosin binding to actin at low Ca2+ levels. I suspect that most of my peers would find that answer to be basically correct although lacking in detail. The purpose of this article is to consider the possibility that steric blocking functions in the reverse manner: increased levels of high affinity myosin molecules force tropomyosin more into the activating position. That is, Ca2+ and high affinity myosin binding are co-activators.
Muscle contraction is driven by the hydrolysis of ATP. Rapid phosphate release can occur only when myosin is bound to actin, ensuring that ATP hydrolysis is coupled to force production (Figure 1). In skeletal and cardiac muscles this process is inhibited at low free Ca2+ by the actin associated proteins tropomyosin, troponin I, troponin T and troponin C.
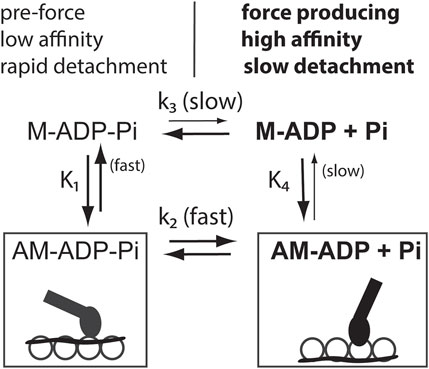
FIGURE 1. Diagram showing that Ca2+ increases both a rate constant between bound actomyosin states and the affinity between actin and myosin. Force producing states are in bold. M is myosin, A is actin, uppercase K’s are equilibrium constants, and lower-case k’s are rate constants. States on the left, called weak binding states, predominate at low free Ca2+. Those on the right, called strong binding states (in bold), predominate at saturating Ca2+ when tropomyosin (wavy lines) has changed its position on actin (open circles). Force production occurs after the transition from pre-force states to force producing attached states. Coupling of rate constants of the upper and lower kinetic pathways and the equilibrium constants for different myosin-nucleotide complexes to regulated actin occurs because detailed balance requires that K1*k2/k-2 = K4*k3/k-3. Rate constants k3 and k-3 are Ca2+ independent. k-2 is unlikely to increase with Ca2+ as that is contrary to force production. Both k2 and K4 increase with Ca2+. These increases are coupled. Such coupling occurs wherever a slow step opposes a step that increases at high Ca2+ levels.
Several laboratories showed in the 1970s that Ca2+ binding to troponin changes the position of tropomyosin relative to actin [1–5]. The mechanism of tropomyosin movement is becoming more clear as a result of high resolution images [6–10]. Two of the three known positions of tropomyosin on actin overlap the binding site for force producing myosin species such as myosin-ADP [9]. Ca2+ alone does not fully stabilize tropomyosin in the fully unblocked state; that also requires binding of myosin-ADP or rigor myosin to actin. The Ca2+-dependent change in tropomyosin orientation led to the proposal that regulation of muscle contraction occurs because tropomyosin sterically blocks the binding site of myosin on actin at low free Ca2+ [2]. However, the winner of any competition depends on the concentrations and affinities of the competing molecules. It is unclear that tropomyosin normally wins that competition. Furthermore, tropomyosin does not appear to compete effectively with myosin that has bound ATP or ADP-Pi.
Tropomyosin-troponin also inhibits the transition between two bound actin-myosin complexes. Figure 1 shows an abbreviated scheme of actin activated ATPase activity of myosin. Inhibition of a step associated with Pi release, k2 in Figure 1, reduces activity independently of the level of myosin binding to actin. As a consequence of this inhibition there is reduction of the high affinity myosin species such as myosin-ADP.
Evidence for an increase in the rate constant of a process associated with Pi release, k2 in Figure 1, will be given in following sections. It will also be shown that the effect of tropomyosin on myosin binding depends on the nucleotide bound to myosin. Refering again to Figure 1, Ca2+ has a greater effect on the binding of myosin-ADP to actin-tropomyosin-troponin (K4) than on binding of myosin-ADP-Pi (K1). The rate constant k3 is not Ca2+ sensitive as this process occurs when myosin is detached from actin. Therefore, increases in both k2 and K4 ensure that detailed balance is maintained (K1*k2/k-2 = K4*k3/k-3).
