 Long Xu
Long Xu Ilia Tutunnikov
Ilia Tutunnikov Yehiam Prior
Yehiam Prior Ilya Sh. Averbukh
Ilya Sh. Averbukh- AMOS and Department of Chemical and Biological Physics, The Weizmann Institute of Science, Rehovot, Israel
Impulsive orientation of symmetric-top molecules excited by two-color femtosecond pulses is considered. In addition to the well-known transient orientation appearing immediately after the pulse and then reemerging periodically due to quantum revivals, we report the phenomenon of field-free long-lasting orientation. Long-lasting means that the time averaged orientation remains non-zero until destroyed by other physical effects, e.g., intermolecular collisions. The effect is caused by the combined action of the field-polarizability and field-hyperpolarizability interactions. The dependence of degree of long-lasting orientation on temperature and pulse parameters is considered. The effect can be measured by means of second (or higher-order) harmonic generation, and may be used to control the deflection of molecules traveling through inhomogeneous electrostatic fields.
1 Introduction
Over the years, diverse optical methods have been developed to align and orient molecules of varying complexity and many applications related to studies of molecular and photon-induced processes are based on the ability to control the absolute orientation of the molecules. For reviews, see [1–6].
There are several laser-based strategies for achieving molecular orientation in the gas phase, including using a combination of intense non-resonant laser and weak electrostatic fields [7–15], and using strong single-cycle terahertz (THz) pulses [16–23], alone or together with optical pulses [24–26]. In addition, laser and THz pulses with twisted polarization were shown to be effective for inducing enantioselective orientation of chiral molecules [27–33].
The techniques listed above rely on the laser-dipole and/or laser-polarizability interactions. Another route to molecular orientation stems from higher-order laser-molecule interactions, e.g., the laser field-hyperpolarizability interaction. Non-resonant phase-locked two-color laser pulses consisting of the fundamental wave (FW) and its second harmonic (SH) were used for inducing molecular orientation by interacting with the molecular hyperpolarizability [34–49].
Here, we investigate the orientation dynamics of symmetric-top molecules excited by single two-color femtosecond laser pulses. In addition to the well-known transient orientation appearing immediately as a response to the laser excitation, we predict the existence of long-lasting orientation. Long-lasting means that the time-averaged orientation remains non-zero, within the model, indefinitely or until destroyed by additional physical effects, e.g., by collisions. The long-lasting orientation induced by a two-color pulse has an intricate dependence on both the molecular polarizability and hyperpolarizability. Related effects have been recently observed in chiral molecules excited by one-color laser pulses with twisted polarization [30, 32] and investigated in non-linear molecules excited by THz pulses [23, 33].
The paper is organized as follows. In the next section, we describe our numerical approaches for simulating the laser-driven molecular rotational dynamics. In Section 3, we present the long-lasting orientation, which is the main result of this work. Section 4 is devoted to a qualitative analysis of the effect, and a derivation of the approximate classical formula for the degree of long-lasting orientation. Additional results are presented in Section 5.
2 Numerical Methods
In this work, the rotational dynamics of symmetric-top molecules is treated within the rigid rotor approximation. We performed both classical and quantum mechanical simulations of molecular rotation driven by two-color laser fields. This section outlines the theoretical approaches used in both cases.
