 János Sarka
János Sarka Bill Poirier
Bill Poirier- Department of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX, United States
In a recent article [AIP Adv. 11, 045033 (2021)], we carried out exact quantum dynamical calculations and computed ro-vibrational energy levels and wave functions for the
1 Introduction
The
On the experimental side, numerous spectroscopic studies have been conducted [6–14]. Of course, the primary challenge with respect to labeling is that experiments provide only spectroscopic transitions, not the ro-vibrational energy levels themselves. Although symmetry and selection rules help, extracting the latter from the former remains a challenge, and has traditionally been something of a “black art.” Recently, more systematic approaches have been developed, based on graph theory and “spectroscopic networks” (SNs) [15, 16], in which the vertices represent rovibrational energy levels, and the lines represent experimentally observed spectroscopic transitions, to extract empirical energy levels directly from experimental data, with well-defined and realistic uncertainties. In particular, the MARVEL code (Measured Active Rotational–Vibrational Energy Levels) [17, 18], has been applied to ro-vibrational spectroscopic data of
On the theoretical side, due to its spectroscopic importance, a variety of
For these reasons, we prefer a fully ab initio computational approach [43], both with regard to the ro-vibrational state calculation itself, as well as the determination of state labels. In particular, to the best of our knowledge, we are the first group to attempt a fully ab initio assignment of ro-vibrational state labels for
As further motivation for adopting a purely ab initio approach, we point out that
Therefore, creating highly accurate line lists can serve as an important tool, from both the experimental and computational points of view. The first such line list was created by [51] with 669 astronomically important lines. This was supplemented by [52], with about three million lines. The newest line list, MiZaTeP [53], contains more than 120 million lines by bringing together the experimental spectroscopic data using MARVEL [13], and theoretical levels computed with the DVR3D code [54–56]. This line list also contains 17 meta-stable states, which are quantum states with very long lifetimes.
In this work, in order to facilitate the expansion of already existing
In all, we provide vibrational ‘v1, v2, |l|’ and rotational ‘J, G, K, U’ quantum labels for more than 2,200 ro-vibrational states, around 1,600 of which are new assignments complementing, and in certain cases arguably correcting, the ∼650 assignments in the MARVEL database [19]. To the best of our knowledge, no previous work has attempted to provide purely ab initio quantum label assignments for computed ro-vibrational states—certainly not to the extent that we have done here, in any event.
2 Materials and methods
In a recent article [43], we carried out the exact ro-vibrational energy level and wave function calculations for the
2.1 ScalIT
The quantum dynamical calculations presented in this article were performed using the ScalIT [58–62] suite of parallel codes. ScalIT is a black-box molecular ro-vibrational spectroscopy code, which for tri- and tetratomic molecules employs an analytical kinetic energy operator expressed in (orthogonal) Jacobi coordinates. The use of direct product basis sets (DPBs) including discrete variable representations (DVRs) results in a Hamiltonian matrix with a sparse structure. For the radial coordinates, phase–space-optimized DVRs (PSO-DVRs) are used [63–68] while for the bend and rotation angles, standard associated Legendre polynomial or Wigner rotation function basis sets are utilized. The Hamiltonian is diagonalized iteratively using sparse Krylov subspace methods together with several different effective numerical optimization strategies, such as the preconditioned inexact spectral transform (PIST) method [69–71], optimal separable basis (OSB) preconditioning [72–75], and the standard iterative quasi-minimal residual (QMR) algorithm [59, 76]. All of these methods working together ensure the effective scaling across massively parallel supercomputing clusters (up to a few thousand cores) and the ability of ScalIT to accurately compute even extremely energetically high-lying quantum states. So far, ScalIT has been used for around a dozen challenging systems, such as Ne4 and HCCH [43, 50, 68, and 77–86], and via extending the capabilities of the ScalIT code through the SwitchIT [87] algorithm to accommodate more complicated Hamiltonians, even CH3CN [87].
The massively parallel capability of ScalIT is based on MPI parallelization, which makes the code uniquely qualified to compute many quantum states with high accuracy for small molecular systems. Other available ro-vibrational spectroscopy codes, e.g., DVR3D [54–56], TROVE [88–90], DOPI [33, 91, 92], DEWE [57, 93–95], GENIUSH [96, 97], and ElVibRot [98–100], in general traditionally only offer single node OPENMP parallelization (although ElVibRot has been made MPI parallel recently [101]). Also, it is worth mentioning that the use of GPUs is spreading slowly to the field of ro-vibrational molecular spectroscopy with a focus on computing ro-vibrational intensities [102].
2.2 Potential energy surface
In this work, we utilized the recently computed
In any event, the
2.3 Previous computational works
For the GLH3P PES [12, 24]—and other earlier PESs, including the PES developed by Cencek and colleagues [20, 21]—a wide range of ro-vibrational calculations have been performed [12, 22–24, 27–29, 31, 32, 34–40, 42, and 43]. Most of these are summarized in a fairly recent review article [40]. In the last couple of years, newer PESs have been also developed, among them, the PES75K+ PES [25] is used here, and also a multi-sheet fit PES including more than one electronic state [26]. By and large, the ro-vibrational studies have focused on increasing the accuracy of numerical convergence, as well as pushing the limits of vibrational/rotational excitation. Indeed, computing ro-vibrational energy levels of
2.4 Symmetry
Jacobi coordinates (denoted as r, R, and θ here) are usually the best choice for describing AB2 triatomic molecules. In such cases, the full G4 permutation–inversion (PI) symmetry of the molecules is fully described. Of course,
Nevertheless, since Jacobi coordinates, in contrast to hyperspherical coordinates [104], do not respect the cyclic permutation operations of the G12 PI group, this poses certain challenges for the ScalIT calculation performed here, which essentially presumes an AB2 structure. More specifically, it becomes necessary to correlate the symmetry labels from the G4 symmetry-adapted ScalIT calculations to the G12/D3h labels, using the Γ(D3h) ↓ G4 correlation table [43]. The “challenge” here actually only concerns the doubly-degenerate D3h irrep pairs, which are computed in different ScalIT calculations corresponding to different G4 irreps. In practice, one looks for identical eigenvalues across two G4 irreps, and identifies those as comprising, in reality, a single doubly-degenerate G12 irrep pair. Better convergence accuracy thus greatly improves the determination of numerically “identical” eigenvalues. Conversely, whatever pair splitting is observed numerically may be taken as an additional, independent measure of the overall numerical convergence accuracy.
The solutions of the AB2 Jacobi Hamiltonian are computed in four separate “symmetry blocks”, corresponding to the four (singly-degenerate) irreps of G4. These irreps can be labeled by two good quantum numbers, p = ±1 (associated with the exchange of any two identical nuclei) and ϵ = ±1 (the total parity). In addition, there are the two good rotational quantum numbers that can be used as completely reliable labels—i.e., the total angular momentum, J, and its projection along the space-fixed Z axis, M. The third rotational quantum number, K, associated with the projection of
Lastly, given the fermionic nature of the H atom nuclei (i.e., protons), it is worth mentioning that the Pauli principle requires the total spin-plus-spatial nuclear wave function to have a totally anti-symmetric or A2 character (in the S3 permutation subgroup of the G12 PI group). For three such particles, the eight-dimensional combined nuclear spin space representation reduces to an irrep direct sum as 4A1⊕2E. The corresponding spatial wave functions (i.e., the ro-vibrational states actually computed) are thus restricted to belonging to either the A2 or E irreps. Therefore, all A1 ro-vibrational states (including what would otherwise be the ground vibrational state), are unphysical, and must be ignored.
3 Results
3.1 Computational details
ScalIT computations were carried out using nuclear masses, just as in our previous article [43]. The computational parameters of this work are summarized in Table 1. In DVR calculations such as those performed here, very often as one increases the basis size in order to improve numerical convergence, one crosses over from a regime where the basis set truncation error dominates, to a regime where the numerical quadrature error dominates. This is indicated by a fast, variational convergence (from above) being replaced by a slow, oscillatory convergence behavior. Usually, when this crossover has occurred, it becomes computationally unfeasible to push the calculation much further through a “brute force” increase in the basis size.
