 Anasua Mukhopadhyay1,2
Anasua Mukhopadhyay1,2 Iliya D. Stoev
Iliya D. Stoev Erika Eiser
Erika Eiser
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Phys., 20 June 2022
Sec. Biophysics
Volume 10 - 2022 | https://doi.org/10.3389/fphy.2022.924864
Prevention of protein aggregation and thus stabilization of proteins has large biological and biotechnological implications. Here we introduce Dynamic Light Scattering (DLS) and DLS-based microrheology to show how native bovine serum albumin (nBSA) forms amyloid fibrils in weakly denaturing conditions as function of time, and how stoichiometric conjugation of BSA with polymer-surfactants (PSpBSA) protects the protein form such aggregation. Employing a combination of Thioflavin-T fluorescence, Fourier transform infrared spectroscopy and other methods, we show that nBSA forms filamentous aggregates with amyloid-like structure, while PSpBSA proteins remain fully dispersed with only minor changes in their folding state, even when continuously heated for up to 5 days in denaturation conditions at 65 °C. Time-resolved DLS-based microrheology studies demonstrate that suspensions of the filamentous nBSA aggregates become viscoelastic for concentrations ≥200 μM. Our results indicate that after 6 days in aggregation conditions, the elastic modulus G′(ω) of nBSA solutions went from zero initially to values of up to 3.6 Pa, indicating that the filaments become long enough to form an entangled, viscoelastic network. Interestingly, heating 200 μM native BSA solutions at 65 °C for 2 days in Eppendorf tubes resulted in self-standing films rather than dispersed filaments. These films exhibited strong ThT-fluorescence intensities and a predominant β-sheet secondary structure in FTIR studies, suggesting that the self-standing microstructure of the film resulted from hierarchical self-assembly of the amyloid fibrils.
Misfolding of proteins and their aggregation into amyloid fibrils is known to be the major cause of various neurodegenerative diseases [1–5]. Such amyloid fibrils typically form due to the hydrophobic interactions between the β-sheets of individual proteins and are generally insoluble [6, 7]. The tendency to form amyloid fibrils was shown to depend on the precise amino acid sequence of proteins, even for proteins that have otherwise an identical native structure [8–10]. The conditions triggering the aggregation process are still not fully understood. Hence, in the past decades much research focused on preventing proteins from self-assembling into aggregates as they are responsible for a number of neurodegenerative diseases. Moreover, finding causes and preventing aggregation of proteins has promoted the engineering of stable biomaterials in pharmaceutical, food and other industries [11–14]. Therapeutic proteins are also of importance in the biopharmaceutical industry, but a major challenge is to avoid formation of aggregates and amyloid-like structures during all stages of the manufacturing process, which can render the protein-based drugs ineffective [15–18]. Side-chain chemical modifications, protein sequence modifications, and covalent attachment of polymeric macromolecules, such as the biocompatible poly(ethylene glycol) (PEG), were some of the approaches to improve protein stability [6, 19–21]. Binding proteins to synthetic molecules non-covalently is another approach to stabilise proteins. The most common additives currently are osmolytes, such as arginine and polyols, which are added in large amounts exceeding the protein-to-additive ratio [22–25]. Addition of polymers such as poly (N-isopropylacrylamide) or poly (propylene oxide), can also stabilise enzymes [26, 27]. It was also reported that poly (4-styrene sulfonate) and poly (vinyl sulfate) can inhibit aggregation of denatured Cytochrome C through Coulomb interactions [28]. The factors involved in the prevention of protein aggregation through polymers are mainly achieved through minimising hydrophobic interactions, strengthening hydrogen bonds, steric interactions and osmotic effects arising from the release of counterions [29–35]. Polyelectrolytes can either enhance or decrease the stability of proteins via Coulomb interactions [36, 37]. Bioconjugation of surface-modified proteins with PEG-based polymer-surfactants, leading to water-less protein liquids that freeze below
Here, we report an experimental study of the aggregation behaviour of native BSA (nBSA) near its denaturation temperature and in the presence of added salt, a condition at which nBSA is known to form fibrils (e.g. for various pH and high temperatures [42–46]). In particular, we present fluorescence spectroscopy, dynamic light scattering (DLS) and FTIR spectroscopy experiments, measured as function of time the nBSA suspensions were exposed to the denaturation conditions, showing the structural changes in nBSA. In parallel, we performed similar experiments on suspensions of bioconjugates of polymer-surfactant modified BSA, named PSpBSA, demonstrating their stability against denaturation and thus fibrillation. We also show that DLS-based microrheology can give new insights into the rheological response of nBSA fibrils in solution. Finally, we report that under certain preparation conditions, we observe film rather than fibril formation in nBSA solutions, while PSpBSA suspensions again remain largely unaffected.
Bovine serum albumin (lyophilized powder, ≥ 96%, A2153), Thioflavin-T (ThT), sodium phosphate monobasic, sodium phosphate dibasic, sodium chloride and glycolic acid ethoxylate lauryl ether were purchased from Sigma-Aldrich. Milli-Q water with a resistivity of about 18.2 MΩ ⋅cm at 25 °C was used for the preparation of all solutions and buffers used in this study. The PBS (saline phosphate buffer, 10 mM, pH 7.0) was freshly prepared before all aggregation experiments and the pH of the solution was maintained by adding 1 M HCl or 1 M NaOH, as deemed necessary. The pH of the solutions was prepared with a precision of ±0.01.
Protein-polymer-surfactant bioconjugates of BSA were prepared in an aqueous solution, involving a three-step process, following the experimental procedure we reported earlier [39, 41]. In short, nBSA carries 100 carboxylic groups stemming from Asp and Glu residues; we covalently linked N, N′-dimethyl-1,3-propanediamine (DMAPA; 2 M; pH 6.5) to these surface-accessible, negatively charged, acidic residues using a carbodiimide (EDC) mediated activation method. The resulting pBSA monomers have a positive Zeta potential due to the 168 positive charges provided by the Lys and Arg residues. These were neutralized by electrostatic coupling of the anionic polymer-surfactant (PS) glycolic acid ethoxylate lauryl ether. Such prepared PSpBSA molecules are sterically stabilized and have a nearly neutral Zeta potential. More details are provided in the Supplementary Material.
nBSA was dissolved in 10 mM phosphate buffer (PB) of pH 7.0 to prepare a 1.2 mM stock solution and stored at 4 °C. Similarly, a 0.8 mM PSpBSA stock solution was prepared, but in Milli-Q water and stored at 4 °C. Protein concentrations were determined by measuring the absorbance of tryptophan at 280 nm with a Thermo Scientific NanoDrop UV-visible spectrophotometer. The molar extinction coefficient of BSA at 280 nm is 43,824 M−1cm−1 [47]. For the aggregation experiments, the stock protein solutions were diluted using a 50 mM PBS buffer (pH 7.0; 50 mM NaCl), containing a final protein concentration of 100, 150 or 200 μM. The protein samples were placed in glass vials and then heated in an oil bath at 65 °C for 2 h, and then quenched on ice to store the samples.
The aggregation of nBSA in the amyloid structures was measured by monitoring the Thioflavin-T (ThT) fluorescence as a function of the time the protein solutions were kept at 65 °C. To this end, we extracted small aliquots (50 μL) from the heated 100 μM protein samples in 10 mM PB of pH 7.0 with increasing incubation times. The extracted aliquots were cooled on ice to stop the denaturation process, and then diluted 10-fold with 10 mM, pH 7.4 PB, containing ThT, such that the final ThT concentration was 18 μM ThT, following a standard procedure reported in literature [45]. In the ThT-fluorescence intensity measurements, we used the standard excitation wavelength λex = 450 nm used for detecting amyloid fibril formation, while the emission was monitored in a wavelength range of 465–650 nm, using a Cary Eclipse fluorescence spectrophotometer. The slit width was 5 nm in both excitation and emission measurements, and all fluorescence measurements were performed at 25 °C. All measurements were repeated three times to get the standard error of the measurement.