2.1 Classical Simulation
In the classical limit, the rotational dynamics of a single rigid top is described by Euler’s equations [50]
where
In the laboratory frame of reference, the electric field of a two-color laser pulse is defined by
where the two terms correspond to the FW and its SH, respectively. ω is the carrier frequency of the FW field, φ is the relative phase of the second harmonic,
where
Here, the overline
To simulate the behavior of an ensemble of non-interacting molecules, we use the Monte Carlo approach. For each molecule, the Euler’s equations [Eq. 1] with the torques in Eqs. 4, 5 are solved numerically using the standard fourth order Runge-Kutta algorithm. In the simulations we used ensembles consisting of
where
2.2 Quantum Simulation
The Hamiltonian describing the rotational degrees of freedom of a molecule and the molecular polarizability and hyperpolarizability couplings to external time-dependent fields can be written as
Here
We use the eigenstates of
where
where
where
In our simulations, the basis set included all the states with
3 Long-Lasting Orientation
We continue to consider the methyl fluoride (

TABLE 1. Molecular properties (in atomic units) of
Figure 1 shows the projection of the dipole moment along the laboratory Z axis,

FIGURE 1. Z-projection of the dipole moment,
In the case of symmetric-top molecules considered here, we observe long-lasting (persistent) orientation, a previously unreported phenomenon in two-color orientation schemes. The inset in Figure 1 demonstrates that after the initial oscillations are washed out, the classical polarization/degree of orientation attains a constant, nonzero value. In the quantum case too, despite its being partially masked by the revivals, the sliding time average of the signal is approximately constant and it persists indefinitely within the adopted model. This long-lasting orientation is one of the main results of this work.
Several comments are in order. Additional physical effects can distort the long-term field-free picture of identical periodically appearing revivals seen in Figure 1. These include the centrifugal distortion and the radiation emission due to rapidly rotating molecular permanent dipole moment. Dephasing of the rotational states caused by the centrifugal distortion leads to the eventual decay of the revivals’ peaks [21, 22]. Nevertheless, the average dipole remains almost unchanged (see [23]). The radiative emission results in the gradual decrease of the rotational energy [21, 22]. However, for a rarefied molecular gas, the estimated relative energy loss during a single revival is very small. The proper description of the behavior on an even longer timescale (nanoseconds), requires the inclusion of collisions and fine structure effects [66, 67], which is beyond the scope of the current work. Furthermore, it should be noted that higher laser pulse intensities lead to higher degree of orientation, but when the intensity is high enough for molecular ionization, another effect kicks in, namely orientation mechanism due to selective molecular ionization of molecules with specific orientation [43, 44]. Considering this kind of orientation is also beyond the scope of this work.
4 Long-Lasting Orientation - A Qualitative Description
An explicit form of the interaction potential [Eq. 7] can be obtained by expressing the electric field vector in the rotating molecular frame of reference. For the sake of the current discussion, this can be done conveniently by using an orthogonal rotation matrix parameterized by the three Euler angles,
We consider the case of a two-color pulse in which both the FW and SH are polarized along Z axis. The interaction potential is obtained by carrying out the summation in Eq. 7 (where all the quantities are expressed in the basis of principal axes of inertia) and we average over the optical cycle. The resulting potential has two contributions,
To facilitate the qualitative discussion in this section, we let
And
The two parts of the interaction potential lead to two distinct effects.
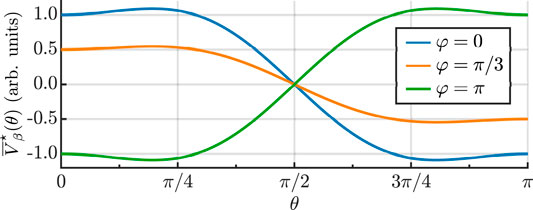
FIGURE 2. Angle dependence of the potential
4.1 Approximate Classical Formula