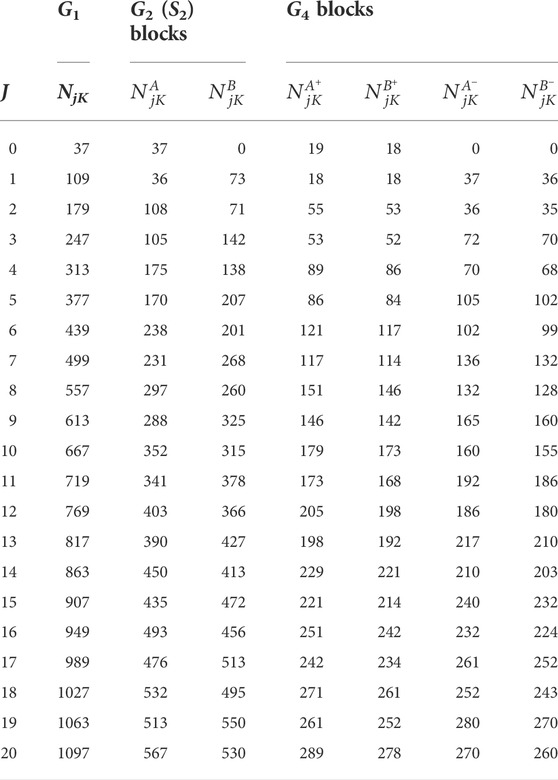
TABLE 1. The total bend-rotation angular basis sizes of each G4 symmetry block,
For extremely accurately converged ro-vibrational calculations, it is therefore necessary to ensure that the quadrature error is minimized. This requires two conditions. First, the PES must be very smooth and well-behaved—which, in the case of PES75K+ (but unlike GLH3P), has already been established. Second, the “primitive basis” calculations used to compute the PSO DVR basis representations must be performed as accurately as possible. To this end, a very large number of 1,001 primitive sinc-DVR grid points were used in the PSO DVR calculations for both of the Jacobi radial coordinates, r and R. The radial ranges used here were also wider than before [43]; here, we used rmin = 0.5 bohr, and rmax = 5.0 bohr for the r coordinate, and Rmin = 0.0 bohr, and Rmax = 5.0 bohr for the R coordinate.
Having put these measures into effect, our next task was to increase the basis sizes for the final calculation as far as possible, in hopes that an extremely high numerical convergence could be achieved prior to crossing over into the quadrature-error-dominated regime. We therefore used significantly larger radial basis sizes than in the previous calculation; i.e., Nr = 300 and NR = 300. The angular dimensions were also increased compared to our previous work [43]; specifically, the number of bend-angle basis functions in the Jacobi coordinate θ was set to jmax = 36 for every J value considered. The resultant total bend-rotation angular basis sizes for each G4 symmetry block calculation, i.e.,
3.2 State Labeling
The ro-vibrational calculations of
Next, we address the vibrational state labels. The
After first determining the D3h irrep labels, we assigned vibrational state labels to the J = 0 pure-vibrational states, which were found to be in complete agreement with earlier studies [9, 10, 12, 13, 24, 32, and 43]. For J > 0, it is advantageous to first determine the vibrational labels, indicating which “vibrational parent” state the ro-vibrational state “belongs to”. This is straightforward to do for low-vibrational and/or rotational excitations, where the energy level spacing is so high that the rotational progressions do not overlap. Higher up in energy, determining the “vibrational parents” becomes much more challenging. For J = 1, the different ro-vibrational progressions start to overlap at the 26th vibration at 10,000 cm−1. Increasing J, this threshold energy value shifts down drastically. For J = 11, even the ro-vibrational states belonging to the zero-point vibration start to overlap with the ro-vibrational progression of the first vibration. Beyond a certain point in both (v1, v2, |l|) and J, it becomes impossible to assign vibrational parents based solely on energy values and D3h symmetry labels.
In order to overcome this difficulty, the GENIUSH code [96, 97] was invoked, which is capable of semi-automatically assigning vibrational parent labels using the rigid rotor decomposition scheme (RRD) [57], based on computing wave function overlaps. To do this, the ScalIT calculations were repeated using GENIUSH, but with greatly reduced accuracy (10−2–10−3 cm−1)—which was nevertheless sufficient to match the energy levels with the ScalIT ones. The RRD overlap matrices were then computed using GENIUSH. In this manner, we were able to assign vibrational parent labels to much more highly excited ro-vibrational states—and for many more such states—than was previously possible.
We next move on to rotational state labels. The
where EJK is the relative energy of the ro-vibrational state corresponding to its vibrational parent. Although for
For l = 0, G = |K|, and so the usual (2J + 1)-fold rotational progression arises. For |l| > 0, however, the±l values double the number of the rotational excited states to 2 (2J + 1). For |l| > J, there is only one rotational progression, where Gmin = (|l| − J) ≤ G ≤ Gmax = (|l| + J). For |l| ≤ J, the rotational excited states can be separated into two distinct rotational progressions [8, 107], with 0 ≤ G ≤ Gmax = (J + |l|), and
As it usually holds that Gmax > J, EJG can become negative—as happens, e.g., for the
The presence of the two progressions requires that in addition to G, a new quantum number, U [8, 107], has to be introduced. U can take the values “u”, “l”, or “m” to distinguish between upper and lower energy levels with the same (v1, v2, |l|, J, G) assignment (note, that U = “l” always refers to levels within the G′ progression). Therefore, the rotational part of the wave function can be unambiguously described by the (J, G, U) quantum labels. This, however, does not mean that we cannot assign K values to each ro-vibrational state, as from the definition of G, we can assign |K| = |G − |l‖ for the unprimed progressions, and |K| = G′ + |l| for the primed progressions. Therefore, in the end, we characterize the rotational part using the (J, G, U, K) quantum label quartet.
For J > 0, D3h irrep labels are also linked to the quantum numbers, but it is actually G which is most directly impacted (for J = 0, |l| = G). The D3h irrep is A1 or A2 if G = 0 (if l = 0, we have A1 for even and A2 for odd J values), and for G > 0, similarly to J = 0, the degenerate pair can be labeled as E unless |l| mod 3 = 0, where the ± l pair splits into A1⊕A2. For large G values, especially for G ≥ 12, the two singly degenerate levels can get closer than 10–5 cm−1; therefore, in certain cases, G values have to be taken into account when assigning D3h labels.
Finally, note that to further aid in the assignment of the rotational labels, the RRD overlaps of GENIUSH also provide insights into the value of K. This occurs through the vibrational parent being assigned (in particular, l) into the G quantum number, as the symmetric top rigid rotor functions are labeled by K. This feature of the approach helped us tremendously in carrying out the task of assigning labels—so long as the RRD overlap values were significantly large.