FTIR analysis of the samples was performed using a Thermo Fisher Scientific Nicolet iS10 FTIR spectrometer. A droplet of 10 μL of protein solution was deposited on a transparent KBr pellet and immediately dried using a mild nitrogen flow. The pellet was then placed in a transmission holder and the IR spectra were acquired in a range of 1,500–1700 cm−1. The background measurement was performed following a similar procedure using pure buffer solution. The dried films of nBSA fibrils were sandwiched between two thin KBr pellets and then placed in the transmission holder for spectra acquisition. For each measurement, 32 scans were collected with a resolution of 2 cm−1 in transmission mode and the corresponding buffer spectrum was subtracted for each measurement. The amide I region (1,600–1700 cm−1) of the FTIR data was deconvoluted with the help of a Fourier self-deconvolution (FSD) method (see Supplementary Material methods for details). The area under each deconvoluted peak was used to estimate the secondary structure in terms of α-helix and β-sheet fractions of the protein.
A Malvern Zetasizer ZSP (633 nm HeNe laser) was used to perform both measurements of the size of the proteins and their aggregates as well as the change in the viscoelastic behavior of the protein samples as function of time. For particle sizing, the Zetasizer was operated in backscattering mode at a scattering angle of 173° to ensure detection of single-scattering events only [48]; measurements were performed on the fresh and heat-set solutions of nBSA and PSpBSA. In DLS-based microrheology, we added 230 nm large, PEG-coated spherical polystyrene-particles (Cambridge Bespoke Colloids, United Kingdom) to the protein solutions, which served as tracers. The volume fraction of probe-particles used was 0.03% in all measurements, ensuring that the dominant scattering (ca. 95%) was due to the colloids and not due to the proteins or their aggregates. In all DLS experiments, we used Malvern ZEN0040 disposable cuvettes (40 μL) and all measurements were performed at 25 °C. The measured intensity-autocorrelation functions g2(q, t) were converted into intermediate scattering functions g1(q, t) using the Siegert relation, g2(q, t) = B(1 + A|g1(q, t)|2), where B is the experimentally determined baseline and A ≈ 1 is a detector-dependent constant. For spherical particles of radius R, the intermediate scattering function writes as g1(q, t) = exp(−q2Dt), where D is the translational particle-diffusion coefficient that is defined through the Stokes-Einstein relation D = kBT(6πηR)−1 with T being the temperature, η the viscosity of the solvent, R is the radius of the scattering particles and kB the Boltzmann constant. The diffusion coefficient of the probe-particles in 3 dimensions is also related to the mean-squared displacement (MSD) through the Einstein relation MSD = ⟨Δr(t)2⟩ = 6Dt. Hence, we extracted the MSDs from our correlation functions g1(q, t) = exp(−q2⟨Δr(t)2⟩/6) using an in-house developed MATLAB routine [48]. The MSDs were then Fourier transformed to obtain the elastic (G′(ω)) and loss (G′′(ω)) moduli of the protein solutions using other, in-house developed MATLAB routines [48]. This transform is based on the Generalized Stokes-Einstein Relation
The protein solution was drop-cast on a carbon-coated copper grid and then dried in air at room temperature for 3 h. Subsequently the grid was put under vacuum for 4 h. The grid was then stained with 0.2% (w/v) aqueous uranyl acetate solution and dried under vacuum for overnight. The TEM analysis was performed using an electron microscope (FEI Tecnai G2) at an operating voltage of 300 kV.
In a typical experiment, 200 μL of a 100 μM nBSA solution in PBS (10 mM, pH 7.0) containing 50 mM NaCl solution was poured into a 600 μL plastic Eppendorf tube (Cliklok microcentrifuge tubes; yellow, T330-6Y) and sealed to minimize evaporation. The sealed tubes were then incubated at 65 °C without agitation in a silicon oil bath for a maximum of 2 days. In the ThT measurements, we extracted a probe after 2 h and quenched it on ice, while FTIR spectroscopy was performed on the actual film that formed after 2 days.
We used the freely available Visual Molecular Dynamics (VMD) software to illustrate the secondary structure of native BSA, using the protein sequence of nBSA that is available on the Protein Databank (pdb: id 3v03).
Here, we studied the aggregation kinetics of three different nBSA and PSpBSA concentrations (100, 150 and 200 μM), which were incubated at 65 °C in glass cuvettes placed in an oil bath. To allow for unperturbed protein aggregation, we avoided agitation or stirring of the samples. All solutions were prepared in PBS buffer with a final pH 7 and 50 mM NaCl in glass vials. First, we investigated the aggregation behavior of different nBSA and PSpBSA concentrations by monitoring the changes in the Thioflavin-T (ThT) fluorescence intensity. ThT is a well-studied fluorescent amyloid marker that exhibits an increase in fluorescence intensity with an emission peak at 485 nm in the presence of β-sheets [49]. A VMD representation of the α-helix-dominated secondary structure of a nBSA molecule with its β-strands, typically displayed in aqueous solutions, is shown in Figure 1A. After incubating 100 μM nBSA samples at 65°C in an oil bath for 2 h, a maximum in the broad absorption peak of their ThT-fluorescence spectra was reached. The spectra were recorded at 25 °C and are shown in Figure 1C: the black dashed line is the averaged spectrum obtained for pure PBS containing ThT, while the solid black line was measured in the presence of freshly prepared nBSA (100 μM). The blue curve shows the averaged spectrum of the sample measured after exposing it for 2 h at the aggregation conditions and subsequent cooling on ice. More detailed, time-resolved ThT-fluorescence measurements for 100, 150 and 200 μM nBSA solutions with the same protein-to-ThT mass ratio are shown in the supporting information (Supplementary Figure S1). These measurements demonstrate that a maximum in the ThT-fluorescence peak was reached after around 90 min for the heated 100 μM nBSA solutions and after 2 h for the higher concentrations. Transmission electron microscopy (TEM) images taken of a 200 μM nBSA solution after exposing it for 2 h to our denaturation conditions revealed filamentous aggregation (Figure 1D). The representative TEM images in Supplementary Figures S2A,B reveal that 200 μM nBSA solutions form fibrillar aggregates of overall length of 54 ± 7 nm (averaged over ∼100 fibrils in 3 different micrographs; Figure 1D and see Supplementary Figure S2) upon heating at 65 °C for 2 h under our aggregation condition. We obtained similar values from DLS measurements, as shown later.