In the case of weak excitation, we can derive an approximate classical formula for the degree of long-lasting orientation. Since the long-lasting orientation manifests itself under field-free conditions, we begin by considering the free motion of a single classical symmetric top. The free motion of the unit vector
where L is the magnitude of angular momentum and
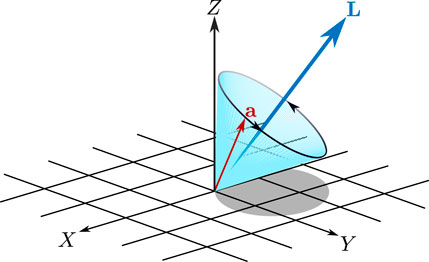
FIGURE 3. Illustration of precession of the vector
The degree of long-lasting orientation (see the inset of Figure 1) can be obtained by considering the ensemble average projection of the molecular axis
where
where
where
For the ensemble averaging, it is advantageous to express all the quantities in the basis of principal axes of inertia. The magnitude of the angular momentum after the pulse,
where
where
and
Here
Finally, we carry out the ensemble average
where
where
For the polarizability interaction alone,
where
According to Eq. 28, the degree of long-lasting orientation scales as
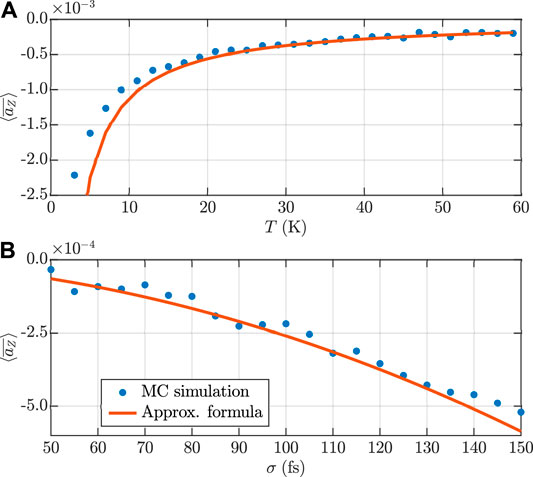
FIGURE 4. (A) Temperature and (B) pulse duration dependence of the degree of long-lasting orientation obtained using the approximate formula in Eq. 28 (red line) and numerically using the impulsive approximation (blue dots). Here
There is a good agreement between the numerical results and the results obtained using the approximate formula, especially at higher temperatures (higher initial angular momenta), where the assumption
The hyperpolarizability part of the interaction potential,
Despite the similarity, the mechanisms behind the long-lasting orientation induced by a femtosecond two-color and a picosecond THz pulse are not the same. It was shown in [23] that in the limit of vanishing THz pulse duration, the induced long-lasting orientation tends to zero. In other words, a δ-kick by a purely orienting potential doesn’t lead to long-lasting orientation. In contrast, here we show that a δ-kick by a combined, aligning and orienting, potentials results in a long-lasting orientation [see the discussion under Eq. 27, also see Figure 5].
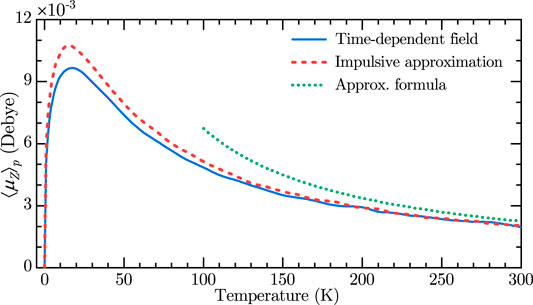
FIGURE 5. Classically calculated permanent values of Z-projection of the dipole moment. The field parameters are similar to Figure 1. Cases of fully time-dependent field (solid blue) and using impulsive approximation (dashed red) are compared. The dotted green line is obtained using the approximate formula in Eq. 28 with
5 Temperature and Polarization Dependence of the Long-Lasting Orientation
Figure 5 depicts the long-lasting orientation of the dipole moment as a function of temperature for the case of collinearly polarized two-color pulse. Due to the short pulse duration (
As an additional example, we consider the case of a cross-polarized two-color pulse in which the polarizations of the FW and SH are along Z and X axes, respectively. Figure 6 shows the dipole signal along the laboratory X axis. Note that the Y and Z-projections of the dipole moment,
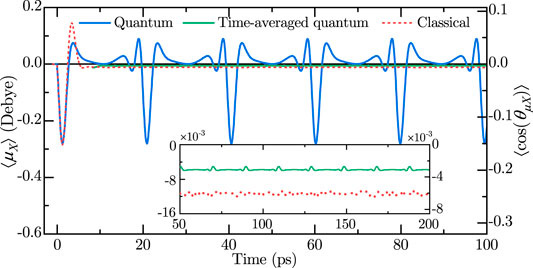
FIGURE 6. X-projection of the dipole moment,
6 Conclusion
We have theoretically demonstrated a new phenomenon of long-lasting (persistent) orientation of symmetric-top molecules excited by a single two-color femtosecond pulse. The residual orientation was shown to last indefinitely (within the adopted model), or until destroyed by other physical effects, e.g., intermolecular collisions. We derived an approximate classical expression revealing several qualitative features of the phenomenon, including the scaling with temperature, pulse duration, and other field and molecular parameters. The predictions of the formula are in full agreement with the results of numerical simulations in the limit of weak excitation. A quick check for different polarizations showed that in the case of cross-polarized FW and SH, the achieved degree of both transient and long-lasting orientation may be higher than in the case of parallel configuration. The magnitude of the long-lasting dipole signal shown in this work is about
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
All the authors participated in formulating the problem and initiating this study. LX and IT equally contributed to the calculations and numerical simulations. All the authors participated in analyzing the results and writing the manuscript. YP and IA supervised and guided the work.