3.3 Ro-vibrational energy levels
The ro-vibrational energy levels reported in this article are presented in cm−1, relative to the zero-point vibrational energy, 4362.1726 cm−1. The levels were obtained for each J, ϵ, and p set of values in a separate ScalIT calculation with a single PIST spectral window, usually including 70 to 120 ro-vibrational states. The number of computed levels and the highest energy level computed is summarized in Table 3 for each J value. Note that these numbers include the unphysical states, and the doubly degenerate states are counted twice. In total, 105 vibrational energy levels were computed, up to 16,500 cm−1, significantly over the isomerization barrier. For J = 1, the computed states (around 350 in all) covered the range up to 17,300 cm−1. For 2
In Table 2, the lowest ro-vibrational energy levels of
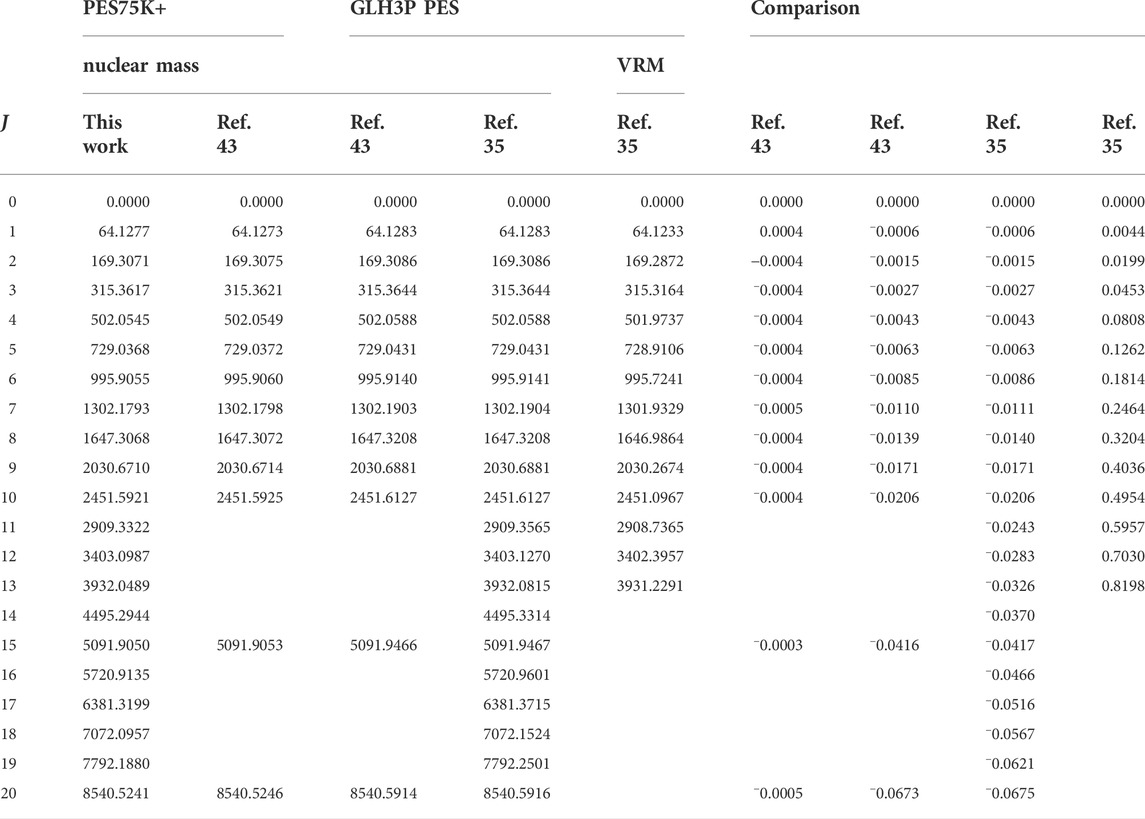
TABLE 2. The lowest ro-vibrational energy levels of
The main focus of this work is, however, to provide vibrational and rotational quantum labels for as many states as possible. In our previous study [43], although D3h irrep labels were provided for selected J values (J = 10, 20, 30, 40, 46) up to J = 46, we only provided a limited number of vibrational and rotational quantum label assignments. Only low J values 0 ≤ J ≤ 5 and J = 10 were considered, and even those were mostly restricted to up to 8,000–9,000 cm−1, so overall, below the isomerization limit. Only for J = 0 and 1 did we go above 10,000 cm−1. Here, we pushed our efforts further with the help of wave function overlaps and provided assignments for almost all states below 10,000 cm−1. In Table 3, we summarize the number of states labeled in this work for each J value separately as well as include the labeling threshold, Elab. As for most J values, not all of the levels were assigned up to the given threshold energy, we separately include the energy value up to which all states are labeled,
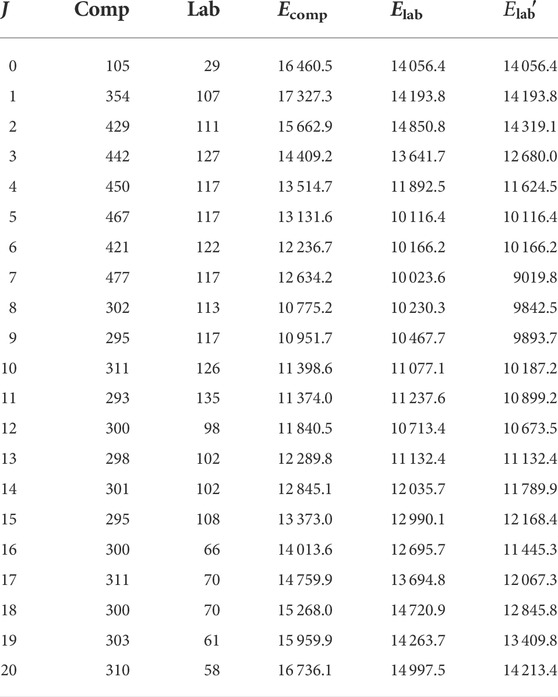
TABLE 3. For each J value, the total number of states computed (comp.) and labeled (lab.). Note that the former number includes the unphysical states as well, and there the doubly degenerate states are counted twice. Ecomp and Elab are the threshold energies for computation and labeling, respectively. As not all the states were labeled up to Elab, a separate threshold was also included, up to which all the states are labeled (