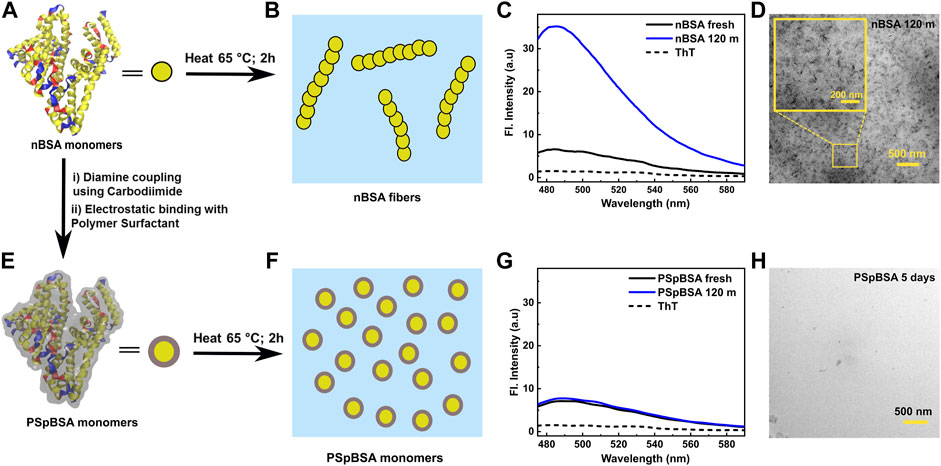
FIGURE 1. (A) Graphical representation of a nBSA monomer (PDB 3V03) using VMD software: α-helixes are yellow, β-sheets are blue and the red stands for random coil; for simplicity the protein is also sketched as yellow disk. (B) Schematic illustration of how nBSA monomers aggregate to form filaments. (C) ThT-emission spectra taken from a fresh 100 μM nBSA solution (pH 7.0, 50 mM NaCl, 25 °C) and the same solution heated for 2 h at 65 °C. For reference, the dashed line was measured for pure PB containing the same final ThT concentration as the protein samples. (D) TEM image of nBSA fibrils formed after heating at 65 °C for 2 h. Inset shows a magnified version of the image. (E) Graphical representation of polymer-surfactant conjugated PSpBSA (more details are given in Supplementary Figure S19 [41]. The grey layer enveloping the protein represents the polymer-surfactant (PS) layer, coating each monomer. (F) Schematic representation of PSpBSA monomers after subjecting them to aggregation conditions. (G) ThT-fluorescence spectra of the fresh and heat-set PSpBSA solutions. As in (C), the dashed line represents the fluorescence activity of ThT in pure PBS. Again, the same protein and ThT concentrations were used (see Experimental Section). (H) TEM images of PSpBSA solution showing aggregate free sample after heating at 65 °C for 5 days.
Our findings agree with results by Holm et al. [44], who reported that nBSA denatures and forms amyloid fibrils at around 65 °C in pH 7.4 Figure 1B. Conducting time-resolved ThT-fluorescence spectroscopy between 60 and 75 °C, they showed that the aggregation process was too fast to follow at higher temperatures, while at 60 °C no significant amyloid formation was observed: Hence, we chose 65 °C as denaturation temperature to follow the aggregation process as function of time both with ThT-fluorescence and in-situ DLS. We used for all three nBSA concentrations the same 1:2 protein-to-ThT molecule ratio, which has been reported to be the optimal ratio to monitor amyloid formation.
Similarly, we studied PSpBSA solutions in aggregation conditions: The absence of any significant increase in our ThT-fluorescence spectra (Figure 1G; Supplementary Figures S3, S4) confirms that the bioconjugation of the protein with a polymer-surfactant (PSpBSA; schematic VMD representation in Figure 1E) provides excellent heat-stability with little structural changes, which was further confirmed in deconvoluted FTIR and Circular Dichroism (CD) spectroscopy (Figure 3; Supplementary Figure S5). These findings also suggests that our polymer-surfactant remains firmly attached to the positively charged BSA monomers and is not released into solution to form micelles, as one could expect for these surfactants, which are also known as Brij 35 [50]. Figure 1F shows a cartoon of the quasi-spherical PSpBSA monomers, while Figures 1G,H show ThT-fluorescence spectra and a TEM image of a dried PSpBSA sample after subjecting their solutions to the same heat treatment and solvent conditions: both measurements show no indication of amyloid filament formation.
The growth of the filamentous aggregates formed by nBSA after heating the samples for up to 120 h was studied with DLS by measuring the intensity-correlation functions g2(q, t) after quenching the samples to room temperature. As the translational diffusion coefficient
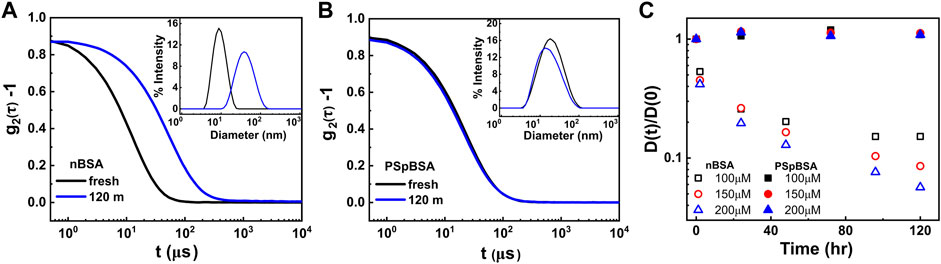
FIGURE 2. (A,B) Intensity-autocorrelation functions of fresh and heat-set 100 μM nBSA and 100 μM PSpBSA solutions, measured using DLS. The insets represent the corresponding intensity distributions as functions of measured particle sizes. (C) Time-evolution of the normalized diffusion coefficients of nBSA (open symbols) and PSpBSA (filled symbols) solutions measured at 65 °C in PBS at pH 7.0, containing 50 mM added NaCl. Here black, red and blue symbols represent the 100, 150 and 200 μM nBSA and PSpBSA concentrations, respectively.
For polarized DLS, the measured g2(q, t) curves of very dilute rod-solutions can still be fitted using the Stokes-Einstein relation for D(t), however, the measured hydrodynamic radius must be replaced by the radius of gyration RG of the rods, reflecting the translational motion of the rods’ centre of mass. Doi and Edwards [51] give an expression for the new translational diffusion coefficient, which depends on the length L and thickness d of the rods as DG = kBT ln(L/d)/(3πηL); here d is simply the diameter of the nBSA monomer.
In Figure 2C, we show the measured translational diffusion coefficients D(t) = DG(theat), normalized by the respective D0, as function of heating times at 65 °C. The individual scattering spectra for 100, 150 and 200 µM nBSA are given in the Supplementary Figures S7–S9, and those for 100 and 200 µM PSpBSA are given in Supplementary Figures S10, S11. For the heat-set nBSA samples, the ratio D(t)/D0 = DG(t)/D0 = ln(L/d)/(L/d) clearly decreases over the heating times we measured, reflecting the slow growth of the protein fibrils. We also observe a faster growth rate for larger nBSA concentrations. All concentrations show a slowing down in growth with time and we observe two quasi-linear regions. This is particularly visible in the 120 h heating study of the 100 µM nBSA sample, where the ratio starts to plateau after about 100 h of heating. The plateau occurs at D(t)/D0 = 0.15, which suggests an average length of the fibrils of about 180 nm, assuming a fibril-diameter of about 3–4 nm corresponding to that of fresh nBSA monomers. We hypothesize that the slow growth of the fibrils follows a special case of the reaction-limited aggregation mechanisms [52, 53] observed in many particulate systems. But unlike systems with dendritic growth, the protein fibrils can only grow linearly, as was observed in TEM images of heat-set nBSA samples [44]. Oozawa and Kasai [54] introduced a theoretical model explaining linear growth in proteins and di Michele et al. [55] reported simulation studies on self-catalytic, linear growth in proteins. Hence, we argue that within the first 2 h of heating all nBSA monomers are converted into dimers or trimers (here called “seed” clusters), showing increased ThT-fluorescence, but the subsequent growth into long filaments occurs by end-to-end fusion of these short dimers to increasingly larger filaments, which explains the plateauing in the ThT-fluorescence peak after about 120 min shown in Supplementary Figures S1, S12. This end-to-end growth of short linear, semi-flexible rods (or oligomers) will be reaction-limited, and it seems to be dependent on a temperature-dependent activation barrier. This hypothesis is supported by the scattering intensity-to-size plots derived from our DLS measurements for the two larger nBSA concentrations that show a remaining fraction of short dimers or trimers coexisting with the much larger fraction of long filaments after 120 h of heating (Supplementary Figure S9).