Funding
We gratefully acknowledge support by the Israel Science Foundation (Grant No. 746/15). IA acknowledges support as the Patricia Elman Bildner Professorial Chair. This research was made possible in part by the historic generosity of the Harold Perlman Family.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2021.689635/full#supplementary-material
References
1. Stapelfeldt H, Seideman T. Colloquium: Aligning Molecules with strong Laser Pulses. Rev Mod Phys (2003) 75:543.
2. Ohshima Y, Hasegawa H. Coherent Rotational Excitation by Intense Nonresonant Laser fields. Int Rev Phys Chem (2010) 29:619.
3. Fleischer S, Khodorkovsky Y, Gershnabel E, Prior Y, Averbukh IS. Molecular Alignment Induced by Ultrashort Laser Pulses and its Impact on Molecular Motion. Isr J Chem (2012) 52:414.
4. Lemeshko M, Krems RV, Doyle JM, Kais S. Manipulation of Molecules with Electromagnetic fields. Mol Phys (2013) 111:1648.
5. Koch CP, Lemeshko M, Sugny D. Quantum Control of Molecular Rotation. Rev Mod Phys (2019) 91:035005.
6. Lin K, Tutunnikov I, Ma J, Qiang J, Zhou L, Faucher O, et al. Spatiotemporal Rotational Dynamics of Laser-Driven Molecules. Adv Photon (2020) 2:024002.
7. Friedrich B, Herschbach D. Enhanced Orientation of Polar Molecules by Combined Electrostatic and Nonresonant Induced Dipole Forces. J Chem Phys (1999) 111:6157.
8. Sakai H, Minemoto S, Nanjo H, Tanji H, Suzuki T. Controlling the Orientation of Polar Molecules with Combined Electrostatic and Pulsed, Nonresonant Laser fields. Phys Rev Lett (2003) 90:083001.
9. Goban A, Minemoto S, Sakai H. Laser-field-free Molecular Orientation. Phys Rev Lett (2008) 101:013001.
10. Ghafur O, Rouzée A, Gijsbertsen A, Siu WK, Stolte S, Vrakking MJJ. Impulsive Orientation and Alignment of Quantum-State-Selected NO Molecules. Nat Phys (2009) 5:289.
11. Holmegaard L, Nielsen JH, Nevo I, Stapelfeldt H, Filsinger F, Küpper J, et al. Laser-induced Alignment and Orientation of Quantum-State-Selected Large Molecules. Phys Rev Lett (2009) 102:023001.
12. Mun JH, Takei D, Minemoto S, Sakai H. Laser-field-free Orientation of State-Selected Asymmetric Top Molecules. Phys Rev A (2014) 89:051402(R).
13. Takei D, Mun JH, Minemoto S, Sakai H. Laser-field-free Three-Dimensional Molecular Orientation. Phys Rev A (2016) 94:013401.
14. Omiste JJ, González-Férez R. Theoretical Description of the Mixed-Field Orientation of Asymmetric-Top Molecules: A Time-dependent Study. Phys Rev A (2016) 94:063408.
15. Thesing LV, Küpper J, González-Férez R. Time-dependent Analysis of the Mixed-Field Orientation of Molecules without Rotational Symmetry. J Chem Phys (2017) 146:244304.