During this work, 2,210 ro-vibrational levels of
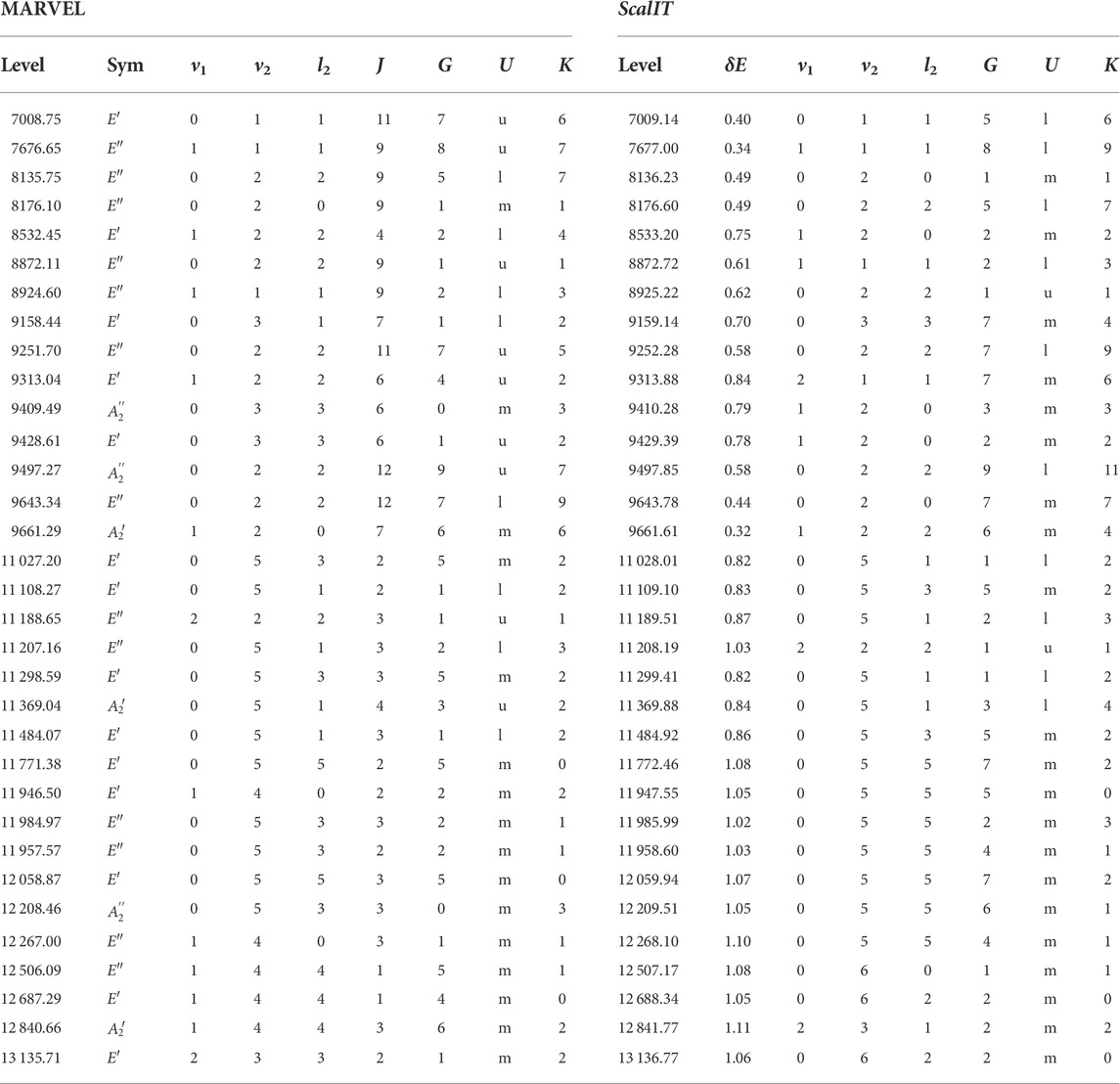
TABLE 4. Suggested vibrational (v1, v2, |l|) and rotational (J, G, U, K) reassignments of
In order to obtain all the quantum labels presented here, our approach has been adapted keeping in mind the difficulties we faced in Ref. [43]. Based on the energy formula of Eqs 2, 3, one would expect a somewhat regular behavior in the shifting of the rotationally excited energy levels belonging to the same vibrational parent. However, this seems to hold only for the l = 0 cases. In Figure 1, the change of B rotational constant is illustrated as J is increased. Each rotational progression belonging to a vibrational state can be characterized by a slightly different B rotational constant, which also shifts slightly by the increase of J. As the vibrational excitation increases, this shift becomes more significant (e.g. in case of 0, 2, 0 and 0, 4, 0). However, for the l > 0 vibrational parents, this shift can be more chaotic (see Figure 2) and it is different for the two distinct rotational progressions, assigned to G and G′ (see Figures 2, 3). For the (1, 3, 1) vibrational parent, e.g., the shift seems to first be positive and then it turns negative as it is in all other cases for both progressions. The G = 0 energy levels of the first 13 vibrational states up to J = 20 are listed in Table 5, while G′ = 0 energy levels of the first 7 l > 0 vibrational states up to J = 16 are included in Table 6.
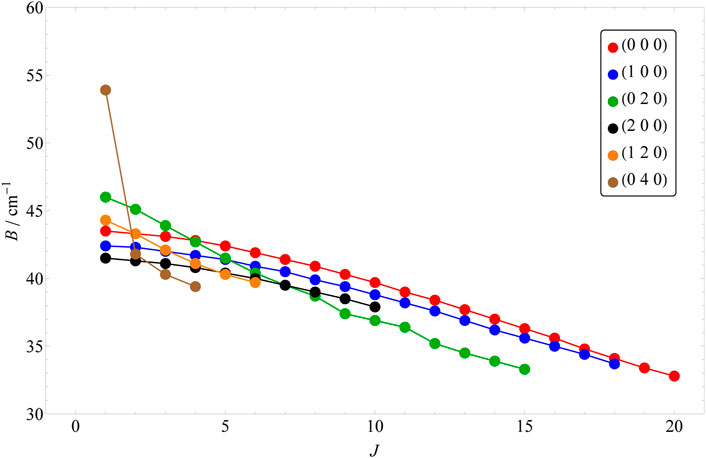
FIGURE 1. The shift of the B rotational constants with the increase of the J value for the first 6 l = 0 vibrational parents.
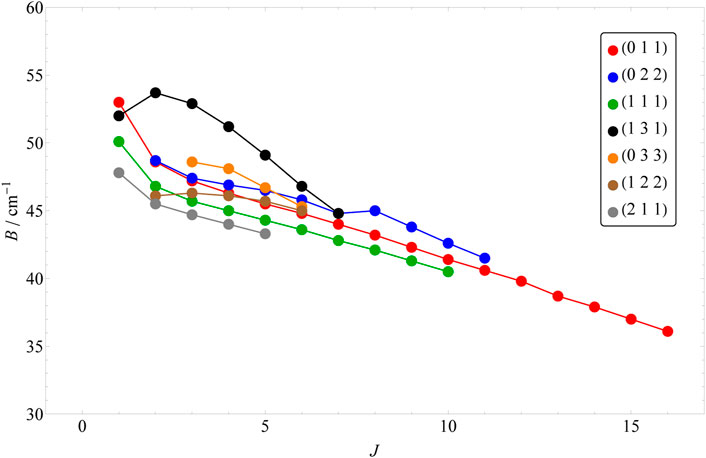
FIGURE 2. The shift of the B rotational constants with the increase of the J value for the first 7 l > 0 vibrational parents. The numbers included here correspond to the G progression.
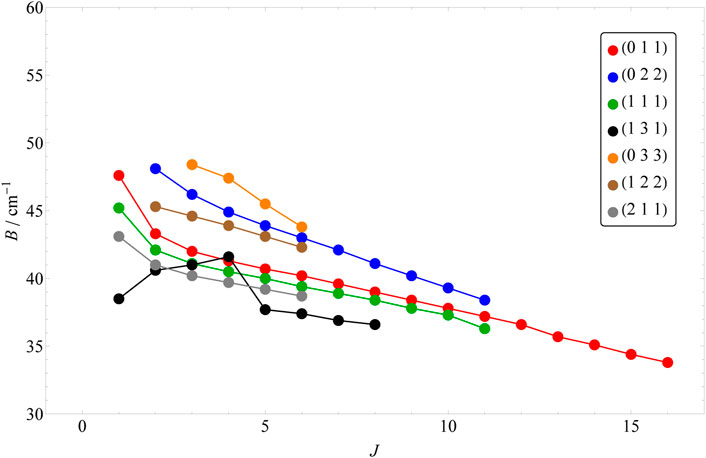
FIGURE 3. The shift of the B rotational constants with the increase of the J value for the first 7 l > 0 vibrational parents. The numbers included here correspond to the G′ progression.