In order to substantiate our hypothesis, we performed control measurements, in which we stopped the heat-induced conformational change in a 100 µM nBSA solution before the ThT-fluorescence peak could reach its plateau value, hence only a fraction can overcome the activation barrier for seed clusters to form and rearrange into amyloid structures. Indeed, the ThT-fluorescence spectra in Supplementary Figure S12 clearly show that stopping the heating after 20 min halts further growth of the fluorescence peak. In addition, DLS measurements of the 20-minute heated samples show a bimodal distribution with more nBSA monomers coexisting first with seed clusters and then with slightly larger rods. DLS measurements of a 200 µM nBSA solution that was heated only for 2 h and then kept at room temperature for days, showed no further growth of the initially formed small clusters (Supplementary Figure S12D), which is in contrast to the fibrillar growth observed for continuous heating (Figure 2C).
To understand the secondary structure of the protein, we performed FTIR studies at different time points during the aggregation. Figure 3 shows the Fourier self-deconvoluted FTIR spectra (see Supplementary Material methods for details) taken in the amide I region (1,600–1700 cm−1) of the fresh nBSA and PSpBSA solutions and after subjecting them for 24 h to our aggregation conditions.
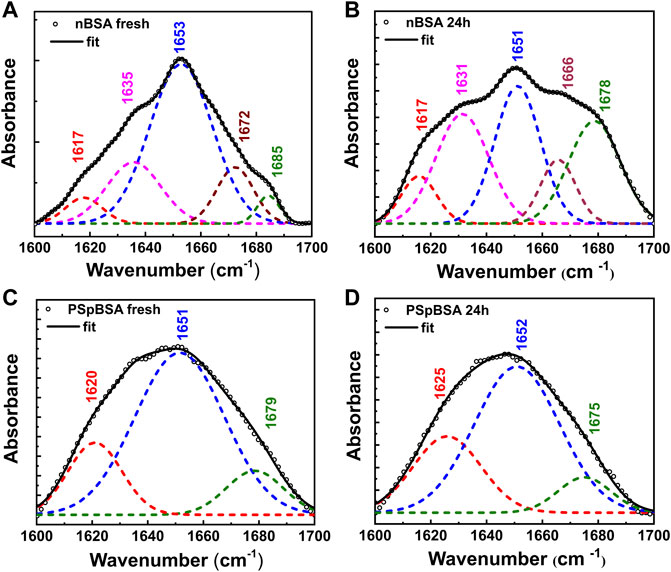
FIGURE 3. Secondary structural changes of nBSA and PSpBSA monitored by FTIR spectra, in the region corresponding to the amide I band (1,600–1700 cm−1) of fresh (A) nBSA and (C) PSpBSA solutions, and (B,D) after heating them at 65 °C for 24 h. The open symbols represent the experimental values, whereas the black solid line shows fits to the experimental data. The dashed lines show the different components, obtained after Fourier self-deconvolution (Table 1).
While ThT-binding fluorescence data provide indirect information of the structure of heat-treated protein solutions, FTIR experiments (Figure 3A) give information on the secondary structure of proteins in terms of α-helix and β-sheet content. Fresh nBSA has a predominant peak at 1,653 cm−1 in the amide I region, which is characteristic of a protein, rich in α-helix. Fourier self-deconvolution of the spectra gives us 66% α-helical content for our fresh nBSA samples (Table 1), which reduces to 39% after 24 h (see Supplementary Material for details). This reduction is accompanied by an increase of the β-sheet content in the BSA fibrils (Figure 3B; Table 1).
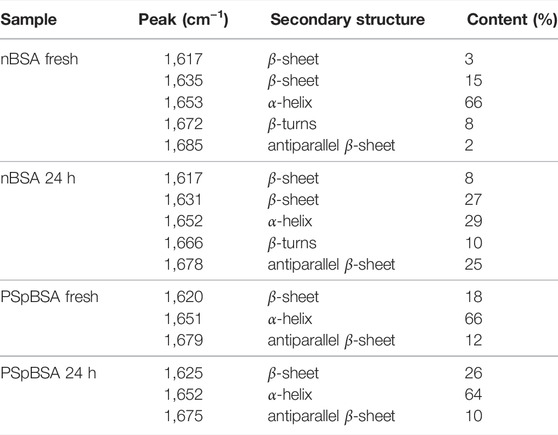
TABLE 1. Secondary structural content analysis from the deconvoluted FT-IR spectra (3) of protein solutions.
We also performed CD studies for nBSA, fresh and sample subjected for 24 h denaturation conditions (Supplementary Figure S5). The secondary structure obtained from fitting and analyzing CD data (Supplementary Table S3) agreed with values from the deconvolution of ATR-FTIR data, which are shown in Table 1. These FTIR studies were similar to those performed for understanding the sol-to-gel transition in regenerated silk fibroin (RSF) protein solutions [56, 57]. Such a transition was associated with the conversion of random coil to β-sheet upon increasing concentration, mechanically vortexing or heating the RSF solution. The formation of amyloid-like structures during gelation was not observed in this case. However, Dubey and coworkers reported [58] the aggregation of RSF, resulting in the formation of amyloid fibrils, having a diameter of 3.2 nm under certain conditions.
Comparing FTIR spectra of fresh nBSA and PSpBSA solutions shows that both are very similar, with a main peak at around 1,652 ± 2 cm−1, indicating a secondary structure, rich in α-helix, and only a small discrepancy between the parallel and antiparallel β-sheet contributions. Upon subjecting PSpBSA to aggregation conditions, we observe very small changes in the FTIR spectrum confirming that heating at around the native protein’s denaturation temperature does not change the bioconjugated counterpart (Figure 3D and Table 1). This is even true when heating the PSpBSA samples at 65 °C for many days or even heating them up to 80 °C for several hours (Supplementary Figures S5, S14; Supplementary Tables S1, S2). Hence, our experimental results infer that stoichiometric conjugation with PEG-based polymer-surfactants renders BSA remarkably inert and prevents it from forming fibrillar aggregates. This opens up an important pathway to develop drugs, foods and household products with much-improved shelf-life and efficacy.
We also studied the viscoelastic properties of the fresh nBSA solutions and under aggregation conditions using DLS-based microrheology. This was achieved by measuring the time-dependent correlation function of the light backscattered from 230 nm large, spherical probe-particles dispersed in the protein sample. From the measured and normalized intensity autocorrelation functions, we first extracted the intermediate correlation function g1(q, t) = exp(−q2Dt) = exp(−q2⟨Δr(t)2⟩/6), which relates the particles’ translational diffusion coefficients to the mean-squared displacements that describe their thermal motion in the viscoelastic medium [48]. The fresh solutions will have viscosities similar to those of the continuous background solvent. The Einstein relation for the viscosity of a dilute suspension of spherical colloids is η = ηw(1 + 2.5ϕ + ⋯), where ηw is the viscosity of water and ϕ is the volume fraction of the colloids. We can assume that nBSA can be approximated as a spherical colloid with a diameter of 3–4 nm, which means ϕ is roughly 2%. This leads to a change in the average viscosity of less than a percent in the present case. Indeed, the measured g1(q, t) curves of all three fresh nBSA concentrations show an almost identical MSDs confirming that the nBSA monomers do not alter the viscosity of pure water even after 7 days at room temperature (Figure 4B).