It is worthwhile noting that while myofibrils and other organized systems have distinct advantages for studying muscle contraction [11], the topic of discussion here is specifically the role of tropomyosin and troponin in regulation. It is now known that the availability of myosin heads can be directly regulated [12]. This involves the interaction of both myosin heads in a pair with each other [13, 14] and the super relaxed state [15, 16]. Emphasis is placed here on solution studies where direct control over myosin availability cannot occur and those fiber studies where the presence of myosin regulation cannot detract from the results.
In the late 1970s several laboratories studied the interaction of soluble subfragments of myosin (S1 with one catalytic unit and HMM with two catalytic units) with actin-tropomyosin-troponin. Two broad classes of interaction species were identified. Weak binding species (containing bound ATP or ADP-Pi) have a low affinity for actin-tropomyosin-troponin that is relatively insensitive to Ca2+. Strong binding species, or force producing species (i.e., containing bound ADP or lacking a nucleotide) have a much higher affinity for actin and their binding to actin is affected to a greater extent by Ca2+ binding to troponin C.
Evidence for low affinity binding of S1 to actin in the absence of tropomyosin or troponin, during steady-state ATP hydrolysis, was observed by rapid light scattering measurements [17]. This binding to actin is weak with an actin affinity, less than 1/400th of that of S1-ADP [18]. Such low affinity binding has a rapid detachment rate from actin as seen in stiffness measurements of single rabbit psoas fibers [19, 20]. This rapid detachment rate [21] contributes to the difficulty of detecting this interaction.
Binding of S1 to actin-tropomyosin-troponin, during steady-state ATP hydrolysis is relatively unaffected by Ca2+ [22–24]. Somewhat larger effects of Ca2+ were reported with the larger myosin fragment, HMM. Ca2+-dependent increases in affinity for actin-tropomyosin-troponin were reported ranging from 1-fold [25] to 5-fold [26] to 10-fold [27]. However, studies on single muscle fibers, containing intact myosin, support a modest effect of Ca2+ on binding of myosin in single rabbit psoas fibers. Thus, pre-force producing cross-bridges have been detected in skinned rabbit psoas muscle fibers even at physiological ionic strength [28]. Furthermore, estimates of the Ca2+ dependency of the association constant for pre-force producing states of myosin was made using the ATP analogue, ATPγS [29]. A modest 2-5-fold increase in association binding constant at saturating Ca2+ was observed. These modest changes in affinity could produce only small changes in myosin binding in muscle because of the high effective actin concentration in muscle [30].
There are qualitative differences in binding of weak binding and strong binding myosin species to actin-tropomyosin-troponin [31]. In one study, the binding constants of strong and weak binding species of S1 were matched by measuring the strong binding species at a higher salt concentration. Increasing concentrations of weak binding species of S1 yielded a noncooperative, hyperbolic binding curve. In contrast, the strong binding species exhibited positive cooperativity consistent with competition with tropomyosin at low S1 concentrations. For that reason, weak binding or pre-force producing states are sometimes called non-activating while strong binding or force producing states are called activating states.
These states have a high affinity [32, 33] and they overlap the inhibitory tropomyosin binding sites on actin. Binding of such myosin states can displace tropomyosin to the active or non-blocked position on actin. Binding of strong states of myosin to pyrene labeled actin results in a fluorescence change not seen with weak binding states [33, 34].
It is instructive to examine equilibrium binding curves of S1-ADP at low and high Ca2+. Figure 2 shows that S1 binding at saturating Ca2+ follows a simple hyperbolic pattern. At very low free Ca2+ levels the binding has a sigmoidal shape characteristic of positive cooperativity [32, 35]; see also [37].
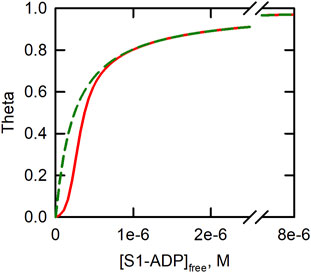
FIGURE 2. Curves describing binding of S1-ADP to actin-tropomyosin-troponin in the absence (red line) and presence (broken green line) of Ca2+. S1 is the soluble, single headed, fragment of myosin. Theta is [S1bound]/[Actintotal] and [S1-ADP]free is the concentration of the unbound myosin-nucleotide complex. Illustrative curves were produced from a model of equilibrium binding to regulated actin [35] using the following parameters: K2 = 4.1 × 106, K1 = K2/80. For Ca2+ L’ = 1, Y = 11.4. For low Ca2+, L’ = 126, Y = 1.16. Parameters are for binding at 180 mM ionic strength [36].