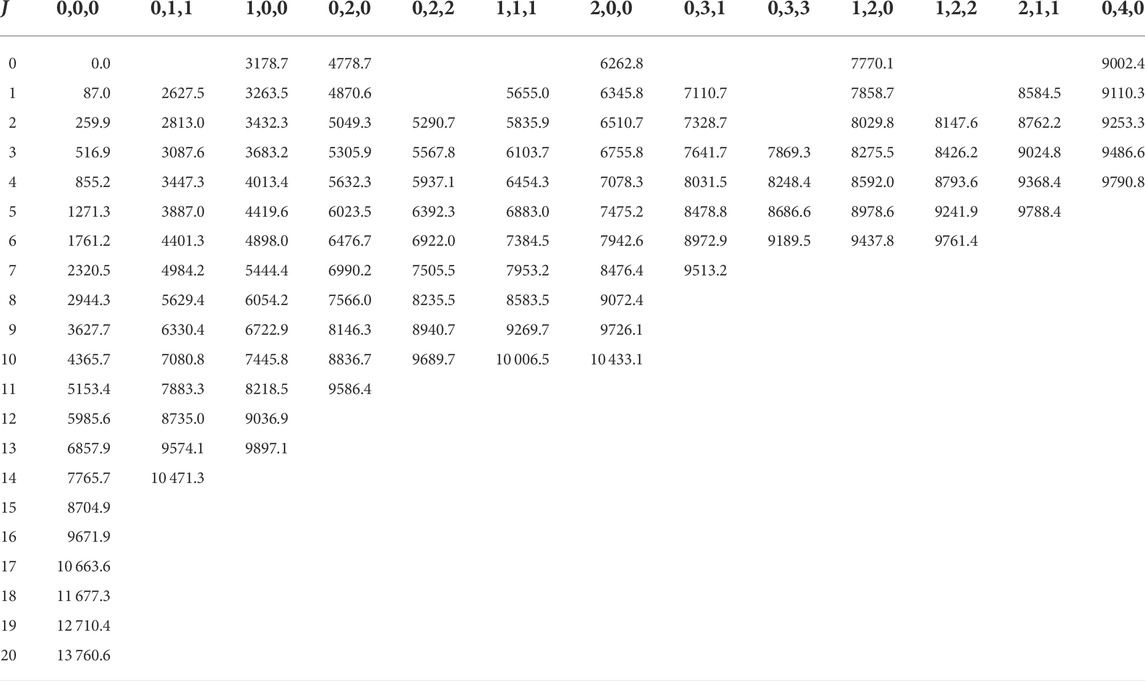
TABLE 5. The G = 0 energy levels of the first 13 vibrational states (v1, v2, |l|) of
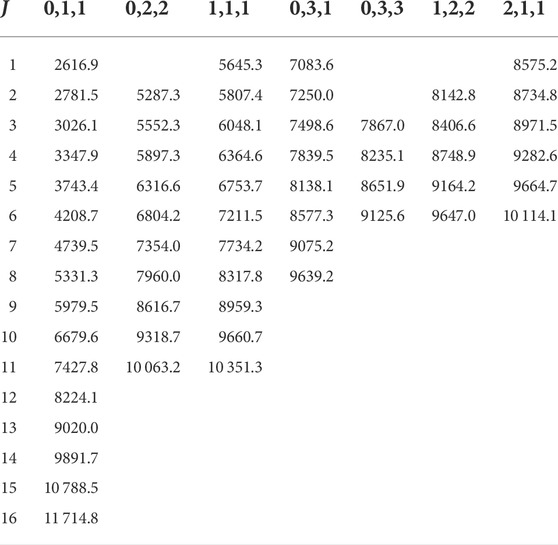
TABLE 6. The G′ = 0 energy levels of the first 7 l > 0 vibrational states (v1, v2, |l|) of
Using the RRD method [57] of GENIUSH to compute wave function overlaps and assign vibrational parents we can push the labeling a lot further than simply relying on the energy progressions. However, after a certain point in the vibrational and/or the rotational energy excitation, the mixing of the wave functions becomes simply too much for the vibrational parents to be unambiguously identified. For low J values, up to J = 6, we were able to get past the isomerization barrier, and in most cases, we could continue on even further. As the rotational excitation increases, the highest vibrational parent we can possibly assign is also decreasing. From J = 7 to 10, we are barely reaching the barrier to linearity, while as J increases further, the rotational energy contribution is also getting bigger, therefore we are again getting past the isomerization barrier (see Table 3). Different vibrational parents also behave differently, e.g. the overlaps of the (1,2,0) state breaks down a lot sooner than those of the next few higher-lying vibrational states. The more spread out progressions are also more difficult to assign fully including all the states within the progression. Although the progression belonging to (1,0,0) can be fully identified up to J = 18, in the progression of (0,1,1) we already start missing levels at J = 16. The ground vibrational state is the only vibrational parent for which all the states were found within the progressions for each J up to J = 20. This, however, might not be possible for J > 20 values.
In certain cases, it can be observed that the G = 0 (or G′ = 0) level is not the lowest level of the progression, which seemingly results in negative rotational excitations [108]. This happens for both higher vibrational excitations [e.g., for (0,5,1) at J = 1], higher rotational excitation (e.g., for (0,1,1) at J = 11), or for both [e.g., for (0,3,3) at J = 3 and for (0,3,1) at J = 6]. In certain cases, this reversing of the energy levels occurs sooner for the G′ progression [e.g., for (0,3,1) at J = 5].
4 Discussion
In this article, we computed ro-vibrational energy levels and wave functions for the
In addition, we carried out vibrational (v1, v2, |l|) and rotational (J, G, U, K) quantum label assignments of ro-vibrational energy levels for more than 2,200 states. To enable this, GENIUSH calculations were also carried out with lower accuracy to obtain ro-vibrational wave functions, which were then used to compute wave function overlaps within the framework of the rigid rotor decomposition method. These RRD overlaps helped greatly to identify the vibrational parents. As part of our efforts, we suggested new vibrational (v1, v2, |l|) and rotational (J, G, U, K) reassignments for certain energy levels within the MARVEL database. We are hoping that the results of this work can be used to further improve previous efforts toward creating spectroscopic line lists (based on both theoretical and experimental data), through the list of all labeled energy levels as provided in the Supplementary Material of this article.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Author contributions
JS and BP designed the research, JS carried out the calculations and analyzed the data, JS and BP discussed the results, JS and BP wrote the manuscript, and BP provided funding.
Funding
This work was supported by the National Science Foundation (CHE-1665370), and the Robert A. Welch Foundation (D-1523).
Acknowledgments
The authors also gratefully acknowledge the Texas Tech University High Performance Computing Center for the use of the Quanah cluster and the Texas Advanced Computing Center for the use of the Lonestar5 and Frontera clusters.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2022.996001/full#supplementary-material
References
1. Miller S, Tennyson J, Geballe TR, Stallard T. Thirty years of H3+ astronomy. Rev Mod Phys (2020) 92:035003. doi:10.1103/revmodphys.92.035003
2. Snow TP, McCall BJ. Diffuse atomic and molecular clouds. Annu Rev Astron Astrophys (2006) 44:367–414. doi:10.1146/annurev.astro.43.072103.150624
3. Miller S, Tennyson J, Lepp S, Dalgarno A. Identification of features due to H3+ in the infrared spectrum of supernova 1987A. Nature (1992) 355:420–2. doi:10.1038/355420a0
4. Miller S, Achilleos N, Ballester GE, Geballe TR, Joseph RD, Prange R, et al. The role of H3+ in planetary atmospheres. Philosophical Trans R Soc Lond Ser A: Math Phys Eng Sci (2000) 358:2485–502. doi:10.1098/rsta.2000.0662
5. Koskinen TT, Aylward AD, Miller S. A stability limit for the atmospheres of giant extrasolar planets. Nature (2007) 450:845–8. doi:10.1038/nature06378
6. Carrington A, Buttenshaw J, Kennedy R. Observation of the infrared spectrum of H3+ at its dissociation limit. Mol Phys (1982) 45:753–8. doi:10.1080/00268978200100591
7. Carrington A, McNab IR, West YD. Infrared predissociation spectrum of the H3+ ion. II. J Chem Phys (1993) 98:1073–92. doi:10.1063/1.464331
8. Lindsay C, McCall BJ. Comprehensive evaluation and compilation of H3+ spectroscopy. J Mol Spectrosc (2001) 210:60–83. doi:10.1006/jmsp.2001.8444
9. Schiffels P, Alijah A, Hinze J. Rovibrational states of H3+. Part 1: The energy region below 9,000 cm−1 and modelling of the non-adiabatic effects. Mol Phys (2001) 101:175. doi:10.1080/00268970210158687
10. Schiffels P, Alijah A, Hinze J. Rovibrational states of H3+. Part 2: The energy region between 9,000 cm-1 and 13,000 cm-1 including empirical corrections for the non-adiabatic effects. Mol Phys (2001) 101:189. doi:10.1080/00268970210158713
11. Asvany O, Hugo E, Schlemmer S, Muller F, Kuhnemann F, Schiller S, et al. Overtone spectroscopy of H2D+ and D2H+ using laser induced reactions. J Chem Phys (2007) 127:154317. doi:10.1063/1.2794331
12. Pavanello M, Adamowicz L, Alijah A, Zobov NF, Mizus II, Polyansky OL, et al. Precision measurements and computations of transition energies in rotationally cold triatomic hydrogen ions up to the midvisible spectral range. Phys Rev Lett (2012) 108:023002. doi:10.1103/physrevlett.108.023002
13. Furtenbacher T, Szidarovszky T, Mátyus E, Fábri C, Csaszár AG. Analysis of the rotational–vibrational states of the molecular ion H3+. J Chem Theor Comput (2013) 9:5471–8. doi:10.1021/ct4004355
14. Furtenbacher T, Szidarovszky T, Fábri C, Császár AG. MARVEL analysis of the rotational–vibrational states of the molecular ions H2D+ and D2H+. Phys Chem Chem Phys (2013) 15:10181. doi:10.1039/c3cp44610g
15. Császár AG, Furtenbacher T. Spectroscopic networks. J Mol Spectrosc (2011) 266:99–103. doi:10.1016/j.jms.2011.03.031
16. Császár AG, Furtenbacher T, Árendás P. Small molecules—big Data. J Phys Chem A (2016) 120:8949–69. doi:10.1021/acs.jpca.6b02293
17. Furtenbacher T, Császár AG, Tennyson J. Marvel: Measured active rotational–vibrational energy levels. J Mol Spectrosc (2007) 245:115–25. doi:10.1016/j.jms.2007.07.005
18. Furtenbacher T, Császár AG. Marvel: Measured active rotational–vibrational energy levels. II. Algorithmic improvements. J Quant Spectrosc Radiat Transf (2012) 113:929–35. doi:10.1016/j.jqsrt.2012.01.005
19.Marvel online (2022). Available from: https://kkrk.chem.elte.hu/marvelonline/index.php (accessed 07 11, 2022).