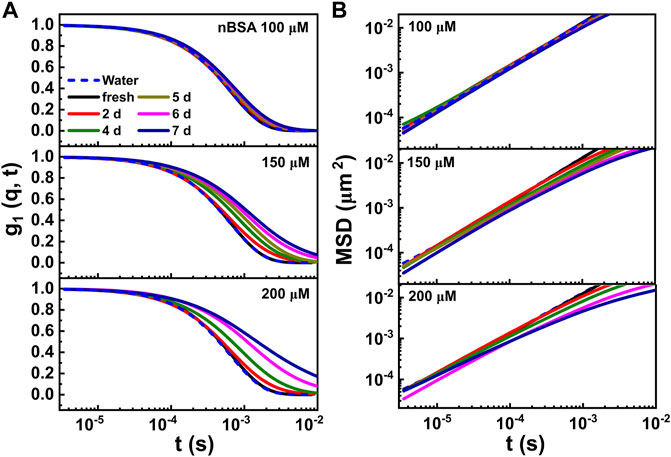
FIGURE 4. (A) Time-dependent intensity autocorrelation curves, measured for the 100, 150 and 200 μM nBSA solutions under the same aggregation conditions mentioned before, containing 0.03 vol/vol% 230 nm PEG-coated polystyrene latex tracer particles. The curves were measured starting from fresh solutions (presented as black curves) up to samples heated at 65 °C for 7 days (navy blue curves). (B) Corresponding MSD data. For comparison, we present calculated MSD curves for the same 230 nm colloids in pure water (blue dashed lines).
In Figure 4A, we show the g1(q, t) curves for a freshly prepared and heat-set nBSA sample measured as function of heating times for up to 7 days for the three different concentrations of nBSA used; for reference, the corresponding MSDs obtained for tracer particles dispersed in pure water are shown in Figure 4B and the corresponding g1(q, t) curves in Figure 4A as dashed blue lines.
When the different nBSA concentrations were exposed to our heating protocol we did observe a shift in the g1(q, t) curves towards lager relaxation times τ = ⟨Δr(t)2⟩/D with progressing heating time. The strongest change was observed for the 200 μM nBSA solution, which developed a weak elasticity plateau in G′(ω) that is shown in Figure 5. Indeed, when proteins aggregate into sufficiently long fibrils, their rotational and translational motion will lead to them intersecting each other, thus giving rise to excluded-volume interactions. These interactions cause a measurable elasticity in the initially Newtonian dispersion of spherical particles. Using the Generalized Stokes–Einstein Relation [59, 60] we can extract the elastic (G′(ω)) and viscous modulus (G′′(ω)) from the MSDs of the weakly elastic, heat-treated nBSA solutions for frequencies up to the MHz range, which conventional rheology cannot provide.
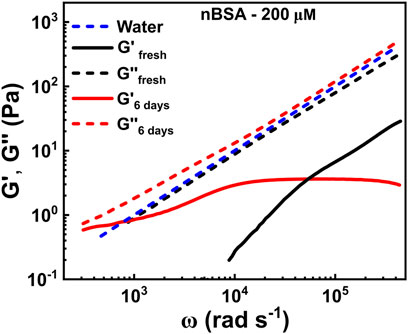
FIGURE 5. Time evolution of the elastic (G′(ω)) and viscous (G′′(ω)) moduli as functions of frequency, extracted from the MSD curves in Figure 4B of 200 μM nBSA in solution under aggregation conditions.
In Figure 5, the dashed blue line represents the loss modulus of water, which is a Newtonian liquid that scales linearly with frequency: G′′(ω) = ηω. The fresh 200 μM nBSA solution (black dashed curve) shows a similar viscosity. As Newtonian fluids have no elasticity, we do not show G′(ω) for water, but the fresh nBSA solution does show some elasticity at very high frequencies, which significantly increases with progressing heating time, while G″(ω) retains the linear frequency behaviour of the fresh sample. After 6 days in aggregation conditions, G′(ω) reaches a plateau at about 3.58 Pa in the intermediate frequency range (Figure 5), corresponding to a possible entanglement length ξ ∼ 105 nm between the rods, assuming the scaling behaviour of the bulk modulus to be Gbulk = kBT/ξ3 [51]. Similarly, we measured plateau values of ∼1.2 Pa and ∼3.0 Pa (Supplementary Figure S15) for 100 and 200 μM concentrations of nBSA fibrils, showing the right trend that lower concentrations will deliver shorter fibrils for the same heating time. Note that these values are only approximate and have an error of 20–30%.
While observing standard fibril formation when heat-setting nBSA solutions, we discovered a completely different aggregation behaviour when incubating the nBSA solutions (here using 200 μM) under aggregation conditions for 2 days in plastic Eppendorf tubes (Figure 6): A stable film of thickness ∼1.6 μm (Figure 6C; Supplementary Figure S16) formed. To understand the nature of the film, we performed ThT-fluorescence experiments in the early stage of film formation. In the first few hours of heating the sample at 65 °C under aggregation conditions, no visible aggregates were detected in the sample. ThT-fluorescence spectra showed a progressively growing maximum in the fluorescence peak at 485 nm (Figure 6D): We observe a fast, initial growth that slows down considerably after about 40 min, similar to the ThT fluorescent behaviour measured for 200 μM nBSA solutions heated in glass cuvettes, demonstrating the formation of amyloid-type filaments. Also 100 and 150 μM nBSA solutions under the same incubation conditions resulted in protein films with similar ThT-fluorescence findings (Supplementary Figures S17, S18). Incubating bioconjugated PSpBSA under aggregation conditions in an Eppendorf tube, on the other hand, showed no change even after 5 days.
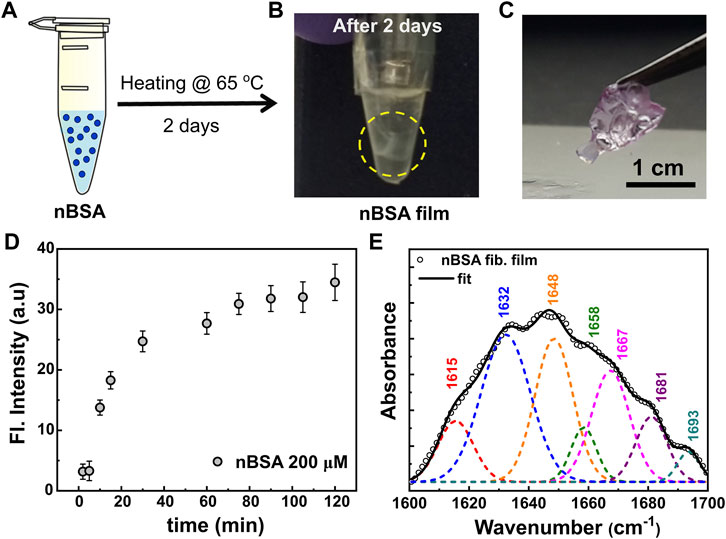
FIGURE 6. (A,B) Scheme of nBSA film formation from fibrils. (A) shows the nBSA monomers in an Eppendorf tube, and the formed film after 2 days of incubation is shown in (B). (C) Pictorial representation of the self-assembled nBSA fibril film. The film is stained with Nile red dye for clarity. (D) Thioflavin-T (ThT) fluorescence intensity curve for the initial 120 min of nBSA film formation. (E) Fourier self-deconvoluted FTIR spectrum of the amide I region of the nBSA film, indicating its predominant β-sheet structure.