This cooperative binding curve has been explained by an equilibrium of regulated actin between states that have low and high affinities, respectively, for strong myosin states [35]. Tropomyosin-troponin reduces binding at low concentrations of S1-ADP. As the concentration of S1-ADP is increased, there is a cooperative transition to a strong binding state of actin-tropomyosin-troponin. Thus, binding of S1-ADP, or other “activating” form of myosin can cause tropomyosin to shift from the blocked state to a state that allows activity. The cooperativity of this process allows myosin to activate binding over a narrow range of myosin concentrations. Tropomyosin-troponin can inhibit binding of myosin in activating states to regulated actin, as in the original Steric Blocking model, but only when the available concentration of myosin heads with bound ADP is very low.
Although the focus of tropomyosin is usually on its effect on myosin binding to actin, the tropomyosin-troponin complex also directly influences the rate of ATP hydrolysis by myosin bound to actin. Several studies have shown that the ATPase activity of myosin subfragment 1 is inhibited at low Ca2+ levels even when most of the myosin S1 is bound to actin [23, 25, 38–40] and when S1 is crosslinked to actin with a zero length crosslinker [41]. The inhibited step was proposed to be associated with the process of Pi release from myosin. The rate of Pi release is very slow when myosin is detached from actin. Therefore, inhibition of Pi release from myosin-ADP-Pi bound to actin has a large impact on the overall rate of ATP hydrolysis [23] (see Figure 1).
Conformation that Ca2+ controlls the rate of a step associated with Pi release was later demonstrated [42]. The rate of Pi release increases 117-fold (relative to saturating skeletal thin filaments at very low Ca2+ or about 1,000-fold relative to myosin in the absence of actin) with both Ca2+ binding to troponin and rigor S1 binding to actin [40]. These extremes are likely lower and upper limits to the regulation by Ca2+. Large increases in the rate of Pi release also occur with native cardiac thin filaments [39]. Evidence for regulation of a transition between two attached actin-myosin states also comes from the Ca2+ dependence of the rate of force redevelopment in psoas muscle fibers [43].
Inhibition of Pi release at low Ca2+ levels has three consequences: inhibition of ATP hydrolysis, reduction in high affinity myosin species (through detailed balance) and inhibition of binding of residual myosin-ADP by tropomyosin in the binding competition shown in Figure 2.
The rate of ATP hydrolysis by myosin S1 in the presence of regulated actin increases during the last 10% or so of the reaction [44]. That increase is striking at low Ca2+ levels where the regulated actin is initially in the inactive state. That activation is driven by the accumulation of S1-ADP that binds tightly to actin [45] and increases ATPase activity [46]. A theoretical model explaining the extra activation of ATPase activity at high concentrations of myosin S1 was proposed some years ago [47]. As with the earlier model of myosin S1 binding to regulated actin [35], the accumulation of myosin-ADP was proposed to stabilize the active state of regulated actin leading to an increase in activity.
At saturating Ca2+ levels, the increase in the ATPase activity, by strong or activating species of myosin, is termed potentiation [45]. Potentiation can be readily demonstrated by doping the ATPase assay with S1 modified with N-ethylmaleimide. This NEM-S1 has little intrinsic ATPase activity but it binds tightly to regulated actin even in the presence of ATP and stabilizes the active state of actin-tropomyosin-troponin [48]. Subsequent research reinforced the idea that Ca2+ does not give full activation of ATPase activity; attachment of “activating” species of myosin was also necessary [49–51].
The 1981 model of regulation of ATPase activity had three “states” of actin-tropomyosin-troponin [47]: inactive-Ca2+-free, inactive-Ca2+-bound and active. Both Ca2+ and an increase in myosin-ADP were deemed essential for stabilizing the active state of actin-tropomyosin-troponin. That model correctly simulated ATPase activity at varying concentrations of both actin and myosin S1.