20. Cencek W, Rychlewski J, Jaquet R, Kutzelnigg W. Sub-microhartree accuracy potential energy surface for H3+ including adiabatic and relativistic effects. I. Calculation of the potential points. J Chem Phys (1998) 108:2831–6. doi:10.1063/1.475702
21. Jaquet R, Cencek W, Kutzelnigg W, Rychlewski J. Sub-microhartree accuracy potential energy surface for H3+ including adiabatic and relativistic effects. II. Rovibrational analysis for H3+ and D3+. J Chem Phys (1998) 108:2837–46. doi:10.1063/1.475703
22. Polyansky O, Prosmiti R, Klopper W, Tennyson J. An accurate, global, ab initio potential energy surface for the H+3 molecule. Mol Phys (2000) 98:261–73. doi:10.1080/00268970009483290
23. Velilla L, Lepetit B, Aguado A, Beswick J, Paniagua M. The H3+ rovibrational spectrum revisited with a global electronic potential energy surface. J Chem Phys (2008) 129:084307. doi:10.1063/1.2973629
24. Pavanello M, Adamowicz L, Alijah A, Zobov NF, Mizus II, Polyansky OL, et al. Calibration-quality adiabatic potential energy surfaces for H3+ and its isotopologues. J Chem Phys (2012) 136:184303. doi:10.1063/1.4711756
25. Mizus II, Polyansky OL, McKemmish LK, Tennyson J, Alijah A, Zobov NF. A global potential energy surface for H3+. Mol Phys (2019) 117:1663–72. doi:10.1080/00268976.2018.1554195
26. Aguado A, Roncero O, Sanz-Sanz C. Three states global fittings with improved long range: Singlet and triplet states of H3+. Phys Chem Chem Phys (2021) 113. doi:10.1039/D0CP04100A
27. Miller S, Tennyson J. Calculation of the high angular momentum dissociation limit for H3+ and H2D+. Chem Phys Lett (1988) 145:117–20. doi:10.1016/0009-2614(88)80161-1
28. Kozin IN, Roberts RM, Tennyson J. Symmetry and structure of rotating H3+. J Chem Phys (1999) 111:140–50. doi:10.1063/1.479260
29. Kostin MA, Polyansky OL, Tennyson J, Mussa HY. Rotation-vibration states of H3+ at dissociation. J Chem Phys (2003) 118:3538–42. doi:10.1063/1.1539034
30. Munro JJ, Ramanlal J, Tennyson J, Mussa HY. Properties of high-lying vibrational states of the molecular ion. Mol Phys (2006) 104:115–25. doi:10.1080/00268970500399648
31. Bachorz RA, Cencek W, Jaquet R, Komasa J. Rovibrational energy levels of H3+ with energies above the barrier to linearity. J Chem Phys (2009) 131:024105. doi:10.1063/1.3167795
32. Alijah A. Accurate calculations and assignments of above-barrier states of up to. J Mol Spectrosc (2010) 264:111–9. doi:10.1016/j.jms.2010.09.009
33. Szidarovszky T, Császár AG, Czakó G. On the efficiency of treating singularities in triatomic variational vibrational computations. The vibrational states of H3+ up to dissociation. Phys Chem Chem Phys (2010) 12:8373. doi:10.1039/c001124j
34. Diniz LG, Alijah JRMA, Pavanello M, Adamowicz L, Polyansky OL, Tennyson J, et al. Vibrationally and rotationally nonadiabatic calculations on H3+ using coordinate-dependent vibrational and rotational masses. Phys Rev A (Coll Park) (2013) 88:032506. doi:10.1103/physreva.88.032506
35. Jaquet R, Carrington T. Using a nondirect product basis to compute J > 0 rovibrational states of H3+. J Phys Chem A (2013) 117:9493–500. doi:10.1021/jp312027s
36. Jaquet R. Investigation of the highest bound ro-vibrational states of H3+, DH2+, HD2+, D3+, and T3+: Use of a non-direct product basis to compute the highest allowed J> 0 states. Mol Phys (2013) 111:2606–16. doi:10.1080/00268976.2013.818727
37. Mátyus E, Szidarovszky T, Császár AG. Modelling non-adiabatic effects in H3+: Solution of the rovibrational Schrödinger equation with motion-dependent masses and mass surfaces. J Chem Phys (2014) 141:154111. doi:10.1063/1.4897566
38. Jaquet R, Khoma MV. Investigation of nonadiabatic effects for the vibrational spectrum of a triatomic molecule: The use of a single potential energy surface with distance-Dependent masses for H3+. J Phys Chem A (2017) 121:7016–30. doi:10.1021/acs.jpca.7b04703
39. Jaquet R, Khoma MV. Investigation of non-adiabatic effects for the ro-vibrational spectrum of H3+: The use of a single potential energy surface with geometry-dependent nuclear masses. Mol Phys (2018) 116:3507–18. doi:10.1080/00268976.2018.1464225
40. Tennyson J, Polyansky OL, Zobov NF, Alijah A, Császár AG. High-accuracy calculations of the rotation-vibration spectrum of H3+. J Phys B: Mol Opt Phys (2017) 50:232001. doi:10.1088/1361-6455/aa8ca6
41. Jaquet R, Lesiuk M. Analysis of QED and non-adiabaticity effects on the rovibrational spectrum of H3+ using geometry-dependent effective nuclear masses. J Chem Phys (2020) 152:104109. doi:10.1063/1.5144293
42. Sarka J, Poirier B, Szalay V, Császár AG. On neglecting Coriolis and related couplings in first-principles rovibrational spectroscopy: Considerations of symmetry, accuracy, and simplicity. II. Case studies for H2O isotopologues, H3+, O3, and NH3. Spectrochim Acta A Mol Biomol Spectrosc (2021) 250:119164. doi:10.1016/j.saa.2020.119164
43. Sarka J, Das D, Poirier B. Calculation of rovibrational eigenstates of H3+ using ScalIT. AIP Adv (2021) 11:045033. doi:10.1063/5.0047823
44. Moss RE. On the adiabatic and non-adiabatic corrections in the ground electronic state of the hydrogen molecular cation. Mol Phys (1996) 89:195–210. doi:10.1080/002689796174083
45. Diniz LG, Alijah A, Mohallem JR. Core-mass nonadiabatic corrections to molecules: H2, H2+, and isotopologues. J Chem Phys (2012) 137:164316. doi:10.1063/1.4762442
46. Huestic DL. Hydrogen collisions in planetary atmospheres, ionospheres, and magnetospheres. Planet Space Sci (2008) 56:1733. doi:10.1016/j.pss.2008.07.012
47. McKemmish LK, Tennyson J. General mathematical formulation of scattering processes in atom–diatomic collisions in the RmatReact methodology. Phil Trans R Soc A (2019) 377:20180409. doi:10.1098/rsta.2018.0409
48. Höveler K, Deiglmayr J, Agner JA, Schmutz H, Merkt F. The H2+ + HD reaction at low collision energies: H3+/H2D+ branching ratio and product-kinetic-energy distributions. Phys Chem Chem Phys (2021) 23:2676–85. doi:10.1039/d0cp06107g
49. Huang X, Schwenke DW, Lee TJ. Highly accurate potential energy surface, dipole moment surface, rovibrational energy levels, and infrared line list for 32S16O2 up to 8,000 cm-1. J Chem Phys (2014) 140:114311. doi:10.1063/1.4868327
50. Kumar P, Jiang B, Guo H, Klos J, Alexander MH, Poirier B. Photoabsorption assignments for the C̃1B2 ← X̃1A1 vibronic transitions of SO2, using new ab initio potential energy and transition Dipole surfaces. J Phys Chem A (2017) 121:1012–21. doi:10.1021/acs.jpca.6b12958