To confirm that the fibril formation in the films is also amyloid-like, we also performed FTIR studies, shown in Figure 6E. Interestingly, they do show resemblance to the results obtained for our heat-set nBSA solutions. In passing, we would like to note that thin film formation through selective non-amyloid type aggregation of polymer-surfactant modified proteins and enzymes has been reported in literature by Sharma et al. [39, 61, 62]. The authors mention that the mechanism of film formation follows alkyl-alkyl based bridging between the polymer-surfactant chains, specifically at low grafting density on the cationized protein surface [63]. This leads to the formation of ∼100–200 nm spheroidal particles in aqueous solutions. Further, in a desiccated environment and in the presence of glutaraldehyde vapours, these spheroidal particles can self-assemble to form crosslinked, micron-sized hierarchical structures, with the near native protein structure still intact. In the current study, we believe that the mechanism of film formation definitely follows a pathway through protein unfolding and fibrillar aggregation. Literature reports also show that heat induced BSA fibrillation can form highly elastic free-standing hydrogel microstructures at low molecular weight fractions through elongated protein fibrils [64]. In this study the authors studied BSA fibrillation in acidic pH, high temperatures (75–80 °C), and much higher protein concentration (1–9 wt%). Moreover, the authors mention that the BSA forms a self-supporting hydrogel at ≥ 3 wt% BSA concentration. In the current study, our experimental conditions are different from the above-mentioned literature; the aggregation behaviour of BSA was investigated at pH 7.0 by incubating the samples at 65 °C. Further, our working concentrations are in the regime 0.66–1.2 wt% BSA (100–200 μM). The stable films that can be seen floating in the Eppendorf-tube solution (Figure 6B), are in sharp contrast to the hydrogel-like network reported by Amdursky et al. [64]. The observed self-assembled fibril-based film formation is induced by the hydrophobic plastic surface and does not occur in glass vials. Moreover, previous reports on Aβ1–40 protein solutions state that amyloid fibrils have a tendency to get adsorbed onto the hydrophobic surfaces of Eppendorf tubes [65]. We believe that similarly, nBSA fibrils in our experiment were adsorbed to the surface of the hydrophobic plastic tube and then self-assembled to form protein films. Remarkably, placing the films in fresh buffer solution and sonicating them did not re-disperse the filaments. Hence, the aggregation process in the Eppendorf tubes must be accompanied by some crosslinking mechanism that still needs to be studied. Interestingly, comparing the FTIR experiments performed on the protein films with the heat-set nBSA fibrils formed in glass cuvettes (Figure 3B) showed that the films contain a 40% β-sheet secondary structural content and an additional peak at a wave number of 1,693 cm−1. This confirms the presence of an additional process occurring simultaneously (Figure 6E and Supplementary Table S4), which may be similar to the aggregation processes observed in regenerated silk fibroin protein solutions [56, 57].
To this end, we note that the structure in fibrillar aggregation of protein solutions does not necessarily lead to amyloid structures. Eiser et al. [66] showed that exposing ovalbumin solutions to pH 12 conditions also leads to an irreversible fibrillar gel formation. In these systems CD measurements revealed that the secondary structure has not changed much demonstrating that β-sheets were also retained, while ThT-spectra indicated that there was no evidence for the existence of amyloid structures. To conclude, the apparent seeding of these robust films formed by amyloid-like fibrils may contribute to the question of what (surface) forces may be key in understanding amyloidosis and other protein aggregation diseases in living organisms.
To summarize, here we demonstrate the exceptional thermal stability of polymer-surfactant conjugated BSA under aggregation conditions. Our combined ThT-fluorescence, DLS and FTIR results give us a clear description of bioconjugated BSA under fibrillation conditions and its comparison with native BSA under similar aggregation conditions. Stability and protection against aggregation for the bioconjugates of BSA (PSpBSA) appear to correlate with the role of PEG-based polymer-surfactant surrounding the native protein. The polymer-surfactant layer surrounding the protein presumably hinders the interprotein contacts and thus prevents the aggregation of protein. DLS measurements also shed light onto the fibril formation of nBSA under varying heating conditions. Further, our microrheological measurements show that the native BSA fibrils form a network of semi-flexible polymers after prolonged aggregation times. But we also show that the nature of aggregation can depend on the environment, such as the container walls. Our study has a huge potential in finding alternative formulations compared to the commonly used osmolytes and detergents that rely on stable protein solutions.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
AM performed the polymer-surfactant conjugation of BSA, and spectroscopic analysis of all data presented here, as well as TEM measurements, supported by KPS; AM and IS did all DLS and DLS-based microscopy measurements, where latter performed all their analysis using Matlab routines developed by the Eiser group. KPS and EE designed the experiments. All authors were equally involved in writing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AM thanks the Commonwealth Scholarship Commission (United Kingdom government), the Department of Chemistry, IITB and KPS to make it possible to spend a research year in the Eiser group at the Cavendish Laboratory. AM acknowledges Commonwealth Scholarship Commission (United Kingdom government) and the Department of Chemistry, IITB for financial support. IS and DK thank EPSRC for financial support. KPS thanks IRCC and SERB-DST India for funding through Grant No. CRG/2020/003706. EE acknowledges the ETN-COLLDENSE (H2020-MCSA-ITN-2014, Grant No. 642774) and support of the Research Council of Norway through its Centres of Excellence funding scheme, project number 262644.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2022.924864/full#supplementary-material
1. Jahn TR, Radford SE. Folding versus Aggregation: Polypeptide Conformations on Competing Pathways. Arch Biochem Biophys (2008) 469:100–17. doi:10.1016/j.abb.2007.05.015