In 1993 a model was proposed for regulation of binding of myosin S1 to actin in the absence of ATP [52]. That binding model has 3 states commonly called B (blocked), C (calcium stabilized), and M (myosin stabilized). It is unclear if these binding states are identical to the states defined from ATPase activities. In much of our work, we estimate the population of the active state of actin-tropomyosin-troponin from the ATPase activity and use binding measurements to estimate the population of the B state [53–55]. We normally assume that the B state is a measure of the inactive-Ca2+-free state but that has not been proven.
Hypertrophic cardiomyopathy causing mutations of troponin often change the distribution of the aforementioned states of actin-tropomyosin-troponin [53, 54]. Mutations that reduce or eliminate positive charges within the C-terminal region of TnT facilitate transitions to the active state [50, 55, 56]. Such mutations increase the activation by Ca2+ from about 30% to about 70% of the maximum rate possible at a low actin concentration. When substituted for wild type TnT, in single muscle fibers, there is a substantial reduction in the Ca2+ concentration required for half maximal activation [57–59]. The C-terminal region of TnT is highly conserved [54] and stabilizes binding of tropomyosin to the non-activating positions of tropomyosin on actin. When wild type TnT is present, both high affinity myosin binding to actin and Ca2+ are needed to move tropomyosin fully to the active position. This is important because deletions of the C-terminal region of TnT, in cardiac muscle, are associated with hypertrophic cardiomyopathy [60]. The conservation of this C-terminal extension in many animal species [54] highlights the significance of displacement of tropomyosin by high affinity myosin, that is steric blocking upside down.
The purpose of this note is to explore the function of tropomyosin-troponin as one component of the overall regulation of striated muscle contraction. It is important to understand the workings of each component to understand the entirety of muscle regulation. Knowledge of the role of each component is particularly important in trying to mitigate dysfunction caused by naturally occurring mutations.
Regulation of contraction has been the object of debate for many years. Following structural evidence for overlapping binding sites for tropomyosin and non-ATP containing myosin for binding to actin, there has been a discussion as to whether regulation occurs by controlling numbers of attached crossbridges or by rate modulation, i.e., controlling the rate of transition between bound actin-myosin states. A stumbling block for many to consider the latter possibility is that the number of attached myosin crossbridges increases following activation by Ca2+. We now know that some of those effects could be due to myosin linked regulatory mechanisms that do not directly involve tropomyosin movement. However, there is certainty competition between binding of myosin-ADP and tropomyosin for actin binding and one needs to explain how this may contribute to changes in myosin binding during Ca2+ activation.
There are several possible mechanisms by which tropomyosin and troponin could produce a Ca2+ dependent increase in myosin binding. One possibility is that these proteins inhibit all myosin binding at low free Ca2+ levels. That view is most often seen in textbooks. McKillop and Geeves introduced a composite two-step binding model in which the first step is assumed to be steric blocking and the second step is a transition between two myosin-actin bound states [52]. That model is widely cited as describing overall activity, but it does not include the transitions shown in Figure 1 that are strictly ATP dependent. It is possible that the second regulated state in the binding model is a measure of the rate of ATP product release in Figure 1. If that is the case, then the effects of Ca2+ on both processes should be comparable to each other.
The mechanism outlined in the present work is totally dependent on ATP as it is based on the great differences in binding of myosin-ATP (or ADP-Pi) and myosin-ADP to actin. Decreases in activity at low Ca2+ levels result in accumulation of low affinity, rapidly detaching myosin species containing ATP or ADP-Pi (Figure 1). Tropomyosin would be situated in the “blocked” state on actin, but it would not block attachment of myosin in these low affinity states.
Ca2+ binding to troponin C creates an equilibrium mixture between an inactive state, where tropomyosin partially overlaps the myosin-ADP binding site (the C state), and the active state where there is no overlap in the binding sites for myosin-ADP and tropomyosin. In solution, the ATPase activity increases from near zero to about 30% of the maximum value. From the argument given in Figure 1, this partial activation decreases levels of myosin containing bound ATP and ADP-Pi and increases myosin-ADP levels. The level of myosin-ADP increase depends on the rate of ADP release from myosin (in muscle varying inversely on the speed of shortening) and the ATP concentration. To the extent that myosin-ADP accumulates, there is an additional increase in activity as the inhibitory effects of the C-terminal region of TnT are overcome.