51. Kao L, Oka T, Miller S, Tennyson J. A table of astronomically important ro-vibrational transitions for the H3+ molecular ion. Astrophys J Suppl Ser (1991) 77:317. doi:10.1086/191606
52. Neale L, Miller S, Tennyson J. Spectroscopic properties of the H3+ molecule: A new calculated line list. Astrophys J (1996) 464:516. doi:10.1086/177341
53. Mizus II, Alijah A, Zobov NF, Lodi L, Kyuberis AA, Yurchenko SN, et al. ExoMol molecular line lists – XX. A comprehensive line list for H3+. Mon Not R Astron Soc (2017) 468:1717–25. doi:10.1093/mnras/stx502
54. Tennyson J, Kostin MA, Barletta P, Harris GJ, Polyansky OL, Ramanlal J, et al. DVR3D: A program suite for the calculation of rotation–vibration spectra of triatomic molecules. Computer Phys Commun (2004) 163:85–116. doi:10.1016/j.cpc.2003.10.003
55. Tennyson J. Perspective: Accurate ro-vibrational calculations on small molecules. J Chem Phys (2016) 145:120901. doi:10.1063/1.4962907
56. Tennyson J, Yurchenko SN. The ExoMol project: Software for computing large molecular line lists. Int J Quan Chem (2017) 117:92–103. doi:10.1002/qua.25190
57. Mátyus E, Fábri C, Szidarovszky T, Czakó G, Allen WD, Császár AG. Assigning quantum labels to variationally computed rotational-vibrational eigenstates of polyatomic molecules. J Chem Phys (2010) 133:034113. doi:10.1063/1.3451075
58. Chen W, Poirier B. Parallel implementation of efficient preconditioned linear solver for grid-based applications in chemical physics. I: Block Jacobi diagonalization. J Comput Phys (2006) 219:185–97. doi:10.1016/j.jcp.2006.04.012
59. Chen W, Poirier B. Parallel implementation of efficient preconditioned linear solver for grid-based applications in chemical physics. II: QMR linear solver. J Comput Phys (2006) 219:198–209. doi:10.1016/j.jcp.2006.03.031
60. Chen W, Poirier B. Parallel implementation of an efficient preconditioned linear solver for grid-based applications in chemical physics. III: Improved parallel scalability for sparse matrix–vector products. J Parallel Distrib Comput (2010) 70:779–82. doi:10.1016/j.jpdc.2010.03.008
61. Chen W, Poirier B. Quantum dynamics on massively parallel computers: efficient numerical implementation for preconditioned linear solvers and eigensolvers. J Theor Comput Chem (2010) 9:825–46. doi:10.1142/s021963361000602x
62. Petty C, Poirier B. Using ScalIT for performing accurate rovibrational spectroscopy calculations for triatomic molecules: A practical guide. Appl Math (Irvine) (2014) 5:2756–63. doi:10.4236/am.2014.517263
63. Poirier B, Light JC. Phase space optimization of quantum representations: Direct-product basis sets. J Chem Phys (1999) 111:4869–85. doi:10.1063/1.479747
64. Poirier B, Light JC. Phase space optimization of quantum representations: Three-body systems and the bound states of HCO. J Chem Phys (2001) 114:6562–71. doi:10.1063/1.1354181
65. Poirier B. Research topic "proton transfer processes in biological reactions: a computational approach" frontiers in chemistry journal. Found Phys (2001) 31:1581–610. doi:10.1023/a:1012642832253
66. Bian W, Poirier B. Accurate and highly efficient calculation of the O(1D)HCl vibrational bound states, using A combination of methods. J Theor Comput Chem (2003) 2:583–97. doi:10.1142/s0219633603000768
67. Light J, Carrington T. Discrete-variable representations and their utilization Adv Chem Phys (2000) 114:263. doi:10.1002/9780470141731.ch4
68. Chen W, Poirier B. Quantum Dynamical calculation of all rovibrational states of HO2 for total angular momentum J = 0–10. J Theor Comput Chem (2010) 9:435–69. doi:10.1142/s0219633610005815
69. Huang S-W, Carrington T. A new iterative method for calculating energy levels and wave functions. J Chem Phys (2000) 112:8765–71. doi:10.1063/1.481492
70. Poirier B, Carrington T. Accelerating the calculation of energy levels and wave functions using an efficient preconditioner with the inexact spectral transform method. J Chem Phys (2001) 114:9254–64. doi:10.1063/1.1367396
71. Poirier B, Carrington T. A preconditioned inexact spectral transform method for calculating resonance energies and widths, as applied to HCO. J Chem Phys (2002) 116:1215–27. doi:10.1063/1.1428752
72. Poirier B, Miller WH. Optimized preconditioners for Green function evaluation in quantum reactive scattering calculations. Chem Phys Lett (1997) 265:77–83. doi:10.1016/s0009-2614(96)01408-x
73. Poirier B. Optimal separable bases and series expansions. Phys Rev A (Coll Park) (1997) 56:120–30. doi:10.1103/physreva.56.120
74. Poirier B. Quantum reactive scattering for three-body systems via optimized preconditioning, as applied to the O+HCl reaction. J Chem Phys (1998) 108:5216–24. doi:10.1063/1.475958
75. Poirier B. Efficient preconditioning scheme for block partitioned matrices with structured sparsity. Numer Linear Algebra Appl (2000) 7:715–26. doi:10.1002/1099-1506(200010/12)7:7/83.0.CO;2-R
76. Freund RW, Nachtigal NM. QMR: A quasi-minimal residual method for non-hermitian linear systems. Numer Math (1991) 60:315–39. doi:10.1007/bf01385726
77. Petty C, Chen W, Poirier B. Quantum Dynamical calculation of bound rovibrational states of HO2 up to largest possible total angular momentum, J ≤ 130. J Phys Chem A (2013) 117:7280–97. doi:10.1021/jp401154m
78. Petty C, Poirier B. Comparison of J-shifting models for rovibrational spectra as applied to the HO2 molecule. Chem Phys Lett (2014) 605–606:16–21. doi:10.1016/j.cplett.2014.05.003
79. Yang B, Chen W, Poirier B. Rovibrational bound states of neon trimer: Quantum dynamical calculation of all eigenstate energy levels and wavefunctions. J Chem Phys (2011) 135:094306. doi:10.1063/1.3630922
80. Brandon D, Poirier B. Accurate calculations of bound rovibrational states for argon trimer. J Chem Phys (2014) 141:034302. doi:10.1063/1.4887459
81. Yang B, Poirier B. Quantum dynamical calculation of rovibrational bound states of Ne2Ar. J Phys B: Mol Opt Phys (2012) 45:135102. doi:10.1088/0953-4075/45/13/135102
82. Yang B, Poirier B. Rovibrational bound states of the Ar2Ne complex. J Theor Comput Chem (2013) 12:1250107. doi:10.1142/s0219633612501076
83. Zhang Z, Li B, Shen Z, Ren Y, Bian W. Efficient quantum calculation of the vibrational states of acetylene. Chem Phys (2012) 400:1–7. doi:10.1016/j.chemphys.2012.01.010