16. Harde H, Keiding S, Grischkowsky D. THz Commensurate Echoes: Periodic Rephasing of Molecular Transitions in Free-Induction Decay. Phys Rev Lett (1991) 66:1834.
17. Averbukh IS, Arvieu R. Angular Focusing, Squeezing, and Rainbow Formation in a Strongly Driven Quantum Rotor. Phys Rev Lett (2001) 87:163601.
19. Fleischer S, Zhou Y, Field RW, Nelson KA. Molecular Orientation and Alignment by Intense Single-Cycle THz Pulses. Phys Rev Lett (2011) 107:163603.
20. Kitano K, Ishii N, Kanda N, Matsumoto Y, Kanai T, Kuwata-Gonokami M, et al. Orientation of Jet-Cooled Polar Molecules with an Intense Single-Cycle THz Pulse. Phys Rev A (2013) 88:061405(R).
21. Damari R, Kallush S, Fleischer S. Rotational Control of Asymmetric Molecules: Dipole- versus Polarizability-Driven Rotational Dynamics. Phys Rev Lett (2016) 117:103001.
22. Babilotte P, Hamraoui K, Billard F, Hertz E, Lavorel B, Faucher O, et al. Observation of the Field-free Orientation of a Symmetric-Top Molecule by Terahertz Laser Pulses at High Temperature. Phys Rev A (2016) 94:043403.
23. Xu L, Tutunnikov I, Gershnabel E, Prior Y, Averbukh IS. Long-lasting Molecular Orientation Induced by a Single Terahertz Pulse. Phys Rev Lett (2020) 125:013201.
24. Daems D, Guérin S, Sugny D, Jauslin HR. Efficient and Long-Lived Field-free Orientation of Molecules by a Single Hybrid Short Pulse. Phys Rev Lett (2005) 94:153003.
25. Gershnabel E, Averbukh IS, Gordon RJ. Orientation of Molecules via Laser-Induced Antialignment. Phys Rev A (2006) 73:061401(R).
26. Egodapitiya KN, Li S, Jones RR. Terahertz-induced Field-free Orientation of Rotationally Excited Molecules. Phys Rev Lett (2014) 112:103002.
27. Yachmenev A, Yurchenko SN. Detecting Chirality in Molecules by Linearly Polarized Laser fields. Phys Rev Lett (2016) 117:033001.
28. Gershnabel E, Averbukh IS. Orienting Asymmetric Molecules by Laser fields with Twisted Polarization. Phys Rev Lett (2018) 120:083204.
29. Tutunnikov I, Gershnabel E, Gold S, Averbukh IS. Selective Orientation of Chiral Molecules by Laser fields with Twisted Polarization. J Phys Chem Lett (2018) 9:1105.
30. Milner AA, Fordyce JAM, MacPhail-Bartley I, Wasserman W, Milner V, Tutunnikov I, et al. Controlled Enantioselective Orientation of Chiral Molecules with an Optical Centrifuge. Phys Rev Lett (2019) 122:223201.
31. Tutunnikov I, Floß J, Gershnabel E, Brumer P, Averbukh IS. Laser-induced Persistent Orientation of Chiral Molecules. Phys Rev A (2019) 100:043406.
32. Tutunnikov I, Floß J, Gershnabel E, Brumer P, Averbukh IS, Milner AA, et al. Observation of Persistent Orientation of Chiral Molecules by a Laser Field with Twisted Polarization. Phys Rev A (2020) 101:021403(R).
33. Tutunnikov I, Xu L, Field RW, Nelson KA, Prior Y, Averbukh IS. Enantioselective Orientation of Chiral Molecules Induced by Terahertz Pulses with Twisted Polarization. Phys Rev Res (2021) 3:013249.
34. Vrakking MJJ, Stolte S. Coherent Control of Molecular Orientation. Chem Phys Lett (1997) 271:209.
35. Dion C, Bandrauk AD, Atabek O, Keller A, Umeda H, Fujimura Y. Two-frequency IR Laser Orientation of Polar Molecules. Numerical Simulations for HCN. Chem Phys Lett (1999) 302:215.