Tropomyosin movement over actin remains a central feature in regulation both by participating in the control of the rate of release of the products of ATP hydrolysis and by overlapping the myosin binding site to allow dual activation by Ca2+ and myosin-ADP. Interesting questions remain such as how the effects produced by tropomyosin and troponin are altered by the other regulatory systems present in muscle. It also appears that there is more to be learned about the reason for conserving the C-terminal region of TnT and the resulting dual activation by Ca2+ and “activating” species of myosin. For example, why does deletion of the C-terminal region of TnT increase the total activation in solution but increase Ca2+ sensitivity in muscle fibers? Understanding the role of tropomyosin-troponin in regulation is a step in answering these questions.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
JC: Conceptualization, Writing–original draft, Writing–review and editing.
The author declares financial support was received for the research, authorship, and/or publication of this article. Funding was provided by a grant from Leepo C. Yu and Victor Yu.
The author thanks Peter Knight and David Heeley for helpful discussions and Victor and Leepo C. Yu for helping to fund this work.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
S1, subfragment 1; HMM, heavy meromyosin; myosin-ADP-Pi, myosin containing the products of ATP hydrolysis; regulated actin, actin containing tropomyosin, troponin T, troponin I and troponin C; strong binding state, having properties similar to myosin with bound ADP; weak binding state, having properties similar to myosin with bound ATP or ADP-Pi.
1. Vibert PJ, Haselgrove JC, Lowy J, Poulsen FR. Structural changes in actin-containing filaments in muscle. J Mol Biol (1972) 716:757–67. doi:10.1016/s0022-2836(72)80036-6
2. Huxley HE. Structural changes in the actin and myosin containing filaments during contraction. Cold Spring Harbor Symp Quant Biol (1972) 37:361–76. doi:10.1101/SQB.1973.037.01.046
3. Parry DAD, Squire JM. Structural role of tropomyosin in muscle regulation: analysis of the X-ray diffraction patterns from relaxed and contracting muscles. J Mol Biol (1973) 75:33–55. doi:10.1016/0022-2836(73)90527-5
4. Wakabayashi T, Huxley HE, Amos LA, Klug A. Three-dimensional image reconstruction of actin-tropomyosin complex and actin-tropomyosin-troponin T-troponin I complex. J Mol Biol (1975) 93:477–97. doi:10.1016/0022-2836(75)90241-7
5. Lehman W, Vibert P, Uman P, Craig R. Steric-blocking by tropomyosin visualized in relaxed vertebrate muscle thin filaments. J Mol Biol (1995) 251(2):191–6. doi:10.1006/jmbi.1995.0425
6. Yamada Y, Namba K, Fujii T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat Commun (2020) 11(1):153. doi:10.1038/s41467-019-14008-1
7. Tobacman LS. Troponin revealed: uncovering the structure of the thin filament on-off switch in striated muscle. Biophys J (2021) 120(1):1–9. doi:10.1016/j.bpj.2020.11.014
8. Risi C, Schafer LU, Belknap B, Pepper I, White HD, Schroder GF, et al. High-resolution cryo-EM structure of the cardiac actomyosin complex. Structure (2021) 29(1):50–60 e4. doi:10.1016/j.str.2020.09.013
9. Poole KJ, Lorenz M, Evans G, Rosenbaum G, Pirani A, Craig R, et al. A comparison of muscle thin filament models obtained from electron microscopy reconstructions and low-angle X-ray fibre diagrams from non-overlap muscle. J Struct Biol (2006) 155(2):273–84. doi:10.1016/j.jsb.2006.02.020
10. Pirani A, Xu C, Hatch V, Craig R, Tobacman LS, Lehman W. Single particle analysis of relaxed and activated muscle thin filaments. J Mol Biol (2005) 346(3):761–72. doi:10.1016/j.jmb.2004.12.013
11. Stehle R, Solzin J, Iorga B, Poggesi C. Insights into the kinetics of Ca2+-regulated contraction and relaxation from myofibril studies. Pflugers Arch (2009) 458(2):337–57. doi:10.1007/s00424-008-0630-2