84. Petty C, Spada RF, Machado FB, Poirier B. Accurate rovibrational energies of ozone isotopologues up to J= 10 utilizing artificial neural networks. J Chem Phys (2018) 149:024307. doi:10.1063/1.5036602
85. Sarka J, Poirier B. Comment on “Calculated vibrational states of ozone up to dissociation” [J. Chem. Phys. 144, 074302 (2016)]. J Chem Phys (2020) 152:177101. doi:10.1063/5.0002762
86. Sarka J, Petty C, Poirier B. Exact bound rovibrational spectra of the neon tetramer. J Chem Phys (2019) 151:174304. doi:10.1063/1.5125145
87. Sarka J, Poirier B. Hitting the trifecta: How to simultaneously push the limits of schrödinger solution with respect to system size, convergence accuracy, and number of computed states. J Chem Theor Comput (2021) 17:7732–44. doi:10.1021/acs.jctc.1c00824
88. Yurchenko SN, Thiel W, Jensen P. Theoretical ROVibrational energies (TROVE): A robust numerical approach to the calculation of rovibrational energies for polyatomic molecules. J Mol Spectrosc (2007) 245:126–40. doi:10.1016/j.jms.2007.07.009
89. Yurchenko SN, Yachmenev A, Ovsyannikov RI. Symmetry-adapted ro-vibrational basis functions for variational nuclear motion calculations: TROVE approach. J Chem Theor Comput (2017) 13:4368–81. doi:10.1021/acs.jctc.7b00506
90. Yurchenko SN, Mellor TM. Treating linear molecules in calculations of rotation-vibration spectra. J Chem Phys (2020) 153:154106. doi:10.1063/5.0019546
91. Czakó G, Furtenbacher T, Császár AG, Szalay V. Variational vibrational calculations using high-order anharmonic force fields. Mol Phys (2004) 102:2411. doi:10.1080/0026897042000274991
92. Furtenbacher T, Czakó G, Sutcliffe BT, Császár AG, Szalay V. The methylene saga continues: Stretching fundamentals and zero-point energy of X˜3B1 CH2. J Mol Struct (2006) 780–781:283–94. doi:10.1016/j.molstruc.2005.06.052
93. Mátyus E, Czakó G, Sutcliffe BT, Császár AG. Vibrational energy levels with arbitrary potentials using the Eckart-Watson Hamiltonians and the discrete variable representation. J Chem Phys (2007) 127:084102. doi:10.1063/1.2756518
94. Mátyus E, Šimunek J, Császár AG. On the variational computation of a large number of vibrational energy levels and wave functions for medium-sized molecules. J Chem Phys (2009) 131:074106. doi:10.1063/1.3187528
95. Fábri C, Mátyus E, Furtenbacher T, Mihály B, Zoltáni T, Nemes L, et al. Variational quantum mechanical and active database approaches to the rotational-vibrational spectroscopy of ketene, H2CCO. J Chem Phys (2011) 135:094307. doi:10.1063/1.3625404
96. Mátyus E, Czakó G, Császár AG. Toward black-box-type full- and reduced-dimensional variational (ro)vibrational computations. J Chem Phys (2009) 130:134112. doi:10.1063/1.3076742
97. Fábri C, Mátyus E, Császár AG. Rotating full- and reduced-dimensional quantum chemical models of molecules. J Chem Phys (2011) 134:074105. doi:10.1063/1.3533950
98. Lauvergnat D, Nauts A. Exact numerical computation of a kinetic energy operator in curvilinear coordinates. J Chem Phys (2002) 116:8560. doi:10.1063/1.1469019
99. Lauvergnat D, Nauts A. Quantum dynamics with sparse grids: A combination of smolyak scheme and cubature. Application to methanol in full dimensionality. Spectrochimica Acta A: Mol Biomol Spectrosc (2014) 119:18–25. doi:10.1016/j.saa.2013.05.068
100. Nauts A, Lauvergnat D. Numerical on-the-fly implementation of the action of the kinetic energy operator on a vibrational wave function: Application to methanol. Mol Phys (2018) 116:3701–9. doi:10.1080/00268976.2018.1473652
101. Chen A, Nauts A, Lauvergnat D. Elvibrot-mpi: Parallel quantum dynamics with smolyak algorithm for general molecular simulation (2021). doi:10.48550/arXiv.2111.13655
102. Al-Refaie AF, Yurchenko SN, Tennyson J. GPU Accelerated intensities MPI (GAIN-MPI): A new method of computing Einstein-A coefficients. Comput Phys Commun (2017) 214:216. doi:10.1016/j.cpc.2017.01.013
103. Zhang JZH. Theory and application of quantum molecular dynamics. Singapore: World Scientific (1999).
104. T Pack R, Parker GA. Quantum reactive scattering in three dimensions using hyperspherical (APH) coordinates. Theory. J Chem Phys (1987) 87:3888–921. doi:10.1063/1.452944
105. Hougen JT. Classification of rotational energy levels for symmetric‐top molecules. J Chem Phys (1962) 37:1433–41. doi:10.1063/1.1733301
106. Watson JKG. Simplification of the molecular vibration-rotation Hamiltonian. Mol Phys (1968) 15:479–90. doi:10.1080/00268976800101381
107. Watson JKG. Higher-order vibration-rotation energies of the X3 molecule. J Mol Spectrosc (1984) 103:350–63. doi:10.1016/0022-2852(84)90062-6
108. Császár AG, Fábri C, Sarka J. Quasistructural molecules. Wires Comput Mol Sci (2020) 10:e1432. doi:10.1002/wcms.1432
109. Fábri C, Sarka J, Császár AG. Communication: Rigidity of the molecular ion H5+. J Chem Phys (2014) 140:051101. doi:10.1063/1.4864360
110. Asvany O, Yamada KMT, Brünken S, Potapov A, Schlemmer S. Experimental ground-state combination differences of CH5+. Science (2015) 347:1346–9. doi:10.1126/science.aaa3304
111. Fábri C, Quack M, Császár AG. On the use of nonrigid-molecular symmetry in nuclear motion computations employing a discrete variable representation: A case study of the bending energy levels of CH5+. J Chem Phys (2017) 147:134101. doi:10.1063/1.4990297
112. Fábri C, Császár AG. Vibrational quantum graphs and their application to the quantum dynamics of CH5+. Phys Chem Chem Phys (2018) 20:16913–7. doi:10.1039/c8cp03019g
113. Sarka J, Császár AG, Mátyus E. Rovibrational quantum dynamical computations for deuterated isotopologues of the methane–water dimer. Phys Chem Chem Phys (2017) 19:15335–45. doi:10.1039/c7cp02061a
Keywords: ab initio, ro-vibrational energy levels, quantum label assignment, high accuracy, ScalIT code
Citation: Sarka J and Poirier B (2022) Assigning quantum labels and improving accuracy for the ro-vibrational eigenstates of
Received: 16 July 2022; Accepted: 12 August 2022;
Published: 04 October 2022.
Edited by:
Breno R. L. Galvão, Federal Center for Technological Education of Minas Gerais, BrazilReviewed by:
Oleg Polyansky, University College London, United KingdomJoseph Abebe Obu, University of Calabar, Nigeria
Copyright © 2022 Sarka and Poirier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: János Sarka, SmFub3MuU2Fya2FAdHR1LmVkdQ==