36. Kanai T, Sakai H. Numerical Simulations of Molecular Orientation Using strong, Nonresonant, Two-Color Laser fields. J Chem Phys (2001) 115:5492.
37. Takemoto N, Yamanouchi K. Fixing Chiral Molecules in Space by Intense Two-Color Phase-Locked Laser fields. Chem Phys Lett (2008) 451:1.
38. De S, Znakovskaya I, Ray D, Anis F, Johnson NG, Bocharova IA, et al. Field-Free Orientation of CO Molecules by Femtosecond Two-Color Laser Fields. Phys Rev Lett (2009) 103:153002.
39. Oda K, Hita M, Minemoto S, Sakai H. All-optical Molecular Orientation. Phys Rev Lett (2010) 104:213901.
40. Wu J, Zeng H. Field-free Molecular Orientation Control by Two Ultrashort Dual-Color Laser Pulses. Phys Rev A (2010) 81:053401.
41. Zhang S, Shi J, Zhang H, Jia T, Wang Z, Sun Z. Field-free Molecular Orientation by a Multicolor Laser Field. Phys Rev A (2011) 83:023416.
42. Frumker E, Hebeisen CT, Kajumba N, Bertrand JB, Wörner HJ, Spanner M, et al. Oriented Rotational Wave-Packet Dynamics Studies via High Harmonic Generation. Phys Rev Lett (2012) 109:113901.
43. Spanner M, Patchkovskii S, Frumker E, Corkum P. Mechanisms of Two-Color Laser-Induced Field-free Molecular Orientation. Phys Rev Lett (2012) 109:113001.
44. Znakovskaya I, Spanner M, De S, Li H, Ray D, Corkum P, et al. Transition between Mechanisms of Laser-Induced Field-free Molecular Orientation. Phys Rev Lett (2014) 112:113005.
45. Mun JH, Sakai H. Improving Molecular Orientation by Optimizing Relative Delay and Intensities of Two-Color Laser Pulses. Phys Rev A (2018) 98:013404.
46. Mun JH, Sakai H, González-Férez R. Orientation of Linear Molecules in Two-Color Laser fields with Perpendicularly Crossed Polarizations. Phys Rev A (2019) 99:053424.
47. Mun JH, Kim DE. Field-free Molecular Orientation by Delay- and Polarization-Optimized Two Fs Pulses. Sci Rep (2020) 10:18875.
48. Mellado-Alcedo D, Quintero NR, González-Férez R. Linear Polar Molecule in a Two-Color Cw Laser Field: A Symmetry Analysis. Phys Rev A (2020) 102:023110.
49. Wang S, Henriksen NE. Optimal Field-Free Molecular Orientation with Nonresonant Two-Color Adiabatic-Turn-On and Sudden-Turn-Off Laser Pulses. Phys Rev A (2020) 102:063120.
51. Coutsias EA, Romero L. The Quaternions with an Application to Rigid Body Dynamics. Sandia Technical Report (2004) SAND2004-0153.
52. Kuipers JB. Quaternions and Rotation Sequences: A Primer with Applications to Orbits, Aerospace and Virtual Reality. Princeton, N.J.: Princeton University Press (1999).
53. Lin K, Tutunnikov I, Qiang J, Ma J, Song Q, Ji Q, et al. All-optical Field-free Three-Dimensional Orientation of Asymmetric-Top Molecules. Nat Commun (2018) 9:5134.
55. Zare RN. Angular Momentum: Understanding Spatial Aspects in Chemistry and Physics. New York: Wiley (1988).
56. Buckingham AD. Advances in Chemical Physics. New York: John Wiley & Sons (2007). p. 107–42. Permanent and Induced Molecular Moments and Long-Range Intermolecular Forces.
57. Sidje RB. Expokit: A Software Package for Computing Matrix Exponentials. ACM Trans Math Softw (1998) 24:130.
58. McDowell RS. Rotational Partition Functions for Symmetric-top Molecules. J Chem Phys (1990) 93:2801.
59. Johnson RD. NIST Computational Chemistry Comparison and Benchmark Database, Release 20. Tech. Rep (2019).
60. Chong DP. Theoretical Calculations of Dipole Moments, Polarizabilities, and Hyperpolarizabilities of HF, OCS, O3, CH3F, and CH3Cl by Local Density Approximation. J Chin Chem Soc (1992) 39:375.