12. Schmid M, Toepfer CN. Cardiac myosin super relaxation (SRX): a perspective on fundamental biology, human disease and therapeutics. Biol Open (2021) 10(2):bio057646. doi:10.1242/bio.057646
13. Wendt T, Taylor D, Messier T, Trybus KM, Taylor KA. Visualization of head-head interactions in the inhibited state of smooth muscle myosin. J Cel Biol (1999) 147(7):1385–90. doi:10.1083/jcb.147.7.1385
14. Scarff CA, Carrington G, Casas-Mao D, Chalovich JM, Knight PJ, Ranson NA, et al. Structure of the shutdown state of myosin-2. Nature (2020) 588(7838):515–20. doi:10.1038/s41586-020-2990-5
15. Stewart MA, Franks-Skiba K, Chen S, Cooke R. Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc Natl Acad Sci U S A (2010) 107(1):430–5. doi:10.1073/pnas.0909468107
16. Hooijman P, Stewart MA, Cooke R. A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys J (2011) 100(8):1969–76. doi:10.1016/j.bpj.2011.02.061
17. Stein LA, Schwarz RP, Chock PB, Eisenberg E. Mechanism of actomyosin adenosine triphosphatase: evidence that adenosine5'-triphosphate hydrolysis can occur without dissociation of theactomyosin complex. Biochemistry (1979) 18:3895–909. doi:10.1021/bi00585a009
18. Eisenberg E, Greene LE. The relation of muscle biochemistry to muscle physiology. Ann Rev Physiol (1980) 42:293–309. doi:10.1146/annurev.ph.42.030180.001453
19. Brenner B, Chalovich JM, Greene LE, Eisenberg E, Schoenberg M. Stiffness of skinned rabbit psoas fibers in MgATP and MgPPi solution. Biophys J (1986) 50:685–91. doi:10.1016/s0006-3495(86)83509-3
20. Schoenberg M, Eisenberg E. Muscle cross-bridge kinetics in rigor and in the presence of ATP analogues. Biophys J (1985) 48:863–71. doi:10.1016/S0006-3495(85)83847-9
21. Brenner B. Muscle mechanics and biochemical kinetics. In: JM Squire, editor. Molecular mechanisms in muscular contraction. Macmillan Press (1990). p. 77–149.
22. Chalovich JM, Chock PB, Eisenberg E. Mechanism of action of troponin-tropomyosin:inhibition of actomyosin ATPase activity without inhibition of myosin binding to actin. J Biol Chem (1981) 256:575–8. doi:10.1016/s0021-9258(19)70009-x
23. Chalovich JM, Eisenberg E. Inhibition of actomyosin ATPase activity by troponin-tropomyosin without blocking the binding of myosin to actin. J Biol Chem (1982) 257:2432–7. doi:10.1016/s0021-9258(18)34942-1
24. Resetar AM, Stephens JM, Chalovich JM. Troponin-tropomyosin: an allosteric switch or a steric blocker? Biophys J (2002) 83(2):1039–49. doi:10.1016/s0006-3495(02)75229-6
25. El-Saleh SC, Potter JD. Calcium-insensitive binding of heavy meromyosin to regulated actin at physiological ionic strength. J Biol Chem (1985) 260:14775–9. doi:10.1016/s0021-9258(17)38640-4
26. Chalovich JM, Eisenberg E. The effect of troponin-tropomyosin on the binding of heavy meromyosin toactin in the presence of ATP. J Biol Chem (1986) 261:5088–93. doi:10.1016/s0021-9258(19)89218-9
27. Wagner PD, Stone DB. Calcium-sensitive binding of heavy meromyosin to regulated actin requires light chain 2 and the head-tail junction. Biochemistry (1983) 22:1334–42. doi:10.1021/bi00275a003
28. Kraft T, Chalovich JM, Yu LC, Brenner B. Parallel inhibition of active force and relaxed fiber stiffness by caldesmon fragments at physiological ionic strength and temperature conditions: additional evidence that weak cross-bridge binding to actin is an essential intermediate for force generation. Biophys J (1995) 68:2404–18. doi:10.1016/s0006-3495(95)80423-6
29. Kraft T, Yu LC, Kuhn HJ, Brenner B. Effect of Ca2+ on weak cross-bridge interaction with actin in the presence of adenosine 5'-[gamma-thio]triphosphate. Proc Natl Acad Sci USA (1992) 89:11362–6. doi:10.1073/pnas.89.23.11362
30. Brenner B. Muscle mechanics and biochemical kinetics. In: JM Squire, editor. Molecular mechanism of muscular contraction. Macmillan Press Ltd. (1989).