2. Luheshi LM, Crowther DC, Dobson CM. Protein Misfolding and Disease: from the Test Tube to the Organism. Curr Opin Chem Biol (2008) 12:25–31. doi:10.1016/j.cbpa.2008.02.011
3. Miranker AD. Unzipping the Mysteries of Amyloid Fiber Formation. Proc Natl Acad Sci U.S.A (2004) 101:4335–6. doi:10.1073/pnas.0401163101
5. Wetzel R. Kinetics and Thermodynamics of Amyloid Fibril Assembly. Acc Chem Res (2006) 39:671–9. doi:10.1021/ar050069h
6. Frokjaer S, Otzen DE. Protein Drug Stability: a Formulation challenge. Nat Rev Drug Discov (2005) 4:298–306. doi:10.1038/nrd1695
8. Williams AD, Portelius E, Kheterpal I, Guo J-t., Cook KD, Xu Y, et al. Mapping Aβ Amyloid Fibril Secondary Structure Using Scanning Proline Mutagenesis. J Mol Biol (2004) 335:833–42. doi:10.1016/j.jmb.2003.11.008
9. Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the Effects of Mutations on Peptide Andprotein Aggregation Rates. Nature (2003) 424:805–8. doi:10.1038/nature01891
10. Khurana R, Ionescu-Zanetti C, Pope M, Li J, Nielson L, Ramírez-Alvarado M, et al. A General Model for Amyloid Fibril Assembly Based on Morphological Studies Using Atomic Force Microscopy. Biophysical J (2003) 85:1135–44. doi:10.1016/s0006-3495(03)74550-0
11. Chi EY, Krishnan S, Randolph TW, Carpenter JF. Physical Stability of Proteins in Aqueous Solution: Mechanism and Driving Forces in Nonnative Protein Aggregation. Pharm Res (2003) 20:1325–36. doi:10.1023/a:1025771421906
12. Mezzenga R, Fischer P. The Self-Assembly, Aggregation and Phase Transitions of Food Protein Systems in One, Two and Three Dimensions. Rep Prog Phys (2013) 76:046601. doi:10.1088/0034-4885/76/4/046601
13. Hauser CAE, Zhang S. Designer Self-Assembling Peptide Materials for Diverse Applications. Macromol Symp (2010) 295:30–48. Wiley Online Library. doi:10.1002/masy.200900171
14. Chiti F, Dobson CM. Protein Misfolding, Functional Amyloid, and Human Disease. Annu Rev Biochem (2006) 75:333–66. doi:10.1146/annurev.biochem.75.101304.123901
15. Dingermann T. Recombinant Therapeutic Proteins: Production Platforms and Challenges. Biotechnol J (2008) 3:90–7. doi:10.1002/biot.200700214
16. Graumann K, Premstaller A. Manufacturing of Recombinant Therapeutic Proteins in Microbial Systems. Biotechnol J (2006) 1:164–86. doi:10.1002/biot.200500051
17. Matasci M, Hacker DL, Baldi L, Wurm FM. Recombinant Therapeutic Protein Production in Cultivated Mammalian Cells: Current Status and Future Prospects. Drug Discov Today Tech (2008) 5:e37–e42. doi:10.1016/j.ddtec.2008.12.003
18. Mahler H-C, Friess W, Grauschopf U, Kiese S. Protein Aggregation: Pathways, Induction Factors and Analysis. J Pharm Sci (2009) 98:2909–34. doi:10.1002/jps.21566
19. Lee LS, Conover C, Shi C, Whitlow M, Filpula D. Prolonged Circulating Lives of Single-Chain Fv Proteins Conjugated with Polyethylene Glycol: a Comparison of Conjugation Chemistries and Compounds. Bioconjug Chem. (1999) 10:973–81. doi:10.1021/bc990076o
20. Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Stability of Protein Pharmaceuticals: an Update. Pharm Res (2010) 27:544–75. doi:10.1007/s11095-009-0045-6
21. Jones MW, Mantovani G, Ryan SM, Wang X, Brayden DJ, Haddleton DM. Phosphine-mediated One-Pot Thiol-Ene "click" Approach to Polymer-Protein Conjugates. Chem Commun (2009) 5272–4. doi:10.1039/b906865a
22. Rudiuk S, Cohen-Tannoudji L, Huille S, Tribet C. Importance of the Dynamics of Adsorption and of a Transient Interfacial Stress on the Formation of Aggregates of Igg Antibodies. Soft Matter (2012) 8:2651–61. doi:10.1039/c2sm07017k
23. Hudson SD, Sarangapani P, Pathak JA, Migler KB. A Microliter Capillary Rheometer for Characterization of Protein Solutions. J Pharm Sci (2015) 104:678–85. doi:10.1002/jps.24201
24. Martin N, Ma D, Herbet A, Boquet D, Winnik FM, Tribet C. Prevention of Thermally Induced Aggregation of IgG Antibodies by Noncovalent Interaction with Poly(acrylate) Derivatives. Biomacromolecules (2014) 15:2952–62. doi:10.1021/bm5005756
25. Ahrer K, Buchacher A, Iberer G, Jungbauer A. Thermodynamic Stability and Formation of Aggregates of Human Immunoglobulin G Characterised by Differential Scanning Calorimetry and Dynamic Light Scattering. J Biochem Biophysical Methods (2006) 66:73–86. doi:10.1016/j.jbbm.2005.12.003
26. Yoshimoto N, Hashimoto T, Felix MM, Umakoshi H, Kuboi R. Artificial Chaperone-Assisted Refolding of Bovine Carbonic Anhydrase Using Molecular Assemblies of Stimuli-Responsive Polymers. Biomacromolecules (2003) 4:1530–8. doi:10.1021/bm015662a
27. Lu S, Song X, Cao D, Chen Y, Yao K. Preparation of Water-Soluble Chitosan. J Appl Polym Sci (2004) 91:3497–503. doi:10.1002/app.13537
28. Sedlák E, Fedunová D, Veselá V, Sedláková D, Antalík M. Polyanion Hydrophobicity and Protein Basicity Affect Protein Stability in Protein−Polyanion Complexes. Biomacromolecules (2009) 10:2533–8. doi:10.1021/bm900480t
29. Seyrek E, Dubin PL, Tribet C, Gamble EA. Ionic Strength Dependence of Protein-Polyelectrolyte Interactions. Biomacromolecules (2003) 4:273–82. doi:10.1021/bm025664a
30. Ladam G, Gergely C, Senger B, Decher G, Voegel J-C, Schaaf P, et al. Protein Interactions with Polyelectrolyte Multilayers: Interactions between Human Serum Albumin and Polystyrene Sulfonate/polyallylamine Multilayers. Biomacromolecules (2000) 1:674–87. doi:10.1021/bm005572q
31. Gong J, Yao P, Duan H, Jiang M, Gu S, Chunyu L. Structural Transformation of Cytochrome C and Apo Cytochrome C Induced by Sulfonated Polystyrene. Biomacromolecules (2003) 4:1293–300. doi:10.1021/bm034090m
32. Cabaleiro-Lago C, Quinlan-Pluck F, Lynch I, Lindman S, Minogue AM, Thulin E, et al. Inhibition of Amyloid β Protein Fibrillation by Polymeric Nanoparticles. J Am Chem Soc (2008) 130:15437–43. doi:10.1021/ja8041806
33. Stogov SV, Izumrudov VA, Muronetz VI. Structural Changes of a Protein Bound to a Polyelectrolyte Depend on the Hydrophobicity and Polymerization Degree of the Polyelectrolyte. Biochem Mosc (2010) 75:437–42. doi:10.1134/s0006297910040061
34. Sinnwell S, Ritter H. Microwave Assisted Hydroxyalkylamidation of Poly(ethylene-Co-Acrylic Acid) and Formation of Grafted Poly(ε-Caprolactone) Side Chains. J Polym Sci A Polym Chem (2007) 45:3659–67. doi:10.1002/pola.22115
35. Xu Y, Mazzawi M, Chen K, Sun L, Dubin PL. Protein Purification by Polyelectrolyte Coacervation: Influence of Protein Charge Anisotropy on Selectivity. Biomacromolecules (2011) 12:1512–22. doi:10.1021/bm101465y