61. Eberly JH, Narozhny NB, Sanchez-Mondragon JJ. Periodic Spontaneous Collapse and Revival in a Simple Quantum Model. Phys Rev Lett (1980) 44:1323.
63. Averbukh IS, Perelman NF. Fractional Revivals: Universality in the Long-Term Evolution of Quantum Wave Packets beyond the Correspondence Principle Dynamics. Phys Lett A (1989) 139:449.
64. Felker PM. Rotational Coherence Spectroscopy: Studies of the Geometries of Large Gas-phase Species by Picosecond Time-Domain Methods. J Phys Chem (1992) 96:7844.
66. Thomas EF, Søndergaard AA, Shepperson B, Henriksen NE, Stapelfeldt H. Hyperfine-structure-induced Depolarization of Impulsively Aligned I2 Molecules. Phys Rev Lett (2018) 120:163202.
67. Thesing LV, Yachmenev A, González-Férez R, Küpper J. The Effect of Nuclear-Quadrupole Coupling in the Laser-Induced Alignment of Molecules. J Phys Chem A (2020) 124:2225.
69. Leibscher M, Averbukh IS, Rabitz H. Molecular Alignment by Trains of Short Laser Pulses. Phys Rev Lett (2003) 90:213001.
70. Leibscher M, Averbukh IS, Rabitz H. Enhanced Molecular Alignment by Short Laser Pulses. Phys Rev A (2004) 69:013402.
71. Lee KF, Litvinyuk IV, Dooley PW, Spanner M, Villeneuve DM, Corkum PB. Two-pulse Alignment of Molecules. J Phys B: Mol Opt Phys (2004) 37:L43.
72. Bisgaard CZ, Poulsen MD, Péronne E, Viftrup SS, Stapelfeldt H. Observation of Enhanced Field-free Molecular Alignment by Two Laser Pulses. Phys Rev Lett (2004) 92:173004.
73. Pinkham D, Mooney KE, Jones RR. Optimizing Dynamic Alignment in Room Temperature CO. Phys Rev A (2007) 75:013422.
74. Zhang S, Lu C, Jia T, Wang Z, Sun Z. Field-free Molecular Orientation Enhanced by Two Dual-Color Laser Subpulses. J Chem Phys (2011) 135:034301.
75. Gershnabel E, Averbukh IS. Electric Deflection of Rotating Molecules. J Chem Phys (2011) 134:054304.
76. Gershnabel E, Averbukh IS. Deflection of Rotating Symmetric Top Molecules by Inhomogeneous fields. J Chem Phys (2011) 135:084307.
Keywords: long-lasting orientation, symmetric-top, two-color laser pulses, polarizability interaction, hyperpolarizability interaction
Citation: Xu L, Tutunnikov I, Prior Y and Averbukh IS (2021) Long-Lasting Orientation of Symmetric-Top Molecules Excited by Two-Color Femtosecond Pulses. Front. Phys. 9:689635. doi: 10.3389/fphy.2021.689635
Received: 01 April 2021; Accepted: 18 June 2021;
Published: 16 July 2021.
Edited by:
Robert Gordon, University of Illinois at Chicago, United StatesReviewed by:
Henrik Stapelfeldt, Aarhus University, DenmarkMonika Leibscher, University of Kassel, Germany
Copyright © 2021 Xu, Tutunnikov, Prior and Averbukh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Long Xu, bG9uZy54dUB3ZWl6bWFubi5hYy5pbA==; Ilia Tutunnikov, aWxpYS50dXR1bm5pa292QHdlaXptYW5uLmFjLmls; Yehiam Prior, eWVoaWFtLnByaW9yQHdlaXptYW5uLmFjLmls; Ilya Sh. Averbukh, aWx5YS5hdmVyYnVraEB3ZWl6bWFubi5hYy5pbA==
†These authors have contributed equally to this work