31. Chalovich JM, Greene LE, Eisenberg E. Crosslinked myosin subfragment 1: a stable analogue of the subfragment- 1.ATP complex. Proc Natl Acad Sci USA (1983) 80(16):4909–13. doi:10.1073/pnas.80.16.4909
32. Greene LE, Eisenberg E. Cooperative binding of myosin subfragment-1 to the actin-troponin-tropomyosin complex. Proc Natl Acad Sci USA (1980) 77(5):2616–20. doi:10.1073/pnas.77.5.2616
33. Taylor EW. Kinetic studies on the association and dissociation of myosin subfragment 1 and actin. J Biol Chem (1991) 266:294–302. doi:10.1016/s0021-9258(18)52434-0
34. Geeves MA, Halsall DJ. Two-step ligand binding and cooperativity. A model to describe the cooperative binding of myosin subfragment 1 to regulated actin. Biophys J (1987) 52(2):215–20. doi:10.1016/s0006-3495(87)83208-3
35. Hill TL, Eisenberg E, Greene LE. Theoretical model for the cooperative equilibrium binding of myosin subfragment 1 to the actin-troponin-tropomyosin complex. Proc Natl Acad Sci USA (1980) 77:3186–90. doi:10.1073/pnas.77.6.3186
36. Gafurov B, Chen YD, Chalovich JM. Ca2+ and ionic strength dependencies of S1-ADP binding to actin-tropomyosin-troponin: regulatory implications. Biophys J (2004) 87(3):1825–35. doi:10.1529/biophysj.104.043364
37. Desai R, Geeves MA, Kad NM. Using fluorescent myosin to directly visualize cooperative activation of thin filaments. J Biol Chem (2015) 290(4):1915–25. doi:10.1074/jbc.m114.609743
38. Tobacman LS, Adelstein RS. Mechanism of regulation of cardiac actin-myosin subfragment 1 by troponin-tropomyosin. Biochemistry (1986) 25(4):798–802. doi:10.1021/bi00352a010
39. Houmeida A, Heeley DH, Belknap B, White HD. Mechanism of regulation of native cardiac muscle thin filaments by rigor cardiac myosin-S1 and calcium. J Biol Chem (2010) 285(43):32760–9. doi:10.1074/jbc.m109.098228
40. Heeley DH, Belknap B, White HD. Mechanism of regulation of phosphate dissociation from actomyosin-ADP-Pi by thin filament proteins. Proc Natl Acad Sci USA (2002) 99(26):16731–6. doi:10.1073/pnas.252236399
41. King RT, Greene LE. Regulation of the adenosinetriphosphatase activity of cross-linked actin-myosin subfragment 1 by troponin-tropomyosin. Biochemistry (1985) 24:7009–14. doi:10.1021/bi00345a039
42. Rosenfeld SS, Taylor EW. The mechanism of regulation of actomyosin subfragment 1 ATPase. J Biol Chem (1987) 262:9984–93. doi:10.1016/s0021-9258(18)61063-4
43. Brenner B, Eisenberg E. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc Natl Acad Sci USA (1986) 83:3542–6. doi:10.1073/pnas.83.10.3542
44. Eisenberg E, Kielley W. Native tropomyosin: effect on the interaction of actin with heavy meromyosin and subfragment-1. Biochem Biophys Res Commun (1970) 40:50–6. doi:10.1016/0006-291x(70)91044-2
45. Bremel RD, Murray JM, Weber A. Manifestations of cooperative behavior in the regulated actin filament during actin-activated ATP hydrolysis in the presence of calcium. Cold Spring Harbor Symp Quant Biol (1972) 37:267–75. doi:10.1101/sqb.1973.037.01.037
46. Bremel RD, Weber A. Cooperation within actin filament in vertebrate skeletal muscle. Nature (1972) 238:97–101. doi:10.1038/newbio238097a0
47. Hill TL, Eisenberg E, Chalovich JM. Theoretical models for cooperative steady-state ATPase activity of myosin subfragment-1 on regulated actin. Biophys J (1981) 35(1):99–112. doi:10.1016/s0006-3495(81)84777-7