36. Wittemann A, Ballauff M. Secondary Structure Analysis of Proteins Embedded in Spherical Polyelectrolyte Brushes by Ft-Ir Spectroscopy. Anal Chem (2004) 76:2813–9. doi:10.1021/ac0354692
37. Schwinté P, Ball V, Szalontai B, Haikel Y, Voegel J-C, Schaaf P. Secondary Structure of Proteins Adsorbed onto or Embedded in Polyelectrolyte Multilayers. Biomacromolecules (2002) 3:1135–43. doi:10.1021/bm025547f
38. Perriman AW, Cölfen H, Hughes RW, Barrie CL, Mann S. Solvent-free Protein Liquids and Liquid Crystals. Angew Chem Int Edition (2009) 48:6242–6. doi:10.1002/anie.200903100
39. Sharma KP, Bradley K, Brogan APS, Mann S, Perriman AW, Fermin DJ. Redox Transitions in an Electrolyte-free Myoglobin Fluid. J Am Chem Soc (2013) 135:18311–4. doi:10.1021/ja4104606
40. Brogan AP, Sharma KP, Perriman AW, Mann S. Enzyme Activity in Liquid Lipase Melts as a Step towards Solvent-free Biology at 150 °C. Nat Commun (2014) 5:5058–8. doi:10.1038/ncomms6058
41. Mukhopadhyay A, Das T, Datta A, Sharma KP. Neat Protein-Polymer Surfactant Bioconjugates as Universal Solvents. Biomacromolecules (2018) 19:943–50. doi:10.1021/acs.biomac.7b01729
42. Militello V, Vetri V, Leone M. Conformational Changes Involved in thermal Aggregation Processes of Bovine Serum Albumin. Biophysical Chem (2003) 105:133–41. doi:10.1016/s0301-4622(03)00153-4
43. Militello V, Casarino C, Emanuele A, Giostra A, Pullara F, Leone M. Aggregation Kinetics of Bovine Serum Albumin Studied by Ftir Spectroscopy and Light Scattering. Biophysical Chem (2004) 107:175–87. doi:10.1016/j.bpc.2003.09.004
44. Holm NK, Jespersen SK, Thomassen LV, Wolff TY, Sehgal P, Thomsen LA, et al. Aggregation and Fibrillation of Bovine Serum Albumin. Biochim Biophys Acta (Bba) - Proteins Proteomics (2007) 1774:1128–38. doi:10.1016/j.bbapap.2007.06.008
45. Vetri V, Librizzi F, Leone M, Militello V. Thermal Aggregation of Bovine Serum Albumin at Different Ph: Comparison with Human Serum Albumin. Eur Biophys J (2007) 36:717–25. doi:10.1007/s00249-007-0196-5
46. Bhattacharya M, Jain N, Mukhopadhyay S. Insights into the Mechanism of Aggregation and Fibril Formation from Bovine Serum Albumin. J Phys Chem B (2011) 115:4195–205. doi:10.1021/jp111528c
48. Stoev ID, Cao T, Caciagli A, Yu J, Ness C, Liu R, et al. On the Role of Flexibility in Linker-Mediated Dna Hydrogels. Soft Matter (2020) 16:990–1001. doi:10.1039/c9sm01398a
49. LeVine H. [18] Quantification of β-sheet Amyloid Fibril Structures with Thioflavin T. Methods Enzymol (1999) 309:274–84. Elsevier. doi:10.1016/s0076-6879(99)09020-5
50. González García Á, Timmers EM, Romijn N, Song S, Sahebali S, Tuinier R, et al. Micellization of a Weakly Charged Surfactant in Aqueous Salt Solution: Self-Consistent Field Theory and Experiments. Colloids Surf A: Physicochemical Eng Aspects (2019) 561:201–8. doi:10.1016/j.colsurfa.2018.10.039
51. Doi M, Edwards SF. The Theory of Polymer Dynamics, 73. New York: Oxford University Press (1988).
52. Ferri F, D’Angelo A, Lee M, Lotti A, Pigazzini MC, Singh K, et al. Kinetics of Colloidal Fractal Aggregation by Differential Dynamic Microscopy. Eur Phys J Spec Top (2011) 199:139–48. doi:10.1140/epjst/e2011-01509-9
53. Wijnen PWJG, Beelen TPM, Rummens CPJ, Van Santen RA. Diffusion- and Reaction-Limited Aggregation of Aqueous Silicate Solutions. J Non-Crystalline Sol (1991) 136:119–25. doi:10.1016/0022-3093(91)90127-r
54. Oosawa F, Kasai M. A Theory of Linear and Helical Aggregations of Macromolecules. J Mol Biol (1962) 4:10–21. doi:10.1016/s0022-2836(62)80112-0
55. Di Michele L, Eiser E, Foderà V. Minimal Model for Self-Catalysis in the Formation of Amyloid-like Elongated Fibrils. J Phys Chem Lett (2013) 4:3158–64. doi:10.1021/jz401600g
56. Matsumoto A, Chen J, Collette AL, Kim U-J, Altman GH, Cebe P, et al. Mechanisms of Silk Fibroin Sol−Gel Transitions. J Phys Chem B (2006) 110:21630–8. doi:10.1021/jp056350v
57. Hu X, Kaplan D, Cebe P. Determining Beta-Sheet Crystallinity in Fibrous Proteins by Thermal Analysis and Infrared Spectroscopy. Macromolecules (2006) 39:6161–70. doi:10.1021/ma0610109
58. Dubey P, Chowdhury PK, Ghosh S. Modulation of Aggregation of Silk Fibroin by Synergistic Effect of the Complex of Curcumin and β-cyclodextrin. Biochim Biophys Acta (Bba) - Proteins Proteomics (2019) 1867:416–25. doi:10.1016/j.bbapap.2019.01.009
59. Mason TG, Weitz DA. Optical Measurements of Frequency-dependent Linear Viscoelastic Moduli of Complex Fluids. Phys Rev Lett (1995) 74:1250–3. doi:10.1103/physrevlett.74.1250
60. Mason TG, Gang H, Weitz DA. Diffusing-wave-spectroscopy Measurements of Viscoelasticity of Complex Fluids. J Opt Soc Am A (1997) 14:139–49. doi:10.1364/josaa.14.000139
61. Farrugia T, Perriman AW, Sharma KP, Mann S. Multi-enzyme cascade Reactions Using Protein-Polymer Surfactant Self-Standing Films. Chem Commun (2017) 53:2094–7. doi:10.1039/c6cc09809f
62. Sharma KP, Harniman R, Farrugia T, Briscoe WH, Perriman AW, Mann S. Dynamic Behavior in Enzyme-Polymer Surfactant Hydrogel Films. Adv Mater (2016) 28:1597–602. doi:10.1002/adma.201504740
63. Sharma KP, Collins AM, Perriman AW, Mann S. Enzymatically Active Self-Standing Protein-Polymer Surfactant Films Prepared by Hierarchical Self-Assembly. Adv Mater (2013) 25:2005–10. doi:10.1002/adma.201204161
64. Amdursky N, Mazo MM, Thomas MR, Humphrey EJ, Puetzer JL, St-Pierre J-P, et al. Elastic Serum-Albumin Based Hydrogels: Mechanism of Formation and Application in Cardiac Tissue Engineering. J Mater Chem B (2018) 6:5604–12. doi:10.1039/c8tb01014e
65. Murray AN, Palhano FL, Bieschke J, Kelly JW. Surface Adsorption Considerations when Working with Amyloid Fibrils in Multiwell Plates and Eppendorf Tubes. Protein Sci (2013) 22:1531–41. doi:10.1002/pro.2339
Keywords: protein aggregation, nBSA, amyloid fibrils, bio-conjugated protein, dynamic light scattering, microrheology, stability against denaturation
Citation: Mukhopadhyay A, Stoev ID, King DA, Sharma KP and Eiser E (2022) Amyloid-Like Aggregation in Native Protein and its Suppression in the Bio-Conjugated Counterpart. Front. Phys. 10:924864. doi: 10.3389/fphy.2022.924864
Received: 20 April 2022; Accepted: 19 May 2022;
Published: 20 June 2022.
Edited by:
Sean D. Chambers, Cook Medical INC., United StatesReviewed by:
Tushar Mukherjee, Indian Institute of Technology Indore, IndiaCopyright © 2022 Mukhopadhyay, Stoev, King, Sharma and Eiser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kamendra P. Sharma, ay5zaGFybWFAY2hlbS5paXRiLmFjLmlu; Erika Eiser, ZXJpa2EuZWlzZXJAbnRudS5ubw==
†ORCID ID: Erika Eiser, orcid.org/0000-0003-2881-8157
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.