48. Pemrick S, Weber A. Mechanism of inhibition of relaxation by N-ethylmaleimide treatment of myosin. Biochemistry (1976) 15(23):5193–8. doi:10.1021/bi00668a038
49. Williams DL, Greene LE, Eisenberg E. Cooperative turning on of myosin subfragment-1 adenosine-triphosphatase activity by the troponin tropomyosin actin complex. Biochemistry (1988) 27(18):6987–93. doi:10.1021/bi00418a048
50. Baxley T, Johnson D, Pinto JR, Chalovich JM. Troponin C mutations partially stabilize the active state of regulated actin and fully stabilize the active state when paired with Δ14 TnT. Biochemistry (2017) 56(23):2928–37. doi:10.1021/acs.biochem.6b01092
51. Heeley DH, Belknap B, White HD. Maximal activation of skeletal muscle thin filaments requires both rigor myosin S1 and calcium. J Biol Chem (2006) 281(1):668–76. doi:10.1074/jbc.m505549200
52. McKillop DFA, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J (1993) 65:693–701. doi:10.1016/s0006-3495(93)81110-x
53. Chalovich JM. Disease causing mutations of troponin alter regulated actin state distributions. J Muscle Res Cel Motil (2012) 33(6):493–9. doi:10.1007/s10974-012-9305-x
54. Chalovich JM, Zhu L, Johnson D. Hypertrophic cardiomyopathy mutations of troponin reveal details of striated muscle regulation. Front Physiol (2022) 13:902079. doi:10.3389/fphys.2022.902079
55. Johnson D, Zhu L, Landim-Vieira M, Pinto JR, Chalovich JM. Basic residues within the cardiac troponin T C terminus are required for full inhibition of muscle contraction and limit activation by calcium. J Biol Chem (2019) 294(51):19535–45. doi:10.1074/jbc.ra119.010966
56. Zhu L, Johnson D, Chalovich JM. C-terminal basic region of troponin T alters the Ca2+-dependent changes in troponin I interactions. Biochemistry (2022) 61(11):1103–12. doi:10.1021/acs.biochem.2c00090
57. Gafurov B, Fredricksen S, Cai A, Brenner B, Chase PB, Chalovich JM. The Δ14 mutation of human cardiac troponin T enhances ATPase activity and alters the cooperative binding of S1-ADP to regulated actin. Biochemistry (2004) 43(48):15276–85. doi:10.1021/bi048646h
58. Lopez Davila AJ, Zhu L, Fritz L, Kraft T, Chalovich JM. The positively charged C-terminal region of human skeletal troponin T retards activation and decreases calcium sensitivity. Biochemistry (2020) 59(43):4189–201. doi:10.1021/acs.biochem.0c00499
59. Johnson D, Landim-Vieira M, Soli SC, Zhu L, Robinson JM, Pinto JR, et al. Eliminating the first inactive state and stabilizing the active state of the cardiac regulatory system alters behavior in solution and in ordered systems. Biochemistry (2020) 59(37):3487–97. doi:10.1021/acs.biochem.0c00430
Keywords: steric blocking, striated muscle regulation, tropomyosin, troponin, weak binding, strong binding, contractile regulatory mechanisms
Citation: Chalovich JM (2024) Steric blocking upside down: a different way of thinking about the competition between myosin and tropomyosin. Front. Phys. 12:1298482. doi: 10.3389/fphy.2024.1298482
Received: 21 September 2023; Accepted: 15 January 2024;
Published: 25 January 2024.
Edited by:
Alexandre Lewalle, King’s College London, United KingdomReviewed by:
Thomas Charles Irving, Illinois Institute of Technology, United StatesCopyright © 2024 Chalovich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph M. Chalovich, Y2hhbG92aWNoakBlY3UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.